

Abstract
Rationale
Endothelial Notch signaling is critical for early vascular development and survival. Yet, previously described mice lacking endothelial ADAM10 (a disintegrin and metalloproteinase 10), a key regulator of Notch signaling, survived into adulthood with organ-specific vascular defects. These findings raised questions about whether these vascular defects were related to Notch signaling or other functions of ADAM10.
Objective
Determine whether compensatory or redundant functions of ADAM17 in Notch signaling can explain the survival of Adam10ΔEC mice, explore the contribution of different Tie2-Cre transgenes to the differences in survival, and establish whether the Adam10ΔEC vascular phenotypes can be recapitulated by inactivation of Notch receptors in endothelial cells.
Methods and Results
Mice lacking ADAM10 and ADAM17 in endothelial cells (Adam10/Adam17ΔEC), which survived postnatally with organ-specific vascular defects, resembled Adam10ΔEC mice. In contrast, Adam10ΔEC mice generated with the Tie2Cre transgene previously used to inactivate endothelial Notch (Adam10ΔECFlv) died by E10.5. qPCR analysis demonstrated that Cre-mediated recombination occurs earlier in Adam10ΔECFlv mice than in the previously described Adam10ΔEC mice. Finally, mice lacking endothelial Notch1 (Notch1ΔEC) share some organ-specific vascular defects with Adam10ΔEC mice, whereas Notch4−/− mice lacking endothelial Notch1 (Notch1ΔEC/Notch4−/−) had defects in all vascular beds affected in Adam10ΔEC mice.
Conclusions
Our results argue against a major role for ADAM17 in endothelial Notch signaling and clarify the difference in phenotypes of previously described mice lacking ADAM10 or Notch in endothelial cells. Most notably, these findings uncover new roles for Notch signaling in the development of organ-specific vascular beds.
Keywords: ADAM10, Notch signaling, ADAM17, Tie2-Cre, vascular biology, vasculature, metalloproteinase, embryonic development
Subject Terms: Angiogenesis, Developmental Biology, Genetically Altered Mice
INTRODUCTION
The proper development of the vascular system is crucial for normal embryogenesis and postnatal development. Blood vessels supply oxygen and nutrients to the developing embryo and provide cues for organ development 1, 2. Essential building blocks of the mouse circulatory system—including the primary intraembryonic vascular plexus, precursors of the aorta and vena cava, and the primary yolk sac vascular plexus—form through the process of vasculogenesis by embryonic day 7.5 (E7.5) 2–5. Vasculogenesis occurs when angioblasts (endothelial cell precursors) coalesce to form a primitive vasculature. After the primary vascular plexus is established, angiogenesis, in which new vessels branch from preexisting ones, is thought to drive most new blood vessel formation later in development 6, 7.
Endothelial Notch signaling is essential for mouse vascular development. Previous studies suggest that little, if any, angiogenic remodeling occurs in the intraembryonic, yolk sac and placenta vasculature of embryos deficient in endothelial Notch1 8, endothelial RBPJ (recombinant binding protein suppressor of hairless) 9, or endothelial Notch ligand Delta-like 4 (Dll4) 10–12. Analysis of intraembryonic vascular development in these mice also revealed defects in the dorsal aortae and/or cardinal veins and an enlarged pericardium 8, 11. Notably, these developmental defects resulted in embryonic lethality by E10.5, suggesting that endothelial Notch signaling is required for survival beyond mid-gestation.
Notch signaling is regulated by cleavage at its extracellular juxtamembrane domain by the metalloprotease ADAM10 (A Disintegrin and Metalloprotease 10) 13, 14. This step is a pre-requisite for γ-secretase processing, which releases the intracellular domain of Notch from its membrane anchor, allowing it to enter the nucleus to regulate gene expression 14–17. Surprisingly, unlike previously described mice lacking Notch1 or RBPJ in endothelial cells, mice deficient in endothelial ADAM10 (Adam10ΔEC) survived into adulthood 18. Adam10ΔEC mice exhibited increased vascular branching in the retinal vasculature 18, reminiscent of other mutants deficient in endothelial Notch signaling 19–21. In addition, adult Adam10ΔEC mice had defects in the vasculature of the heart, liver, diaphragm, kidneys, small intestine, and long bones 18.
Studies with inducible endothelial knockout mice have provided crucial insights into the role of Notch signaling in retinal angiogenesis, a widely used model of developmental angiogenesis beginning after birth 19, 22–24. However, the early lethality of other Notch pathway knockout mice has limited study of the role of Notch signaling in vascular development following E10.5—including the development of organ-specific vascular beds that arise in mid- to late gestation. Consequently, the survival of Adam10ΔEC mice and their distinct vascular abnormalities raised questions about why their phenotype differed so strongly from that of previously generated Notch1ΔEC and RbpjΔEC mice, which die by E10.5 with defects in early vascular development 8, 9, 18. Consistent with a principal role for ADAM10 in regulating Notch signaling, mice lacking ADAM10—either systemically or in specific tissues—typically strongly resemble mice lacking components of the Notch pathway 25–27.
Here, we explored possible mechanisms underlying the difference in lethality and vascular phenotypes in Adam10ΔEC mice and previously described endothelial Notch-pathway knockout mice. One possibility was that the related metalloprotease ADAM17, which has been implicated in Notch signaling, has compensatory or redundant functions with ADAM10, allowing it to support sufficient Notch signaling during development to circumvent embryonic lethality 15, 16, 28, 29. To investigate this possibility, we generated mice lacking ADAM10 and ADAM17 in endothelial cells (Adam10/17ΔEC mice). A second possibility was that differences in the activity of the endothelial-specific Cre line (Tie2-Cre) used to inactivate ADAM10 and the Tie2-Cre line used previously to inactivate endothelial RBPJ or Notch1 could explain the observed differences in lethality 8, 9. To test this possibility, we generated Adam10ΔEC mice with the Tie2-Cre previously used to generate Notch1ΔEC and RbpjΔEC mice 8, 9, 30. Finally, to determine whether the organ-specific vascular defects in Adam10ΔEC mice are related to a deficiency in endothelial Notch signaling or to other functions of ADAM10, we generated Notch1ΔEC and Notch1ΔEC/Notch4−/− mice with the Tie2-Cre used to generate Adam10ΔEC mice 18, 31 and tested whether expression of the Notch1 intracellular domain in endothelial cells could rescue the defects observed in Adam10ΔEC mice. Our findings reveal previously uncharacterized roles for Notch1 and Notch4 in the embryonic and postnatal development of organ-specific vascular structures.
METHODS
Please see the Online Supplement for details.
Mice
To generate mice lacking endothelial ADAM10 and ADAM17 (Adam10/Adam17ΔEC mice), previously described Adam10flox/flox 18, 32, Adam17flox/flox 33, 34, and Tie2-Cre 31 mice were crossed. In addition, we used two independent Tie2-Cre lines (Tie2-Cre(Ywa) 31; Tie2-Cre(Flv) 30), to generate mice lacking endothelial ADAM10 [Adam10ΔEC(Ywa) or Adam10ΔEC(Flv)]. Endothelial Notch1-deficient mice (Notch1ΔEC(Ywa)) were generated by crossing Notch1flox/flox 35 with Tie2-Cre(Ywa) mice 31. For Notch1ΔEC/Notch4−/− compound knockout mice, Notch1flox/flox mice, Tie2-Cre(Ywa), and Notch4−/− mice 9, 36 were mated. Adam10ΔEC(Ywa mice carrying a transgene expressing the intracellular domain of Notch1 (NICD) under control of the ubiquitous GT(ROSA)26Sor promoter with an upstream floxed stop codon were generated by matings with (Gt(ROSA)26Sortm1(Notch1)Dam/J) mice to produce Adam10ΔEC-NICD+ animals. All mice were of mixed genetic background, and littermates served as controls (unless otherwise indicated). All animal studies were IACUC approved.
Histopathology
Histopathological analysis was performed on 8-week-old mice (unless otherwise indicated). Organs were collected and processed for hematoxylin and eosin staining 18, or for immunohistochemistry, as indicated.
RESULTS
Mice lacking ADAM10 and ADAM17 in endothelial cells
To address whether ADAM17 has compensatory or redundant functions in endothelial Notch signaling in the absence of ADAM10, we generated mice lacking both ADAM10 and ADAM17 with the Tie2-Cre used previously in Adam10ΔEC mice 31. Like Adam10ΔEC mice, Adam10/17ΔEC were born at a Mendelian ratio (n=165, Adam10/17ΔEC: 78 [47%], Adam10flox/floxAdam17flox/flox: 87 [53%]) and survived into adulthood with a median life span of ~14 weeks (Figure 1A). The developing retinal vasculature of Adam10/17ΔEC mice showed increased density and looping at the leading edge at postnatal day P5, with similar vascular front progression compared to controls (Figure 1B-H).
Figure 1. A10/17ΔEC mice survive into adulthood and exhibit defects in retinal vascular development.
