

Abstract
Rationale
Compound 21 (C-21) is a highly selective non-peptide angiotensin AT2 receptor (AT2R) agonist.
Objective
To test the hypothesis that chronic AT2R activation with C-21 induces natriuresis via an action at the renal proximal tubule (RPT) and lowers blood pressure (BP) in experimental angiotensin II (Ang II)-dependent hypertension.
Methods and Results
In rats, Ang II infusion increased both sodium (Na+) retention and BP on Day 1 and BP remained elevated throughout the 7 day infusion period. Either intrarenal or systemic administration of C-21 prevented Ang II-mediated Na+ retention on Day 1, induced continuously negative cumulative Na+ balance compared with Ang II alone, and reduced BP chronically. The effects of C-21 are likely to be mediated by action on the RPT as acute systemic C-21-induced natriuresis was additive to that induced by chlorothiazide and amiloride. At 24h of Ang II infusion, AT2R activation with C-21, both intrarenally and systemically, translocated AT2Rs from intracellular sites to the apical plasma membranes of RPT cells without altering the total cellular pool of AT2Rs and internalized/inactivated major RPT Na+ transporters Na+-H+-exchanger-3 (NHE-3) and Na+/K+ATPase (NKA). C-21 lowered BP to a similar degree whether administered before or subsequent to the establishment of Ang II-dependent hypertension.
Conclusions
Chronic AT2R activation initiates and sustains receptor translocation to RPT apical plasma membranes, internalizes/inactivates NHE-3 and NKA, prevents Na+ retention resulting in negative cumulative Na+ balance, and lowers BP in experimental Ang II-induced hypertension. Acting uniquely at the RPT, C-21 is a promising candidate for the treatment of hypertension and Na+-retaining states in humans.
Subject Terms: Ion Channels/Membrane Transport, Pathophysiology, Nephrology and Kidney, Animal Models of Human Disease, ACE/Angiotensin Receptors/Renin Angiotensin System
Keywords: Blood pressure; natriuresis; hypertension, kidney; natriuretic hormone; sodium channels; angiotensin receptor; antihypertensive therapy/diuretics; hypertension
INTRODUCTION
The renin-angiotensin system (RAS) is a complex hormonal system composed of multiple enzymes, peptides, and receptors controlling sodium (Na+) excretion and blood pressure (BP) (1,2). Angiotensin II (Ang II), its major effector peptide, acts at two major angiotensin receptors, the type-1 (AT1R) and type-2 (AT2R) receptors. The vast majority of Ang II biological actions are mediated by AT1Rs, including vasoconstriction, antinatriuresis, cellular dedifferentiation and growth, aldosterone secretion and sympathetic nervous system activation; each of which can increase BP. In contrast, AT2Rs generally induce the opposite effects, including vasodilation, natriuresis, cellular differentiation, and growth inhibition (1). Because AT1Rs are expressed at a higher level than AT2Rs in most adult tissues, including vasculature and the kidney, AT1R actions usually predominate in vivo, especially under baseline unstimulated conditions. However, upon RAS stimulation or during AT1R blockade, when unblocked AT2Rs are exposed to high levels of angiotensin, AT2R-mediated vasodilation and natriuresis can readily be demonstrated (3–5).
According to the Guyton hypothesis (6,7), the kidneys play a critical role in the control of BP by regulating Na+ excretion. Under normal physiological conditions, a primary increase in renal Na+ retention expands extracellular fluid volume and increases BP. However, the increase in BP in turn is offset by pressure-induced natriuresis, returning BP to or towards its original baseline level. The development of hypertension requires an abnormality in pressure-natriuresis, wherein the increase in BP is accompanied by a natriuretic response that is insufficient to lower BP to normal.
The renal proximal tubule (RPT) is a critical nephron segment in the development of hypertension induced by Ang II (8,9). Coffman and colleagues (9) have demonstrated in cross-transplantation studies in renal tubule-selective AT1R knockout mice that AT1Rs must be present in the RPT to sustain a hypertensive response to continuous systemic Ang II infusion. Li and Zhuo (10) recently have confirmed the critical importance of RPT AT1Rs to control BP through their actions to increase Na+ reabsorption by demonstrating in AT1R-null mice that transfer of AT1Rs into RPT cells (RPTC) enables a hypertensive response to Ang II.
AT2Rs are expressed in the adult kidney RPT (11,12). Recently, we demonstrated that acute systemic administration of the highly selective non-peptide AT2R agonist Compound 21 (C-21) induces natriuresis by activating and recruiting AT2Rs to the apical plasma membranes of RPTCs in vivo (13–15). C-21-induced AT2R activation evoked a bradykinin- nitric oxide (NO)-cyclic 3′,5′ guanosine monophosphate (cGMP) signaling cascade that stimulated downstream signaling mediators Src kinase and extracellular signal-related kinase (ERK), internalizing/inactivating major RPT Na+ transporters Na+-H+ exchanger-3 (NHE-3) and Na+/K+ATPase (NKA) resulting in natriuresis (15). Preliminary data indicated that AT2R activation might prevent Na+ retention and lower BP by improving the pressure-natriuresis relationship in Ang II-dependent hypertension (15).
The present study was designed to explore in depth the therapeutic potential of AT2R activation in the Ang II infusion model of experimental hypertension in the rat. We hypothesized that AT2R activation would increase Na+ excretion and prevent or reverse hypertension chronically in this model via an action in the RPT. Here we show that both systemic and intrarenal AT2R activation with C-21 reduces BP and augments cumulative Na+ excretion chronically in a model of Ang II-dependent hypertension. Our results suggest that long term AT2R activation is a potential new approach in the treatment of Na+/fluid-retaining states and hypertension in humans.
METHODS
Please see the Online Data Supplement for detailed methods [telemetric BP probe and systemic and intrarenal mini-pump implantation for chronic systemic and intrarenal C-21 infusion studies, respectively; standard protocol for acute in vivo studies, renal cortical interstitial infusion and BP measurements for acute in vivo studies; total renal cortical homogenate preparation and Western blot analysis; RPTC apical membrane isolation and Western blot analysis; in vivo kidney perfusion and fixation procedure; confocal immunofluorescence microscopy; and specific experimental protocols].
Animal preparation
All experimental protocols were approved by the Animal Care and Use Committee at the University of Virginia and performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The experiments were conducted on 12-week-old female Sprague-Dawley rats (Harlan; N=182). Female animals were used to maximize the renal expression of AT2Rs (16). All animals were housed in a vivarium under controlled conditions (temperature, 21±1°C; humidity, 60±10%; light, 8:00–20:00) and fed a normal Na+ diet (0.3% Na+; Harlan).
Urine Na+ and plasma potassium measurements
Urinary Na+ and plasma potassium concentrations were measured using a flame photometer (Instrumentation Laboratory-943) and reported as μmol/min and mmol/L respectively. Cumulative Na+ balance was measured as the amount of Na+ consumed minus UNaV and reported as mEq.
Pharmacological agents
C-21 (60 ng/kg/min for chronic intrarenal studies, 300 ng/kg/min for chronic systemic studies, and 100, 200, and 300 ng/kg/min for acute systemic studies; Vicore Pharma), a highly selective, non-peptide AT2R agonist (Ki=0.4 mol/L) was employed to activate intrarenal and systemic AT2Rs. C-21 demonstrates 25,000-fold selectivity at AT2Rs compared with AT1Rs (14). C-21 was administered systemically at doses that are selective for AT2Rs in the rat (14). The intrarenal dose of C-21 was chosen as 20% of the systemic dose based on renal blood flow. Ang II (200 ng/kg/min; Bachem) was used to induce Ang II–dependent hypertension. PD-123319 (PD; 10 μg/kg/min; Parke-Davis) a specific AT2R antagonist (IC50=2×10−8 mol/L and >1×10−4 mol/L for AT2R and AT1Rs, respectively), was used to block intrarenal AT2Rs. Amiloride (0.8 μg/kg/min; Tocris) was used to inhibit intrarenal ENaC activity. Chlorothiazide (0.1 μg/kg/min; Sigma) was used to inhibit intrarenal NCC activity. Amiloride and chlorothiazide doses were administered on the basis of dose-ranging studies with target incremental natriuretic responses of 1–2 μmol/min.
Statistical analysis
Data are presented as mean ± 1 SE. Statistical significance was determined using 1-way ANOVA followed by multiple comparisons testing with the Student–Newman–Keuls test with 95% confidence. The level of significance was set at P<0.05.
