

Abstract
Objective
To carry out a deep characterisation of the main androgen-responsive tissues involved in spinal and bulbar muscular atrophy (SBMA).
Methods
73 consecutive Italian patients underwent a full clinical protocol including biochemical and hormonal analyses, genitourinary examination, bone metabolism and densitometry, cardiological evaluation and muscle pathology.
Results
Creatine kinase levels were slightly to markedly elevated in almost all cases (68 of the 73; 94%). 30 (41%) patients had fasting glucose above the reference limit, and many patients had total cholesterol (40; 54.7%), low-density lipoproteins cholesterol (29; 39.7%) and triglyceride (35; 48%) levels above the recommended values. Although testosterone, luteinising hormone and follicle-stimulating hormone values were generally normal, in one-third of cases we calculated an increased Androgen Sensitivity Index reflecting the presence of androgen resistance in these patients. According to the International Prostate Symptom Score (IPSS), 7/70 (10%) patients reported severe lower urinal tract symptoms (IPSS score >19), and 21/73 (30%) patients were moderately symptomatic (IPSS score from 8 to 19). In addition, 3 patients were carriers of an indwelling bladder catheter. Videourodynamic evaluation indicated that 4 of the 7 patients reporting severe urinary symptoms had an overt prostate-unrelated bladder outlet obstruction. Dual-energy X-ray absorptiometry scan data were consistent with low bone mass in 25/61 (41%) patients. Low bone mass was more frequent at the femoral than at the lumbar level. Skeletal muscle biopsy was carried out in 20 patients and myogenic changes in addition to the neurogenic atrophy were mostly observed.
Conclusions
Our study provides evidence of a wide non-neural clinical phenotype in SBMA, suggesting the need for comprehensive multidisciplinary protocols for these patients.
Introduction
Spinal and bulbar muscular atrophy (SBMA), also known as Kennedy's disease (KD), is a rare, slowly progressive, neuromuscular disorder. It is caused by expansion of a polymorphic CAG repeat sequence encoding a polyglutamine (polyQ) tract in the first exon of the androgen steroid hormone receptor (AR) gene on chromosome X.1 The AR CAG repeat ranges in size from 9 to 36 in healthy respondents but from 38 to 68 in patients with SBMA.2–4
SBMA is characterised by loss of lower motor neurons in the spinal cord and brainstem, leading to progressive limb and bulbar muscle weakness and atrophy. SBMA affects adult males with onset usually between 30 and 50 years.5 6 PolyQ tract length negatively correlates with age at onset without affecting disease progression.2 7 Degeneration of posterior columns can lead to loss of vibratory sensation in distal extremities.8
AR is expressed ubiquitously.9 On binding of testosterone or dihydrotestosterone, AR translocates to the nucleus where it regulates the expression of a subset of genes. In SBMA, polyQ-AR accumulates in the nucleus, causing cell toxicity which is considered a major pathogenic mechanism.10
Nuclear inclusions of the mutated protein are a pathological hallmark of polyQ diseases. Diffuse nuclear and cytoplasmic polyQ-AR accumulation is found in residual motor neurons, as well as in several neural and non-neural tissues in SBMA.5 11 Along with these pathological findings, non-neurological symptoms are common in SBMA. The majority of patients show endocrine dysfunctions mainly consisting of partial androgen insensitivity.2 3 12 These include gynaecomastia, testicular atrophy, erectile dysfunction (ED) and decreased fertility. Abdominal obesity, dyslipidemia and glucose intolerance are also detected in some patients. Elevated serum creatine kinase (CK), higher than expected for a purely neurogenic disease, and myogenic changes on muscle biopsy indicate an underlying myopathy.13 14 Even if overt cardiomyopathy has not been recognised in SBMA,15 an increased incidence of Brugada-type ECG changes has been recently reported in a large Japanese population with SBMA.16
Non-neurological clinical features have not been extensively investigated in previous reports. The aim of the present study was a deep characterisation of the involvement of the main androgen-responsive tissues in a large collection of patients with SBMA. The findings highlight novel non-neural dysfunctions and a wide phenotypic spectrum, suggesting the need for a comprehensive, multidisciplinary approach to SBMA.
Patients and methods
Patients and neurological evaluation
Seventy-three consecutive patients diagnosed with SBMA were recruited after obtaining written informed consent. All were Italian and followed in different centres across Italy. Development of muscular weakness was considered the onset of disease. Since there are no validated functional scales for SBMA, neurological disability was evaluated according to the activity of daily living (ADL) scale17 and the revised Amyotrophic Lateral Sclerosis Functional Rating Scale-Revised (ALSFRS-R).18 Functional status was also assessed using the 6 Min Walk Test (6MWT),19 which is considered a biomarker of motor impairment in SBMA.20 The 6MWT was performed in patients with and without devices.
Biochemical and hormonal analyses
Blood tests included complete blood count, total CK, fasting glucose, glycated haemoglobin (HbA1c), total cholesterol, low-density lipoproteins (LDL), high-density lipoproteins (HDL) and triglycerides. Bone metabolism was investigated by serum calcium, phosphorus, alkaline phosphatase, parathyroid hormone (PTH) and 25-OH vitamin D determination in the same period of the year between March and September. The reproductive hormonal profile included serum concentrations of total testosterone, luteinising hormone (LH) and follicle-stimulating hormone (FSH).21 Testosterone values were multiplied for LH to determine the Androgen Sensitivity Index (ASI) (normal values <138 U×nmol/L2).22 Prostate-specific antigen (PSA) was also measured.
Genitourinary examination
According to the International Prostate Symptom Score (IPSS),23 lower urinary tract symptoms (LUTS) were classified as mild (IPSS 0–7), moderate (8–19) and severe (>19) (see online supplementary material). Seven patients reporting severe LUTS (IPSS>19) were further characterised by prostate volume analysis by transabdominal ultrasonography24 and videourodynamic evaluation performed according to the International Continence Society (ICS) recommendations.25 The results of the pressure-flow study were classified according to ICS normograms.25 Images were obtained to evaluate morphology of the bladder, bladder neck and urethra, and vesicourethral reflux. The presence and severity of ED was investigated with the International Index of Erectile Function (IIEF-15) questionnaire.26 ED was considered as IIEF<26.26 The presence of gynaecomastia was also recorded.
