

Abstract
Electrical stimulation of sub-cortical brain regions (the basal ganglia), known as deep brain stimulation (DBS), is an effective treatment for Parkinson’s disease (PD). Chronic high frequency (HF) DBS in the subthalamic nucleus (STN) or globus pallidus interna (GPi) reduces motor symptoms including bradykinesia and tremor in patients with PD, but the therapeutic mechanisms of DBS are not fully understood. We developed a biophysical network model comprising of the closed loop cortical-basal ganglia-thalamus circuit representing the healthy and parkinsonian rat brain. The network properties of the model were validated by comparing responses evoked in basal ganglia (BG) nuclei by cortical (CTX) stimulation to published experimental results. A key emergent property of the model was generation of low-frequency network oscillations. Consistent with their putative pathological role, low-frequency oscillations in model BG neurons were exaggerated in the parkinsonian state compared to the healthy condition. We used the model to quantify the effectiveness of STN DBS at different frequencies in suppressing low-frequency oscillatory activity in GPi. Frequencies less than 40 Hz were ineffective, low-frequency oscillatory power decreased gradually for frequencies between 50 Hz and 130 Hz, and saturated at frequencies higher than 150 Hz. HF STN DBS suppressed pathological oscillations in GPe/GPi both by exciting and inhibiting the firing in GPe/GPi neurons, and the number of GPe/GPi neurons influenced was greater for HF stimulation than low-frequency stimulation. Similar to the frequency dependent suppression of pathological oscillations, STN DBS also normalized the abnormal GPi spiking activity evoked by CTX stimulation in a frequency dependent fashion with HF being the most effective. Therefore, therapeutic HF STN DBS effectively suppresses pathological activity by influencing the activity of a greater proportion of neurons in the output nucleus of the BG.
Keywords: Deep brain stimulation, Parkinson’s disease, 6-OHDA lesioned rat model, Subthalamic nucleus, Computational model, Pathological oscillatory activity
1 INTRODUCTION
Parkinson’s disease (PD) is a neurological disorder caused by degeneration of dopaminergic neurons in the substantia nigra pars compacta (SNc) (Agid et al., 1987; Hornykiewicz, 1998). The primary motor symptoms of PD are rest tremor, akinesia/bradykinesia, rigidity, postural instability and gait disorders (Jankovic et al., 2000; Quinn et al., 1989; Rajput et al., 2008). Levodopa, a dopamine precursor, is used as a first-line therapy for treating PD. However, patients treated with levodopa can develop debilitating dyskinesias (Marsden et al., 1982), after which surgical interventions are often recommended. Chronic high frequency stimulation in the subthalamic nucleus (STN) is effective in suppressing PD motor symptoms (Moro et al., 2010; Weaver et al., 2009). However, despite the clinical effectiveness of STN deep brain stimulation (DBS), its mechanisms are not fully understood.
6-OHDA-lesioned rats and MPTP-treated non-human primates are widely used animal models to study the pathophysiology of PD (Blesa & Przedborski, 2014). Although animal models are rendered parkinsonian by a common mechanism (loss of dopaminergic neurons), there is considerable variation in the neuronal activity underlying the pathophysiology, including differences in firing rates, firing patterns, responses to cortical stimulation, and neuronal synchronization across different basal ganglia (BG) structures (Kita & Kita, 2011; Nambu et al., 2000). Computational models of the BG play an important role in helping to understand both PD pathophysiology and the therapeutic mechanism of DBS. Neural activity in several existing computational models of the BG closely matches neural acitivity in MPTP-treated primates (Hahn & McIntyre, 2010; Humphries & Gurney, 2012; Kang & Lowery, 2013; Rubin & Terman, 2004; So, Kent, et al., 2012), but no current computational model adequately represents the 6-OHDA lesioned rat model of PD.
The objective of the present study was to develop a computational model representing the parkinsonian state in 6-OHDA lesioned rats, and, following validation, use the model to investigate the therapeutic mechanisms of STN DBS in alleviating parkinsonian symptoms. We implemented a biophysical model with Hodgkin-Huxley type neurons to represent the closed loop cortex-basal ganglia-thalamus-cortex circuit, and used the model to study the effectiveness of STN DBS at different frequencies in suppressing pathological low-frequency oscillatory neural activity. Pathological low-frequency oscillatory activity across different BG nuclei is correlated with motor symptoms of Parkinson’s disease (Brocker et al., 2013; Kühn et al., 2008; Levy et al., 2002), and thereby serves as a model-based proxy for the efficacy of DBS.