RESULTS
Effects of chronic intrarenal and systemic C-21 infusion on systolic BP (SBP), diastolic BP (DBP), activity levels, cardiac output, and total peripheral vascular resistance in Ang II-dependent hypertension
We first determined whether direct intrarenal administration of C-21 would lower BP in the Ang II infusion model. As demonstrated in Figure 1 Panel A, systemic Ang II infusion (200 ng/kg/min) increased SBP from 126 ± 3 to 156 ± 4 mmHg on Day 1 (F=35.6; P<0.0001) and to 187 ± 2 mmHg at the end of the 7 day infusion period (F= 23.2; P<0.0001). As shown in Online Figure I, Panel A, Ang II induced a parallel increase in DBP from 91 ± 1 to 122 ± 3 mmHg (F=85.6, P<0.0001) on Day 1 and to 134 ± 7 mmHg (F=7.7, P<0.0001) on Day 7. Concurrent intrarenal administration of AT2R antagonist PD for 7 days did not significantly change the pressor response to Ang II (Figures 1, Panel A and Online Figure I, Panel A), although PD did cause a nonsignificant increase in SBP. Chronic intrarenal administration of the non-peptide AT2R agonist C-21abolished the systolic pressor response to systemic Ang II on Day 1 (F=7.03; P<0.01) and throughout the 7 day infusion period (F=4.54; P<0.001) with a parallel reduction in DBP (Online Figure I, Panel A).
Figure 1.
Panel A. Mean systolic blood pressure (SBP) in response to chronic systemic infusion of 5% dextrose in water (D5W; control;
 ), systemic infusion of ANG II (sANG II; 200 ng/kg/min;
), systemic infusion of ANG II (sANG II; 200 ng/kg/min;
 ), sANG II + systemic infusion of C-21 (sC-21; 300 ng/kg/min ; ) sANG II + intrarenal (IR) infusion of C-21 (60 ng/kg/min;
), sANG II + systemic infusion of C-21 (sC-21; 300 ng/kg/min ; ) sANG II + intrarenal (IR) infusion of C-21 (60 ng/kg/min;
 ), and sANG II + IR infusion of PD (10 μg/kg/min;
), and sANG II + IR infusion of PD (10 μg/kg/min;
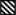 ). Results are reported as mm Hg. Panel B. Consecutive 24h urine Na+ excretion (UNaV) in response to conditions in Panel A. Results are reported as μmol/min. Data represent mean ± 1 SE. Panel A. Day 1: sANG II vs. sANG II + sC-21, F=18.54, P<0.001. sANG II vs. sANG II + IR C-21, F=7.03, P<0.01. All Days: sANG II vs. sANG II + sC-21, F=7.33, P<0.001. sANG II vs. sANG II + IR C-21, F=4.54, P<0.001. Panel B. Day 1: sANG II vs. sANG II + sC-21, F=7.18, P<0.05. sANG II vs. sANG II + IR C-21, F=5.05, P<0.05. All Days: sANG II vs. sANG II + sC-21, F=5.10, P<0.001. sANG II vs. sANG II + IR C-21, F=4.71, P<0.01. sANG II vs. sANG II + IR PD, F=6.76, P<0.001.
). Results are reported as mm Hg. Panel B. Consecutive 24h urine Na+ excretion (UNaV) in response to conditions in Panel A. Results are reported as μmol/min. Data represent mean ± 1 SE. Panel A. Day 1: sANG II vs. sANG II + sC-21, F=18.54, P<0.001. sANG II vs. sANG II + IR C-21, F=7.03, P<0.01. All Days: sANG II vs. sANG II + sC-21, F=7.33, P<0.001. sANG II vs. sANG II + IR C-21, F=4.54, P<0.001. Panel B. Day 1: sANG II vs. sANG II + sC-21, F=7.18, P<0.05. sANG II vs. sANG II + IR C-21, F=5.05, P<0.05. All Days: sANG II vs. sANG II + sC-21, F=5.10, P<0.001. sANG II vs. sANG II + IR C-21, F=4.71, P<0.01. sANG II vs. sANG II + IR PD, F=6.76, P<0.001.
Next, we determined whether systemic C-21 infusion would induce a similar depressor action as intrarenal infusion. Systemic C-21 again abolished the increase in SBP in response to Ang II on infusion Day 1 (F=18.54; P<0.001) and throughout the 7 day infusion period (F=7.33, P<0.001) with a parallel reduction of DBP (Online Figure I, Panel A). There was no significant difference in hypotensive responses to intrarenal vs. systemic C-21 administration. Activity levels (Online Figure I, Panel B) were not significantly changed by Ang II, C-21, or PD. Similarly, cardiac output and total peripheral vascular resistance measurements (Online Figure II) were not significantly influenced by any of the experimental agents.
Effects of chronic intrarenal and systemic C-21 infusion on 24h UNaV and cumulative Na+ balance in Ang II-dependent hypertension
As shown in Figure 1, Panel B, systemic Ang II administration reduced 24h UNaV from 0.92 ± 0.04 to 0.33 ± 0.06 μmol/min (F= 61.89; P<0.0001) on Day 1. Subsequently, there was no significant difference in UNaV responses to Ang II or vehicle control. While the antinatriuretic effects of systemic Ang II in the presence or absence of PD were similar during the first 24h, intrarenal PD administration enhanced the antinatriuretic response to systemic Ang II (F=6.76; P<0.001) during the entire 6 day collection period. Intrarenal administration of C-21 abolished the antinatriuretic response to systemic Ang II on Day 1 (F=5.05; P<0.05) and reduced Ang II-mediated Na+ retention during the entire 6 day period (F=4.71; P<0.01).
Cumulative Na+ balance is depicted in Online Figure III. In response to systemic Ang II infusion, cumulative Na+ balance increased from 0 to 0.32 ± 0.36 mEq on Day 1 after which Na+ balance returned to control levels for the remaining 5 days of study. Concurrent administration of intrarenal PD induced a similar degree of positive Na+ balance on Day 1, but resulted in a more positive Na+ balance for the entire study period (F=2.5; P<0.05) than Ang II alone. Intrarenal C-21 abolished the Ang II-induced positive Na+ balance on Day 1 (F=10.78; P<0.01) and resulted in a sustained negative Na+ balance throughout the study (F=4.35; P<0.01). Similar to intrarenal C-21, systemic C-21 prevented the positive Na+ balance induced by Ang II alone on Day 1 (F=16.14; P<0.001), and induced a negative Na+ balance state for the entire study compared with that induced by Ang II alone (F=3.13; P<0.05).
Effects of chronic systemic C-21 infusion on plasma renin activity, aldosterone, potassium, and creatinine in Ang II-dependent hypertension (Online Figure IV)
Plasma renin activity (Panel A), potassium (Panel B), and creatinine (Panel D) were not significantly changed by Ang II or systemic C-21 administration although there was a nonsignificant decrease in creatinine with Ang II that was not present when C-21 was co-administered with Ang II. Plasma aldosterone (Panel C), as expected, was markedly increased by Ang II alone either in the absence or presence of systemic C-21.
Effects of acute systemic C-21 infusion ± intrarenal infusion of ENaC inhibitor amiloride or NCC inhibitor chlorothiazide on UNaV and MAP in volume expanded rats
We then determined whether the natriuretic actions of C-21, which occur largely in the RPT would be additive to those of diuretics acting in the distal tubule and cortical collecting duct. In acute experiments in anesthetized rats, systemic C-21 administration induced a dose-dependent natriuretic response (F= 19.3; P<0.001; Figure 2, Panel A). ENaC inhibitor amiloride administered intrarenally induced a similar dose-dependent natriuretic response (F=7.6; P<0.001). Combination of systemic C-21 and intrarenal amiloride induced a greater natriuretic response compared with either systemic C-21 (F=5.6; P<0.01) or intrarenal amiloride (F=7.1; P<0.001) alone. The UNaV response to the combination of systemic C-21 and intrarenal amiloride was additive. Similarly, intrarenal chlorothiazide induced a dose-dependent natriuretic response (F=6.1; P<0.001). The combination of systemic C-21 and intrarenal chlorothiazide also induced a greater increase in UNaV than either C-21 (F=7.9; P<0.001) or chlorothiazide (F=4.0; P≤0.01) alone. Again, the natriuretic response to C-21 and chlorothiazide was additive. As shown in Figure 2, Panel B, there was no significant change in mean arterial pressure from control with any of these agents in these acute experiments.
Figure 2.