Bone densitometry
Femur and lumbar (L1–L4) dual-energy X-ray absorptiometry (DXA) scans were performed by the same technician, with spine phantom calibration before each examination. The mean Bone Mineral Density (BMD) index and mean T-scores were considered. The total BMD coefficient of variation was 1%. Osteopenia was defined as T-score <−1 SD, and osteoporosis as ≤−2.5 SD.27
Cardiological evaluation
Cardiological evaluation included clinical history, standard 12-lead ECG and standard two-dimensional Doppler echocardiography. Additionally, all patients underwent a modified ECG recording with right precordial leads placed to the third intercostal space, to increase the sensitivity for Brugada-like abnormalities. Brugada-like ST-segment and T-wave (ST-T) abnormalities in right precordial leads were classified according to the second consensus conferences on the Brugada syndrome.28 The sodium channel blocker test was not performed. All ECGs were evaluated by two experienced electrophysiologists (DC and MS).
Muscle pathology
Twenty patients underwent muscle biopsy as part of the diagnostic workup. Serial cryostat sections of fresh frozen tissue were stained with routine histochemical reactions.29 Muscle tissue involvement was classified according to our previous study13: type I, neurogenic atrophy (ie, type-grouping, atrophic and target/targetoid fibres); type II, neurogenic atrophy plus one or two myopathic changes (>3% centrally nucleated fibres, vesicular nuclei, basophilic degenerating fibres, lobulated fibres, fibre splitting); type III, neurogenic atrophy plus three or more myopathic changes. Age and ADL score at biopsy were recorded for each patient.
Genetic analysis
Genomic DNA was extracted from peripheral blood leucocytes using the standard salting out procedure. CAG repeats were amplified by PCR as previously described7 and repeats fragment sizing was performed on an ABI PRISM 3700 DNA Sequencer (Applied Biosystems, Foster City, California, USA). The specific length of CAG repeats was further verified via Sanger sequencing.
Statistical analysis
Linear correlation analysis was performed using the rank linear regression of Spearman. A Student t test and χ2 test were used to compare patients with SBMA with control data from 60 age-matched healthy males. The significance level was set at p<0.05.
The study was approved by the local Ethics Committee.
Results
Patients’ ages ranged from 27 to 78 years (mean 57.3 years; SD=10.2). The mean disease duration since disease onset, which occurred on average at 42.26±9.4 years (range 29–64; median=46), was 13.1±6.9 years (range 0–29; median=13). CAG repeat numbers ranged from 43 to 52 (mean 45.87; SD=2.56). At the time of examination, 12 (16%) patients had no muscular weakness (ADL scale grade=0), 41 (56%) patients had mild weakness (grade 1); 13 (18%) mild-to-moderate weakness (grade 2); and 7 (10%) moderate-to-severe weakness (grade 3). Weakness-free patients reported muscle cramps, fasciculations, fatigability, or had elevated serum CK. Mean ALSFRS-R was 41±3.7 (range 30–48; median=42). Eleven patients were unable to complete the 6MWT due to severe walking impairment or because they were wheelchair users. The average 6MWT distance was 356±127 m (range 107–578; median=322).
Biochemical and hormonal data are shown in tables 1 and 2. CK levels were slightly-to-markedly elevated in 68/73 cases (94%). Thirty (41%) patients had fasting glucose above reference; of these, 20 also had increased Hb1Ac. Many patients had total cholesterol (40; 54.7%), LDL cholesterol (29; 39.7%) and triglycerides (35; 48%) above recommended levels. HDL was below recommended levels in 50/73 (68.4%) patients, of whom 24 had high total cholesterol. Concomitant elevated fasting glucose and total cholesterol were observed in 15 patients (20.5%). Seven patients were on hyperlipidaemia-correcting medications and 10 on oral antidiabetics. No correlations were observed between biochemical data and age, or polyQ length.
Table 1.
Biochemical profile of patients with spinal and bulbar muscular atrophy
| Mean±SD (range; median) | Reference range | Out of reference range | ||
|---|---|---|---|---|
| High | Low | |||
| Biochemical analyses | ||||
| Haematocrit (%) | 0.43±0.02 (0.37–0.50; 0.43) | 0.41–0.507 | 1/73 | 0/73 |
| Creatine kinase (UI/L) | 1095.8±823 (119–4406; 823) | 20–180 | 68/73 | 0/73 |
| Fasting glucose (nmol/L) | 5.9±1.7 (3.7–14.1; 5.4) | 3.7–5.6 | 29/73 | 0/73 |
| Glycated haemoglobin (nmol/L) | 38.9±8.2 (25–75; 37) | 20–38 | 25/73 | 0/73 |
| Triglycerides (mg/dL) | 1.94±1.26 (0.51–9.28; 1.66) | <2.11 | 35/73 | – |
| Total cholesterol (nmol/L/L) | 5.3±1.0 (3.32–8; 5.33) | <5.18 | 40/73 | – |
| High-density lipoprotein (nmol/L) | 1.42±0.5 (0.32–2.86; 1.34) | >1.55 | – | 50/73 |
| Low-density lipoprotein (nmol/L) | 3.3±0.87 (1.78–4.68; 3.2) | <3.34 | 29/73 | – |
| Prostate-specific antigen (µg/L) | 1.17±1.18 (0.11–21.7; 0.5) | <4 | 3/73 | – |
Table 2.
