

Abstract
Histone acetylation, including acetylated H3K14 (H3K14ac), is generally linked to gene activation. Monomethylated histone H3 lysine 4 (H3K4me1), together with other gene-activating marks, denotes active genes. In contrast to usual gene-activating can function of H3K14ac and H3K4me1, we here show that the dual histone modification mark H3K4me1-H3K14ac is recognized by ZMYND8 (also called RACK7) and can function to counteract gene expression. We identified ZMYND8 as a transcriptional corepressor of the H3K4 demethylase JARID1D. ZMYND8 antagonized the expression of metastasis-linked genes, and its knockdown increased the cellular invasiveness in vitro and in vivo. The plant homeodomain (PHD) and Bromodomain cassette in ZMYND8 mediated the combinatorial recognition of H3K4me1-H3K14ac and H3K4me0-H3K14ac by ZMYND8. These findings uncover an unexpected role for the signature H3K4me1-H3K14ac in attenuating gene expression and reveal a previously unknown metastasis-suppressive epigenetic mechanism in which ZMYND8’s PHD-Bromo cassette couples H3K4me1-H3K14ac with downregulation of metastasis-linked genes.
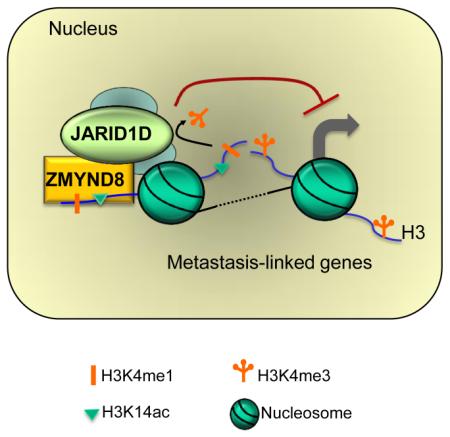
Graphical abstract
Introduction
Covalent modifications of histones play a central role in epigenetically and transcriptionally regulating gene expression (Jenuwein and Allis, 2001; Strahl and Allis, 2000). One of the well-studied histone modifications is histone acetylation, which is catalyzed by histone acetyltransferases and can be reversed by histone deacetylases (Berger, 2002). Histone acetylation affects chromatin architecture by neutralizing the positive charge of lysine residues and serving as a docking site for binding proteins, and it is generally associated with gene activation (Wang et al., 2008). For instance, acetylated histone H3 lysine 9 (H3K9ac) and H3K14ac are highly correlated with gene activation (Pokholok et al., 2005; Wang et al., 2008).
In addition to acetylation, histone lysine methylation is a key mark for the epigenetic and transcriptional regulation of gene expression. Unlike histone acetylation, this modification is associated with either gene activation or silencing, dependent on the sites of methylation (Barski et al., 2007). For example, methylated H3K4 is commonly linked to gene activation (Barski et al., 2007; Pokholok et al., 2005; Ruthenburg et al., 2007), whereas methylated H3K27 represents a repressive mark. In addition to the methylation sites, methylation states are important for different modes of gene regulation (Barski et al., 2007; Gu and Lee, 2013). For instance, the distribution profiles of monomethylated H3K4 (H3K4me1), H3K4me2, and H3K4me3 highly overlap but are often distinct (Wang et al., 2009). H3K4me3 is generally located at the transcription start sites (TSSs) (Bernstein et al., 2002) and is positively correlated with the transcription frequency of active genes (Barski et al., 2007; Pokholok et al., 2005). Strikingly, this mark occupies as much as 75% of all human gene-regulatory regions, spanning the TSSs in several cell types, including embryonic stem cells, primary hepatocytes, and B cells (Guenther et al., 2007). Of interest, H3K4me3 and H3K27me3 can form bivalent domains, which are known to keep the genes poised (activatable but repressed) (Bernstein et al., 2006). Most H3K4me2, along with H3K4me3, co-occupies around TSSs (Schneider et al., 2004), but its subset can be found in enhancers and the bodies of active genes. H3K4me1, together with acetylated H3K27 (H3K27ac), signifies enhancers, which modulate the temporal and spatial expression of genes (Heintzman et al., 2009; Visel et al., 2009).
It has been well documented that the effects of histone acetylation and methylation on gene expression can be mediated by specific binding proteins called “readers” (Kutateladze, 2011; Musselman et al., 2012; Yun et al., 2011). Acetylation marks can be recognized by Bromodomain (Bromo), Pleckstrin homology domain, and plant homeodomains (PHDs). Recently, it has been shown that Yeats domains read this mark (Li et al., 2014; Shanle et al., 2015). Lysine methylation interacts with multiple domains, including PHDs, chromodomains, tudor domains, and PWWP domains (Kutateladze, 2011; Musselman et al., 2012; Yun et al., 2011). In particular, PHDs contain the Cys4-His-Cys3 motif with dual zinc finger structures. The PHDs, including those in BHC80, TRIM24, TRIM33, and KDM5B, interact with unmethylated H3K4 (Klein et al., 2014; Lan et al., 2007; Tsai et al., 2010; Xi et al., 2011). Other PHDs read methylated lysines, methylated arginines, or acetyl lysines in histones (Dhar et al., 2012; Musselman and Kutateladze, 2011; Yun et al., 2011). PHDs are linked to several biological processes, including H3K4 trimethylation, IgG class switching, and hematopoietic malignancies (Musselman and Kutateladze, 2011).
We have previously showed that JARID1D (also known as KDM5D and SMCY), a male-specific protein expressed on the Y chromosome, represses its target gene by demethylating H3K4me3 and H3K4me2 (Lee et al., 2007). Our recent study has demonstrated that the H3K4 demethylase JARID1D is an anti-invasive factor that acts against prostate cancer metastasis (Li et al., 2016). In the present study, we sought to gain new insight into JARID1D-mediated gene regulation. We identified ZMYND8 (also called RACK7) as a transcriptional corepressor for the JARID1D-mediated downregulation of metastasis-linked genes. We also found that the PHD/Bromo domain cassette of ZMYND8 is a reader for the dual histone mark H3K4me1-H3K14ac and that the signature H3K4me1-H3K14ac can act as a gene-repressive signature for metastasis-linked genes.
Results
ZMYND8 is a JARID1D-interacting protein
To better understand how JARID1D regulates gene expression, we searched for new JARID1D-interacting proteins. Affinity purification and a mass spectrometric analysis of JARID1D-associated proteins led us to identify the previously unknown JARID1D-interacting proteins, such as ZMYND8, Mga, and DBC1, in addition to the already reported Ring6a (Lee et al., 2007) (Figure 1A). In particular, ZMYND8 contains a PHD, a Bromo, a PWWP, and a zinc finger-harboring MYND domain (Figure 1B). We have recently reported that Bromo-PWWP in the ZMYND8-homologous protein ZMYND11 interacts with H3K36me3 in H3.3 but not in H3.1 (Wen et al., 2014). Because of a putative chromatin-binding function of ZMYND8, we focused on understanding the role of ZMYND8 in JARID1D-mediated gene repression. Our cell fractionation experiment demonstrated that ZMYND8 was localized exclusively in the nucleus in DU145 prostate cancer cells (Figure 1C).
Figure 1. ZMYND8 is a JARID1D-associated protein.