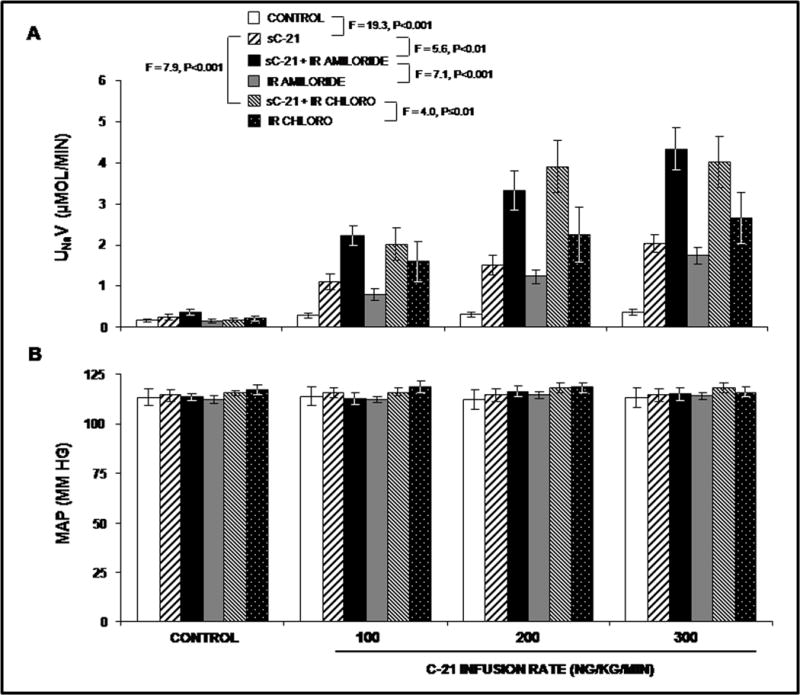
Panel A. Urine Na+ excretion (UNaV) in volume-expanded rats in response to the following acute conditions:(
 ) control, (
) control, (
 ) systemic infusion of C-21 (sC-21; 100, 200, and 300 ng/kg/min), (
) systemic infusion of C-21 (sC-21; 100, 200, and 300 ng/kg/min), (
 ) sC-21 + intrarenal (IR) infusion of amiloride (0.8 μg/kg/min), (
) sC-21 + intrarenal (IR) infusion of amiloride (0.8 μg/kg/min), (
 ) IR infusion of amiloride, (
) IR infusion of amiloride, (
 ) sC-21 + IR infusion of chlorothiazide (CHLORO; 0.1 μg/kg/min), and (
) sC-21 + IR infusion of chlorothiazide (CHLORO; 0.1 μg/kg/min), and (
 ) IR infusion of CHLORO. Panel B. Mean arterial pressure (MAP) in response to the conditions in Panel A. Results are reported as mm Hg. Data represent mean ± 1 SE. Panel A. control vs. sC-21, F=19.3, P<0.001. sC-21 vs. sC-21 + IR amiloride, F=5.6, P<0.01. sC-21 + IR amiloride vs. IR amiloride, F=7.1, P<0.001. sC-21 vs. sC-21 + IR CHLORO, F=7.9, P<0.001. sC-21 + IR CHLORO vs. IR CHLORO, F=4.0, P≤0.01.
) IR infusion of CHLORO. Panel B. Mean arterial pressure (MAP) in response to the conditions in Panel A. Results are reported as mm Hg. Data represent mean ± 1 SE. Panel A. control vs. sC-21, F=19.3, P<0.001. sC-21 vs. sC-21 + IR amiloride, F=5.6, P<0.01. sC-21 + IR amiloride vs. IR amiloride, F=7.1, P<0.001. sC-21 vs. sC-21 + IR CHLORO, F=7.9, P<0.001. sC-21 + IR CHLORO vs. IR CHLORO, F=4.0, P≤0.01.
Effects of systemic or intrarenal C-21 infusion on 24h total cortical homogenate and RPTC Apical plasma membrane AT2R density in Ang II-induced hypertension
To determine whether AT2R activation with C-21 in the presence of Ang II still induces receptor translocation to the apical plasma membranes of RPTCs, we employed confocal immunofluorescence microscopy and Western blot analysis. Figure 3, Panel A, depicts the subcellular distribution of AT2Rs as determined by confocal immunofluorescence microscopy in a representative set of rat RPTCs in response to systemic vehicle (control), systemic Ang II infusion, systemic Ang II + systemic C-21, and systemic Ang II + intrarenal C-21. The RPTC apical plasma membrane marker phalloidin (red), subapical membrane marker adaptor protein-2 (AP2; blue) and the AT2R (green) are depicted sequentially left-to-right for all four conditions. As shown in the merged, enlarged merged, and enlarged AT2R photomicrographs (Figure 3, Panel A with quantifications shown in Panel B), administration of Ang II alone reduced AT2R intensity in the apical plasma membrane from control levels (P<0.001). Systemic C-21 markedly increased AT2R fluorescence intensity in the apical plasma membrane as indicated by the increased yellow fluorescence in the merged and enlarged merged panels and as green fluorescence in the enlarged AT2R panel (P<0.001). Similar to systemic C-21 infusion, intrarenal administration of C-21 increased the RPTC apical plasma membrane AT2R fluorescence intensity (P<0.01).
Figure 3.
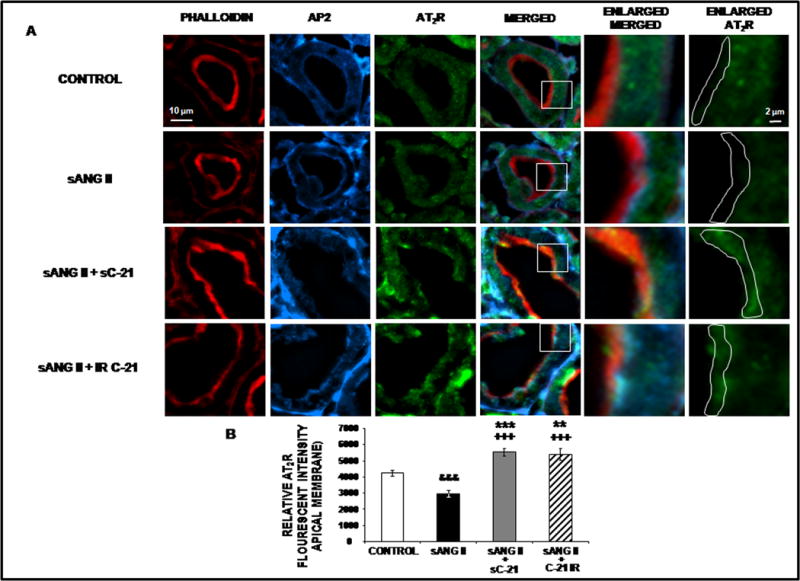
Confocal micrographs (600X) of renal proximal tubule cell (RPTC) thin sections (5–8 μm) AT2R protein in response to 1 day of chronic systemic infusion of 5% dextrose in water (D5W; control), systemic infusion of ANG II (sANG II; 200 ng/kg/min), sANG II + systemic infusion of C-21 (sC-21; 300 ng/kg/min), and sANG II + intrarenal (IR) infusion of C-21 (60 ng/kg/min). Panel A. The first row of images represents control treatment, the second row represents sANG II treatment, the third row represents sANG II + sC-21 treatment, and the fourth row represents sANG II + IR C-21 treatment from a representative set of RPTCs. The first column depicts brush border membrane staining with phalloidin. The second column depicts subapical membrane staining with adaptor protein-2 (AP2). The third column depicts AT2R staining. The fourth column depicts a merged image. The fifth column depicts an enlarged image of the square section in the merged image. The sixth column depicts the enlarged image with AT2R staining only. The white line encompasses the brush border apical membrane that was quantified for AT2R intensity. The scale bars in the first and sixth columns represent 10 and 2 μm, respectively. Panel B. The quantification of RPTC apical membrane AT2R fluorescence intensity performed on 20 independent measurements of RPTCs from a representative rat following control (
 ), sANG II (
), sANG II (
 ), sANG II + sC-21 (
), sANG II + sC-21 (
 ), and sANG II + IR C-21 (
), and sANG II + IR C-21 (
 ) treatments. Data represent mean ± 1 SE. **P<0.01 and ***P<0.001 from control. +++P<0.001 from sANG II. &&&P<0.001from control.
) treatments. Data represent mean ± 1 SE. **P<0.01 and ***P<0.001 from control. +++P<0.001 from sANG II. &&&P<0.001from control.
As shown in Figure 4, Panel A, there was no significant difference among control, systemic Ang II, or systemic Ang II + systemic C-21 in total cortical homogenate AT2R protein by Western blot analysis, although there was a non-significant trend to reduced levels in Ang II-infused animals. To complement the immunofluorescence studies on AT2R recruitment, we isolated RPTC apical plasma membranes using the lectin pull-down method. Western blot analysis clearly demonstrated AT2R recruitment to the RPTC apical plasma membranes in response to systemic C-21 (P<0.01) (Figure 4, Panel B). There was a non-significant decrease in RPTC apical plasma membrane AT2Rs in response to Ang II alone. Similarly, intrarenal C-21 increased apical membrane expression of AT2R protein without changing total AT2R levels as determined in cortical homogenates (Figure 4, Panels C & D; P<0.01). Ang II reduced total cortical AT2R protein levels (P<0.05) in these experiments.
Figure 4.