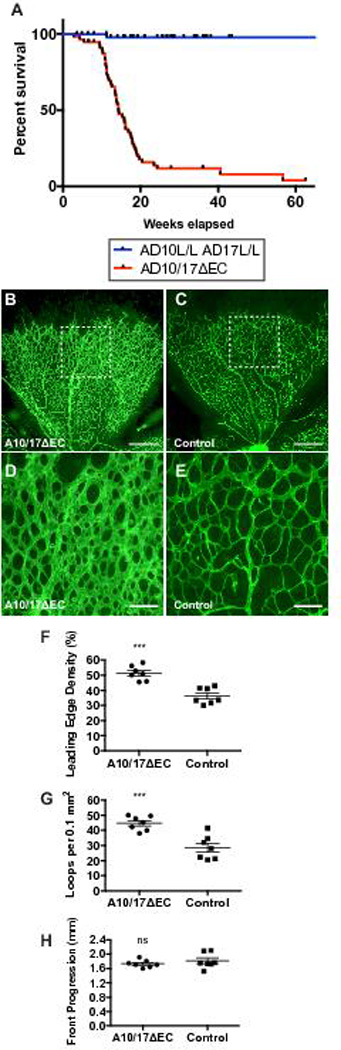
A, Kaplan-Meier survival curve for 57 A10/17ΔEC mice and 54 AD10flox/flox/AD17flox/flox littermate controls. Median survival of A10/17ΔEC mice was ~14 weeks. B to E, representative isolectin-B4-stained retinas of P5 A10/17ΔEC mice (B) and A10flox/flox/A17flox/flox controls without Tie2-Cre (C). Higher magnification image of representative area of the A10/17ΔEC retina (D) shows increased isolectin-B4 stained vascular coverage at the leading edge compared to a control retina (E). F to G, Quantification of vascular density (F) and vascular loops per 0.1 mm2 (G) at the leading edge shows an increase in both parameters in A10/17ΔEC retinas (n=7) compared to controls (n=7). H, Retinal progression from the optic disk to the retina periphery was not significantly affected in A10/17ΔEC compared to controls. Data shown as mean ± SEM. *** signifies p<0.001, “ns” indicates no significant difference, two-tailed student’s t-test. Scale bars, 500µm (B-C), and 100µm (D-E).
Histopathological analysis of Adam10/17ΔEC mice revealed abnormal vascular and tissue morphology in a limited set of organs. Adam10/17ΔEC livers contained large, abnormal subcapsular vessels not present in controls (Figure 2A,B). Surrounding these atypical vessels, mutant livers showed increased CD31 staining and sinusoidal dilatation (Figure 2C,D). Adam10/17ΔEC hearts had enlarged subepicardial vessels, and an increase in myocardial endothelial cell number, confirmed by increased CD31 staining compared to controls (Figure 2E-H). In addition, there was an increase in glomerular size, cellularity, and endothelial staining in Adam10/17ΔEC kidneys over controls (Figure 2I-L, see supplemental Figure II for quantification of Adam10/17ΔEC vascular defects). Moreover, hyperplastic polyps populated the small intestine of Adam10/17ΔEC mice. These polyps consisted of abnormal dense nests of CD31+ cells and were occasionally associated with dilated, fluid-filled vascular spaces (Figure 2M-P; supplemental Figure IA-B). The number of Mac2+ macrophages in close proximity to the affected vessels in the heart, liver and glomeruli was increased in Adam10/17ΔEC over controls, as in Adam10ΔEC animals (supplemental Figure III). No obvious vascular defects were evident in other tissues, such as the brain and lungs (supplemental Figure IV).
Figure 2. A10/17ΔEC mice exhibit defects in organ-specific vascular beds.

Histopathological analysis of hematoxylin and eosin (H&E) or CD31 (endothelial cell marker) stained specimens reveal defects in vascular beds of the liver, heart, kidney, and small intestine of A10/17ΔEC mice. A to D, H&E-stained A10/17ΔEC liver (A) shows enlarged vessels (arrow) near the liver surface not present in controls (B). The enlarged vessels are CD31+ (C, arrow) and are associated with changes in surrounding sinusoidal endothelium not seen in controls (D). E to H, an A10/17ΔEC heart (E) shows an enlarged subepicardial vessel and myocardial hypercellularity compared to a control (F). The enlarged vessel in the A10/17ΔEC heart (G, arrow) is CD31+ and myocardial hypercellularity is associated with increased CD31 staining compared to the control (H). I to L, an H&E stained A10/17ΔEC kidney shows larger, more hypercellular glomeruli (I, arrow) than a control (J). CD31 staining is increased in A10/17ΔEC glomeruli (K, arrow) compared to control glomeruli (L). M to P, small intestine of an A10/17ΔEC mouse contains hyperplastic polyps (M, arrow) not present in controls (N). Polyps contain abnormal nests of CD31+ cells (O, black arrows) and are occasionally fluid-filled (red arrow) in contrast to the regular architecture of control villi (P). H&E micrographs shown are representative of 8-week old animals analyzed for each genotype (n=3 A10/17ΔEC, n=3 A10flox/flox/A17flox/flox). See supplemental Figure II for quantification. Scale bars, 100µm (A-H), 50µm (I-L), and 200µm (M-P).
Adam10/17ΔEC mice also had long bone defects, with significantly shorter femurs, tibias, and humeri than littermate controls (Figure 3A-D, supplemental Figure V). H&E staining of Adam10/17ΔEC femurs showed dysplastic and frequently discontinuous growth plates characterized by disorganized hypertrophic chondrocytes (Figure 3E,F). CD31 staining revealed enlarged, irregularly shaped, improperly oriented, and frequently, fluid filled metaphyseal vessels at the chondro-osseus junction, as well as solid nests of CD31+ cells without lumens (Figure 3G,H, supplemental Figures IE,F, VI). In littermate controls, metaphyseal vessels were small and perpendicular to the growth plate. Micro-CT analysis showed that the disruption of the growth plate had not closed by 40 weeks of age in an Adam10/17ΔEC mouse, just as in older Adam10ΔEC mice (supplemental Figure VII). Moreover, we observed evidence for osteopenia in femurs (supplemental Figure VIII). There were no evident craniofacial abnormalities or defects in suture patency in Adam10/17ΔEC and Adam10ΔEC mice, suggesting that intramembranous bone formation is not affected (supplemental Figure IX, additional analyses of the bones of Adam10/17ΔEC and Adam10ΔEC mice are shown in supplemental Figures X-XIII). Collectively, postnatal survival of Adam10/17ΔEC mice was similar to that of Adam10ΔEC mice, and the vascular defects in Adam10/17ΔEC mice were present in the small intestine of Adam10ΔEC mice, and were also present, albeit with somewhat reduced severity, in the liver, heart and kidney 18 (supplemental Figure XIV).
Figure 3. A10/17ΔEC long bone growth is disrupted.
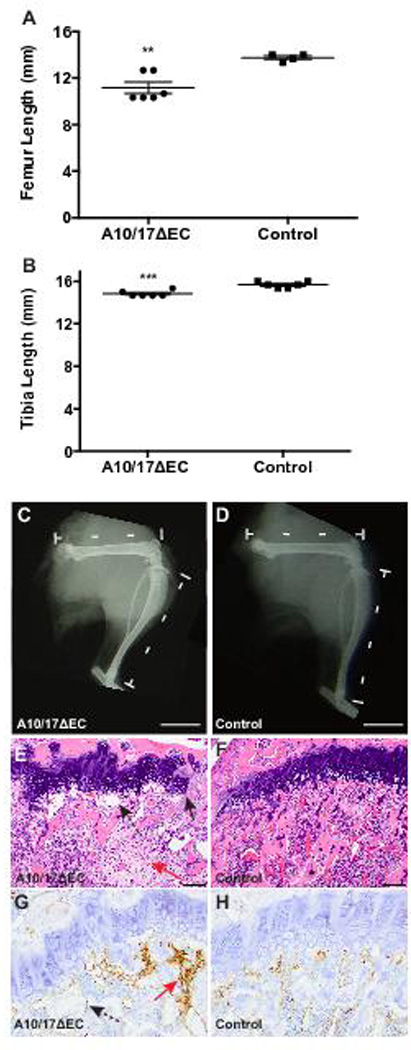
A to B, The femurs (A) and tibiae (B) of 8-week old A10/17ΔEC mice are significantly shorter than in littermate controls. C,D, representative radiographs of the hindlimbs show a shorter femur and tibia in an A10/17ΔEC mouse (C) than in a A10flox/flox/A17flox/flox control (D). E to H. H&E and CD31 staining of femurs shows disruptions in the growth plate of A10/17ΔEC mice (E, arrow-solid line) and enlarged, abnormally oriented vessels under the growth plate (E and G, arrows-dashed line), and solid nests of CD31+ cells not forming vascular spaces (E and G, red arrows) in contrast to continuous growth plate and small, regularly spaced vessels adjacent to the control growth plate (F,H). Data shown as mean ± SEM. ** signifies p<0.01, *** signifies p<0.001, two-tailed student t-test. Radiographs shown are representative of 8-week old animals analyzed for each genotype (n=3 A10/17ΔEC, n=3 A10flox/flox/A17flox/flox). Scale bars, 4mm (C-D), 100µm (E-F), and 50µm (G-H).