(A) Immunoaffinity purification and mass spectrometric analysis of JARID1D-associated proteins. JARID1D-associated proteins from nuclear extracts from FLAG-JARID1D-expressing stable H1299 cells were immunopurified with anti-FLAG (α-FLAG) affinity resins. The proteins bands were analyzed by mass spectrometry. Asterisks indicate breakdowns or non-specific polypeptides. (B) Schematic representation of human JARID1D, ZMYND8, and ZMYND8 deletion mutants. (C) Nuclear localization of JARID1D and ZMYND8 in DU145 cells. Nuclear and cytoplasmic fractions of DU145 cells were blotted with the indicated antibodies. p84 and β-actin were used as a nuclear marker and a cytoplasmic marker, respectively. WCE, whole cell extracts. (D) Co-immunoprecipitation between ectopically expressed FLAG-tagged JARID1D and endogenous ZMYND8 protein. Anti-FLAG-immunoprecipitated samples were blotted with anti-FLAG and anti-ZMYND8 (α-ZMYND8) antibodies. (E) Coimmunoprecipitation between endogenous JARID1D and ZMYND8 in DU145 cells. Anti-JARID1D (α-JARID1D)-immunoprecipitated samples were blotted with anti-JARID1D and anti-ZMYND8 antibodies. (F) Mapping of the ZMYND8 region responsible for the interaction with JARID1D. FLAG-JARID1D and HA-ZMYND8 (or its truncated mutants) were ectopically expressed in 293T cells. Expression levels were examined using anti-HA and anti-FLAG antibodies (Left panel). Following co-immunoprecipitation (IP), the eluates were examined by western blot analysis using anti-HA (α-HA) and anti-FLAG antibodies (Right panel). Open rectangular triangles denote specific bands, and asterisks indicate nonspecific polypeptides. (G and H) Mapping of the JARID1D region responsible for the interaction with ZMYND8. Recombinant full-length JARID1D and its deletion mutants were analyzed using anti-JARID1D antibody (G). A Co-IP assay was performed using recombinant JARID1D, JARID1D mutants, and ZMYND8 that were isolated from Sf9 or Sf21 insect cells (H). See also Figure S1.
To validate the association between ZMYND8 and JARID1D, we ectopically expressed FLAG-JARID1D and immunoprecipitated it using anti-FLAG beads. This co-immunoprecipitation (Co-IP) experiment showed that JARID1D interacted with ZMYND8 (Figure 1D). In addition, our additional Co-IP experiments confirmed the endogenous association between JARID1D and ZMYND8 (Figure 1E). In contrast, the interaction of ZMYND8 with other JARID1 family members was undetectable in our assay condition (Figures S1A and S1B). Consistent with the previous report (Gong et al., 2015), ZMYND8 interacted with LSD1 (Figures S1A and S1B). To map which region is accountable for the interaction between ZMYND8 and JARID1D, we generated different deletion constructs of ZMYND8 and JARID1D. Our results demonstrated that the PHD2 domain in JARID1D and a middle region (410–730aa) in ZMYND8 were required for their interaction (Figures 1B and 1F–1H).
Chromatin locations of ZMYND8 highly overlap those of JARID1D
To assess the chromatin co-occupancy of ZMYND8 and JARID1D, we determined their chromatin landscapes using chromatin immunoprecipitation (ChIP) coupled with high throughput sequencing (ChIP-Seq). ZMYND8 peaks were located at the promoter (51.36%), intron (21.95%) and intergenic (23.74%) regions, and JARID1D peaks were localized at the promoter (32.48%), intron (26.46%), and intergenic (38.37%) regions (Figure 2A). In genic regions, average signals of ZMYND8 and JARID1D peaks were highly enriched near the TSSs (Figure 2B). Consistent with the physical interaction between ZMYND8 and JARID1D, peak locations and target genes of ZMYND8 greatly overlapped those of JARID1D (Figures 2C–2E). Gene expression programs that were co-occupied by ZMYND8 and JARID1D were associated with diverse cellular processes, including signaling transduction, transcription, cell adhesion, cell-matrix adhesion, and cell junction assembly (Figure 2F).
Figure 2. The chromatin landscape of ZMYND8 greatly overlaps that of JARID1D.

(A) The genomic distribution of ChIP-seq peaks of JARID1D and ZMYND8. ChIP-Seq was performed using DU145 prostate cancer cells. (B) Average enrichment of JARID1D and ZMYND8 in the genic regions, including TSS and TTS. TSS, transcriptional start site; TTS, transcriptional termination site. (C) Heat maps of genomic co-localization of JARID1D and ZMYND8 in DU145 cells. (D) Venn diagram of the overlap between JARID1D and ZMYND8 peaks. (E) Venn diagram of genes co-occupied by JARID1D and ZMYND8. (F) Gene ontology analysis of genes co-occupied by JARID1D and ZMYND8.
ZMYND8 suppresses the invasiveness of prostate cancer cells in vitro and in vivo
Because our recent study has showed that JARID1D is an anti-invasive factor that acts against prostate cancer metastasis (Li et al., 2016), we determined whether ZMYND8 regulates cellular invasion using two androgen-independent and moderately invasive human prostate cancer cell lines DU145 and CWR22Rv1 (Nemeth et al., 1999; Sramkoski et al., 1999). ZMYND8 depletion using two independent shRNAs had a negligible effect on cell proliferation and migration abilities (Figures 3A–3C). Because the cell migration assay measures only the migratory abilities of cells, we additionally examined the invasive abilities of cells using cell invasion assay in which cells digest and invade a Matrigel matrix that mimics the in vivo extracellular matrix. Similar to JARID1D knockdown (Figures S1C and S1D) (Li et al., 2016), ZMYND8 knockdown increased the invasiveness of DU145 and CWR22Rv1 cells (Figures 3D, S1E, and S1F).
Figure 3. ZMYND8 knockdown increases the invasive abilities of prostate cancer cells in vitro and in vivo.

(A) Western blot and quantitative RT-PCR analysis of ZMYND8 levels following the treatment of DU145 cells with shZMYND8s (shZMYND8-95 and shZMYND8-97). (B–D) Effects of ZMYND8 knockdown on the proliferation (B), migration (C) and invasion (D) of DU145 cells. Following the cell migration and invasion assays, cells were stained with crystal violet and counted in at least five fields. (E–G) The effect of ZMYND8 knockdown on the in vivo metastatic abilities of DU145 cells in an intravenous mouse xenograft model. DU145 cells with stably expressing firefly luciferase were infected with lentiviruses containing scramble shRNA (shScramble) or shZMYND8-97. The representative bioluminescent images of mice (shScramble, n=6; shZMYND8, n=6) 8-10 weeks after tail vein injection are shown (E), and their quantified bioluminescent signals were individually plotted (F). Representative images of hematoxylin and eosin–stained lung tissues (G). Data are presented as the mean ± SEM (error bars). See also Figure S1.
To determine the effect of ZMYND8 knockdown on the invasiveness of prostate cancer cells in vivo, we treated luciferase-expressing DU145 cells using control shRNA (shScramble) or shZMYND8 and injected cells into mice via tail veins. We then monitored the in vivo abilities of cancer cells that invade into a distant organ (e.g., the lung as a primary site) from the blood system and form tumors using a luciferase imaging system. Our results demonstrated that the 8th week after injection, luciferase signals were about 5-fold higher in the shZMYND8 group (n = 6) than in the control shRNA group (n = 6) (Figures 3E and 3F). Our hematoxylin and eosin staining confirmed tumor development in shZMYND8 group (Figure 3G). These results indicate that ZMYND8 suppresses the in vivo invasiveness of prostate cancer cells.
ZMYND8 acts as a transcriptional corepressor of JARID1D at metastasis-linked genes
To assess how ZMYND8 regulates cellular invasiveness, we first determined the effect of ZMYND8 knockdown on whole genome mRNA expression using RNA-Seq. We then compared ZMYND8-regulated genes with JARID1D-regulated genes, because JARID1D also impedes cellular invasiveness (Li et al., 2016). Interestingly, 45.8% of shJARID1D-upregulated genes (889/1,940) overlapped 33.0% of shZMYND8-upregulated genes (889/2,696) (Figure 4A). In contrast, relatively fewer genes were coincidentally downregulated by JARID1D and ZMYND8 knockdown (Figure 4B). Consistent with the increased cellular invasiveness by ZMYND8 knockdown, gene expression programs that were co-upregulated by shZMYND8 and shJARID1D included metastasis-linked processes, such as cell adhesion (Figure 4C). In contrast, gene expression programs that were co-downregulated by ZMYND8 knockdown and JARID1D knockdown did not include metastasis-linked processes (Figure 4D). Some co-upregulated genes (190) were co-occupied by ZMYND8 and JARID1D (Figures S2A and S2B).