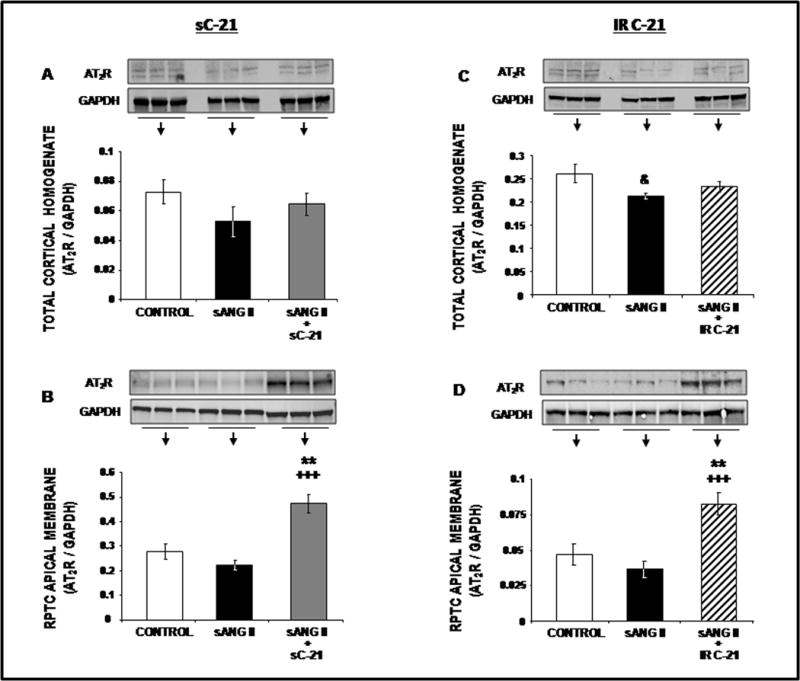
Western blot analysis of total cortical homogenate (Panels A and C) and RPTC apical membrane (Panels B and D) AT2R protein expression following 1 day of control (
 ), systemic ANG II (sANG II;
), systemic ANG II (sANG II;
 ), sANG II + systemic C-21 (sC-21;
), sANG II + systemic C-21 (sC-21;
 ), and sANG II + intrarenal (IR) C-21 (
), and sANG II + intrarenal (IR) C-21 (
 ) treatments. All blots are normalized to GAPDH. Data represent mean ± 1 SE. **P<0.01from control. +++P<0.001 from sANG II. &P<0.05 from control.
) treatments. All blots are normalized to GAPDH. Data represent mean ± 1 SE. **P<0.01from control. +++P<0.001 from sANG II. &P<0.05 from control.
Effects of systemic or intrarenal C-21 infusion on 24h total cortical homogenate and RPTC Apical plasma membrane NHE-3 and phospho-NHE-3 in Ang II-induced hypertension
The next experiments were aimed at determining whether chronic C-21-induced natriuresis is related to internalization and inactivation of the major RPTC apical membrane Na+ transporter NHE-3. Figure 5 demonstrates the subcellular distribution for NHE-3 in response to systemic or intrarenal C-21 in Ang II-infused rats. Representative confocal micrographs are shown in Panel A and quantitation of the relative fluorescence intensity in Panel B. Ang II alone did not alter total NHE-3 fluorescence intensity or distribution in RPTCs. However, systemic C-21 + Ang II infusion increased the retraction of NHE-3 to the RPTC subapical membranes (P<0.01; yellow color in the subapical layer marked by AP2). Similarly to systemic C-21 infusion, intrarenal C-21 increased NHE-3 translocation into the subapical membranes of RPTCs (P<0.01).
Figure 5.
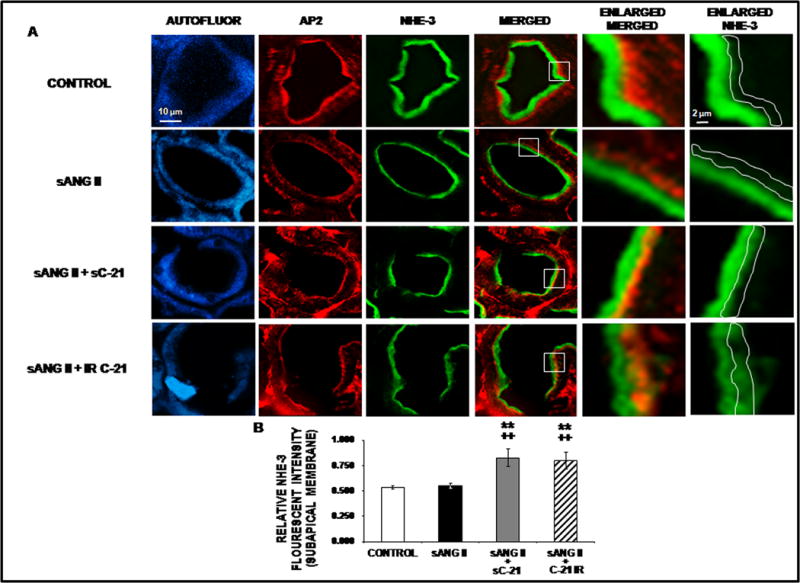
Confocal micrographs (600X) of renal proximal tubule cell (RPTC) thin sections (5–8 μm) NHE-3 protein in response to 1 day of chronic systemic infusion of 5% dextrose in water (D5W; control), systemic infusion of ANG II (sANG II; 200 ng/kg/min), sANG II + systemic infusion of C-21 (sC-21; 300 ng/kg/min), and sANG II + intrarenal (IR) infusion of C-21 (60 ng/kg/min). Panel A. The first row of images represents control treatment, the second row represents sANG II treatment, the third row represents sANG II + sC-21 treatment, and the fourth row represents sANG II + C-21 IR treatment from a representative set of RPTCs. The first column depicts confocal autofluorescence. The second column depicts subapical membrane staining with adaptor protein-2 (AP2). The third column depicts NHE-3 staining. The fourth column depicts a merged image. The fifth column depicts an enlarged image of the square section in the merged image. The sixth column depicts the enlarged image with NHE-3 staining only. The white line encompasses the subapical membrane that was quantified for NHE-3 intensity. The scale bars in the first and sixth columns represent 10 and 2 μm, respectively. Panel B. The quantification of RPTC subapical membrane NHE-3 fluorescence intensity performed on 20 independent measurements of RPTCs from a representative rat following control (
 ), sANG II (
), sANG II (
 ), sANG II + sC-21 (
), sANG II + sC-21 (
 ), and sANG II + C-21 IR (
), and sANG II + C-21 IR (
 ) treatments. Data represent mean ± 1 SE. **P<0.01from control. ++P<0.001 from sANG II.
) treatments. Data represent mean ± 1 SE. **P<0.01from control. ++P<0.001 from sANG II.
Western blot analysis of NHE-3 is shown in Figure 6. While Ang II or Ang II + systemic or intrarenal C-21 did not alter total cortical homogenate NHE-3 expression (Panels A & C), systemic C-21 reduced RPTC apical plasma membrane localization of NHE-3 in isolated membranes (P<0.001; Panel B) consistent with C-21-induced NHE-3 retraction shown by immunofluorescence.
Figure 6.
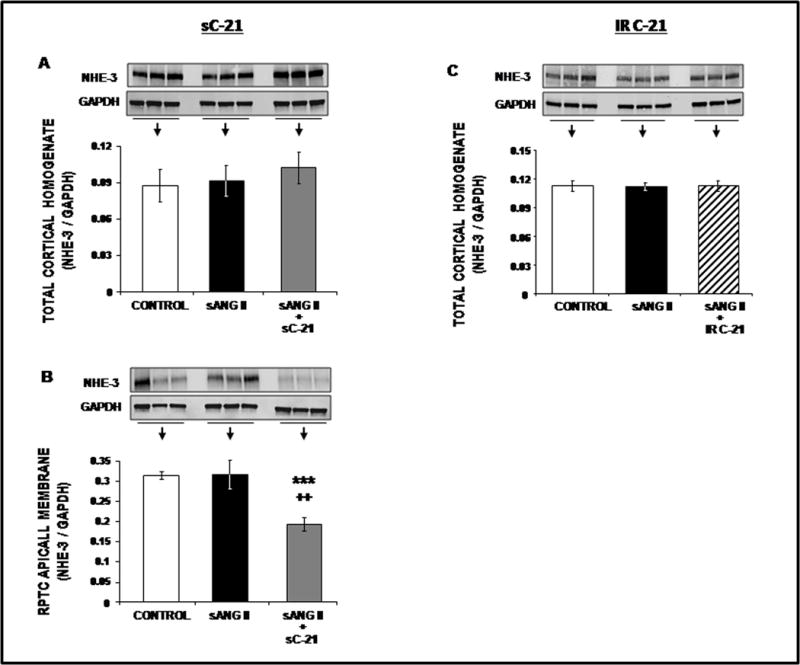
Western blot analysis of total cortical homogenate (Panels A and C) and RPTC apical membrane (Panels B) NHE-3 protein expression following 1 day of control (
 ), systemic ANG II (sANG II;
), systemic ANG II (sANG II;
 ), sANG II + systemic C-21 (sC-21;
), sANG II + systemic C-21 (sC-21;
 ), and sANG II + intrarenal (IR) C-21 (
), and sANG II + intrarenal (IR) C-21 (
 ) treatments. All blots are normalized to GAPDH. Data represent mean ± 1 SE. ***P<0.01from control. ++P<0.001 from sANG II.