Hormonal profile of patients with SBMA
| Mean±SD (range; median) | Reference range | Out of reference range | Controls Mean±SD |
p Value of patients vs controls | ||
|---|---|---|---|---|---|---|
| High | Low | |||||
| Total testosterone (nmol/L) | 19.2±7.1 (6.79–41.88; 18) | 10–29 | 7/73 | 5/73 | 16.2±4.4 | 0.005 |
| Follicle-stimulating hormone (IU/L) | 7.2±5.1 (1.2–26; 6) | 1–8 | 23/73 | 0/73 | 6.1±3.2 | ns |
| LH (IU/L) | 6.84±3.6 (1.97–25.5; 5.9) | 1–8 | 20/73 | 0/73 | 4.8±3.6 | 0.0015 |
| ASI (LH×testosterone) (U×nmol/L2) | 135.17±99.6 (21.7–619.39; 109.2) | <138 | 26/73 | – | 82.7±43.8 | 0.0001 |
ASI, Androgen Sensitivity Index; LH, luteinising hormone; ns, not significant; SBMA, spinal and bulbar muscular atrophy.
Testosterone was decreased in five cases (7%) and elevated in seven (9%). Above reference testosterone concomitant with elevated LH/FSH was observed in two cases; none had concomitant reduced LH/FSH. In all cases with decreased testosterone, LH and FSH values were normal. Both testosterone and LH values were significantly higher in patients than in controls (p=0.005 and p=0.0015, respectively). The average ASI value (LH×testosterone) in our cohort of patients was within the normal range (135.17±99.6 U×nmol/L2) and, as expected, higher than in controls (p=0001). However, 26 patients (35%) showed a slightly-to-markedly increased ASI. No relations between testosterone/ASI values and polyQ length, age at onset, functional scores and biochemical data were found.
Genitourinary tract details are summarised in online supplementary table S1. Serum PSA was increased in three patients (4%). Based on IPSS, 21 patients (30%) were moderately symptomatic and 7 (10%) reported severe LUTS. Forty-two (60%) patients were asymptomatic/mildly symptomatic. The IPSS score directly correlated with age (p=0.0051; see online supplementary figure S1), and inversely with ADL scale grade (p=0.018; see online supplementary figure S2); it was otherwise unrelated to ASI, polyQ length and biochemical tests. Three patients carrying an indwelling bladder catheter (aged 69–78 years) did not fill in the questionnaire. One had a diagnosis of underactive detrusor function in the absence of cervical-urethral obstruction, while a prostate-unrelated obstruction of the bladder outlet was reported in the remaining two patients. Patients with severe LUTS underwent transabdominal ultrasonography and videourodynamic evaluation to better define underlying mechanisms. In this group, the median age was 66 years (52–69); all patients had normal PSA and had not undergone prostate surgery. Two patients were taking α-blockers. Ultrasonography demonstrated normal or moderately hypertrophic prostate volume (median 35 cc, range 25–50). Videourodynamic evaluation indicated that four patients had overt bladder outlet obstruction, two were in the equivocal area and one was unobstructed. In all patients, the cystourethrography was normal, with a competent bladder neck regularly opening during micturition, and no vesicoureteral reflux, bladder diverticulum or detrusor-external sphincter dyssynergia.
Ten (13.6%) patients refused to fill in the IIEF questionnaire. For all remaining patients, IIEF questionnaire results pointed to a mild-to-severe ED (mean 15.9±7.6; range 0–25). IIEF scores were inversely correlated with age (r=−0.46; p=0.0001) but were unrelated to ASI and polyQ length.
In 57/73 (78%) patients, a bilateral gynaecomastia was noted. Of them, two had undergone surgery for breast reduction. Also, the presence of gynaecomastia was unrelated to ASI and polyQ length.
Bone metabolism and DXA scan data are summarised in table 3. We observed 22/61 (36%) cases of lumbar and/or femoral osteopenia, and 3 (5%) cases of femoral osteoporosis. Low BMD was more frequent at the femoral level (20 patients) than lumbar level (1 patient). Lumbar and femoral decreased BMD was present in four patients. Compared with controls, a higher number of patients with SBMA showed low BMD at the femoral level (p=0.022). Conversely, patients with SBMA had higher BMD and T-scores (p=0.0019 and p=0.0001, respectively) at the lumbar level. Serum 25-hydroxyvitamin D deficiency (<50 nmol/L) was observed in 40/61 (65%) patients, while PTH, calcium, phosphorus and alkaline phosphatase were normal in all respondents. No difference in BMD was observed between patients with normal LH-testosterone levels and those with hypogonadism (low testosterone and/or elevated testosterone/LH), nor between patients with normal versus deficient 25-hydroxyvitamin D. Similarly, no difference was observed between patients with normal and high ASI. DXA data were also unrelated to functional measures, polyQ length and age.
Table 3.
Bone metabolism and DXA findings of patients with SBMA
| Mean±SD (range; median) | Controls Mean±SD |
p Value of patients vs controls | |
|---|---|---|---|
| Bone metabolism analyses | |||
| Serum calcium (mmol/L) (reference range 2.1–2.8) | 2.4±0.1 (2.19–2.69; 2.39) | 2.3±0.1 | ns |
| Serum phosphorus (mmol/L) (reference range 1–1.5) | 0.97±0.14 (0.64–1.29; 0.98) | 1.0±0.2 | ns |
| Alkaline phosphatase (UI/L) (reference range 44–147) | 55.7±16.5 (34–111; 52) | – | – |
| Parathyroid hormone (pg/mL) (reference range 10–60) | 28.7±12.0 (5.8–63; 27.1) | 33.3±15.6 | ns |
| 25-OH vitamin D (nmol/L) (reference range >50) | 43.3±23.0 (8.9–133; 40.84) | 37.6±28.5 | ns |
| DXA parameters | |||
| Lumbar BMD (g/cm2) | 1.19±0.19 (0.79–1.42; 1.14) | 1.08±0.21 | 0.0019 |
| Lumbar T-score | 0.66±1.38 (−1.5–5.2; 0.45) | −0.33±0.95 | 0.0001 |
| Femoral BMD (g/cm2) | 0.97±0.22 (0.26–1.9; 0.95) | 1.02±0.19 | ns |
| Femoral T-score | −0.65±1.16 (−4.31–1.9; −0.6) | −0.41±0.72 | ns |
| Low BMD | Patient number | Control number | p patients vs controls |
| Lumbar level | 1/61 | 6/60 | 0.045 |
| Femoral level | 20/61 | 9/60 | 0.022 |
| Femoral/lumbar level | 4/61 | 3/60 | ns |
BMD, Bone Mineral Density; DXA, dual-energy X-ray absorptiometry; ns, not significant; SBMA, spinal and bulbar muscular atrophy.