Figure 4. ZMYND8 is required for the JARID1D-mediated downregulation of metastasis-linked genes.

(A and C) Venn diagrams (A) and gene ontology analysis (C) of genes upregulated by JARID1D and ZMYND8 knockdown. Whole genome expression levels were measured by the RNA-seq. (B and D) Venn diagrams (B) and gene ontology analysis (D) of genes downregulated by JARID1D and ZMYND8 knockdown. (E) The effect of JARID1D or ZMYND8 knockdown on expression levels of multiple metastasis-linked genes in DU145 cells. A quantitative RT-PCR analysis was performed using samples from at least three independent experiments (i.e., n ≥ 3). (F–I) The effect of ZMYND8 knockdown on JARID1D and H3K4me3 levels at Slug (F), CD44 (G), VEGFA (H), and EGFR (I) genes in DU145 cells. Chromatin levels of ZMYND8, JARID1D, and H3K4me3 were measured by quantitative ChIP that was performed using samples from at least three independent experiments (i.e., three biological replicates). IgG was used as a negative control. Data are presented as the mean ± SEM (error bars). ChIP PCR amplicons are indicated by the small blue lines in Figures 7A-7D. See also Figure S2.
Because JARID1D and ZMYND8 suppress cellular invasiveness, we focused on determining the role of JARID1D and ZMYND8 on regulating metastasis-linked genes. Our analysis of ChIP-Seq data, including manual examination, indicate that ZMYND8 and JARID1D co-occupied multiple metastasis-linked genes, including Slug, CD44, vascular endothelial growth factor A (VEGFA), and epidermal growth factor receptor (EGFR) (as listed in Figures S2C and S2D). We examined the effects of ZMYND8 and JARID1D knockdown on expression of these genes using quantitative RT-PCR. Our results showed that most of these metastasis-linked genes were upregulated by ZMYND8 or JARID1D knockdown (Figures 4E, S2C, and S2D). Our quantitative ChIP experiments confirmed that ZMYND8 and JARID1D were co-localized at Slug, CD44, VEGFA, and EGFR genes (Figures 4F–4I). Our ChIP results also showed that ZMYND8 repressed and occupied other JARID1D target genes, such as the matrix metalloproteinase 1 (MMP1) and MMP3, that we previously reported (Li et al., 2016) (Figures 4E, S2E, and S2F). CD44, a single-chain transmembrane glycoprotein, promotes metastasis (Hiraga et al., 2013), and Slug induces epithelial-mesenchymal transition to facilitate metastasis (Shih and Yang, 2011; Yang and Weinberg, 2008). EGFR overexpression is linked to invasion and metastasis of multiple types of cancer (Lu et al., 2001). VEGFA facilitates tumor metastasis (Liu et al., 2011). MMPs enhance cell invasion and metastasis by digesting extracellular matrix (Kessenbrock et al., 2010).
To determine whether ZMYND8 plays a role in regulating JARID1D occupancy and H3K4me3 levels, we determined the effect of ZMYND8 knockdown on JARID1D and H3K4me3 levels at Slug, CD44, VEGFA, EGFR, MMP1, and MMP3 genes. Our quantitative ChIP experiments demonstrated that ZMYND8 knockdown downregulated JARID1D levels while increasing H3K4me3 levels at these genes (Figures 4F–4I, S2E, and S2F). These results indicate that ZMYND8 acts as a transcriptional corepressor of JARID1D.
PHD-Bromo in ZMYND8 reads H3K4me0-H3K14ac and H3K4me1-H3K14ac
To further understand the role of ZMYND8 in gene regulation, we assessed the molecular characteristics of its chromatin-binding modules, such as PHD and Bromo. Our in vitro peptide pull-down assays showed that ZMYND8-PHD/Bromo (ZMYND8-PB) interacted with unmethylated H3K4 (H3 [1-21aa]; referred to as “H3K4me0”) and to a lesser extent H3K4me1 (Figures 5A and 5B). In contrast, the interaction of ZMYND8-PB with other methyl marks was insignificant in the same washing condition (300 mM NaCl), although the association of ZMYND8-PB with H3K36me1 and H3K4me2 was weakly detectable in a low salt (150 mM NaCl) wash (Figures 5B and S3A). Our additional assays showed that ZMYND8-PHD alone did not interact with H3K4me0 and that the H3-binding activity of PHD/Bromo was abolished by partial deletions of Bromo in ZMYND8-PB (Figures 5A, 5C, and S3B). These results indicate that both PHD and Bromo are required for the recognition of H3K4me0 and H3K4me1 by ZMYND8.
Figure 5. ZMYND8 recognizes H3K4me0-H3K14ac and H3K4me1-H3K14ac via the PHD/Bromo cassette.

(A) Schematic representation of ZMYND8-PHD/Bromo/PWWP (ZMYND8-PBP), ZMYND8-PHD/Bromo (ZMYND8-PB), and ZMYND8-PB deletion mutants. (B) Peptide pull-down assays using the indicated biotinylated peptides and the GST-tagged ZMYND8-PB. (C) Peptide pull-down assays using H3 (1-21) peptide and either the recombinant GST-ZMYND8-PB or its deletion mutants. (D and E) Isothermal titration calorimetry (ITC) assays to measure the interaction of the indicated peptides with ZMYND8-PB (D) or ZMYND8-PBP (E). (F) Peptide pull-down assays using the indicated biotinylated peptides and recombinant GST-ZMYND8-PBP. (G) ITC assays to measure the interaction of the indicated peptides with the recombinant ZMYND8-PBP. See also Figure S3.
Because a PWWP domain is located adjacent to Bromo, we examined the effect of PWWP on the interaction between H3K4me0 and ZMYND8-PB. H3K4me0 peptide interacted more strongly with ZMYND8-PHD/Bromo/PWWP (ZMYND8-PBP; KD ≈ 7.2 μM) than with ZMYND8-PB (KD ≈ 15 μM) (Figures 5D and 5E). Interestingly, the binding activities of ZMYND8-PB and ZMYND8-PBP to H3K4me3 were negligible (Figures 5B and 5E). Because many Bromo domains are known to bind to acetylated lysines, we also determined whether ZMYND8-Bromo recognizes a specific acetylated lysine. Our assays using H2Aac, H2Bac, H3ac, and H4ac peptides showed that ZMYND8-PBP specifically interacted with H3K14ac (Figures 5F and S3C). Our Isothermal titration calorimetry (ITC) assay demonstrated that ZMYND8-PBP interacted with H3K4me0K14ac peptide at a high affinity (KD ≈ 1.0 μM) (Figure 5G). In contrast, ZMYND8-PBP did not have any significant KD values for H3K36me2 and H4K16ac (Figures S3D and S3E), although it was reported that ZMYND8 interacted with these modifications (Adhikary et al., 2016; Gong et al., 2015). However, it is possible that ZMYND8 may recognize these modifications during DNA damage response and retinoic acid response (Adhikary et al., 2016; Gong et al., 2015). Interestingly, we observed that ZMYND8-PBP bound to H3K4me1K14ac peptide at a considerably high affinity (KD ≈ 5.4 μM) (Figure 5G).