) treatments. All blots are normalized to GAPDH. Data represent mean ± 1 SE. ***P<0.01from control. ++P<0.001 from sANG II.
Phosphorylation of NHE-3 at serines 552 and 605 after activation of cyclic adenosine monophosphate (cAMP)-dependent protein kinase A is required for maximum inhibition of NHE-3 since the mutation of these amino acids individually reduced the inhibitory effect on NHE-3 promoted by cAMP (18,19). Thus, phosphorylated NHE-3 is an established indicator of NHE-3 retraction/internalization and activation. Subcellular distribution for phosphorylated NHE-3 [Ser 552] (pSer552-NHE-3) is shown in Figure 7. Panel A shows representative confocal micrographs and Panel B quantitative fluorescence intensity for pSer552-NHE-3. Ang II alone reduced pSer552-NHE-3 in subapical membranes of RPTCs (P<0.05). Combination of C-21 + Ang II markedly increased pSer552-NHE-3 in the RPTC subapical membranes (P<0.001) suggesting NHE-3 internalization. As shown in Figure 8, Ang II did not alter pSer552-NHE-3, but C-21 + Ang II increased it (P<0.01). Similar results are shown for phosphorylated NHE-3 [Ser 605] (pSer605-NHE-3) in Online Figure V & VI. Similar to systemic C-21 infusion, intrarenal C-21 administration increased both pSer552-NHE-3 and pSer605-NHE-3 (Online Figures V & VI).
Figure 7.
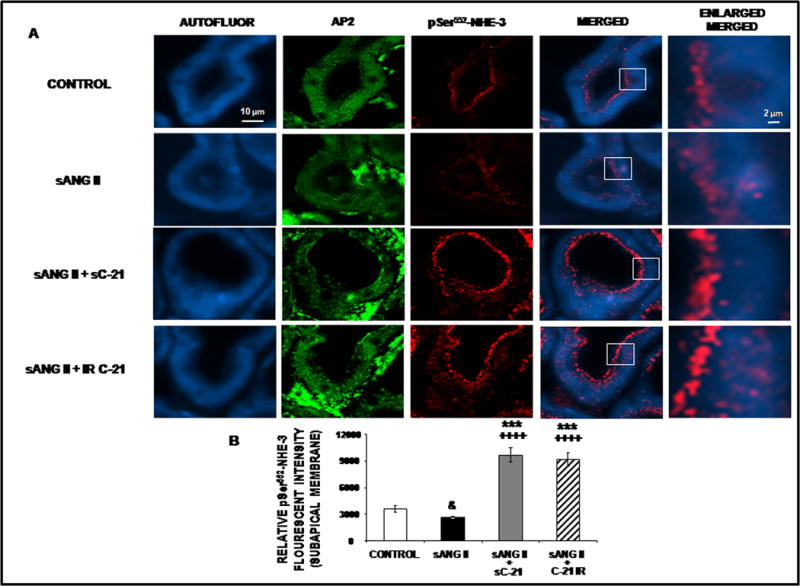
Confocal micrographs (600X) of renal proximal tubule cell (RPTC) thin sections (5–8 μm) pSer552-NHE-3 protein in response to 1 day of chronic systemic infusion of 5% dextrose in water (D5W; control), systemic infusion of ANG II (sANG II; 200 ng/kg/min), sANG II + systemic infusion of C-21 (sC-21; 300 ng/kg/min), and sANG II + intrarenal (IR) infusion of C-21 (60 ng/kg/min). Panel A. The first row of images represents control treatment, the second row represents sANG II treatment, the third row represents sANG II + sC-21 treatment, and the fourth row represents sANG II + IR C-21 treatment from a representative set of RPTCs. The first column depicts confocal autofluorescence. The second column depicts subapical membrane staining with adaptor protein-2 (AP2). The third column depicts pSer552-NHE-3 staining. The fourth column depicts a merged image of confocal autofluorescence and pSer552-NHE-3. The fifth column depicts an enlarged image of the square section in the merged image. The scale bars in the first and fifth columns represent 10 and 2 μm, respectively. Panel B. The quantification of RPTC subapical pSer552-NHE-3 fluorescence intensity performed on 20 independent measurements of RPTCs from a representative rat following control (
 ), sANG II (
), sANG II (
 ), sANG II + sC-21(
), sANG II + sC-21(
 ), and sANG II + IR C-21 (
), and sANG II + IR C-21 (
 ) treatments. Data represent mean ± 1 SE. ***P<0.001 from control. ++++P<0.0001 from sANG II. &P<0.05 from control.
) treatments. Data represent mean ± 1 SE. ***P<0.001 from control. ++++P<0.0001 from sANG II. &P<0.05 from control.
Figure 8.
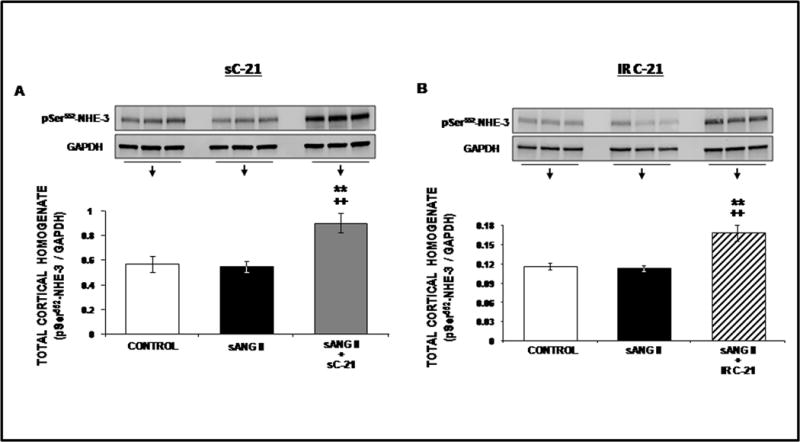
Western blot analysis of total cortical membrane pSer552-NHE-3 protein expression following 1 day of control (
 ), systemic ANG II (sANG II;
), systemic ANG II (sANG II;
 ), sANG II + systemic C-21 (sC-21;
), sANG II + systemic C-21 (sC-21;
 ), and sANG II + intrarenal (IR) C-21 (
), and sANG II + intrarenal (IR) C-21 (
 ) treatments. All blots are normalized to GAPDH. Data represent mean ± 1 SE. **P<0.01from control. ++P<0.001 from sANG II.
) treatments. All blots are normalized to GAPDH. Data represent mean ± 1 SE. **P<0.01from control. ++P<0.001 from sANG II.
Effects of systemic or intrarenal C-21 infusion on 24h total cortical homogenate and RPTC basolateral membrane NKA during Ang II-induced hypertension
We next determined whether C-21 also induces internalization and inactivation of αNKA. Systemic Ang II infusion reduced basolateral intracellular fluorescence intensity of αNKA and concurrent systemic C-21 infusion reversed this effect (P<0.0001; Online Figure VII, Panels A & B). However, there was no significant effect on total αNKA protein with systemic Ang II alone or combined with systemic C-21 (Online Figure VIII, Panel A). Similar results were demonstrated when C-21 was administered directly into the kidney instead of systemically (Online Figure VII, Panels A & B and Online Figure VIII, Panel B). Phosphorylated NKA [Ser 23] (pSer23-NKA), an established marker for NKA internalization, was not significantly increased (pSer23-NKA; Online Figure IX) in response to Ang II administration but was markedly reduced in RPTCs in the presence of systemic C-21 (P<0.0001). A similar reduction in pSer23-NKA in RPTCs was observed when systemic Ang II was combined with direct intrarenal administration of C-21 (P<0.0001).
Effects of systemic C-21 infusion on total cortical homogenate ERK 1/2, Phospho ERK 1/2, Src, and Phospho Src in Ang II-induced hypertension
To determine whether AT2R activation stimulates the Src/ERK signaling pathway, we performed Western blot analysis of total renal cortical homogenate. Western blot analyses of phospho-Src [Tyr 416] (pTyr416-Src), Src, phospho-ERK 1/2 [Thr 202/Tyr 204} (pThr/Tyr202/204-ERK 1/2), and total ERK 1/2 are shown in Online Figure X. C-21 infusion did not significantly alter Src or ERK levels during these experiments.