Twenty-seven patients had hypertension and three had ischaemic heart disease. There was no evidence of specific structural heart muscle disease. Three patients (4%) had Brugada-like ECG changes. One patient showed a type 2 ‘saddleback’ pattern in the standard V1-V2 precordial leads (fourth intercostal space; figure 1A), which became type 1 ‘coved type’ in upward right precordial leads (third intercostal space); in the other two patients, Brugada ECG changes were detected only in the right precordial leads placed in the third intercostal space and both were non-diagnostic ‘saddleback’ patterns (types 2 and 3, respectively).28 Other ECG alterations were identified in 15 (20.5%) patients, mostly consisting of left ventricular hypertrophy, pathological Q-waves (4 cases) and intraventricular conduction abnormalities (3 cases).
Figure 1.
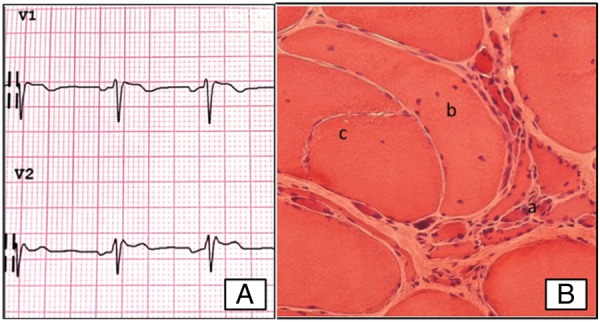
(A) One patient showed a type 2 ‘saddleback’ pattern in the standard V1-V2 precordial leads (fourth intercostal space). (B) Representative skeletal muscle pathology of a SBMA patient. A cluster of atrophic, angulated fibres (a), several fibres with central nuclei (b).
Muscle biopsies were obtained from quadriceps femoris in 19 patients and from biceps brachii in 1 (table 4). All patients who underwent muscle biopsy showed weakness, and/or fasciculations, and/or muscle atrophy. Eighteen of the 20 biopsies showed myopathic changes together with neurogenic atrophy (figure 1B), graded type I–III as explained in the Methods section. Myopathic grading was unrelated to age at onset, nor to disease duration, ADL scale or age at biopsy.
Table 4.
Clinical and muscle pathology features of patients with spinal and bulbar muscular atrophy
| Patient | Age at onset (year) | Age at biopsy (year) | CAGs (nr) | ADL scale grade at biopsy | Muscle pathology |
|---|---|---|---|---|---|
| 1 | 45 | 61 | 47 | 2 | II |
| 2 | 46 | 43 | 42 | 1 | I |
| 3 | 50 | 57 | 45 | 2 | II |
| 4 | 64 | 71 | 44 | 1 | III |
| 5 | 41 | 45 | 47 | 0 | II |
| 6 | 37 | 44 | 50 | 1 | III |
| 7 | 46 | 55 | 48 | 1 | III |
| 8 | 24 | 28 | 44 | 1 | I |
| 9 | 29 | 39 | 47 | 1 | III |
| 10 | 49 | 55 | 45 | 2 | III |
| 11 | 48 | 51 | 44 | 1 | II |
| 12 | 35 | 38 | 50 | 0 | III |
| 13 | 50 | 52 | 44 | 1 | III |
| 14 | 54 | 54 | 44 | 0 | II |
| 15 | 52 | 56 | 41 | 2 | III |
| 16 | 58 | 70 | 45 | 2 | II |
| 17 | 70 | 80 | 42 | 1 | III |
| 18 | 68 | 68 | 45 | 1 | II |
| 19 | 45 | 57 | 46 | 2 | III |
| 20 | 50 | 59 | 47 | 2 | I |
ADL scale, activity of daily living scale (0: normal; 1: mild weakness of limb muscles, climbs stairs easily but aware of weakness; 2: mild-to-moderate weakness, climbs stairs with difficulty and generally uses a cane; 3: moderate-to-severe weakness, uses a wheelchair most of the time or mostly recumbent); muscle pathology (type I, only signs of neurogenic atrophy; type II, signs of neurogenic atrophy plus 1 or 2 myopathic changes; type III, signs of neurogenic atrophy plus 3 or more myopathic changes); nr, number of triplets.
Discussion
The observation that polyQ-AR expression is elevated in lower motor neurons—the vulnerable neuronal cell type in SBMA—had led to the assumption that SBMA is a motor neuron disease. However, several lines of evidence, ranging from cell biology to clinical features, support SBMA as a multisystem disorder. We show a wider than known clinical involvement of non-neural androgen-responsive tissues in patients with SBMA.