ADAM10ΔEC mice with Tie2-Cre (Flv) die early in embryogenesis
Our analysis of Adam10/17ΔEC mice argued against major compensatory or redundant functions of ADAM17 in Adam10ΔEC mice 18. We therefore tested the alternative hypothesis that a difference in the Tie2-Cre transgene used in our studies (Yanagisawa Tie2-Cre, Ywa 31) and in previous studies (Flavell Tie2-Cre, Flv 30) could explain the difference in survival of Adam10ΔEC (postnatal survival) and previously described Notch1ΔEC or RbpjΔEC (embryonic lethality) mice 8, 9, 18. Matings of Adam10flox/wt Tie2-Cre(Flv) males with Adam10flox/flox females produced no live Adam10ΔEC(Flv) offspring (Table 1). In utero, Adam10ΔEC(Flv) embryos were present at Mendelian ratios between E7.5 and E8.5 (Table 1). At E10.5, embryos were present at an approximately Mendelian ratio, but 11 out of 12 Adam10ΔEC(Flv) were dead. The surface of E10.5 Adam10ΔEC(Flv) yolk sacs lacked the large branching vessels seen in controls (Figure 4A-D). All E10.5 Adam10ΔEC(Flv) embryos had enlarged pericardial sacs (Fig. 4E, arrow) and appeared smaller than Adam10flox/+ Tie2-Cre(Flv) littermate controls (Fig. 4F). At E11.5- E13.5, Adam10ΔEC(Flv) genetic material was only found in resorption sites. Thus, Adam10ΔEC(Flv) mice suffer from embryonic lethality by E10.5 with defects resembling those previously described in Notch1ΔEC and RbpjΔEC mice that were also generated with Tie2-Cre(Flv) 8, 9, 36.
Table 1.
Offspring of matings of Adam10flox/+Tie2-Cre males with Adam10flox/flox females at different developmental stages.
| Stage | Total | Adam10flox/+ | Adam10flox/flox |
Adam10flox/+ Tie2Cre+/−(Flv) |
Adam10flox/flox Tie2Cre+/−(Flv) |
|---|---|---|---|---|---|
| E7.5-E8.5 | 42 | 11 (11) | 9 (11) | 12 (11) | 10 (11) |
| E10.5-E13.5 | 111 | 29 (28) | 24 (28) | 31 (28) | 27*(28) |
| Postnatal | 108 | 43 (27) | 27 (27) | 38 (27) | 0 (27) |
Dead embryos or resorption remnants
Figure 4. Deletion of ADAM10 in endothelial cells using a different Tie2-Cre transgenic driver results in abnormal embryonic development and earlier Cre-mediated recombination.
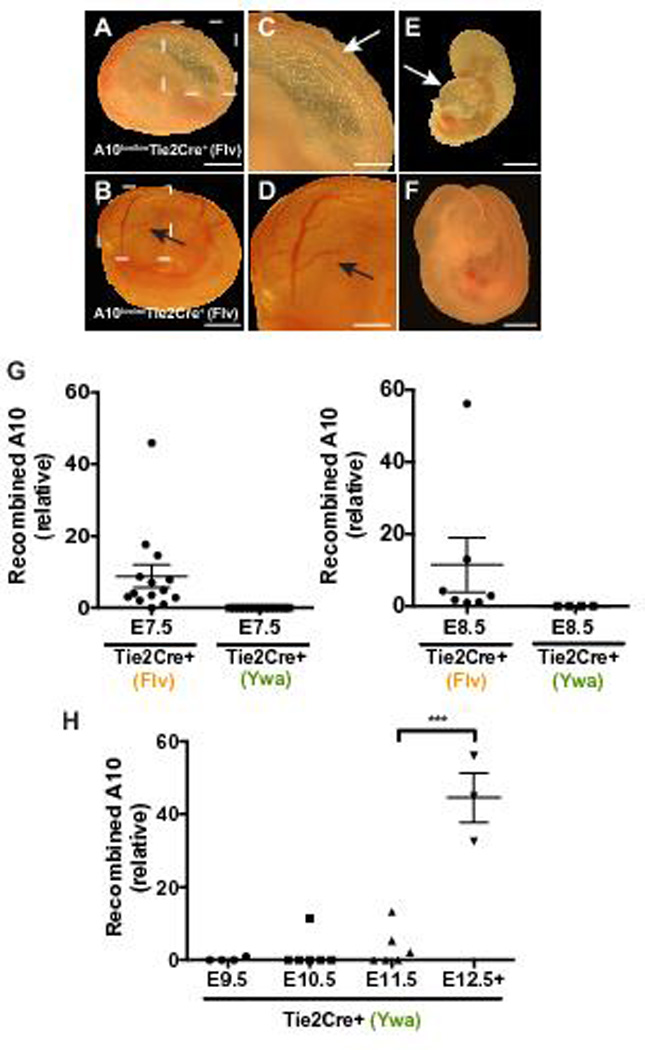
A to F, Representative images of whole mount preparations of embryos and yolk sacs harvested at E10.5. A to D, a yolk sac isolated from an E10.5 A10flox/flox-Tie2-Cre+(Flv) embryo appeared wrinkled (A,C) and devoid of the large vessels seen in the yolk sac of the A10flox/wt-Tie2-Cre+(Flv) littermate control (B,D, arrows). The A10flox/flox-Tie2-Cre+(Flv) embryo is smaller (E) than the normally developing littermate control (F). A10flox/flox-Tie2-Cre+(Flv) embryos had an enlarged pericardial sac (E, arrow), and were dead as determined by the absence of a heartbeat (11 out of 12 embryos). G, Total DNA from E7.5-E8.5 Tie2-Cre+(Flv) embryos or from E7.5-E8.5 Tie2-Cre+(Ywa) embryos were subjected to qPCR to determine levels of recombined Adam10 in embryos. 13 out of 14 E7.5 Tie2-Cre+(Flv) embryos (n=6 A10flox/wtTie2-Cre+(Flv); n=8, A10flox/floxTie2-Cre+(Flv)) showed detectable recombined Adam10 product while no recombined Adam10 was detected in E7.5 Tie2-Cre+(Ywa) embryos (n=7, A10flox/floxTie2-Cre+(Ywa)). 7 out of 7 E8.5 Tie2-Cre+(Flv) embryos (n=6 A10flox/wt Tie2-Cre+(Flv); n=1, A10flox/flox Tie2-Cre+(Flv)) had detectable recombined Adam10 while no recombined Adam10 was detected in 4 A10flox/flox Tie2-Cre+(Ywa) embryos. Samples with no detectable signal are plotted as zero. H, Total DNA prepared from A10flox/floxTie2-Cre+(Ywa) embryos between E9.5 and E13.5 were analyzed as described for G by qPCR. Recombined Adam10 was detected in 1 of 4 E9.5 embryos, 1 of 6 E10.5 embryos, and 3 of 6 E11.5 embryos. Recombined Adam10 was detectable in all embryos between E12.5 and E13.5 (n=3). Expression levels in G and H were normalized to Gapdh or the promoter region of Mrc1. Data shown as mean ± SEM. *** signifies p<0.001 in two-tailed student’s t-test. Scale bars, 1mm (A,B,E,F), 500µm (C,D).
Next, we focused on whether a difference in the timing of Tie2-Cre-mediated recombination might contribute to differences in survival of Adam10ΔEC(Flv) and Adam10ΔEC(Ywa) mice. While qPCR analysis showed recombined Adam10 in E7.5 Adam10ΔEC(Flv) conceptuses, no recombination was detectable in E7.5 Adam10ΔEC(Ywa) embryos prepared under identical conditions (Figure 4G). Adam10 recombination in Adam10ΔEC(Ywa) mice was only consistently detectable by E12.5 (Figure 4H, supplemental Figure XV). These results suggest that ADAM10 excision occurs earlier in Adam10ΔEC(Flv) than in Adam10ΔEC(Ywa) embryos, providing a plausible explanation for their difference in survival. In addition, we found a higher extent of recombination of floxed Adam10 alleles in some, but not all tissues examined in young adult Adam10flox/wt (Flv) versus Adam10flox/wt(Ywa) mice (supplemental Figure XVI).