Effects of chronic systemic AT2R treatment on SBP following establishment of Ang II-induced hypertension
As shown in Online Figure XI, systemic administration of C-21, initiated on Day 3 following the establishment of Ang II-induced hypertension, reduced both SBP (F=3.5; P<0.01) and DBP (F=3.21; P<0.01) to baseline control levels by Day 7.
DISCUSSION
Our previous study in normal Sprague Dawley rats and genetically engineered mice documented that acute systemic AT2R activation with C-21 induces natriuresis by translocating AT2Rs to the apical plasma membranes of RPTCs and internalizing/inhibiting major RPTC transporters NHE-3 and NKA (15). The present study builds on these findings by demonstrating in an Ang II infusion model of experimental hypertension that chronic AT2R activation with C-21 prevents initial renal Na+ retention and lowers BP over a 7 day period. In addition, C-21 was an effective natriuretic and antihypertensive agent whether administered systemically or directly into the kidney in this experimental model. We also demonstrated that continuous C-21 administration, both systemically and intrarenally, induced sustained negative cumulative Na+ balance accompanied by AT2R recruitment from intracellular sites to the apical plasma membranes of RPTCs and internalization/inactivation of major RPT Na+ transporters NHE-3 and NKA. Importantly, C-21-induced natriuresis was related to inhibition of Na+ transport in the RPT as it was additive to that observed with diuretics acting at either the distal tubule or the cortical collecting duct. We further demonstrated that C-21 was equally effective in lowering BP whether administered before or after Ang II-dependent hypertension had been established. Taken together, these results strongly support a role for AT2R agonists as natriuretic/diuretic agents that improve the pressure natriuresis relationship and are potential candidates for the treatment of hypertension and disorders of Na+ and fluid retention in humans.
In the Ang II infusion model of experimental hypertension, major Na+ retention occurred within the first 24h of initiating the infusion, as previously reported (15), and Na+ excretion returned to approximately normal levels after 24h and beyond. Although the major effect of C-21 was to prevent Na+ retention at 24h, a continuing effect of C-21 to promote negative cumulative Na+ balance was observed throughout the study. Our data are highly consistent with a primary RPT action of C-21. Since we did not perform formal renal function studies we cannot rule out a small renal hemodynamic effect as contributing to the natriuresis and reduced BP level. However, the action of C-21 was purely tubular in our previous acute study (15). Similarly, we cannot exclude a systemic vascular effect of C-21 as contributing, at least in part, to the chronic reduction in BP in these animals. However, the fact that C-21 prevented Ang II-induced hypertension in equivalent fashion whether the C-21 was administered systemically or intrarenally argues strongly for the primacy of the kidneys in causing the reduction in BP.
Importantly, concurrent use of AT1R antagonist was not required to uncover the chronic natriuretic and hypotensive actions of AT2R agonist administration in this study. Past studies indicated that cardiovascular and renal responses to AT2R activation are only observed when the RAS is activated, as is the case in this study, or when AT1Rs are concurrently blocked (1,3,4). Indeed, the hypotensive action of chronic C-21 was somewhat unexpected because most studies have failed to demonstrate BP-lowering effects in the absence of AT1R blockade, albeit in spontaneously hypertensive rats (SHR) or stroke-prone SHR (17,18). However, further studies on the effects of the combination of AT1R antagonist and AT2R agonist administration are clearly indicated to clarify the therapeutic utility of AT2R agonist/AT1R antagonist combination in this model.
In the present study, AT2R activation in the presence of Ang II induced translocation of AT2Rs from intracellular sites to the apical plasma membranes of RPTCs 24h after initiation of C-21 infusion without any alteration in total cellular AT2R expression, as validated by both confocal immunofluorescence studies and Western blot analysis of isolated RPTC apical membranes and total renal cortical cells. This is consistent with our previous demonstration that systemic C-21 administration acutely (at 30–90 min) recruits AT2Rs to the apical plasma membranes of RPTCs (15). Our previous studies have shown that natriuretic responses to endogenous renal AT2R agonist Ang III are also accompanied by translocation of AT2Rs along microtubules to the apical plasma membranes of RPTCs (19). Furthermore, dopamine D1 receptor activation with selective receptor agonist fenoldopam induces natriuresis in an AT2R-dependent manner. Fenoldopam translocates AT2Rs to the apical plasma membranes of RPTCs via a cAMP- and protein phosphatase 2A-dependent signaling pathway (19). SHR fail to develop either AT2R recruitment or natriuretic responses to Ang III and both can be restored by increasing Ang III levels via blockade of aminopeptidase N. Together with the present observations, the evidence indicates that AT2R recruitment to the apical plasma membranes of RPTCs is likely a common mechanism initiating and sustaining long term natriuretic responses to endogenous AT2R agonists dopamine and Ang III, as well as exogenous agonist C-21 (19–21). Because AT2Rs do not internalize in renal epithelial cells (22,23), their recruitment and continuous apical plasma membrane expression may stabilize and reinforce the natriuretic response.
Of the Na+ filtered into the nephron, approximately 67% is reabsorbed isotonically in the RPT (24,25). The principal apical membrane Na+ transporter in the RPT is NHE-3, which also participates in supporting flow-dependent glomerulo-tubular balance (25,26). NHE-3-null mice are hypovolemic and hypotensive, exhibit metabolic acidosis, reduced reabsorption of Na+, HCO3−, and fluid, and increased mortality when subjected to low-Na+ intake (27,28). In light of the major physiological role of NHE-3 in the RPT, relatively small changes in its activity could have significant pathophysiological consequences. Indeed, NHE-3 is among the most highly regulated transport proteins of cell membranes, modulated by multiple physiological and pathological conditions (27). NHE-3 is expressed along the microvilli of the RPTC brush border but can also be detected in subapical, intracellular, and vesicular compartments, consistent with the regulation of its activity by membrane trafficking (29,30).
Acute regulation of NHE-3 occurs via changes in phosphorylation, membrane trafficking and/or membrane localization acting on the existing cellular NHE-3 pool (31). The cytoplasmic loop of NHE-3 contains several phosphorylation sites that are targeted by different kinases. The cAMP-dependent protein kinase A phosphorylates NHE-3 in its carboxy-terminus. Phosphorylation of serines 552 and 605 is required for maximum inhibition of NHE-3 by cAMP because mutation of these serines individually reduces the inhibitory effect on NHE-3 promoted by cAMP (31,32).
Retraction/internalization of NHE-3 is a marker of reduced NHE-3 activity (33,34). In our previous studies, we showed that acute systemic C-21 administration (30–90 min) retracts NHE-3 from the tips to the bases of apical microvilli and into the subapical membranes of RPTCs as a mechanism to reduce RPTC Na+ transport (15). In the present study, we demonstrated chronic (24h) C-21 administration induced retraction/internalization of NHE-3 by confocal fluorescence microscopy and Western blot analysis. We further demonstrated increased total cortical homogenate pSer552-NHE-3 and pSer605-NHE-3 levels, as established indicators of NHE-3 retraction/internalization (34,35).
In addition to NHE-3, we demonstrated a parallel internalization and inactivation of NKA, the major Na+ transporter across the basolateral membranes of RPTCs. We showed that Ang II markedly reduced intracellular NKA fluorescence intensity while C-21 reversed this response without change in the total cellular pool of NKA. In the rat RPT, the α-1 subunit of NKA is phosphorylated at serine 23 by the action of protein kinase C (36). NKA phosphorylation at this site plays a major role both in the regulation of NKA enzyme activity and subcellular distribution, as demonstrated by mutation of this phosphorylation site (37–39). In the present study, we demonstrated that C-21 induced NKA dephosphorylation at serine 23, indicating retraction and inactivation of NKA.
However, we found no evidence of chronic activation of Src or ERK signaling molecules downstream of AT2Rs. This contrasts with C-21-induced Src and ERK activation following acute systemic administration of C-21 in our previous study that is likely explained by the early response of these pathways to AT2R activation and cGMP, as demonstrated previously (15,40).
The results of this study encourage the evaluation of acute and chronic AT2R activation in the control of Na+ excretion and BP, with and without concurrent AT1R blockade, in the TGR(mRen2)27 rat, a model of human primary hypertension in which the endogenous tissue RAS is activated, as well as a volume-expanded hypertensive model (DOCA/salt hypertension) (41,42). Future investigations will explore the potential role for AT2R agonists as natriuretic/diuretic agents in hypertension and disorders of Na+ and fluid retention in humans.
Supplementary Material
Novelty and Significance.
What Is Known?
Angiotensin II (Ang II) activates two major receptors, type-1 (AT1R) and type-2 (AT2R).
AT2R activation generally opposes the actions of Ang II via AT1Rs.
Within the kidney proximal tubule, acute AT2R activation with non-peptide agonist Compound-21 (C-21) recruits AT2Rs to apical plasma membranes and increases sodium (Na+) excretion in a bradykinin - nitric oxide - cyclic guanosine 3′,5′-monophosphate (cyclic GMP) -dependent manner in normal Sprague-Dawley rats.