Molecular basis for the recognition of H3K4me1-H3K14ac and H3K4me0-H3K14ac by the ZMYND8-PHD/Bromo
To understand the molecular basis by which ZMYND8 recognizes H3K4me0-H3K14ac (and H3K4me1-H3K14ac), we solved the crystal structure of ZMYND8-PBP (93-426aa) at 1.8 Å (Table 1). However, the electron densities of the histone peptide were not detected, although the peptide had been added for crystallization. Interestingly, ZMYND8’s PHD shared considerable sequence homology with H3K4me0-interacting PHDs in TRIM24, TRIM33, and BHC80 and was superimposed well with PHDs in TRIM24 and TRIM33 (Tsai et al., 2010; Xi et al., 2011) (Figures S4A and S4B). We performed structural modelling for H3K4me0-H3K14ac recognition by ZMYND8 by aligning our free ZMYND8-PBP structure with the reported H3 peptide-bound TRIM33-PB structures (Xi et al., 2011) (Figure S4C). The modelled H3(1-14)K4me0K14ac peptide fitted nicely to the conserved H3 binding surface of ZMYND8-PHD/Bromo with no discernible steric clash (Figure 6A). The H3 binding surface of ZMYND8 is electrostatically negative (Figure 6A), which is favorable for its interaction with the positive H3 tail. Aside from anti-parallel β–sheet formation between the H3 backbone and PHD finger, H3 binding to PHD/Bromo may be facilitated by several charge-stabilized hydrogen bonds (i.e., H3R2, ZMYND8-E124; H3K9, ZMYND8-E117; and H3K14ac, ZMYND8-N248) (Figures 6B–6D). Because ZMYND8-PBP interacted with H3K4me1K14ac peptide, we also performed structural modeling for H3K4me1-H3K14ac recognition by ZMYND8-PHD/Bromo. As shown in Figures 6E, 6F, and S4D, ZMYND8-PHD/Bromo may read H3K4me1K14ac in an almost identical manner to H3K4me0K14ac peptide.
Table 1.
Data collection and refinement statistics (Related to Figure 6)
| Crystal | ZMYND893-426-ZN* | ZMYND893-426-Native |
|---|---|---|
| Data collection | ||
| Space group | P212121 | P212121 |
| Cell dimensions | ||
| a, b, c (Å) | 67.1, 68.5, 70.5 | 67.1, 68.0, 70.3 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 |
| Wavelength (Å) | 1.2821 | 0.9790 |
| Resolution (Å) | 39.6-2.00 (2.03-2.00)† | 39.5-1.80 (1.87-1.80)† |
| Unique reflections | 42958 (2171) | 30158 (2901) |
| Rsym (%) | 9.0 (67.2) | 7.9 (67.3) |
| I/σ(I) | 29.1 (2.1) | 22.4 (2.6) |
| Completeness (%) | 99.8 (98.3) | 99.3 (97.7) |
| Redundancy | 4.5 (3.4) | 4.7 |
| Refinement (F>0) | ||
| Resolution (Å) | 39.5-1.80 | |
| No. of reflections | 28650 | |
| Rwork/Rfree (%) | 18.7/22.4 | |
| No. atoms | ||
| Protein | 2530 | |
| Ion | 3 | |
| Water | 179 | |
| B factors (Å2) | ||
| Protein | 37.3 | |
| Ion | 31.1 | |
| Water | 42.1 | |
| r.m.s. deviations | ||
| Bond lengths (Å) | 0.013 | |
| Bond angles (°) | 1.34 | |
| Ramachandran plot | ||
| Most favored (%) | 97 | |
| Allowed (%) | 3 |
Values in this column are based on anomalous scaling.
Values in parentheses refer to the highest resolution shell.
Figure 6. Molecular basis for the interaction of ZMYND8-PHD/Bromo with H3K4me0-H3K14ac and H3K4me1-H3K14ac.

(A) Electrostatic surface view of the ZMYND8-PBP in complex with modeled H3K4me0K14ac peptide. Electrostatic potential is shown as a spectrum ranging from red (negative charge) to blue (positive charge). (B) Structural modelling of ZMYND8-PBP bound to unmodified H3 (1-6aa) peptides. ZMYND8-PBP is in ribbon view. The invisible N-terminal fragment (D96-G105) of ZMYND8-PHD is shown as a dotted line. Close-up view shows recognition of H3 peptide by ZMYND8-PHD (blue). Znf (pink), zinc finger motif between Bromo (green) and PWWP (orange); Magenta dashes, hydrogen bonds. (C) Structural modeling of ZMYND8-PBP bound to H3K4me0K14ac (1-14aa) peptides. Blue spheres, Zinc atoms; Magenta dashes, hydrogen bonds. (D) Close-up view of structural modeling for the recognition of H3K14ac by ZMYND8 Bromo (green). The side chains of two residues, Y247 and N248, are shown in magenta sticks, with blue for nitrogen and red for oxygen. Cyan dashes, hydrogen bonds. (E) Electrostatic surface view of the ZMYND8-PBP in complex with modelled H3K4me1K14ac peptide. Electrostatic potential is shown as a spectrum ranging from red (negative charge) to blue (positive charge). (F) Structural modelling of ZMYND8-PBP bound to H3K4me1 (1-6aa) peptides. ZMYND8-PBP is in ribbon view. The invisible N-terminal fragment (D96-G105) of ZMYND8-PHD is shown as dotted line. Close-up view shows recognition of H3 peptide by ZMYND8-PHD (blue). Znf (pink), zinc finger motif between Bromo (green) and PWWP (orange); Magenta dashes, hydrogen bonds. (G) ITC assays to measure the interaction of H3K4me0 (1-25aa) with ZMYND8-PBP or its PHD mutants. (H) ITC assays to measure the interaction of H3K14ac (1-15aa) with ZMYND8-PBP or its Bromo mutants (N248A or Y247A/N248A). For A–F, the modeled H3 peptide is depicted as sticks with yellow for carbon, blue for nitrogen, and red for oxygen. See also Figure S4.
To support our structure model, we performed ITC assays using several ZMYND8-PBP mutants and H3 peptide. Alanine mutations of D96, D99, D104, D108, and E124 as well as an N-terminal truncated version of ZMYND8-PBP (residue 103-426) resulted in significant losses in the H3K4me0-binding activity of ZMYND8-PBP, suggesting the importance of these residues in mediating electrostatic interactions (Figure 6G). The acidic residue D108, which is conserved as glutamic acid (E) among TRIM24, TRIM33, and BHC80 (Figure S4A), is located at the PHD-Bromo interface and may contribute to PHD-Bromo association rather than to direct H3-binding (Figures 6B and S4E). Thus, D108A likely destabilized the PHD-Bromo interaction, compromising its histone binding activity. F109D dramatically enhanced PBP’s binding activity by ~11-fold (Figure 6G). The enhanced binding activity of ZMYND8-PBP by F109D suggests that F109 spatially overlaps the acidic residues that participate in unmodified H3K4 recognition in other H3K4me0-binding PHD fingers (Figures S4A and S4B), in line with our structural model (Figures 6B and 6C). We also performed ITC assays using K14ac pocket mutants of ZMYND8-Bromo (i.e., N248A and Y247A/N248A) (Figure 6C). In support of our structural analysis, these mutations decreased the binding activities of ZMYND8-PBP (Figure 6H).
ZMYND8 antagonizes the expression of metastasis-linked genes by recognizing the dual histone mark H3K4me1-H3K14ac
Because the above in vitro results showed that H3K4me1-H3K14ac was recognized by the JARID1D corepressor ZMYND8, we tested the possibility that the chromatin locations of ZMYND8 and JARID1D are associated with those of H3K4me1 and H3K14ac. We performed ChIP-Seq for H3K4me1 and H3K14ac and compared their ChIP-Seq profiles with those of ZMYND8 and JARID1D. The heatmap of ChIP-Seq profiles showed that genomic location profiles of ZMYND8 and JARID1D near the TSSs might be associated, at least in part, with those of H3K4me1 and H3K14ac but not with those of H3K27me3 (Figure S5A). In line with the possibility that ZMYND8 acts as a JARID1D corepressor by recognizing H3K4me1 and H3K14ac at metastasis-linked genes, ZMYND8 and JARID1D peaks were co-localized with H3K4me1 and H3K14ac peaks (albeit less extensively with H3K14ac peaks) at multiple metastasis-linked genes, including Slug, CD44, EGFR, and VEGFA (Figures 7A–7D and S5B–S5I). H3K4me3 peaks at these genes were largely located at positions that were different from co-localized sites of H3K4me1, H3K14ac, ZMYND8, and JARID1D peaks (Figures 7A–7D and S5B–S5I). These findings are consistent with the previous report showing that H3K4me1 pattern near the TSSs is different from H3K4me3 pattern (Cheng et al., 2014).