Analysis of endothelial, pericyte and lymphatic markers
In order to gain insight into the potential mechanisms underlying the vascular phenotypes observed in Adam10ΔEC(Ywa) mice, we performed qPCR analysis on CD31+ endothelial cells isolated from the lungs, liver, heart, and kidneys of Adam10ΔEC mice. The Notch target gene and marker for arterial endothelial cells Ephrin B2 (EfnB237) showed significantly reduced expression relative to the endothelial cell marker CD31 in endothelial cells from the lungs and liver of Adam10ΔEC(Ywa) mice compared to controls. Furthermore, we found a trend towards reduced expression of EfnB2 in heart endothelial cells from Adam10ΔEC(Ywa) mice compared to controls and no significant difference in endothelial cells isolated from the kidney (supplemental Figure XVII). There was also no significant difference between the expression of the venous endothelial cell marker Ephrin B4 (EphB4) in Adam10ΔEC(Ywa) and control mice in any of the endothelial cells isolated from the different tissues examined here (supplemental Figure XVII). In addition, immunohistochemistry for the lymphatic marker LYVE-1 revealed no evident difference in staining in the kidney, small intestine, and heart of Adam10ΔEC(Ywa) mice compared to controls, but increased staining of Adam10ΔEC(Ywa) liver sinusoidal vasculature compared to controls (supplemental figure XVIII), consistent with the known expression of LYVE-1 in sinusoidal vessels of the liver 38, 39. Finally, when we stained several of the abnormal vascular structures in Adam10ΔEC(Ywa) mice for PDGFRβ or NG2 to assess pericyte recruitment, we found a similar staining pattern in Adam10ΔEC(Ywa) glomeruli compared to controls and clear staining of the intestinal polyps of Adam10ΔEC(Ywa) mice, as in the villi of control small intestines (supplemental figure XIX). Moreover, the abnormal subcapsular liver vessels were PDGFRβ+ while abnormal subepicardial vessels were both PDGFRβ+ and NG2+ positive (supplemental figure XIX).
Notch1ΔEC mice generated with Ywa Tie2-Cre share some organ-specific vascular defects with Adam10ΔEC mice
The difference in survival of Adam10ΔEC(Ywa) mice, which live into adulthood, and Adam10ΔEC(Flv) mice, which die during embryogenesis, raised the possibility that Notch1ΔEC mice generated with the Tie2-Cre(Ywa) might develop beyond E10.5. We therefore generated Notch1ΔEC(Ywa) mice, which were found at approximately Mendelian ratios between late gestation and birth (Supplemental Table I). About 30% of Notch1ΔEC(Ywa) mice (from crosses of Notch1flox/flox-Tie2Cre+/− males with Notch1flox/flox females) displayed early postnatal lethality (supplemental Figure XX). However, over 50% of Notch1ΔEC(Ywa) mice survived at least 8 weeks and several were subjected to histopathological analysis. The Notch1ΔEC(Ywa) livers contained abnormal subcapsular vessels, as seen in Adam10ΔEC(Ywa) and Adam10/17ΔEC(Ywa) mice (Figure 5A-D). Notch1ΔEC(Ywa) kidneys contained somewhat larger, hypercellular glomeruli with increased CD31 staining compared to controls (Figure 5E-H). In addition, Notch1ΔEC(Ywa) mice had significantly shorter long bones than littermate controls (supplemental Figure V). While growth plates of Notch1ΔEC(Ywa) mice were generally intact and continuous (Figure 5I), the shape and size of vessels adjacent to the femoral growth plate appeared different than in controls (Fig. 5M-N), which was further corroborated by CD31 staining (Figure 5K-P, see also supplemental Figure VI).
Figure 5. Several organ-specific vascular beds are affected in N1ΔEC mice.

A to L, Histopathological analysis of H&E- and CD31-stained specimen from N1ΔEC mice revealed defects in vascular beds of the liver, kidney, and femur. A to D, the livers of N1ΔEC mice contain enlarged surface vessels (A, arrow) not present in controls (B). The enlarged vessels are CD31+ (C, arrow) and associated with mild changes in nearby sinusoidal endothelium not seen in the control (D). E to H, glomeruli of an N1ΔEC mouse (E, arrow) are larger than control glomeruli (F). N1ΔEC glomeruli show increased CD31 staining (G, arrow) compared to controls (H). Abnormally enlarged vessels are seen under the femoral growth plate of an N1ΔEC mouse (I, arrow) in contrast to the small vessels seen in a control mouse (J, arrow). The enlarged vessels in N1ΔEC femurs are CD31+ (K) and are immediately adjacent to growth plate, and are smaller in control mice (L, see supplemental figure VI for higher magnification images and quantification). H&E micrographs are representative of 8-week old animals analyzed for each genotype (n=4 N1ΔEC, n=4 N1flox/flox), see supplemental Figure XX for quantification. Scale bars, 100µm (A-D, I-L), 50µm (E-H).
Interestingly, unlike Adam10ΔEC(Ywa) and Adam10/17ΔEC(Ywa) mice, Notch1ΔEC(Ywa) mice lacked polyps in their small intestine and only had relatively mild defects in the heart and diaphragm vasculature (supplemental Figures XXI, XXII). Moreover, P5 Notch1ΔEC(Ywa) retinas had only small areas with increased vascular density at the developing vasculature’s leading edge (Figure 6A–D), although the average vascular density and vascular looping was not significantly different from controls (Figure 6E-F). Interestingly, the progression of the retinal vascular front in Notch1ΔEC(Ywa) mice was delayed compared to littermate controls (Figure 6G). By P12, development of the deep retinal vascular plexus was also not significantly affected in Notch1ΔEC(Ywa) mice compared to controls (supplemental Figure XXIII). Notch1ΔEC(Ywa) mice thus exhibit some, but not all, of the vascular changes observed in Adam10ΔEC(Ywa) and Adam10/17ΔEC(Ywa) mice (for quantification, see supplemental Figure XXVI).
Figure 6. Notch1 and Notch4 control retinal vascular development.
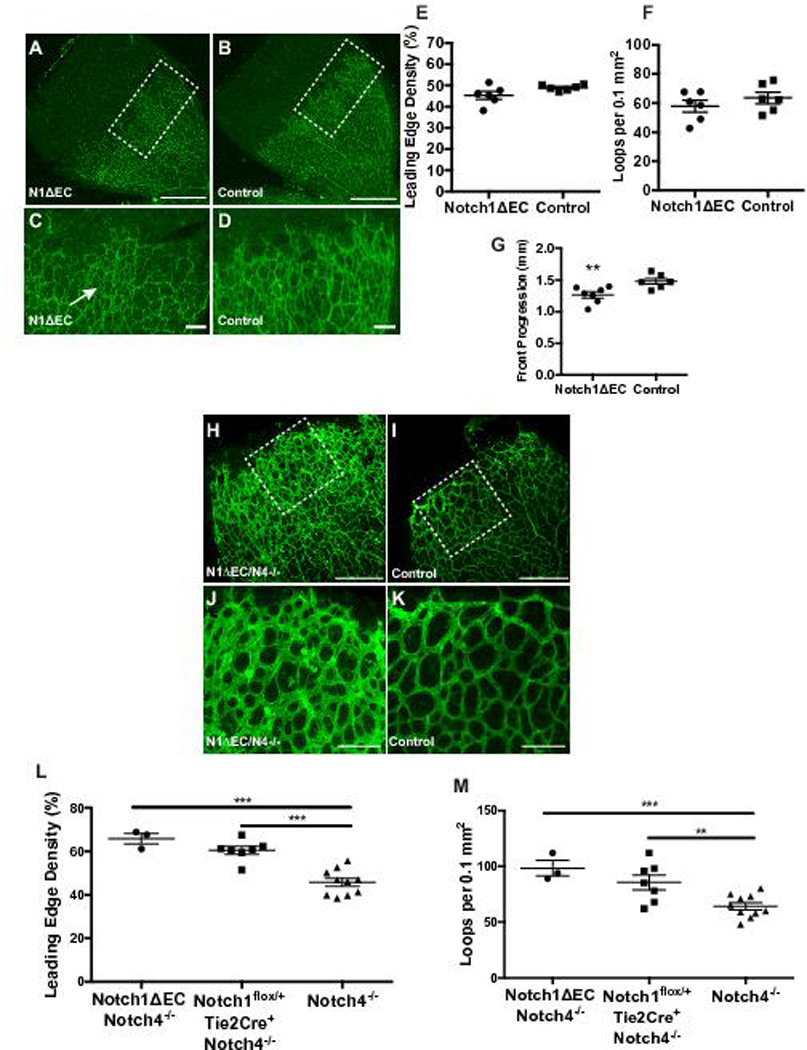
A to D, representative isolectin-B4 stained retinas of P5 N1ΔEC (A) and N1flox/+-Tie2Cre+ (B) control mice. Enlarged image of representative N1ΔEC (C) retina shows uneven isolectin-B4 stained vessels at the leading edge with small patches of increased vascular coverage (arrow) compared to a control (D). E to G, Quantification of vascular density (E) and vascular loops per 0.1 mm2 (F) at leading edge of the retinal vasculature shows no significant difference in vascular density (p=0.08) and vascular loops per field in N1ΔEC retinas (n=7) compared to controls (n=7). G, Retinal progression from the optic disk to the retina periphery was decreased in N1ΔEC (n=7) compared to controls (n=6). H to K, representative isolectin-B4 stained retinas of P5 N1ΔEC/N4−/− (H) and N4−/− (I) control mice. Enlarged image of representative N1ΔEC/N4−/− (J) retina shows increased isolectin-B4 stained vascular coverage at the leading edge compared to a control retina (K). L,M. N1ΔEC/N4−/− retinas (n=3) showed increased vascular density (L) and increased vascular loops (M) at the developing front of the retina. In N1ΔEC/N4−/− retinas, progression from the optic disk to the retina periphery was decreased (N) compared to N4−/− controls. Data shown as mean ± SEM. ** signifies p<0.01 and *** signifies p<0.001 in two-tailed student t-test. Scale bars, 500µm (A-B), 100µm (C-D, J-K), 300µm (H-I).