What New Information Does This Article Contribute?
Chronic AT2R activation (systemically or intrarenally) with C-21 increased Na+ excretion and normalized blood pressure (BP) in the Ang II-infusion model of experimental hypertension.
Chronic AT2R activation translocated AT2Rs to apical plasma membranes, internalized and inactivated sodium transporters sodium-hydrogen exchanger-3 (NHE-3) and Na+/K+ATPase (NKA) and functionally reduced sodium transport in renal proximal tubule cells.
C-21 normalized BP whether administered before or after Ang II-dependent hypertension had been established.
Classically, Ang II induces hypertension by promoting antinatriuresis and vasoconstriction. On the other hand, acute AT2R activation induces natriuresis by increasing renal bradykinin, nitric oxide and cyclic GMP, recruiting AT2Rs to apical plasma membranes and internalizing and inactivating major Na+ transporter molecules (NHE-3 and NKA) in the renal proximal tubules (RPT) of normal rats.
Here we report that chronic (intrarenal or systemic) AT2R activation with C-21 induces sustained reduction in cumulative Na+ balance and reverses Ang II-dependent hypertension by acting at the RPT. C-21, administered chronically, recruited AT2R to apical plasma membranes and internalized/inactivated NHE-3 and NKA in this nephron segment. C-21 normalized BP whether administered prior or subsequent to the establishment of Ang II-dependent hypertension, and the natriuresis engendered by C-21 was additive to that of diuretics acting at the distal tubule (chlorothiazide) and cortical collecting duct (amiloride), indicating a primary RPT effect.
These findings indicate that AT2R activation can lower BP chronically under conditions when the renin-angiotensin system is stimulated; thus, AT2Rs are predicted to be legitimate therapeutic targets for hypertension in humans. Currently no effective diuretic/natriuretic agent is available that acts in the RPT. AT2R activation, therefore, may provide a complimentary nephron-specific site for diuresis/natriuresis in humans.
Acknowledgments
We thank Vicore Pharmaceuticals for proving C-21 and Dr. Peter Aronson (Yale School of Medicine) for generously providing the NHE-3 antibody used in Western blot experiments in this manuscript. We thank Dr. Mark Conaway (University of Virginia School of Medicine) for performing statistical analysis.
SOURCES OF FUNDING
This work was supported by National Institutes of Health grant R01-HL-128189 to Robert M. Carey.
Nonstandard Abbreviations and Acronyms
- Ang II
angiotensin II
- Ang III
des-aspartyl1- angiotensin II
- AP2
adaptor protein 2
- ATP
adenosine triphosphate
- AT1R
angiotensin type-1 receptor
- AT2R
angiotensin type-2 receptor
- BP
blood pressure
- C-21
Compound 21
- cAMP
adenosine cyclic 3′ 5 ′-monophosphate
- cGMP
guanosine cyclic 3′ 5′-monophosphate
- DBP
diastolic blood pressure
- ENaC
epithelial sodium channel
- ERK
extracellular signal-related kinase
- MAP
mean arterial pressure
- NCC
sodium-chloride channel
- NHE-3
sodium-hydrogen exchanger-3
- NKA
sodium/potassium ATPase
- NO
nitric oxide
- PD
PD-123319
- Phospho-NHE-3
phosphorylated soiumd-hydrogen exchanger-3
- RAS
renin-angiotensin system
- RPT
renal proximal tubule
- RPTC
renal proximal tubule cell
- SBP
systolic blood pressure
- SHR
spontaneously hypertensive rats
- Src
Src family kinase
- UNaV
urinary sodium excretion
Footnotes
DISCLOSURES
None.
References
- 1.Carey RM, Siragy HM. Newly recognized components of the renin-angiotensin system: potential roles in cardiovascular and renal regulation. Endocr Rev. 2003;24:261–271. doi: 10.1210/er.2003-0001. [DOI] [PubMed] [Google Scholar]
- 2.Riet LT, Van Esch JHM, Roks AJM, Ven den Meiracker AH, Danser AHJ. Hypertension: Renin-angiotensin system alterations. Circ Res. 2015;116:960–975. doi: 10.1161/CIRCRESAHA.116.303587. [DOI] [PubMed] [Google Scholar]
- 3.Jones ES, Vinh A, McCarthy CA, Gaspari TA, Widdop RE. AT2 receptors: functional relevance in cardiovascular disease. Pharmacol Ther. 2008;120:292–316. doi: 10.1016/j.pharmthera.2008.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carey RM. Cardiovascular and renal regulation by the angiotensin type 2 receptor: the AT2 receptor comes of age. Hypertension. 2005;45:840–844. doi: 10.1161/01.HYP.0000159192.93968.8f. [DOI] [PubMed] [Google Scholar]
- 5.Padia SH, Carey RM. AT2 receptors: beneficial counter-regulatory role in cardiovascular and renal function. Pflugers Arch. 2013;465:99–110. doi: 10.1007/s00424-012-1146-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guyton AC, Coleman TG, Cowley AW., Jr Arterial pressure regulation. Overriding dominance of the kidneys in long-term regulation and in hypertension. Am J Med. 1972;52:584–594. doi: 10.1016/0002-9343(72)90050-2. [DOI] [PubMed] [Google Scholar]
- 7.Guyton AC. The surprising kidney-fluid mechanism for pressure control: its infinite gain. Hypertension. 1990;16:725–730. doi: 10.1161/01.hyp.16.6.725. [DOI] [PubMed] [Google Scholar]
- 8.Li H, Weatherford ET, Davis DR, Sigmund CJ. Renal proximal tubule angiotensin AT1A receptors regulate blood pressure. Am J Physiol Regul Integr Comp Physiol. 2011;301:R1067–R1077. doi: 10.1152/ajpregu.00124.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crowley SD, Gurley SB, Oliverio MI, Coffman T. Angiotensin II causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proc Natl Acad Sci USA. 2006;103:17985–17990. doi: 10.1073/pnas.0605545103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gurley SB, Riquier-Brison AD, Schnermann J, Coffman T. AT1A angiotensin receptors in the renal proximal tubule regulate blood pressure. Cell Metab. 2011;13:469–475. doi: 10.1016/j.cmet.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li XC, Zhuo JL. Proximal tubule-dominant transfer of AT1a receptors induces blood pressure responses to intracellular angiotensin II in AT1a receptor-deficient mice. Am J Physiol Regul Integr Comp Physiol. 2013;304:R588–R598. doi: 10.1152/ajpregu.00338.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ozono R, Wang ZQ, Moore AF, Inagami T, Siragy HM, Carey RM. Expression of the subtype 2 angiotensin (AT2) receptor protein in the rat kidney. Hypertension. 1997;30:1238–1246. doi: 10.1161/01.hyp.30.5.1238. [DOI] [PubMed] [Google Scholar]
- 13.Miyata N, Park F, Li XF, Cowley AW., Jr Distribution of angiotensin AT1 and AT2 receptor subtypes in the rat kidney. Am J Physiol Renal Physiol. 1999;277:F437–F446. doi: 10.1152/ajprenal.1999.277.3.F437. [DOI] [PubMed] [Google Scholar]
- 14.Wan Y, Wallinder C, Plouffe B, Beaudry H, Mahalingam AK, Wu X, Johansson B, Holm M, Botoros M, Karlen A, Pettersson A, Nyberg F, Fandriks L, Gallo-Payet N, Hallberg A, Alerman M. Design, synthesis and biological evaluation of the first selective nonpeptide AT2 receptor agonist. J Med Chem. 2004;47:5995–6008. doi: 10.1021/jm049715t. [DOI] [PubMed] [Google Scholar]
- 15.Bosnyak S, Jones ES, Christopoulos A, Aguilar M-I, Thomas WG, Widdop RE. Relative affinity of angiotensin peptides and novel ligands at AT1 and AT2 receptors. Clin Sci. 2011;121:297–303. doi: 10.1042/CS20110036. [DOI] [PubMed] [Google Scholar]
- 16.Kemp BA, Howell NL, Gildea JJ, Keller SR, Padia SH, Carey RM. AT2 receptor activation induces natriuresis and lowers blood pressure. Circ Research. 2014;115:388–399. doi: 10.1161/CIRCRESAHA.115.304110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hilliard LM, Mirabito KM, Denton KM. Unmasking the potential of the angiotensin AT2 receptor as a therapeutic target in hypertension in men and women: what we know and what we still need to find out. Clin Exp Pharmacol Physiol. 2013;40:542–550. doi: 10.1111/1440-1681.12067. [DOI] [PubMed] [Google Scholar]