Figure 7. ZMYND8 downregulates metastasis-linked genes by reading the signature H3K4me1-H3K14ac.

(A-D) Genome browser view of ChIP-Seq peaks for input (a negative control), ZMYND8, JARID1D, H3K14ac, H3K4me1, H3K4me3, and H3K27me3 at CD44 (A), Slug (B), VEGFA (C), and EGFR (D) genes in DU145 cells. The blue lines indicate ChIP PCR amplicons for E-H. ChIP-Seq signals of JARID1D, ZMYND8, H3K4me1, H3K14ac, H3K4me3, and H3K27me3 in the tracks represent their remainder signals after input signals were subtracted from their original signals. (E-H) The effect of JARID1D knockdown on ZMYND8 occupancy at CD44 (E), Slug (F), VEGFA (G), and EGFR (H) genes in DU145 cells. Chromatin levels of ZMYND8 and JARID1D were measured by quantitative ChIP (n ≥ 3). PCR values were normalized to input. (I) Western blot analysis of ectopic expression of WT FLAG-ZMYND8, its PHD mutants (D108A, F109D), and its Bromo mutant (YN/AA). DU145 cells infected with shZMYND8-97 virus were transfected with control vector, WT FLAG-ZMYND8 or its point mutants. shLuc-treated DU145 cells that were transfected with control vector were used as a control. (J) The effects of ectopic expression of WT FLAG-ZMYND8, its PHD mutants (D108A, F109D), and its Bromo mutant (YN/AA) on expression levels of CD44, Slug, VEGFA, and EGFR genes in ZMYND8-depleted DU145 cells (n = 5). (K and L) The effects of ectopic expression of WT FLAG-ZMYND8, its PHD mutants (D108A, F109D), and its Bromo mutant (YN/AA) on invasive ability in ZMYND8-depleted DU145 cells. Cells were stained with crystal violet (K) and counted in at least five different fields (L). Data are presented as the mean ± SEM (error bars). See also Figures S5 and S6.
We determined the effect of JARID1D knockdown on ZMYND8 occupancy using ChIP. Consistent with the notion that the interaction of H3K4me1-H3K14ac with ZMYND8-PBP is inhibited by H3K4me3, our ChIP data showed that ZMYND8 occupancy at Slug, CD44, EGFR, and VEGFA genes was decreased by JARID1D knockdown (Figures 7E– 7H). In addition, our ChIP-Seq results showed that JARID1D knockdown resulted in slightly increased H3K4me3 levels and moderately decreased H3K4me1 levels at these genes and other metastasis-linked genes (Figures S6A–S6H). Similarly, JARID1D knockdown upregulated average H3K4me3 level and downregulated average H3K4me1 level at genes that were co-occupied and co-repressed by JARID1D and ZMYND8 (Figures S6I and S6J). It should be noted that small increases in H3K4me3 levels are correlated with large increases in gene expression (Pokholok et al., 2005). Nevertheless, these results are consistent with the notion that JARID1D primarily demethylates H3K4me3 and H3K4me2 (Lee et al., 2007) and may thus establish a steady-state level of H3K4me1 that can be recognized by ZMYND8.
We also determined the effect of ZMYND8 knockdown on H3K4me3 and H3K4me1 levels using ChIP-Seq. Similar to JARID1D knockdown, ZMYND8 knockdown increased H3K4me3 levels and decreased H3K4me1 levels at multiple metastasis-linked genes, including Slug, CD44, EGFR, and VEGFA (Figures S6A–S6H). In addition, ZMYND8 knockdown slightly upregulated average H3K4me3 level and moderately downregulated average H3K4me1 level at genes that were co-occupied and co-repressed by JARID1D and ZMYND8 (Figures S6I and S6J). Average levels of H3K4me3 and H3K4me1 at co-target genes of ZMYND8 and JARID1D were less changed by ZMYND8 knockdown than by JARID1D knockdown. This may be because of the following possible reasons: JARID1D’s chromatin levels are likely reduced more by JARID1D knockdown than by ZMYND8 knockdown; and JARID1D directly regulates H3K4me3 and H3K4me1 levels at co-target genes of ZMYND8 and JARID1D, whereas ZMYND8 may indirectly modulate their levels by recruiting JARID1D.
To further examine whether the interaction of ZMYND8-PHD/Bromo with H3K4me1-H3K14ac plays a role in downregulating gene expression, we examined the effect of the expression of FLAG-ZMYND8, its PHD mutants, and its Bromo mutant on co-target genes of JARID1D and ZMYND8 in ZMYND8-depleted DU145 cells. Ectopic expression of FLAG-ZMYND8 significantly downregulated the expression levels of Slug, CD44, EGFR, and VEGFA genes (Figures 7I and 7J). F109D mutation, which enhanced the binding activity of ZMYND8-PHD, also repressed the expression of these genes (Figures 7I and 7J). In contrast, D108A and Y247A/N248A mutations, which decreased the binding activity of ZMYND8-PHD and ZMYND8-Bromo, respectively, did not repress gene expression (Figures 7I and 7J). Moreover, the effects of ectopic expression of these mutants on the invasion of ZMYND8-depleted DU145 cells were consistent with their effects on gene expression (Figures 7K and 7L). These results suggest that the binding activity of ZMYND8 PHD-Bromo to H3K4me1-H3K14ac is required for the ZMYND8-mediated transcriptional downregulation of Slug, CD44, EGFR, and VEGFA genes.
Discussion
Both H3K4me1 and H3K14ac are usually associated with gene activation, although a subset of H3K4me1 mark, along with H3K27me3 and H4K20me1, may be linked to gene repression (Cheng et al., 2014). In the present study, our results revealed that the dual histone mark H3K4me1-H3K14ac is recognized by the PHD-Bromo cassette in ZMYND8. In addition, we demonstrated that a combinatorial reading of H3K4me1-H3K14ac by ZMYND8 antagonizes the expression of metastasis-linked genes. Therefore, our findings indicate that H3K4me1-H3K14ac that associates with ZMYND8 acts as a gene-repressive histone signature to counteract the expression of metastasis-linked genes.
There are multiple dual module cassettes that can read two histone marks (Wang and Patel, 2011). The PHD-Bromo cassette in TRIM24 binds to the dual mark H3K4me0-H3K23ac to activate ERα target genes (Tsai et al., 2010). The PHD-Bromo cassette in TRIM33 interacts with the dual mark H3K9me3-H3K18ac, which may denote a poised chromatin state of TGF-β-responsive genes (Xi et al., 2011). The PHD-Bromo cassette in BPTF reads H3K4me3 and H4K16ac marks on different tails in a single nucleosome and is associated with gene activation (Ruthenburg et al., 2011). In addition to PHD-Bromo cassettes, there are other dual module cassettes, including PHD-PHD (Wang and Patel, 2011). For example, PHD-PHD in DPF3b recognizes the dual mark H3K4me0-H3K23ac on a single H3 tail to activate gene expression (Zeng et al., 2010). In the current study, we demonstrated that ZMYND8-PHD/Bromo recognizes the dual marks H3K4me1-H3K14ac and H3K4me0-H3K14ac in vitro. Our ChIP-Seq analysis showed that ZMYND8 peaks, along with JARID1D peaks, were aligned with H3K4me1 and H3K14ac peaks, indicating that ZMYND8 interacts with H3K4me1-H3K14ac in vivo. Intriguingly, the in vivo recognition of H3K4me1-H3K14ac by ZMYND8 may be dependent on JARID1D, because JARID1D knockdown decreased ZMYND8 occupancy at metastasis-linked genes (Figures 7E–7H). This recognition may also rely on JARID1D’s demethylation activity, because decreased levels of ZMYND8 occupancy by JARID1D knockdown coincide with increased H3K4me3 levels (Figures 7E–7H and S6A–S6H). In this respect, ZMYND8-PHD/Bromo is an unprecedented dual module cassette whose binding activity to H3K4me1-H3K14ac may be dependent on the demethylation of H3K4me3 and H3K4me2 by JARID1D.