Additional inactivation of Notch4 in Notch1ΔEC mice results in vascular defects resembling those in Adam10ΔEC(Ywa) mice
Notch4 has been reported to be highly expressed in the heart, moderately in the lung and placenta and at low levels in the liver, skeletal muscle, kidney, pancreas, spleen, lymph node, thymus, bone marrow and fetal liver (http://source-search.princeton.edu/). Previous work showed that Notch4 is predominantly expressed in endothelial cells 36, 40, 41. To determine whether Notch4 has redundant or compensatory functions in the organ beds that are affected in Adam10ΔEC(Ywa) but not Notch1ΔEC(Ywa) mice, we generated mice lacking endothelial Notch1 that also lacked Notch4 systemically (Notch1ΔEC/Notch4−/−). These animals were present at the expected Mendelian ratio late in gestation and at birth (Supplemental Table II). However, most Notch1ΔEC/Notch4−/− mice died shortly after birth (supplemental Figure XXB), with few surviving into adulthood.
Like Adam10ΔEC(Ywa) mice, the retinal vasculature of P5 Notch1ΔEC/Notch4−/− mice revealed an increase in vascular density and looping compared to Notch4−/− littermate controls (Figure 6H-K, Notch4−/− mice appeared normal compared to age-matched wild type controls, see supplemental methods and supplemental Figure XXV). Several surviving Notch1ΔEC/Notch4−/− mice (n=4) were subjected to histopathological analysis. In addition to the vascular defects seen in the liver (Fig. 7A-D), long bones (Figure 7E-H, supplemental Figure VI), and kidney (supplemental Figure XXVI), which were also present in Notch1ΔEC(Ywa) mice, Notch1ΔEC/Notch4−/− mice exhibited enlarged subepicardial vessels and myocardial endothelial hypercellularity (Fig. 7I-L), intestinal polyps with abnormal CD31+ nests (Figure 7M-P), and increased endothelial staining in the diaphragm (supplemental Figure XXII). Thus, Notch1ΔEC/Notch4−/− mice have the full range of vascular defects (in the retina, liver, kidney, long bones, heart, small intestine, and diaphragm) seen in Adam10ΔEC(Ywa) and Adam10/17ΔEC(Ywa) mice (quantification in supplemental Figure XXVII).
Figure 7. All organ-specific vascular beds affected in A10ΔEC mice are altered in N1ΔEC/N4−/− mice.

H&E- and CD31-staining of specimens from N1ΔEC/N4−/− mice revealed vascular defects in all of the organ beds affected in A10ΔEC 18 and A10/17ΔEC mice. A to D, N1ΔEC/N4−/− livers (A) have enlarged surface vessels (arrows) absent in controls (B). Enlarged vessels are CD31+ (C, arrows) and are associated with pronounced changes in surrounding sinusoidal endothelium not seen in controls (D). E to G. Vessels directly abutting the femoral growth plate are abnormally large in N1ΔEC/N4−/− mice (E) compared to controls (F). Enlarged vessels in N1ΔEC/N4−/− femurs are CD31+ (G) and are abnormally oriented in contrast to perpendicularly oriented vessels seen in controls (H, see supplemental Figure VI for higher magnification images and quantification). I to J, an N1ΔEC/N4−/− heart (I) shows an increase in myocardial cellularity and enlarged subepicardial vascular spaces (I, arrow) compared to N4−/− controls (J). Enlarged vessels in N1ΔEC/N4−/− hearts are CD31+ (K). There is a pronounced increase in CD31 staining of N1ΔEC/N4−/− myocardium compared to N4−/− control myocardium (L). M to P, like A10ΔEC 18 and A10/17ΔEC mice, N1ΔEC/N4−/− mice develop hyperplastic polyps in the small intestine (M), never seen in controls (N). N1ΔEC/N4−/− polyps are hypercellular with abnormal CD31+ structures (O, arrow) not seen in control villi (P). H&E micrographs shown are representative of 6- to 8-week old animals analyzed for each genotype (n=4 N1ΔEC/N4−/− mice, n=3 N4−/− controls), see supplemental Figure XXII for quantification. Scale bars, 100µm (A-H, I-L, M-P).
Since Tie2-Cre also drives recombination in hematopoietic cell lineages and since Tie2-mediated disruption of Notch signaling is known to affect erythropoiesis 42, 43, we evaluated the red blood cell count, the hemoglobin levels and the percentage of reticulocytes in several mutant strains generated in this study (supplemental Figures XXVIII and XXIX). As previously described for A10ΔEC mice, we observed a significant increase in reticulocytes and decreased RBC and hemoglobin in Adam10/17ΔEC mice and Notch1ΔEC/Notch4−/− mice. The anemia observed in Adam10/17ΔEC was significantly worse compared to previously described A10ΔEC mice and to Notch1ΔEC and Notch1ΔEC/Notch4−/− mice, possibly due to additional effects dependent on ADAM17 and independent of Notch signaling. When we performed a mouse phenotype database search (http://www.informatics.jax.org/mp/annotations/MP:0001577), we did not find evidence for an independent role for anemia in causing the unique combination of vascular phenotypes described here, further supporting the interpretation that they are caused by defects in endothelial ADAM10/Notch signaling.
Finally, we found that Tie2-Cre(Ywa)-dependent expression of the Notch1 intracellular domain (NICD), which is generated through sequential processing of Notch1 by ADAM10 and presenilin, rescued the vascular defects in Adam10ΔEC(Ywa) mice (n=3), further supporting the interpretation that a lack of ADAM10/presenilin-generated NICD and downstream Notch signaling is responsible for the observed vascular defects in Adam10ΔEC animals (see supplemental Figures XXX and XXXI).
DISCUSSION
Notch signaling in endothelial cells is essential for the earliest stages of vascular development 8–12, 36, but little is currently known about endothelial Notch function later in embryogenesis. Our previous observation that Adam10ΔEC mice survived into adulthood with defects in several organ-specific vascular beds raised the question of whether these defects were caused by a lack of Notch signaling. Here we demonstrated that the organ-specific vascular phenotypes in Adam10ΔEC mice were recapitulated in mice deficient in endothelial Notch receptors, and could be rescued by endothelial-specific expression of the Notch1 intracellular domain in Adam10ΔEC mice. In addition, we found no evidence for redundant or compensatory function of the related ADAM17 in endothelial Notch signaling. Instead, our results suggest that the timing of Tie2-Cre-dependent excision accounts for the postnatal survival with organ-specific vascular defects observed in Adam10ΔEC mice.
ADAM10 is considered the principal Notch processing enzyme during physiological, ligand-induced Notch signaling; yet ADAM17 can also cleave Notch in cell-based assays under non-physiological conditions 15, 16, 28. We found that mice lacking endothelial ADAM17 and ADAM10 survived in postnatal life and had all of the vascular phenotypes seen in Adam10ΔEC mice. In contrast, previously described Adam17ΔEC mice, generated with Tie2-Cre(Ywa), had no evident defects in vascular development 34. These findings argue against a major compensatory or redundant function for ADAM17 in endothelial cells lacking ADAM10 during organ-specific vascular development.
Instead, we found that differences in the temporal expression of the Tie2-Cre(Ywa) used here and the Tie2-Cre(Flv) used to inactivate Notch1 and Rbpj in previous studies most likely account for the survival of the previously described Adam10ΔEC mice 18. When ADAM10 was deleted with Tie2-Cre(Flv), the resulting Adam10ΔEC(Flv) mice died by E10.5 and appeared morphologically similar to previously described Notch1ΔEC(Flv) mice 8. Moreover, we found that Tie2-Cre(Ywa) induced Adam10 recombination later than Tie2-Cre(Flv), potentially explaining the postnatal survival of Adam10ΔEC(Ywa) mice and the embryonic lethality in Adam10ΔEC(Flv) mice. Earlier inactivation of ADAM10 by Tie2-Cre(Flv) (by E7.5) may disrupt the development of the primary intraembryonic vascular network, dorsal aortae and cardinal veins, and yolk sac vasculature 8, 36. Any of these defects would presumably be incompatible with life beyond mid-gestation. In contrast, later deletion of ADAM10 (starting around E12.5) by Tie2-Cre(Ywa) apparently allows these animals to circumvent early embryonic lethality and survive—likely because the yolk sac and early embryonic vasculature are not detectably affected.