We found that more than 40% of patients with SBMA in our cohort reported moderate or severe LUTS, three of them carrying indwelling bladder catheters. This frequency figure is definitely higher than expected since the prevalence of LUTS has been estimated to amount up to no more than 30% in a general male population of the same age range.30 Videourodynamic evaluation mostly pointed to a bladder outlet obstruction in patients with severe urinary symptoms. While the age of these respondents (median 66 years) would suggest benign prostatic hyperplasia,31 these observations argue against such a diagnosis: ultrasonography ruled out prostate enlargement; PSA values were normal; cystourethrography was always unremarkable, with a competent bladder neck regularly opening during micturition; two carriers of an indwelling bladder catheter had a prostate-unrelated obstruction. The mechanisms through which polyQ-AR could cause bladder obstruction is unclear, but androgen insensitivity may be postulated. Low androgen levels have been associated with a higher risk of bladder outlet obstruction,32 and hypogonadism represents a key factor bridging metabolic syndrome and urinary symptoms.33 The finding of urinary tract obstruction in SBMA is quite intriguing since an analogous mechanism leading to renal failure and death has been observed in the murine SBMA model, in which altered excitability of elevator ani/bulbocavernosus muscles could contribute to urinary obstruction.34 Since motor neurons to bulbocavernosus muscles, originating from Onuf's nucleus, are protected from neurodegeneration,11 a similar cell-autonomous dysfunction of pelvic floor muscles could be considered in patients with SBMA, given the increasing literature supporting the idea of skeletal muscle as a site of mutant AR toxicity.13 35–38 An autonomic dysfunction might be also involved. Patients affected with SBMA usually do not report symptoms that may suggest autonomic dysfunction; nevertheless, the subclinical involvement of both the orthosympathetic and parasympathetic nervous systems has been reported.39 Moreover, one single patient, carrying the longest CAG repeat ever found (68 triplets), reported autonomic dysfunctions such as decreased sweating and difficulties with ejaculation.4 In our SBMA cohort, we have studied Sympathetic Skin Response (SSR) and our preliminary findings are consistent with a prolonged latency of the potential (unpublished data).
Androgens act on bone metabolism, promoting periosteal bone formation mostly during puberty,40 and reducing bone resorption mostly during adult life.41 Indeed, hypogonadism was found in about 15% of men with osteoporosis,40 and patients with androgen insensitivity syndrome had decreased BMD.42 43 About 40% of patients with SBMA in our cohort showed low BMD mainly consistent with osteopenia. This prevalence was not unexpected as osteopenia is reported in at least 30% of men aged 50 years or older.44 45 In addition, osteopenia and fractures are common in patients with muscular atrophy,46–48 and increased bone resorption was detected in patients with amyotrophic lateral sclerosis.49 We were surprised by the relatively well-preserved spinal bone density compared with the increased frequency of low femoral BMD, this latter being even higher than in controls. A possible explanation might be that reduced mechanical load secondary to muscle weakness in the lower limbs—the most frequent site of disease onset—had led to femoral bone loss.50 However, the lack of correlation between DXA measures and functional scores argues against this hypothesis. Moreover, it would remain unexplained why bone density is preserved at the lumbar level compared with age-matched male patients.44 45 The potential to evaluate contributions of 25-hydroxyvitamin D deficiency is limited, since no difference in BMD was observed between patients with normal versus deficient serum levels. Taken together, these findings suggest the need for a careful bone health follow-up in SBMA.
As previously described,15 there was no sign of structural cardiomyopathy in our patients. A recent study reported a high prevalence (11.8%) of Brugada-like ECG in a Japanese population with SBMA, ascribed to a downregulation of the SCN5A gene leading to sodium current reduction in the myocardium.16 Of note, two patients had symptomatic Brugada syndrome and died suddenly during follow-up. In our study, we found non-diagnostic Brugada ECG abnormalities in three patients (4%), mostly recorded in non-conventional upward right precordial leads (third intercostal space), and not associated with any relevant symptom. This prevalence is significantly lower than that reported in the Japanese study: the discrepancy may be explained by the different ethnic background, the Brugada syndrome more frequent in the Asian than the Caucasian population. Although no sudden cardiac death events were observed in our cohort, our findings confirm the need for accurate and serial ECG evaluation to identify Brugada-like repolarisation abnormalities. Thus, our suggestion is to record lead V1-V2 over the III and II intercostal space, to enhance the sensitivity for detection of Brugada like ECG-changes. Patients presenting with a Brugada-like ECG should be prudently advised to prevent and correct hypokalaemia, to promptly treat a fever >38° by antipyretic therapy, and to use caution in taking antiarrhythmic drugs known to worsen ECG abnormalities and potentially trigger ventricular tachyarrhythmias in Brugada syndrome.
Twenty patients in our cohort underwent skeletal muscle biopsy during their diagnostic workup. As in our previous study conducted on a smaller series,13 we detected myogenic changes in addition to neurogenic atrophy in the majority of biopsies. We did not confirm any relationship of motor disability with the amount of myogenic pathology, even if, interestingly, the highest CAG repeat number (50) was measured in two mildly symptomatic patients with type III muscle pathology.
Hormonal and biochemical profiles were overall consistent with previous reports.2 3 36 51 52 We confirmed elevated CK levels in nearly all cases. Although testosterone, LH and FSH were generally normal, in one-third of cases we calculated a slightly-to-markedly increased ASI, reflecting androgen resistance. Our patients with SBMA frequently had elevated total cholesterol, triglycerides and fasting glucose, underlying a partial metabolic syndrome. Given the age range of our patients, we cannot establish to what extent androgen insensitivity rather than age per se contributes to these metabolic abnormalities. Previously, Rhodes et al.3 found mean cholesterol and glucose levels in their cohort of patients with SBMA similar to a national sample of age-matched men. In our patients, we did not note any age-dependent change of biochemical parameters. Whatever the mechanism, the need to treat diabetes/glucose intolerance and hyperlipidaemia raises some questions. For instance, the use of statins may be challenging. Statin-induced muscle toxicity is well documented,53 and there are no published data on safety of statins in SBMA. A similar therapeutic issue may be recognised for ED, which was observed at variable degrees in every patient in our cohort. The lack of scientific evidence supporting the efficacy and safety of medications for ED or other disorders related to SBMA highlights the need for clinical trials of symptomatic agents along with those for disease-modifying treatments.
We provided evidence of a wide non-neural clinical phenotype in SBMA. While the mechanism of the polyQ-AR damage remains to be established, that is, whether polyQ expansion in AR causes disease through a loss of function or a gain of toxic function, more effort should be made to set up comprehensive, multidisciplinary protocols for patients affected with SBMA and, possibly, for symptomatic female carriers.