2 METHODS
The model included 10 single compartment model neurons in each of the cortex (CTX), striatum (Str), STN, globus pallidus externa (GPe), globus pallidus interna (GPi; or, in the rat, the homologous entopeduncular nucleus, EP), and thalamus (TH) interconnected with model synapses to form a functional network (Fig. 1(A),(B)). The intra-cortical and intra-striatal wiring configurations were stochastic, while all other connectivity followed a structured/deterministic pattern based on prior computational models (Rubin & Terman, 2004; So, Kent, et al., 2012). All model equations are provided in Appendix A. Simulations were implemented in Matlab R2014a with equations solved using the forward Euler method with a time step of 0.01 ms. We ran additional simulations with both shorter and longer time steps (0.005, 0.025 ms), and the model results (peak oscillatory frequencies) were robust across different time steps.
Fig. 1.
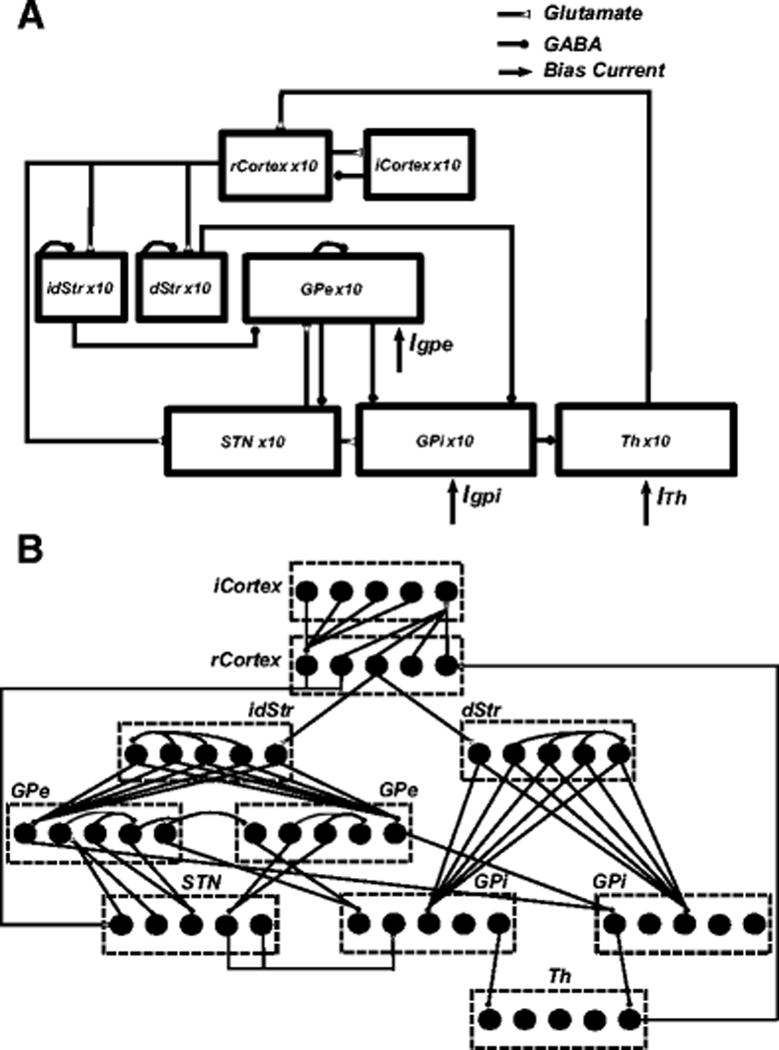
Cortical-basal ganglia-thalamus network model. (A) Model schematic showing connections within the network. (B) Details of synaptic connections within the network model. Each rCortex neuron receives excitatory input from one TH neuron and inhibitory input from four randomly selected iCortex neurons. Each iCortex neuron receives excitatory input from four randomly selected rCortex neurons. Each dStr neuron receives excitatory input from one rCortex neuron and inhibitory axonal collaterals from three randomly selected dStr neurons. Each idStr neuron receives excitatory input from one rCortex neuron and inhibitory axonal collaterals from four randomly selected idStr neurons. Each STN neuron receives inhibitory input from two GPe neurons and excitatory input from two rCortex neurons. Each GPe neuron receives inhibitory axonal collaterals from any two other GPe neurons and inhibitory input from all idStr neurons. Each GPi neuron receives inhibitory input from two GPe neurons and inhibitory input from all dStr neurons. Some GPe/GPi neurons receive excitatory input from two STN neurons, while others do not. Each TH neuron receives inhibitory input from one GPi neuron
2.1 CTX Model Neuron
The cortical network comprised reciprocally connected regular spiking (RS) excitatory neurons and fast-spiking inhibitory interneurons (FSI), both based on the model developed by Izhikevich (Izhikevich, 2003). Cortical neurons in the model were quiescent at resting membrane potential. The membrane potential, vrs, of a regular spiking cortical neuron was calculated using
where Iie is the synaptic current from FSI to RS neuron (each RS neuron received synaptic input from four FSI), and Ithco is the synaptic input received from the TH (each RS neuron received synaptic input from a single TH neuron). An alpha synapse was used to model the synaptic dynamics,
where is the maximal synaptic conductance, td is the synaptic transmission delay, and τ is the time constant. All synaptic transmission delays are shown in Table 1.