Interestingly, Tie2-Cre(Ywa)-driven recombination also allowed Notch1ΔEC(Ywa) mice to survive into postnatal life, with abnormal vessels under the liver surface, enlarged glomeruli and malformed metaphyseal vessels in long bones. This suggest that these phenotypes, which resemble those previously reported for Adam10ΔEC(Ywa) mice, are attributable to ADAM10-dependent Notch1 signaling. However, unexpectedly, several vascular beds with defects in Adam10ΔEC(Ywa) mice appeared normal or only mildly affected in Notch1ΔEC(Ywa) mice (specifically the vasculature of the small intestine, retina, heart, diaphragm and liver sinusoids).
The generally normal appearance of Notch1ΔEC(Ywa) retinal vasculature was particularly unexpected since previous studies reported that defective Notch signaling results in abnormal developmental retinal angiogenesis 19–21, 23. However, in these studies, Notch signaling was targeted using gamma-secretase inhibitors or endothelial Notch ligand knockdown, which can block signaling through multiple Notch receptors 19–21. Interestingly, similar to what we observed in Notch1ΔEC(Ywa) retinas, postnatal inactivation of Notch1 alone led to increased vascular sprouting in specific regions of the retina vasculature 19. However, when Notch1 is blocked with a specific antibody for a short time after birth, this results in strongly increased retinal vascular density 44. Since our study relied on constitutive inactivation of endothelial Notch1 during development, this could have allowed for Notch4 to compensate for the absence of Notch1. Notably, when we also inactivated Notch4 in mice lacking Notch1 in endothelial cells, the resulting Notch1ΔEC/Notch4−/−(Ywa) mice had all the vascular abnormalities previously identified in Adam10ΔEC(Ywa) and Adam10/17ΔEC(Ywa) mice—including increased vascular density in the developing retina 18–21, 23, 44. These findings suggest that Notch4 has redundant or compensatory functions with endothelial Notch1 during normal retinal vascular development. Our cumulative findings suggest that Notch1 and Notch4 cooperate to support the normal development of several organ-specific vascular beds. In addition, our observation that the defects in Adam10ΔEC(Ywa) mice can be rescued by Tie2-Cre(Ywa)-dependent overexpression of the intracellular domain of Notch1 (NICD) suggests that, when ADAM10 is deleted from endothelial cells, the NICD can support signaling through both Notch1 and 4 during development of organ-specific vascular structures.
Interestingly, elements of the heart and liver vascular phenotypes described here and previously 18 have also been observed when other components of Notch signaling were targeted in the endothelium of postnatal mice. Inactivation of the Notch ligand Dll4 in endothelial cells for 10 weeks postnatally led to abnormal subcapsular liver vessels and dilated sinusoids 45. Meanwhile, knockdown of endothelial RBPJ starting from birth resulted in abnormally large vessels near the heart surface in 2-week old mutant mice 46. Finally, inactivation of RBPJ in endothelial cells at 2 weeks resulted in defects in the femoral and tibial growth plates and in metaphyseal vessels 47. However, no defects in other specialized vascular beds were reported in these animals. The vascular phenotypes described in Notch1ΔEC, Notch1ΔEC/Notch4−/−, and Adam10ΔEC(Ywa) mice point to previously unappreciated roles for ADAM10-dependent Notch signaling in organ-specific vascular development. In contrast to previously described Notch1ΔEC(Flv) 8 mice, the aorta, vena cava, and the yolk sac vasculature appear to develop normally in the Notch1ΔEC, Notch1ΔEC/Notch4−/−, and Adam10ΔEC embryos described here. Perhaps ADAM10/Notch signaling must be disrupted during the specific mid- to late gestational window (~E11.5+) targeted in this study to elicit the particular combination of vascular defects observed here. Such a scenario could explain why only some organ-specific vascular phenotypes are observed upon postnatal inactivation of components of the Notch signaling pathway 45–47. Further studies will be necessary to determine whether the organ-specific vascular phenotypes vary with the timing or duration of inactivation of ADAM10/Notch signaling.
Since it appears that specialized, organ-specific vessel beds are primarily affected in the knockout animals described here, we will briefly outline potential models for how ADAM10/Notch signaling could orchestrate the development of these specialized vascular networks. In each case, we propose that one or more ADAM10/Notch-dependent endothelial cell fate decision(s) are required to assemble the individual affected vascular beds. In one scenario, a specialized endothelial cell would be induced to branch off an existing vessel within the organ and differentiate, as in angiogenesis. Like the endothelial tip-stalk cell fate decision in the developing retina, this specialized endothelial cell—perhaps analogous to a tip cell—would coordinate proper development of the vasculature by preventing adjacent cells from assuming its cell fate through ADAM10/Notch-dependent lateral inhibition. In the absence of ADAM10/Notch signaling, there could be an increase in the number of one specialized cell type at the expense of another, leading to changes in the vascular organization of glomeruli, intestinal villi, and other affected structures. Importantly, unlike in the retinal vascular tree, which eventually remodels to acquire a normal appearance in adult Adam10ΔEC(Ywa) mice18, other affected vascular beds presumably lack the ability to remodel and restore normal vascular appearance.
The observed phenotypes may also arise from defects in artery-vein cell fate decisions in the nascent vasculature 48, perhaps favoring venous over arterial differentiation. This is consistent with the decreased expression of the arterial marker EfnB2 in Adam10ΔEC(Ywa) endothelial cells from some affected vessel beds; further studies will be necessary to gain a better understanding of underlying mechanisms. Alternatively, or in addition, the defects could also affect vessel maturation and specialization 49, 50. Interestingly, staining of select organs in Adam10ΔEC(Ywa) mice revealed PDGFRβ+ and/or NG2+cell populations in affected organ-specific vascular structures, suggesting that pericytes, which are key mediators in vessel stabilization and maturation 51, 52, are still found in the vasculature of several affected vessel beds in A10ΔEC mice. Further study will be required to determine if normal pericyte ensheathment of endothelial cells occurs in these vessel beds. Finally, differentiation of nascent endothelium into fenestrated or sinusoidal endothelium 53, 54 could be compromised in developing organ-specific vascular beds. In each scenario, ADAM10/Notch signaling would be required for selection between specialized endothelial cell fates and to control specialized vessel development in response to organ-specific cues. An intriguing third possibility is that the nascent vascular networks in affected vessel beds could form through de novo vasculogenesis, from endothelial progenitors arising from local, organ-specific precursors or progenitors recruited from the bone marrow, depending on developmental stage 55. The nascent endothelium could then form or mature abnormally as described in the models above.
In conclusion, our results support a model in which Notch1 and Notch4 control the development of several organ-specific vascular beds in an ADAM10-dependent manner. Importantly, the timing of the inactivation of Notch signaling during development appears to profoundly affect the resulting developmental phenotypes—with early inactivation leading to the previously described early embryonic lethality and later inactivation allowing postnatal survival of animals with a unique set of vascular defects. Future studies of the mutant mouse strains generated with the Tie2-Cre (Ywa) transgene will provide unique opportunities to explore how ADAM10/Notch signaling control the development and specialization of organ-specific vascular compartments.
Supplementary Material
Novelty and Significance.
What Is Known?
The development of the specialized, organ-specific vasculature of structures such as the kidney glomeruli, liver sinusoids, and coronary vessels is poorly understood.
Inactivation of the metalloproteinase ADAM10 (a disintegrin and metalloprotease 10) in endothelial cells specifically affects organ specific vascular structures.
ADAM10 typically regulates cell specialization through a cell surface receptor called Notch, yet inactivation of Notch in blood vessels results in lethality well before these vascular structures develop, raising questions about the cause of the vascular defects in mice lacking ADAM10 in endothelial cells.
What New Information Does This Article Establish?
We report that the timing of the inactivation of Notch and ADAM10 in endothelial cells (ΔEC) is crucial.
Early inactivation of ADAM10 results in early lethality, like previously described NotchΔEC mice, whereas later inactivation of Notch1 with Notch4 affects all the specialized vascular structures that are abnormal in Adam10ΔEC mice.
This work uncovers an essential role for the ADAM10/Notch signaling pathway in the development of organ specific vascular structures such as coronaries, kidney glomeruli and liver sinusoids.
Pathological changes in specialized vasculature such as kidney glomeruli and coronary vessels are a major cause of human disease. The desire to repair and rebuild these vascular structures provides a strong incentive to understand the molecular pathways that orchestrate their development. We knew from our previous studies that inactivation of the metalloprotease ADAM10 in endothelial cells causes a particularly interesting combination of defects in specialized vascular beds, including glomeruli, liver sinusoids and cardiac vessels. Here we show for the first time that these defects were caused by loss of signaling through a receptor called Notch, a master regulator of cellular specialization. Importantly, we show that the timing of blocking ADAM10/Notch signaling is crucial to eliciting these vascular defects. Early inactivation was known to cause lethality by blocking the very first steps of vessel development, but we now show that later inactivation selectively affects the development of specialized vascular beds, allowing survival. This opens up unique opportunities to learn more about the development of specialized, organ-specific vascular structures. Ultimately, we hope that this knowledge will help guide and inform efforts to re-build or repair these vessels in order to combat organ-specific vascular diseases such as diabetic nephropathy, glomerulonephritis, and myocardial infarction.