The present study indicates that ZMYND8 acts as JARID1D’s transcriptional corepressor. This is supported by our following findings: 1) ZMYND8 directly interacted with JARID1D; 2) ZMYND8’s target genes highly overlapped JARID1D’s target genes; 3) knockdown of ZMYND8 or JARID1D upregulated the expression of their co-target genes; and 4) knockdown of ZMYND8 or JARID1D decreased the chromatin occupancy of each other in metastasis-linked genes, suggesting that ZMYND8 and JARID1D are, to some extent, inter-dependent for their association with chromatin. Interestingly, ZMYND8 can also interact with estrogen receptor (ER), a transcriptional activator (Malovannaya et al., 2011). The ZMYND8-ER interaction is hormone-dependent, as transcriptional activation by ZMYND8-ER is enhanced by the estrogen (Malovannaya et al., 2011). ZMYND8-JARID1D interaction, albeit male-specific, may not be dependent on androgen, because it occurs in the androgen-independent cell line DU145 (Chlenski et al., 2001). The interaction between ZMYND8 and ER results in gene activation (Malovannaya et al., 2011), whereas ZMYND8 and JARID1D cooperate for gene repression. For these reasons, ZMYND8 may be a Ying-Yang factor that activates or represses gene expression in a hormone-specific, partner-dependent manner.
Knockdown of ZMYND8 or JARID1D upregulated multiple co-target genes of ZMYND8 and JARID1D (Figures 4E, S2C and S2D), indicating their gene-repressive functions. Interestingly, the chromatin profiles of the transcriptional corepressors ZMYND8 and JARID1D near the TSSs are somewhat associated with many active genes with H3K4me3 in the absence of H3K27me3 (Figures 7A–7D and S5A-S5I). This is not surprising, because transcriptional corepressors often occupy active genes to antagonize transcriptional coactivators. For example, the transcriptional co-repressor EZH2 counteracts the transcriptional coactivator ARID1A, a component of SWI/SNF2 chromatin remodeling complex, at active genes (Bitler et al., 2015). Polycomb proteins also antagonize Trithorax proteins (Schuettengruber et al., 2007). Then, how are JARID1D and ZMYND8 recruited to active genes? There are at least two mutually non-exclusive possibilities: 1) ZMYND8 recognizes pre-existing H3K4me1-H3K14ac peaks and may bridge JARID1D to co-target genes of JARID1D and ZMYND8; and 2) JARID1D and ZMYND8 may be recruited to their target genes via the DNA-binding transcription factors; In this regard, anti-FLAG affinity purification and mass spectrometric analysis of FLAG-JARID1D eluates resulted in the identification of the transcription factor Mga.
Cancer metastasis is devastating and clinically challenging. Metastasis is a complicated process in which cancer cells penetrate into blood systems and depart the blood vessels to colonize in distant organs (Chaffer and Weinberg, 2011). For metastasis, cancer cells acquire increased migratory and invasive abilities, which are in principle subject to epigenetic control. Our recent study showed that JARID1D loss (about 25%–30%) as well as decreased JARID1D levels frequently occur in metastatic prostate cancer (Li et al., 2016). JARID1D knockdown increased H3K4me3 levels at metastasis-linked genes and decreased ZMYND8 levels at the same genes, resulting in increased gene expression (Figures 7E–7H, S2C, and S6A–S6H) (Li et al., 2016). Our in vitro results showed that the interaction between ZMYND8 and H3K4me3 was undetectable. These findings suggest that transcriptional downregulation of metastasis-linked genes by JARID1D and ZMYND8 is released by increased H3K4me3 levels that result from the downregulation or loss of JARID1D in metastatic prostate cancer. Therefore, our results provide epigenetic insights into how the downregulation or loss of JARID1D may increase expression of metastasis-linked genes to enhance the invasive ability of cancer cells.
The epigenetic switch plays a critical role in regulating gene expression. Many key differentiation-associated genes undergo an epigenetic switch during cellular differentiation. For example, differentiation-associated genes are poised and marked by bivalency comprising H3K4me3 and H3K27me3 in stem cells but can be activated during differentiation via bivalency resolution to H3K4me3-containing monovalency (Bernstein et al., 2006; Dhar et al., 2016). In this regard, it is tempting to speculate that H3K4me1-H3K14ac acts as a poised epigenetic signature that is converted to the active dual mark H3K4me3-H3K14ac during cancer metastasis. Taken together, our work uncovers a previously unknown invasion-suppressive mechanism in which the PHD/Bromo-containing H3K4me1-H3K14ac reader ZMYND8, together with a histone H3K4 demethylase, epigenetically suppresses cellular invasiveness by antagonizing the expression of metastasis-linked genes.
Experimental Procedures
Antibodies, plasmids, and cell lines
DU145 and CWR22Rv1 prostate cancer cells were purchased from ATCC. Cell culture reagents were obtained from Invitrogen/Gibco. The anti-JARID1D antibodies were purchased from Bethyl (A310-624A, A301-751A) and Santa Cruz (sc-83944). Anti-ZMYND8 antibodies were from Bethyl (A310-739A) and Santa Cruz (sc-100824). Anti-H3K4me3 (07-473), anti-H3K27me3 (07-449) and anti-H3K14ac (MABE351) antibodies were from Millipore. Anti-H3K4me1 (ab8895) antibody was from Abcam. Anti-GST was from Santa Cruz. Anti-HA was from Covance. Anti-JARID1A, JARID1B, JARID1C antibodies were from Bethyl. Anti-LSD1 antibody was from Novus. Anti-p84 antibody was from GeneTex. Anti- FLAG mouse monoclonal M2 (F3165), anti-β-Actin (A5441), shZMYND8-95 (TRCN0000037995) shZMYND8-97 (= shZMYND8) (TRCN0000037997), and control shRNA (shLuciferase) were from Sigma Aldrich. shJARID1D-4 (= shJARID1D, GCCAACCATGTGCAATGTAAC) and shScramble were cloned into the PLKO.1 vector
The cDNAs encoding ZMYND8-PHD/Bromo/PWWP, its point mutants, its deletion mutants were cloned to the bacterial expression vector pGEX6P-1 (Amersham Biosciences). To express JARID1D, ZMYND8 and ZMYND8 mutants in mammalian cell lines, their cDNAs were cloned into the pFLAG -CMV2 vector (Sigma). HA-ZMYND8 and its truncated mutants were generated by replacing the FLAG tag with the HA tag in the pFLAG-CMV2 vector. All point mutations were generated by site-directed mutagenesis according to the manufacturer’s instructions (Stratagene).
ChIP and ChIP-Seq assays
ChIP assays were carried out as previously described (Dhar et al., 2016; Dhar et al., 2012). To compare H3K4me1 and H3K4me3 levels between shLuciferase-, shJARID1D-, and shZMYND8-treated DU145, ChIP with reference exogenous genome (ChIP-Rx) was performed as previously described (Orlando et al., 2014). In brief, two biological replicates for each group were used for ChIP-Rx experiments. Drosophila melanogaster chromatin and Drosophila-specific H2Av antibody were spiked-in to each ChIP reaction as a minor fraction. The spike-in chromatin (53083), Drosophila-specific H2Av antibody (61686), spike-in ChIP positive control primer set (71037), and negative control primer set (71028) were purchased from Active Motif. The immunoprecipitated DNA were analyzed by Next-Generation Sequencing (NGS) (UCI GHTF).