Supplementary Material
Footnotes
Correction notice: This article has been corrected since it was published Online First. The Figure 1B footnote has been corrected to “(B) Representative skeletal muscle pathology of a SBMA patient. A cluster of atrophic, angulated fibres (a), several fibres with central nuclei (b)”.
Collaborators: The Italian Study Group on Kennedy's disease: Jessica Mandrioli, MD and Giuliana Galasso, MD, Department of Neurosciences, Sant'Agostino-Estense Hospital, University of Modena and Reggio Emilia, Modena, Italy; Letizia Mazzini, MD, ALS Centre Department of Neurology, Eastern Piedmont University, Maggiore della Carità Hospital, Novara, Italy; Silvia Romito, MD, Neurology Unit, Verona Hospital, Verona, Italy; Paola Tonin, MD and Mauro Scarpelli, MD, Department of Neurological Sciences and Vision, Section of Neurology, University of Verona, Verona, Italy; Giulia Ricci, MD and Gabriele Siciliano, MD, Department of Clinical and Experimental Medicine, Neurological Clinic, University of Pisa, Pisa, Italy; Antonio Petrucci, MD, Neurology Unit, San Camillo Hospital, Rome, Italy; Roberto Massa, MD, Institute of Neurology, Tor Vergata University, Rome, Italy; Alberto Polo, MD, Neurology Unit, Legnago Hospital, Legnago (VR), Italy; Caterina Mariotti, MD, Unit of Genetics of Neurodegenerative and Metabolic Diseases—IRCCS Foundation, “C. Besta” Neurological Institute, Milan, Italy; Anna Sagnelli, MD, Clinic of Central and Peripheral Degenerative Neuropathies Unit, Department of Clinical Neurosciences—IRCCS Foundation, “C. Besta” Neurological Institute, Milan, Italy; Arianna Palmieri, PhD, Department of Philosophy, Sociology, Pedagogy and Applied Psychology, University of Padova, Padova, Italy; Chiara Briani, Neuromuscular Center, Department of Neurosciences, University of Padova, Padova, Italy.
Contributors: GS had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. GS and GQ contributed in the study concept and design. GQ, GS, VS, Mandrioli, Galasso, Mazzini, Dr Romito, Dr Tonin, Dr Scarpelli, Dr Ricci, Dr Siciliano, Dr Petrucci, Dr Massa, Dr Polo, Dr Mariotti, Dr Sagnelli and Dr Briani were involved in the patient recruitment. GQ, LB, CS, EDR, MV, GZ, EP and AG carried out the neurological evaluation. DC and MS took part in the cardiological evaluation. CF, AF, NC, LA and MI performed the genitourinary assessment. Palmieri contributed in the IIEF administration. AF and LS participated in the bone studies. CB and MP conducted the genetic analyses. ME carried out the statistical analysis. All the authors were involved in the critical revision of the manuscript for important intellectual content. GS and GQ acted as the study supervisors.
Funding: This research was supported in part by research grants 18722 from AFM Telethon (GS), RF-2011-02350097 from the Italian Ministry of Health (GS), and GTB12001D from Telethon Biobank (EP).
Competing interests: None declared.
Ethics approval: Ethics Committee of “Azienda Ospedaliera di Padova”, Padova, Italy.
Provenance and peer review: Not commissioned; externally peer reviewed.
Contributor Information
Collaborators: Jessica Mandrioli, Giuliana Galasso, Letizia Mazzini, Silvia Romito, Paola Tonin, Mauro Scarpelli, Giulia Ricci, Gabriele Siciliano, Antonio Petrucci, Roberto Massa, Alberto Polo, Caterina Mariotti, Anna Sagnelli, Arianna Palmieri, and Chiara Briani
References
- 1.La Spada AR, Wilson EM, Lubahn DB, et al. . Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature 1991;352:77–9. 10.1038/352077a0 [DOI] [PubMed] [Google Scholar]
- 2.Atsuta N, Watanabe H, Ito M, et al. . Natural history of spinal and bulbar muscular atrophy (SBMA): a study of 223 Japanese patients. Brain 2006;129:1446–55. 10.1093/brain/awl096 [DOI] [PubMed] [Google Scholar]
- 3.Rhodes LE, Freeman BK, Auh S, et al. . Clinical features of spinal and bulbar muscular atrophy. Brain 2009;132:3242–51. 10.1093/brain/awp258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grunseich C, Kats IR, Bott LC, et al. . Early onset and novel features in a spinal and bulbar muscular atrophy patient with a 68 CAG repeat. Neuromuscul Disord 2014;24:978–81. 10.1016/j.nmd.2014.06.441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Adachi H, Katsuno M, Minamiyama M, et al. . Widespread nuclear and cytoplasmic accumulation of mutant androgen receptor in SBMA patients. Brain 2005;128:659–70. 10.1093/brain/awh381 [DOI] [PubMed] [Google Scholar]
- 6.Katsuno M, Tanaka F, Adachi H, et al. . Pathogenesis and therapy of spinal and bulbar muscular atrophy (SBMA). Prog Neurobiol 2012;99:246–56. 10.1016/j.pneurobio.2012.05.007 [DOI] [PubMed] [Google Scholar]
- 7.Fratta P, Nirmalananthan N, Masset L, et al. . Correlation of clinical and molecular features in spinal bulbar muscular atrophy. Neurology 2014;82:2077–84. 10.1212/WNL.0000000000000507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Polo A, Teatini F, D'Anna S, et al. . Sensory involvement in X-linked spino-bulbar muscular atrophy (Kennedy's syndrome): an electrophysiological study. J Neurol 1996;243:388–92. 10.1007/BF00868997 [DOI] [PubMed] [Google Scholar]