Acknowledgments
We thank Sarah Loh for excellent technical assistance, Robert Frawley for his MATLAB code, Lyudmila Lukashova and Adele Boskey for micro CT assistance, Katia Manova, Ning Fan, Mesruh Turkekul, and Afsar Barlas for help with immunohistochemistry, and Yurii Chinenov and Thorsten Maretzky for helpful discussions.
SOURCES OF FUNDING
This work was supported by NIH GM64750 (CPB), T32GM007739 (Tri-Institutional MD-PhD program, ROA), T32EY00713820 (Tri-Institutional Vision Research Training Program, ROA), and T32HD606006 (Weill Cornell Medicine Developmental Biology Program, ROA) and NIH NCI CA008748 (SM, KM).
Nonstandard Abbreviations and Acronyms
- ADAM
a disintegrin and metalloprotease
- Dll4
Delta-like 4
- Tie2
angiopoietin-1 receptor
- Tie2-Cre
Cre-recombinase controlled by the Tie2 promoter
- ΔEC
deleted by Tie2-Cre in endothelial cells
- RBPJ
recombinant binding protein suppressor of hairless
- CD31
cluster of differentiation 31 protein, also termed
- PECAM1
platelet endothelial cell adhesion molecule 1
- LYVE-1
lymphatic vessel endothelial hyaluronan receptor 1
- NG2
NG2 Chondroitin Sulfate Proteoglycan
- PDGFRβ
platelet-derived growth factor receptor β
- NICD
Notch1 intracellular domain.
Footnotes
DISCLOSURES
None.
REFERENCES
- 1.Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005;438:932–936. doi: 10.1038/nature04478. [DOI] [PubMed] [Google Scholar]
- 2.Coultas L, Chawengsaksophak K, Rossant J. Endothelial cells and VEGF in vascular development. Nature. 2005;438:937–945. doi: 10.1038/nature04479. [DOI] [PubMed] [Google Scholar]
- 3.Drake CJ, Fleming PA. Vasculogenesis in the day 6.5 to 9.5 mouse embryo. Blood. 2000;95:1671–1679. [PubMed] [Google Scholar]
- 4.Chong DC, Koo Y, Xu K, Fu S, Cleaver O. Stepwise arteriovenous fate acquisition during mammalian vasculogenesis. Dev Dyn. 2011;240:2153–2165. doi: 10.1002/dvdy.22706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lindskog H, Kim YH, Jelin EB, Kong Y, Guevara-Gallardo S, Kim TN, Wang RA. Molecular identification of venous progenitors in the dorsal aorta reveals an aortic origin for the cardinal vein in mammals. Development. 2014;141:1120–1128. doi: 10.1242/dev.101808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Risau W. Mechanisms of angiogenesis. Nature. 1997;386:671–674. doi: 10.1038/386671a0. [DOI] [PubMed] [Google Scholar]
- 7.Roca C, Adams RH. Regulation of vascular morphogenesis by Notch signaling. Genes Dev. 2007;21:2511–2524. doi: 10.1101/gad.1589207. [DOI] [PubMed] [Google Scholar]
- 8.Limbourg FP, Takeshita K, Radtke F, Bronson RT, Chin MT, Liao JK. Essential role of endothelial Notch1 in angiogenesis. Circulation. 2005;111:1826–1832. doi: 10.1161/01.CIR.0000160870.93058.DD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krebs LT, Shutter JR, Tanigaki K, Honjo T, Stark KL, Gridley T. Haploinsufficient lethality and formation of arteriovenous malformations in Notch pathway mutants. Genes Dev. 2004;18:2469–2473. doi: 10.1101/gad.1239204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Copeland JN, Feng Y, Neradugomma NK, Fields PE, Vivian JL. Notch signaling regulates remodeling and vessel diameter in the extraembryonic yolk sac. BMC Dev Biol. 2011;11:12. doi: 10.1186/1471-213X-11-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Duarte A, Hirashima M, Benedito R, Trindade A, Diniz P, Bekman E, Costa L, Henrique D, Rossant J. Dosage-sensitive requirement for mouse Dll4 in artery development. Genes Dev. 2004;18:2474–2478. doi: 10.1101/gad.1239004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gale NW, Dominguez MG, Noguera I, Pan L, Hughes V, Valenzuela DM, Murphy AJ, Adams NC, Lin HC, Holash J, Thurston G, Yancopoulos GD. Haploinsufficiency of delta-like 4 ligand results in embryonic lethality due to major defects in arterial and vascular development. Proc Natl Acad Sci U S A. 2004;101:15949–15954. doi: 10.1073/pnas.0407290101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kopan R, Cagan R. Notch on the cutting edge. Trends Genet. 1997;13:465–467. doi: 10.1016/s0168-9525(97)01318-8. [DOI] [PubMed] [Google Scholar]
- 14.Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137:216–233. doi: 10.1016/j.cell.2009.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van Tetering G, van Diest P, Verlaan I, van der Wall E, Kopan R, Vooijs M. Metalloprotease ADAM10 is required for Notch1 site 2 cleavage. J Biol Chem. 2009;284:31018–31027. doi: 10.1074/jbc.M109.006775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bozkulak EC, Weinmaster G. Selective use of ADAM10 and ADAM17 in activation of Notch1 signaling. Mol Cell Biol. 2009;29:5679–5695. doi: 10.1128/MCB.00406-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Groot AJ, Habets R, Yahyanejad S, Hodin CM, Reiss K, Saftig P, Theys J, Vooijs M. Regulated proteolysis of NOTCH2 and NOTCH3 receptors by ADAM10 and presenilins. Mol Cell Biol. 2014;34:2822–2832. doi: 10.1128/MCB.00206-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Glomski K, Monette S, Manova K, De Strooper B, Saftig P, Blobel CP. Deletion of Adam10 in endothelial cells leads to defects in organ-specific vascular structures. Blood. 2011;118:1163–1174. doi: 10.1182/blood-2011-04-348557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hellstrom M, Phng LK, Hofmann JJ, Wallgard E, Coultas L, Lindblom P, Alva J, Nilsson AK, Karlsson L, Gaiano N, Yoon K, Rossant J, Iruela-Arispe ML, Kalen M, Gerhardt H, Betsholtz C. Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature. 2007;445:776–780. doi: 10.1038/nature05571. [DOI] [PubMed] [Google Scholar]
- 20.Lobov IB, Renard RA, Papadopoulos N, Gale NW, Thurston G, Yancopoulos GD, Wiegand SJ. Delta-like ligand 4 (Dll4) is induced by VEGF as a negative regulator of angiogenic sprouting. Proc Natl Acad Sci U S A. 2007;104:3219–3224. doi: 10.1073/pnas.0611206104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Suchting S, Freitas C, le Noble F, Benedito R, Breant C, Duarte A, Eichmann A. The Notch ligand Delta-like 4 negatively regulates endothelial tip cell formation and vessel branching. Proc Natl Acad Sci U S A. 2007;104:3225–3230. doi: 10.1073/pnas.0611177104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stahl A, Connor KM, Sapieha P, Willett KL, Krah NM, Dennison RJ, Chen J, Guerin KI, Smith LE. Computer-aided quantification of retinal neovascularization. Angiogenesis. 2009;12:297–301. doi: 10.1007/s10456-009-9155-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Benedito R, Rocha SF, Woeste M, Zamykal M, Radtke F, Casanovas O, Duarte A, Pytowski B, Adams RH. Notch-dependent VEGFR3 upregulation allows angiogenesis without VEGF-VEGFR2 signalling. Nature. 2012;484:110–114. doi: 10.1038/nature10908. [DOI] [PubMed] [Google Scholar]
- 24.Ehling M, Adams S, Benedito R, Adams RH. Notch controls retinal blood vessel maturation and quiescence. Development. 2013;140:3051–3061. doi: 10.1242/dev.093351. [DOI] [PubMed] [Google Scholar]
- 25.Hartmann D, de Strooper B, Serneels L, Craessaerts K, Herreman A, Annaert W, Umans L, Lubke T, Lena Illert A, von Figura K, Saftig P. The disintegrin/metalloprotease ADAM 10 is essential for Notch signalling but not for alpha-secretase activity in fibroblasts. Hum Mol Genet. 2002;11:2615–2624. doi: 10.1093/hmg/11.21.2615. [DOI] [PubMed] [Google Scholar]