Mouse xenograft experiments
DU145 cells were transfected with pGL4.51[luc2/CMV/Neo] plasmid (Promega) and selected with 500 μg/ml G418. Two weeks later, single cell colonies that stably expressed luciferase and showed chemiluminescence signals upon luciferin treatment were selected using the IVIS 100 Imaging System (Xenogen). The clone DU145-Luc2 #9 was used in mouse xenograft studies. Male nu/nu mice (6-8 weeks old) were purchased from The University of Texas MD Anderson Cancer Center (Houston, Texas). DU145-Luc2 #9 cells were infected with viruses containing shScramble or shZMYND8-97. Cells were selected for at least 3 days using puromycin. Cells (1 × 106 per mouse) were injected via the tail veins. Mice were monitored using the IVIS 100 System after implantation. Eight weeks later, they were euthanized, and lung tissues were collected for standard histological examination. All animals used and studied were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Texas MD Anderson Cancer Center.
Statistical analyses
The statistical significance was analyzed by Student’s t-test. p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***) indicate statistically significant changes.
Supplementary Material
Highlights.
▶ ZMYND8 acts as a JARID1D corepressor for JARID1D-mediated gene repression
▶ PHD-Bromo in ZMYND8 reads H3K4me1-H3K14ac and H3K4me0-H3K14ac
▶ ZMYND8 has an anti-metastasis function
▶ H3K4me1-H3K14ac is associated with repression of metastasis-linked genes
eTOC Blurb.
Li et al. reveal that ZMYND8 recognizes the dual histone mark H3K4me1-H3K14ac and acts as a transcriptional corepressor of the H3K4 demethylase JARID1D. ZMYND8-mediated recognition of H3K4me1-H3K14ac and JARID1D-catalyzed H3K4 demethylation cooperate to antagonize the expression of metastasis-linked genes.
Acknowledgments
We are thankful to Dr. Zhenbo Han and Mr. Su Zhang for their technical assistance and to Drs. Chunru Lin and Liuqing Yang for their reagents. We thank the staff at beamline BL17U of the Shanghai Synchrotron Radiation Facility and the China National Center for Protein Sciences Beijing for providing facility support. This study was supported by grants to M.G.L. from the NIH (R01 CA157919 and R01 GM095659) and the Cancer Prevention and Research Institute of Texas (CPRIT) (RP110183), to H.L. from the Ministry of Science and Technology of China (2016YFA0500700) and the Tsinghua University Initiative Scientific Research Program, to X.S. from the Welch foundation (G1719) and CPRIT (RP110471 and RP140323), to Z. W. from the NIH (ES025761), to Y.L. from the National Natural Science Foundation of China (31400633) and China Postdoctoral Science Foundation (2014T70069), to W.L. from the NIH (R01 HG007538) and CPRIT (RP110471), and to R.S. from the NIH (R01 GM078455 and R01 GM105754) and Sylvester Comprehensive Cancer Center at University of Miami Health System. N.L. was supported by a postdoctoral fellowship from the Center for Cancer Epigenetics at the MD Anderson Cancer Center. Y.L. was supported by a Tsinghua Advanced Innovation fellowship from the Beijing Advanced Innovation Center for Structural Biology at Tsinghua University.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
N.L. designed and performed the biochemical, cell biological, and mouse xenograft experiments. Y.L. designed and carried out structural and biophysical experiments. J.L. and K.C. performed the bioinformatics analysis. X.Z. carried out biophysical experiments. N.L., H.W., G.Z., and H.S. performed the ChIP-seq experiments. T.C., S.D., and P.K. prepared reagents and provided technical assistance. X.S. coordinated peptide array screening. H.L., K.C., Z.W., F.L., R.S. W.L., and X.S. provided expertise and critical input. M.G.L., N.L., H.L., X.Z., Y.L., W.L. and J.L. analyzed data and wrote the manuscript. M.G.L. conceived, initiated, and designed the study. M.G.L. H.L., and W.L. supervised the study.
Accession numbers
The accession number for the structure data reported in this paper is PDB 5B73. The GEO accession number for the ChIP-Seq and RNA-Seq data is GSE82260.
References
- Adhikary S, Sanyal S, Basu M, Sengupta I, Sen S, Srivastava DK, Roy S, Das C. Selective Recognition of H3.1K36 Dimethylation/H4K16 Acetylation Facilitates the Regulation of All-trans-retinoic Acid (ATRA)-responsive Genes by Putative Chromatin Reader ZMYND8. The Journal of biological chemistry. 2016;291:2664–2681. doi: 10.1074/jbc.M115.679985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Berger SL. Histone modifications in transcriptional regulation. Curr Opin Genet Dev. 2002;12:142–148. doi: 10.1016/s0959-437x(02)00279-4. [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Humphrey EL, Erlich RL, Schneider R, Bouman P, Liu JS, Kouzarides T, Schreiber SL. Methylation of histone H3 Lys 4 in coding regions of active genes. Proc Natl Acad Sci U S A. 2002;99:8695–8700. doi: 10.1073/pnas.082249499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- Bitler BG, Aird KM, Garipov A, Li H, Amatangelo M, Kossenkov AV, Schultz DC, Liu Q, Shih Ie M, Conejo-Garcia JR, et al. Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A-mutated cancers. Nature medicine. 2015;21:231–238. doi: 10.1038/nm.3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331:1559–1564. doi: 10.1126/science.1203543. [DOI] [PubMed] [Google Scholar]
- Cheng J, Blum R, Bowman C, Hu D, Shilatifard A, Shen S, Dynlacht BD. A role for H3K4 monomethylation in gene repression and partitioning of chromatin readers. Mol Cell. 2014;53:979–992. doi: 10.1016/j.molcel.2014.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chlenski A, Nakashiro K, Ketels KV, Korovaitseva GI, Oyasu R. Androgen receptor expression in androgen-independent prostate cancer cell lines. The Prostate. 2001;47:66–75. doi: 10.1002/pros.1048. [DOI] [PubMed] [Google Scholar]
- Dhar SS, Lee SH, Chen K, Zhu G, Oh W, Allton K, Gafni O, Kim YZ, Tomoiga AS, Barton MC, et al. An essential role for UTX in resolution and activation of bivalent promoters. Nucleic acids research. 2016;44:3659–3674. doi: 10.1093/nar/gkv1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhar SS, Lee SH, Kan PY, Voigt P, Ma L, Shi X, Reinberg D, Lee MG. Trans-tail regulation of MLL4-catalyzed H3K4 methylation by H4R3 symmetric dimethylation is mediated by a tandem PHD of MLL4. Genes & development. 2012;26:2749–2762. doi: 10.1101/gad.203356.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong F, Chiu LY, Cox B, Aymard F, Clouaire T, Leung JW, Cammarata M, Perez M, Agarwal P, Brodbelt JS, et al. Screen identifies bromodomain protein ZMYND8 in chromatin recognition of transcription-associated DNA damage that promotes homologous recombination. Genes & development. 2015;29:197–211. doi: 10.1101/gad.252189.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu B, Lee MG. Histone H3 lysine 4 methyltransferases and demethylases in self-renewal and differentiation of stem cells. Cell Biosci. 2013;3:39. doi: 10.1186/2045-3701-3-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenther MG, Levine SS, Boyer LA, Jaenisch R, Young RA. A chromatin landmark and transcription initiation at most promoters in human cells. Cell. 2007;130:77–88. doi: 10.1016/j.cell.2007.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459:108–112. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiraga T, Ito S, Nakamura H. Cancer stem-like cell marker CD44 promotes bone metastases by enhancing tumorigenicity, cell motility, and hyaluronan production. Cancer Res. 2013;73:4112–4122. doi: 10.1158/0008-5472.CAN-12-3801. [DOI] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141:52–67. doi: 10.1016/j.cell.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein BJ, Piao L, Xi Y, Rincon-Arano H, Rothbart SB, Peng D, Wen H, Larson C, Zhang X, Zheng X, et al. The histone-H3K4-specific demethylase KDM5B binds to its substrate and product through distinct PHD fingers. Cell Rep. 2014;6:325–335. doi: 10.1016/j.celrep.2013.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutateladze TG. SnapShot: Histone readers. Cell. 2011;146:842–842. doi: 10.1016/j.cell.2011.08.022. e841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan F, Collins RE, De Cegli R, Alpatov R, Horton JR, Shi X, Gozani O, Cheng X, Shi Y. Recognition of unmethylated histone H3 lysine 4 links BHC80 to LSD1-mediated gene repression. Nature. 2007;448:718–722. doi: 10.1038/nature06034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MG, Norman J, Shilatifard A, Shiekhattar R. Physical and functional association of a trimethyl H3K4 demethylase and Ring6a/MBLR, a polycomb-like protein. Cell. 2007;128:877–887. doi: 10.1016/j.cell.2007.02.004. [DOI] [PubMed] [Google Scholar]