- 18.Bosnyak S, Welungoda IK, Hallberg A, Alterman M, Widdop RE, Jones ES. Stimulation of angiotensin AT2 receptors by the non-peptide agonist, Compound 21, evokes vasodepressor effects in conscious spontaneously hypertensive rats. Brit J Pharmacol. 2010;159:709–716. doi: 10.1111/j.1476-5381.2009.00575.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rehman A, Leibowitz A, Yamamoto N, Rartureau Y, Paradis P, Schiffrin EL. Angiotensin type 2 receptor agonist Compound 21 reduces vascular injury and myocardial fibrosis in stroke-prone spontaneously hypertensive rats. Hypertension. 2011:291–299. doi: 10.1161/HYPERTENSIONAHA.111.180158. [DOI] [PubMed] [Google Scholar]
- 20.Padia SH, Kemp BA, Howell NL, Keller SR, Gildea JJ, Carey RM. Mechanism of dopamine (D1) and angiotensin type-2 receptor interaction in natriuresis. Hypertension. 2012;59:437–445. doi: 10.1161/HYPERTENSIONAHA.111.184788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Padia SH, Howell NL, Kemp BA, Fournie-Zaluski MC, Roques BP, Carey RM. Intrarenal aminopeptidase N inhibition restores defective angiotensin II type-2 receptor-mediated natriuresis in spontaneously hypertensive rats. Hypertension. 2010;55:474–480. doi: 10.1161/HYPERTENSIONAHA.109.144956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Padia SH, Kemp BA, Howell NL, Gildea JJ, Keller SR, Carey RM. Intrarenal angiotensin III infusion induces natriuresis and angiotensin type 2 receptor translocation in Wistar-Kyoto but not spontaneously hypertensive rats. Hypertension. 2009;53:338–343. doi: 10.1161/HYPERTENSIONAHA.108.124198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hein L, Meinel L, Pratt RE, Dzau VJ, Koblika BK. Intracellular trafficking of angiotensin and its AT1 and AT2 receptors: evidence for selective sorting of receptor and ligand. Mol Endocrinol. 1997;11:1266–1277. doi: 10.1210/mend.11.9.9975. [DOI] [PubMed] [Google Scholar]
- 24.Gwathmey TM, Shaltout HA, Pendergrass KD, Pirro NT, Figueroa JP, Rose JC, Diz DI, Chappell MC. Nuclear angiotensin II type 2 (AT2) receptors are functionally linked to nitric oxide production. Am J Physiol Renal Physiol. 2009;296:F1484–F1493. doi: 10.1152/ajprenal.90766.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Presig PA, Rector FG., Jr Role of Na+-K+ antiport in rat proximal tubule NaCl absorption. Am J Physiol Renal Physiol. 1988;255:F461–F465. doi: 10.1152/ajprenal.1988.255.3.F461. [DOI] [PubMed] [Google Scholar]
- 26.Aronson PS. Role of ion exchange in mediating NaCl transport in the proximal tubule. Kidney Int. 1996;49:1665–1670. doi: 10.1038/ki.1996.243. [DOI] [PubMed] [Google Scholar]
- 27.Wang T. Flow-activated transport events along the nephron. Curr Opin Nephrol Hypertens. 2006;15:530–536. doi: 10.1097/01.mnh.0000242180.46362.c4. [DOI] [PubMed] [Google Scholar]
- 28.Schultheis PJ, Clarke LL, Meneton P, Miller ML, Soleimani M, Gawenis LR, Riddle TM, Duffy JJ, Doetschman T, Wang T, Giebisch G, Aronson PS, Lorenz JN, Shull GE. Renal and intestinal absorptive defects in mice lacking the NHE3 Na+/K+ exchanger. Nat Genet. 1998;19:282–285. doi: 10.1038/969. [DOI] [PubMed] [Google Scholar]
- 29.Ledoussal C, Lorenz JN, Nieman ML, Soleimani M, Schultheis PJ, Shull GE. Renal salt wasting in mice lacking NHE3 Na+/H+ exchanger but not in mice lacking NHE2. Am J Physiol Renal Physiol. 2001;281:F718–F727. doi: 10.1152/ajprenal.2001.281.4.F718. [DOI] [PubMed] [Google Scholar]
- 30.Girardi AC, Di Sole F. Deciphering the mechanisms of the Na+/H+ exchanger-3 regulation in organ dysfunction. Am J Physiol Cell Physiol. 2012;302:C1569–C1587. doi: 10.1152/ajpcell.00017.2012. [DOI] [PubMed] [Google Scholar]
- 31.Biemesderfer D, Pizzonia J, Abu-Alfa A, Exner M, Reilly R, Igarashi P, Aronson PS. NHE3: a Na+/H+ exchanger isoform of renal brush border. Am J Physiol Renal Physiol. 1993(265):F736–F742. doi: 10.1152/ajprenal.1993.265.5.F736. [DOI] [PubMed] [Google Scholar]
- 32.Riquier AD, Lee DH, McDonough AA. Renal NHE3 and NaPi2 partition into distinct membrane domains. Am J Physiol Cell Physiol. 2009;296:C900–C910. doi: 10.1152/ajpcell.00526.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Donowitz M, Li X. Regulatory binding partners and complexes of NHE3. Physiol Rev. 2007;87:825–872. doi: 10.1152/physrev.00030.2006. [DOI] [PubMed] [Google Scholar]
- 34.Moe OW, Amemiya M, Yamaji Y. Activation of protein kinase A acutely inhibits and phosphorylates N/H exchanger NHE-3. J Clin Invest. 1995;96:2187–2194. doi: 10.1172/JCI118273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao H, Wiederkehr MR, Fan L, Collazo RL, Crowder LA, Moe OW. Acute inhibition of Na/H exchanger NHE-3 by cAMP. Role of protein kinase A and NHE-3 phosphoserines 552 and 605. J Biol Chem. 1999;274:3978–3987. doi: 10.1074/jbc.274.7.3978. [DOI] [PubMed] [Google Scholar]
- 36.Kocinsky HS, Girardi ACC, Biemesderfer D, Nguyen T, Mentone SA, Orlowski J, Aronson PS. Use of phosphor-specific antibodies to determine the phosphorylation of endogenous Na+/K+ exchanger NHE3 at PKA consensus sites. Am J Physiol Renal Physiol. 2005;289:F249–F258. doi: 10.1152/ajprenal.00082.2004. [DOI] [PubMed] [Google Scholar]
- 37.Kurashima K, Yu FH, Cabado AG, Szabo EZ, Grinstein S, Orlowski J. Identification of sites required for down-regulation of Na+/H+ 3 exchanger NHE3 activity by cAMP-dependent protein kinase. J Biol Chem. 1997;45:28672–28679. doi: 10.1074/jbc.272.45.28672. [DOI] [PubMed] [Google Scholar]
- 38.Belusa R, Wang Z-M, Matsubara T, Sahlgren B, Dulubova I, Nairn AC, Ruoslahti E, Greengard P, Aperia A. Mutation of the protein kinase C phosphorylation site on rat α1 Na+,K+-ATPase alters regulation of intracellular Na+ and pH and influences cell shape and adhesiveness. J Biol Chem. 1997;272:20179–20184. doi: 10.1074/jbc.272.32.20179. [DOI] [PubMed] [Google Scholar]
- 39.Therien AG, Blostein R. Mechanisms of sodium pump regulation. Am J Physiol Cell Physiol. 2000;279:C541–C566. doi: 10.1152/ajpcell.2000.279.3.C541. [DOI] [PubMed] [Google Scholar]
- 40.Kava L, Rossi NF, Mattingly R, Yingst DR. Increased expression of Na,K-ATPase and a selective increase in phosphorylation at Ser-11 in the cortex of the 2-kidney, 1-clip hypertensive rat. Am J Hypertens. 2012;25:487–491. doi: 10.1038/ajh.2011.247. [DOI] [PubMed] [Google Scholar]
- 41.Nascimento NR, Kemp BA, Howell NL, Gildea JJ, Santos CF, Harris TE, Carey RM. Role of Src family kinase in extracellular renal cyclic guanosine 3′,5′-monophosphate- and pressure-induced natriuresis. Hypertension. 2011;58:107–113. doi: 10.1161/HYPERTENSIONAHA.110.168708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blendea MC, Jacobs D, Stump CS, McFarlane SI, Ogrin C, Bahtyiar G, Stas S, Kumar P, Sha Q, Ferrario CM, Sowers JR. Abrogation of oxidative stress improves insulin sensitivity in the Ren-2 rat model of tissue angiotensin II overexpression. Am J Physiol Endocrinol Metab. 2005;288:E353–359. doi: 10.1152/ajpendo.00402.2004. [DOI] [PubMed] [Google Scholar]
- 43.Wenceslau CF, Rossoni LV. Rostafuroxin ameliorates endothelial dysfunction and oxidative stress in resistance arteries from deoxycorticosterone acetate-salt hypertensive rats: the role of Na+K+-ATPase/cSRC pathway. J Hypertens. 2014;32:542–554. doi: 10.1097/HJH.0000000000000059. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.