- 26.Swiatek PJ, Lindsell CE, del Amo FF, Weinmaster G, Gridley T. Notch1 is essential for postimplantation development in mice. Genes Dev. 1994;8:707–719. doi: 10.1101/gad.8.6.707. [DOI] [PubMed] [Google Scholar]
- 27.Huppert SS, Le A, Schroeter EH, Mumm JS, Saxena MT, Milner LA, Kopan R. Embryonic lethality in mice homozygous for a processing-deficient allele of Notch1. Nature. 2000;405:966–970. doi: 10.1038/35016111. [DOI] [PubMed] [Google Scholar]
- 28.Brou C, Logeat F, Gupta N, Bessia C, LeBail O, Doedens JR, Cumano A, Roux P, Black RA, Israel A. A novel proteolytic cleavage involved in Notch signaling: the role of the disintegrin-metalloprotease TACE. Mol Cell. 2000;5:207–216. doi: 10.1016/s1097-2765(00)80417-7. [DOI] [PubMed] [Google Scholar]
- 29.Mumm JS, Kopan R. Notch signaling: from the outside in. Dev Biol. 2000;228:151–165. doi: 10.1006/dbio.2000.9960. [DOI] [PubMed] [Google Scholar]
- 30.Koni PA, Joshi SK, Temann UA, Olson D, Burkly L, Flavell RA. Conditional vascular cell adhesion molecule 1 deletion in mice: impaired lymphocyte migration to bone marrow. J Exp Med. 2001;193:741–754. doi: 10.1084/jem.193.6.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, Yanagisawa M. Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev Biol. 2001;230:230–242. doi: 10.1006/dbio.2000.0106. [DOI] [PubMed] [Google Scholar]
- 32.Jorissen E, Prox J, Bernreuther C, Weber S, Schwanbeck R, Serneels L, Snellinx A, Craessaerts K, Thathiah A, Tesseur I, Bartsch U, Weskamp G, Blobel CP, Glatzel M, De Strooper B, Saftig P. The disintegrin/metalloproteinase ADAM10 is essential for the establishment of the brain cortex. J Neurosci. 2010;30:4833–4844. doi: 10.1523/JNEUROSCI.5221-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Horiuchi K, Kimura T, Miyamoto T, Takaishi H, Okada Y, Toyama Y, Blobel CP. Cutting Edge: TNF-{alpha}-Converting Enzyme (TACE/ADAM17) Inactivation in Mouse Myeloid Cells Prevents Lethality from Endotoxin Shock. J Immunol. 2007;179:2686–2689. doi: 10.4049/jimmunol.179.5.2686. [DOI] [PubMed] [Google Scholar]
- 34.Weskamp G, Mendelson K, Swendeman S, Le Gall S, Ma Y, Lyman S, Hinoki A, Eguchi S, Guaiquil V, Horiuchi K, Blobel CP. Pathological Neovascularization Is Reduced by Inactivation of ADAM17 in Endothelial Cells but Not in Pericytes. Circ Res. 2010;106:932–940. doi: 10.1161/CIRCRESAHA.109.207415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang X, Klein R, Tian X, Cheng HT, Kopan R, Shen J. Notch activation induces apoptosis in neural progenitor cells through a p53-dependent pathway. Dev Biol. 2004;269:81–94. doi: 10.1016/j.ydbio.2004.01.014. [DOI] [PubMed] [Google Scholar]
- 36.Krebs LT, Xue Y, Norton CR, Shutter JR, Maguire M, Sundberg JP, Gallahan D, Closson V, Kitajewski J, Callahan R, Smith GH, Stark KL, Gridley T. Notch signaling is essential for vascular morphogenesis in mice. Genes Dev. 2000;14:1343–1352. [PMC free article] [PubMed] [Google Scholar]
- 37.Swift MR, Weinstein BM. Arterial-venous specification during development. Circ Res. 2009;104:576–588. doi: 10.1161/CIRCRESAHA.108.188805. [DOI] [PubMed] [Google Scholar]
- 38.Mouta Carreira C, Nasser SM, di Tomaso E, Padera TP, Boucher Y, Tomarev SI, Jain RK. LYVE-1 is not restricted to the lymph vessels: expression in normal liver blood sinusoids and down-regulation in human liver cancer and cirrhosis. Cancer Res. 2001;61:8079–8084. [PubMed] [Google Scholar]
- 39.Nonaka H, Tanaka M, Suzuki K, Miyajima A. Development of murine hepatic sinusoidal endothelial cells characterized by the expression of hyaluronan receptors. Dev Dyn. 2007;236:2258–2267. doi: 10.1002/dvdy.21227. [DOI] [PubMed] [Google Scholar]
- 40.Villa N, Walker L, Lindsell CE, Gasson J, Iruela-Arispe ML, Weinmaster G. Vascular expression of Notch pathway receptors and ligands is restricted to arterial vessels. Mech Dev. 2001;108:161–164. doi: 10.1016/s0925-4773(01)00469-5. [DOI] [PubMed] [Google Scholar]
- 41.Uyttendaele H, Marazzi G, Wu G, Yan Q, Sassoon D, Kitajewski J. Notch4/int-3, a mammary proto-oncogene, is an endothelial cell-specific mammalian Notch gene. Development. 1996;122:2251–2259. doi: 10.1242/dev.122.7.2251. [DOI] [PubMed] [Google Scholar]
- 42.Tang Y, Harrington A, Yang X, Friesel RE, Liaw L. The contribution of the Tie2+ lineage to primitive and definitive hematopoietic cells. Genesis. 2010;48:563–567. doi: 10.1002/dvg.20654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Venkatesh DA, Park KS, Harrington A, Miceli-Libby L, Yoon JK, Liaw L. Cardiovascular and hematopoietic defects associated with Notch1 activation in embryonic Tie2-expressing populations. Circ Res. 2008;103:423–431. doi: 10.1161/CIRCRESAHA.108.177808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu Y, Cain-Hom C, Choy L, Hagenbeek TJ, de Leon GP, Chen Y, Finkle D, Venook R, Wu X, Ridgway J, Schahin-Reed D, Dow GJ, Shelton A, Stawicki S, Watts RJ, Zhang J, Choy R, Howard P, Kadyk L, Yan M, Zha J, Callahan CA, Hymowitz SG, Siebel CW. Therapeutic antibody targeting of individual Notch receptors. Nature. 2010;464:1052–1057. doi: 10.1038/nature08878. [DOI] [PubMed] [Google Scholar]
- 45.Djokovic D, Trindade A, Gigante J, Badenes M, Silva L, Liu R, Li X, Gong M, Krasnoperov V, Gill PS, Duarte A. Combination of Dll4/Notch and Ephrin-B2/EphB4 targeted therapy is highly effective in disrupting tumor angiogenesis. BMC Cancer. 2010;10:641. doi: 10.1186/1471-2407-10-641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nielsen CM, Cuervo H, Ding VW, Kong Y, Huang EJ, Wang RA. Deletion of Rbpj from postnatal endothelium leads to abnormal arteriovenous shunting in mice. Development. 2014;141:3782–3792. doi: 10.1242/dev.108951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ramasamy SK, Kusumbe AP, Wang L, Adams RH. Endothelial Notch activity promotes angiogenesis and osteogenesis in bone. Nature. 2014;507:376–380. doi: 10.1038/nature13146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim YH, Hu H, Guevara-Gallardo S, Lam MT, Fong SY, Wang RA. Artery and vein size is balanced by Notch and ephrin B2/EphB4 during angiogenesis. Development. 2008;135:3755–3764. doi: 10.1242/dev.022475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jain RK. Molecular regulation of vessel maturation. Nat Med. 2003;9:685–693. doi: 10.1038/nm0603-685. [DOI] [PubMed] [Google Scholar]
- 50.LeBlanc AJ, Krishnan L, Sullivan CJ, Williams SK, Hoying JB. Microvascular repair: post-angiogenesis vascular dynamics. Microcirculation. 2012;19:676–695. doi: 10.1111/j.1549-8719.2012.00207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bergers G, Song S. The role of pericytes in blood-vessel formation and maintenance. Neuro Oncol. 2005;7:452–464. doi: 10.1215/S1152851705000232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Armulik A, Abramsson A, Betsholtz C. Endothelial/pericyte interactions. Circ Res. 2005;97:512–523. doi: 10.1161/01.RES.0000182903.16652.d7. [DOI] [PubMed] [Google Scholar]
- 53.Aird WC. Phenotypic heterogeneity of the endothelium: II. Representative vascular beds. Circ Res. 2007;100:174–190. doi: 10.1161/01.RES.0000255690.03436.ae. [DOI] [PubMed] [Google Scholar]
- 54.Aird WC. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ Res. 2007;100:158–173. doi: 10.1161/01.RES.0000255691.76142.4a. [DOI] [PubMed] [Google Scholar]
- 55.Schmidt A, Brixius K, Bloch W. Endothelial precursor cell migration during vasculogenesis. Circ Res. 2007;101:125–136. doi: 10.1161/CIRCRESAHA.107.148932. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.