- Li N, Dhar SS, Chen TY, Kan PY, Wei Y, Kim JH, Chan CH, Lin HK, Hung MC, Lee MG. JARID1D Is a Suppressor and Prognostic Marker of Prostate Cancer Invasion and Metastasis. Cancer Res. 2016;76:831–843. doi: 10.1158/0008-5472.CAN-15-0906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Wen H, Xi Y, Tanaka K, Wang H, Peng D, Ren Y, Jin Q, Dent SY, Li W, et al. AF9 YEATS domain links histone acetylation to DOT1L-mediated H3K79 methylation. Cell. 2014;159:558–571. doi: 10.1016/j.cell.2014.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Xu J, Wang M, Wang Q, Bi Y, Han M. Tumor-derived vascular endothelial growth factor (VEGF)-a facilitates tumor metastasis through the VEGF-VEGFR1 signaling pathway. International journal of oncology. 2011;39:1213–1220. doi: 10.3892/ijo.2011.1138. [DOI] [PubMed] [Google Scholar]
- Lu Z, Jiang G, Blume-Jensen P, Hunter T. Epidermal growth factor-induced tumor cell invasion and metastasis initiated by dephosphorylation and downregulation of focal adhesion kinase. Molecular and cellular biology. 2001;21:4016–4031. doi: 10.1128/MCB.21.12.4016-4031.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malovannaya A, Lanz RB, Jung SY, Bulynko Y, Le NT, Chan DW, Ding C, Shi Y, Yucer N, Krenciute G, et al. Analysis of the human endogenous coregulator complexome. Cell. 2011;145:787–799. doi: 10.1016/j.cell.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musselman CA, Kutateladze TG. Handpicking epigenetic marks with PHD fingers. Nucleic acids research. 2011;39:9061–9071. doi: 10.1093/nar/gkr613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musselman CA, Lalonde ME, Cote J, Kutateladze TG. Perceiving the epigenetic landscape through histone readers. Nature structural & molecular biology. 2012;19:1218–1227. doi: 10.1038/nsmb.2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemeth JA, Harb JF, Barroso U, Jr., He Z, Grignon DJ, Cher ML. Severe combined immunodeficient-hu model of human prostate cancer metastasis to human bone. Cancer Res. 1999;59:1987–1993. [PubMed] [Google Scholar]
- Orlando DA, Chen MW, Brown VE, Solanki S, Choi YJ, Olson ER, Fritz CC, Bradner JE, Guenther MG. Quantitative ChIP-Seq normalization reveals global modulation of the epigenome. Cell reports. 2014;9:1163–1170. doi: 10.1016/j.celrep.2014.10.018. [DOI] [PubMed] [Google Scholar]
- Pokholok DK, Harbison CT, Levine S, Cole M, Hannett NM, Lee TI, Bell GW, Walker K, Rolfe PA, Herbolsheimer E, et al. Genome-wide map of nucleosome acetylation and methylation in yeast. Cell. 2005;122:517–527. doi: 10.1016/j.cell.2005.06.026. [DOI] [PubMed] [Google Scholar]
- Ruthenburg AJ, Allis CD, Wysocka J. Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Molecular cell. 2007;25:15–30. doi: 10.1016/j.molcel.2006.12.014. [DOI] [PubMed] [Google Scholar]
- Ruthenburg AJ, Li H, Milne TA, Dewell S, McGinty RK, Yuen M, Ueberheide B, Dou Y, Muir TW, Patel DJ, et al. Recognition of a mononucleosomal histone modification pattern by BPTF via multivalent interactions. Cell. 2011;145:692–706. doi: 10.1016/j.cell.2011.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider R, Bannister AJ, Myers FA, Thorne AW, Crane-Robinson C, Kouzarides T. Histone H3 lysine 4 methylation patterns in higher eukaryotic genes. Nat Cell Biol. 2004;6:73–77. doi: 10.1038/ncb1076. [DOI] [PubMed] [Google Scholar]
- Schuettengruber B, Chourrout D, Vervoort M, Leblanc B, Cavalli G. Genome regulation by polycomb and trithorax proteins. Cell. 2007;128:735–745. doi: 10.1016/j.cell.2007.02.009. [DOI] [PubMed] [Google Scholar]
- Shanle EK, Andrews FH, Meriesh H, McDaniel SL, Dronamraju R, DiFiore JV, Jha D, Wozniak GG, Bridgers JB, Kerschner JL, et al. Association of Taf14 with acetylated histone H3 directs gene transcription and the DNA damage response. Genes & development. 2015;29:1795–1800. doi: 10.1101/gad.269977.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih JY, Yang PC. The EMT regulator slug and lung carcinogenesis. Carcinogenesis. 2011;32:1299–1304. doi: 10.1093/carcin/bgr110. [DOI] [PubMed] [Google Scholar]
- Sramkoski RM, Pretlow TG, 2nd, Giaconia JM, Pretlow TP, Schwartz S, Sy MS, Marengo SR, Rhim JS, Zhang D, Jacobberger JW. A new human prostate carcinoma cell line, 22Rv1. In vitro cellular & developmental biology Animal. 1999;35:403–409. doi: 10.1007/s11626-999-0115-4. [DOI] [PubMed] [Google Scholar]
- Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- Tsai WW, Wang Z, Yiu TT, Akdemir KC, Xia W, Winter S, Tsai CY, Shi X, Schwarzer D, Plunkett W, et al. TRIM24 links a non-canonical histone signature to breast cancer. Nature. 2010;468:927–932. doi: 10.1038/nature09542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F, et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009;457:854–858. doi: 10.1038/nature07730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Patel DJ. Combinatorial readout of dual histone modifications by paired chromatin-associated modules. The Journal of biological chemistry. 2011;286:18363–18368. doi: 10.1074/jbc.R111.219139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Schones DE, Zhao K. Characterization of human epigenomes. Current opinion in genetics & development. 2009;19:127–134. doi: 10.1016/j.gde.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, Cui K, Roh TY, Peng W, Zhang MQ, et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nature genetics. 2008;40:897–903. doi: 10.1038/ng.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen H, Li Y, Xi Y, Jiang S, Stratton S, Peng D, Tanaka K, Ren Y, Xia Z, Wu J, et al. ZMYND11 links histone H3.3K36me3 to transcription elongation and tumour suppression. Nature. 2014;508:263–268. doi: 10.1038/nature13045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi Q, Wang Z, Zaromytidou AI, Zhang XH, Chow-Tsang LF, Liu JX, Kim H, Barlas A, Manova-Todorova K, Kaartinen V, et al. A poised chromatin platform for TGF-beta access to master regulators. Cell. 2011;147:1511–1524. doi: 10.1016/j.cell.2011.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell. 2008;14:818–829. doi: 10.1016/j.devcel.2008.05.009. [DOI] [PubMed] [Google Scholar]
- Yun M, Wu J, Workman JL, Li B. Readers of histone modifications. Cell research. 2011;21:564–578. doi: 10.1038/cr.2011.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng L, Zhang Q, Li S, Plotnikov AN, Walsh MJ, Zhou MM. Mechanism and regulation of acetylated histone binding by the tandem PHD finger of DPF3b. Nature. 2010;466:258–262. doi: 10.1038/nature09139. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.