- 9.Poletti A. The polyglutamine tract of androgen receptor: from functions to dysfunctions in motor neurons. Front Neuroendocrinol 2004;25:1–26. 10.1016/j.yfrne.2004.03.001 [DOI] [PubMed] [Google Scholar]
- 10.Beitel LK, Alvarado C, Mokhtar S, et al. . Mechanisms mediating spinal and bulbar muscular atrophy: investigations into polyglutamine-expanded androgen receptor function and dysfunction. Front Neurol 2013;4:53 10.3389/fneur.2013.00053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li M, Miwa S, Kobayashi Y, et al. . Nuclear inclusions of the androgen receptor protein in spinal and bulbar muscular atrophy. Ann Neurol 1998;44:249–54. 10.1002/ana.410440216 [DOI] [PubMed] [Google Scholar]
- 12.Dejager S, Bry-Gauillard H, Bruckert E, et al. . A comprehensive endocrine description of Kennedy's Disease revealing androgen insensitivity linked to CAG repeat length. J Clin Endocrinol Metab 2002;87:3893–901. [DOI] [PubMed] [Google Scholar]
- 13.Sorarù G, D'Ascenzo C, Polo A, et al. . Spinal and bulbar muscular atrophy: skeletal muscle pathology in male patients and heterozygous females. J Neurol Sci 2008;264:100–5. 10.1016/j.jns.2007.08.012 [DOI] [PubMed] [Google Scholar]
- 14.Sorenson EJ, Klein CJ. Elevated creatine kinase and transaminases in asymptomatic SBMA. Amyotroph Lateral Scler 2007;8:62–4. 10.1080/17482960600765040 [DOI] [PubMed] [Google Scholar]
- 15.Querin G, Melacini P, D'Ascenzo C, et al. . No evidence of cardiomyopathy in spinal and bulbar muscular atrophy. Acta Neurol Scand 2013;128:30–2. 10.1111/ane.12140 [DOI] [PubMed] [Google Scholar]
- 16.Araki A, Katsuno M, Suzuki K, et al. . Brugada syndrome in spinal and bulbar muscular atrophy. Neurology 2014;82:1813–21. 10.1212/WNL.0000000000000434 [DOI] [PubMed] [Google Scholar]
- 17.Doyu M, Sobue G, Mukai E, et al. . Severity of X-linked recessive bulbospinal neuronopathy correlates with the size of the tandem CAG repeat in androgen receptor gene. Ann Neurol 1992;32:707–10. 10.1002/ana.410320517 [DOI] [PubMed] [Google Scholar]
- 18.Cedarbaum JM, Stambler N, Malta E, et al. . The ALSFRS-R: a revised ALS functional rating scale that incorporates assessments of respiratory function. BDNF ALS Study Group (Phase III). J Neurol Sci 1999;169:13–21. 10.1016/S0022-510X(99)00210-5 [DOI] [PubMed] [Google Scholar]
- 19.ATS Committee on Proficiency Standards for Clinical Pulmonary Function Laboratories. ATS statement: guidelines for the Six-Minute Walk Test. Am J Respir Crit Care Med 2002;166:111–17. 10.1164/ajrccm.166.1.at1102 [DOI] [PubMed] [Google Scholar]
- 20.Takeuchi Y, Katsuno M, Banno H, et al. . Walking capacity evaluated by the 6-Minute Walk Test in spinal and bulbar muscular atrophy. Muscle Nerve 2008;38:964–71. 10.1002/mus.21077 [DOI] [PubMed] [Google Scholar]
- 21.Aiman J, Griffin JE, Gazak JM, et al. . Androgen insensitivity as a cause of infertility in otherwise normal men. N Engl J Med 1979;300:223–7. 10.1056/NEJM197902013000503 [DOI] [PubMed] [Google Scholar]
- 22.Zuccarello D, Ferlina A, Vinanzi C, et al. . Detailed functional studies on androgen receptor mild mutations demonstrate their association with male infertility. Clin Endocrinol 2008;68:580–8. 10.1111/j.1365-2265.2007.03069.x [DOI] [PubMed] [Google Scholar]
- 23.Plante M, Corcos J, Gregoire I, et al. . The International Prostate Symptom Score: physician versus self-administration in the quantification of symptomatology. Urology 1996;47:326–8. 10.1016/S0090-4295(99)80447-6 [DOI] [PubMed] [Google Scholar]
- 24.Kälkner KM, Kubicek G, Nilsson J, et al. . Prostate volume determination: differential volume measurements comparing CT and TRUS. Radiother Oncol 2006;81:179–83. 10.1016/j.radonc.2006.10.003 [DOI] [PubMed] [Google Scholar]
- 25.Schäfer W, Abrams P, Liao L, et al. , International Continence Society. Good urodynamic practices: uroflowmetry, filling cystometry, and pressure-flow studies. Neurourol Urodyn 2002;21:261–74. 10.1002/nau.10066 [DOI] [PubMed] [Google Scholar]
- 26.Rosen RC, Riley A, Wagner G, et al. . The International Index of Erectile Function (IIEF): a multidimensional scale for assessment of erectile dysfunction. Urology 1997;49:822–30. 10.1016/S0090-4295(97)00238-0 [DOI] [PubMed] [Google Scholar]
- 27.Leib ES, Lewiecki EM, Binkley N, et al. , International Society for Clinical Densitometry. Official positions of the International Society for Clinical Densitometry. J Clin Densitom 2004;7:1–6. 10.1385/JCD:7:1:1 [DOI] [PubMed] [Google Scholar]
- 28.Antzelevitch C, Brugada P, Borggrefe M, et al. . Brugada syndrome: report of the second consensus conference: endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation 2005;111:659–70. 10.1161/01.CIR.0000152479.54298.51 [DOI] [PubMed] [Google Scholar]
- 29.Dubowitz V. Histological and histochemical stains and reactions. In: Dubowitz V, ed. Muscle biopsy. A practical approach. London, UK: Bailliere Tindall, 1985:19–40. [Google Scholar]
- 30.Verhamme KM, Dieleman JP, Bleumink GS, et al. . Incidence and prevalence of lower urinary tract symptoms suggestive of benign prostatic hyperplasia in primary care—the Triumph project. Eur Urol 2002;42:323–8. 10.1016/S0302-2838(02)00354-8 [DOI] [PubMed] [Google Scholar]
- 31.Corona G, Vignozzi L, Rastrelli G, et al. . Benign prostatic hyperplasia: a new metabolic disease of the aging male and its correlation with sexual dysfunctions. Int J Endocrinol 2014;2014:32945 10.1155/2014/329456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koritsiadis G, Stravodimos K, Mitropoulos D, et al. . Androgens and bladder outlet obstruction: a correlation with pressure-flow variables in a preliminary study. BJU Int 2008;101:1542–6. 10.1111/j.1464-410X.2008.07521.x [DOI] [PubMed] [Google Scholar]
- 33.Hammarsten J, Peeker R. Urological aspects of the metabolic syndrome. Nat Rev Urol 2011;8:483–94. 10.1038/nrurol.2011.112 [DOI] [PubMed] [Google Scholar]
- 34.Yu Z, Dadgar N, Albertelli M, et al. . Androgen-dependent pathology demonstrates myopathic contribution to the Kennedy disease phenotype in a mouse knock-in model. J Clin Investigation 2006;116:2663–72. 10.1172/JCI28773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Monks DA, Johansen JA, Mo K, et al. . Overexpression of wild-type androgen receptor in muscle recapitulates polyglutamine disease. Proc Natl Acad Sci U S A 2007;104:18259–64. 10.1073/pnas.0705501104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Palazzolo I, Stack C, Kong L, et al. . Overexpression of IGF-1 in muscle attenuates disease in a mouse model of spinal and bulbar muscular atrophy. Neuron 2009;63:316–28. 10.1016/j.neuron.2009.07.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cortes CJ, Ling SC, Guo LT, et al. . Muscle expression of mutant androgen receptor accounts for systemic and motor neuron disease phenotypes in spinal and bulbar muscular atrophy. Neuron 2014;82:295–307. 10.1016/j.neuron.2014.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lieberman AP, Yu Z, Murray S, et al. . Peripheral androgen receptor gene suppression rescues disease in mouse models of spinal and bulbar muscular atrophy. Cell Rep 2014;7:774–84. 10.1016/j.celrep.2014.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rocchi C, Greco V, Urbani A, et al. . Subclinical autonomic dysfunction in spinobulbar muscular atrophy (Kennedy disease). Muscle Nerve 2011;44:737–40. 10.1002/mus.22159 [DOI] [PubMed] [Google Scholar]
- 40.Jackson GL. Beliefs about osteoporosis. Arch Int Med 1987;147:1845 10.1001/archinte.1987.00370100159027 [DOI] [PubMed] [Google Scholar]
- 41.Dupree K, Dobs A. Osteopenia and male hypogonadism. Rev Urol 2006;6:30–4. [PMC free article] [PubMed] [Google Scholar]
- 42.Ferlin A, Schipilliti M, Di Mambro A, et al. . Osteoporosis in Klinefelter's syndrome. Mol Hum Reprod 2010;16:402–10. 10.1093/molehr/gaq026 [DOI] [PubMed] [Google Scholar]
- 43.Danilovic DL, Correa PH, Costa EM, et al. . Height and bone mineral density in androgen insensitivity syndrome with mutations in the androgen receptor gene. Osteoporos Int 2010;18:369–74. 10.1007/s00198-006-0243-6 [DOI] [PubMed] [Google Scholar]
- 44.Donaldson MG, Cawthon PM, Lui LY, et al. . Estimates of the proportion of older white men who would be recommended for pharmacologic treatment by the new US National Osteoporosis Foundation guidelines. J Bone Miner Res 2010;25:1506–11. 10.1002/jbmr.55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang J, Morgan SL, Saag KG. Osteopenia: debates and dilemmas. Curr Rheumatol Rep 2013;15:384 10.1007/s11926-013-0384-5 [DOI] [PubMed] [Google Scholar]
- 46.Rufo A, Del Fattore A, Capulli M, et al. . Mechanisms inducing low bone density in Duchenne muscular dystrophy in mice and humans. J Bone Miner Res 2011;26:1891–903. 10.1002/jbmr.410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vestergaard P, Glerup H, Steffensen BF, et al. . Fracture risk in patients with muscular dystrophy and spinal muscular atrophy. J Rehabil Med 2001;33:150–5. 10.1080/165019701750300609 [DOI] [PubMed] [Google Scholar]
- 48.Joyce NC, Hache LP, Clemens PR. Bone health and associated metabolic complications in neuromuscular diseases. Phys Med Rehabil Clin N Am 2012;23:773–99. 10.1016/j.pmr.2012.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ishizaki F, Koyama T, Sunayashiki T, et al. . Control of bone remodeling by nervous system. Bone metabolic changes in neurological diseases. Clin Calcium 2010;20:1841–9. doi:CliCa101218411849 [PubMed] [Google Scholar]
- 50.Lloyd SA, Lang CH, Zhang Y, et al. . Interdependence of muscle atrophy and bone loss induced by mechanical unloading. Bone Miner Res 2014;29:1118–30. 10.1002/jbmr.2113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mariotti C, Castellotti B, Pareyson D, et al. . Phenotypic manifestations associated with CAG-repeat expansion in the androgen receptor gene in male patients and heterozygous females: a clinical and molecular study of 30 families. Neuromusc Disord 2000;10:391–7. 10.1016/S0960-8966(99)00132-7 [DOI] [PubMed] [Google Scholar]
- 52.Ni W, Chen S, Qiao K, et al. . Genotype-phenotype correlation in Chinese patients with spinal and bulbar muscular atrophy. PloS ONE 2015;10:e0122279 10.1371/journal.pone.0122279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Armitage J. The safety of statins in clinical practice. Lancet 2007;370:1781–90. 10.1016/S0140-6736(07)60716-8 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.