Table 1.
Synaptic Connection Parameters
| Synaptic connection | Transmission delay (td) |
Source |
|---|---|---|
| CTX-dStr | 5.1 ms | (Kita & Kita, 2011) |
| CTX-idStr | 5.1 ms | (Kita & Kita, 2011) |
| CTX-STN | 5.9 ms | (Kita & Kita, 2011) |
| dStr-GPi | 4 ms | (Nakanishi et al., 1987) |
| idStr-GPe | 5 ms | (Kita & Kitai, 1991) |
| STN-GPi | 1.5 ms | (Nakanishi et al., 1987) |
| STN-GPe | 2 ms | (Kita & Kitai, 1991) |
| GPe-STN | 4 ms | (Fujimoto & Kita, 1993) |
| GPe-GPi | 3 ms | (Nakanishi et al., 1991) |
| GPi-TH | 5 ms | (Xu et al., 2008) |
| TH-CTX | 5.6 ms | (Walker et al., 2012) |
The membrane potential, vfsi, of a FSI was calculated using
where Iei is the synaptic current from RS to FSI neuron (each FSI received synaptic inputs from four RS neurons). In both equations, u is a state variable that represents the recovery of membrane potential.
2.2 Str Model Neuron
Medium spiny neurons (MSN) comprise 90–95% of all striatal neurons in rodents (Chang & Kitai, 1985; Chang et al., 1982), and MSN neurons of the direct and indirect pathways are modulated by D1 and D2 dopamine receptors, respectively (Nicola et al., 2000). The striatal network included medium spiny neurons (MSN) from both the direct and indirect pathways, as developed previously (McCarthy et al., 2011), that were quiescent at rest. The membrane potential vstr of direct and indirect MSNs was calculated using
where INa, IK and Il are voltage-dependent sodium and potassium ionic currents and a nonspecific leakage current, Im is an outward potassium current modulated by acetylcholine through M1 muscarinic receptors, and Igaba is recurrent inhibitory synaptic current (each direct and indirect MSNs received inhibitory axonal collaterals from 30% and 40% of the remaining MSNs, respectively (Taverna et al., 2008), modeled using an exponential synapse), and Icostr is the synaptic input from the CTX (each MSN received excitatory input from one RS CTX neuron, modeled using an alpha synapse). The exponential synapse was modeled using
where is the maximal synaptic conductance, td is the synaptic transmission delay, and τ is the time constant.
2.3 STN Model Neuron
STN neurons were adopted from a previous model (Otsuka et al., 2004) and were spontaneously active with firing rates in the range of 2–10 spikes/s, which is comparable to rates observed in rat in vivo. The membrane potential of a STN neuron, vstn, was calculated using
where INa, IK and Il are voltage-gated sodium and potassium ionic currents and a non-specific leakage current, IL is a L-type calcium current, IT is a T-type calcium current, ICaK is a calcium-dependent potassium current that is dependent upon the intracellular calcium concentration, and Igesn is the inhibitory synaptic current from GPe with dynamics modeled using a bi-exponential synapse,
Here, is the maximal synaptic conductance, td is the synaptic transmission delay, τr is the rise time, and τd is the decay time. Rise and decay times of τr = 1.1 ms and τd = 7.8 ms respectively were used for IPSCs elicited at GPe-STN synapses (Baufreton et al., 2009). Each STN neuron received inhibitory input from two GPe neurons. Model STN neurons included both AMPA and NMDA glutamate receptors with the AMPA/NMDA receptor ratio equal to one (Farries et al., 2010). Icosn,ampa and Icosn,nmda are the CTX-STN synaptic currents mediated by AMPA-R and NMDA-R, respectively (each STN neuron received excitatory input from two cortical neurons). Rapid rise (τr = 0.5 ms) and decay (τd = 2.49 ms) times were used for AMPA-R EPSCs, while NMDA-R EPSCs (τr = 2 ms and τd = 90 ms) were slower.
2.4 GP Model Neurons
The GPe and GPi/EP neurons were modified from those in a previous model (So, Kent, et al., 2012). The constant applied bias current representing the striatal input to GPe was replaced by the synaptic current from indirect MSNs. The membrane potential of a GPe neuron, vGPe, was calculated using
The ionic currents are similar to STN neurons, as described above, except for the addition of a high threshold calcium current, ICa, and the absence of a L-type calcium current. Pallidal neurons receive differential innervation from STN and this is believed to be the origin of the dichotomous firing behavior of GP neurons in rodents (Mallet, Pogosyan, Márton, et al., 2008). Consistent with this observation, the model included two types of GPe neurons, with some receiving excitatory input from two STN neurons (Isnge,ampa and Isnge,nmda), while others did not (Fig. 1(B)). The STN-GPe synaptic connections were mediated by both AMPA and NMDA glutamate receptors (Götz et al., 1997). The decay time of GPe NMDA-R was slightly faster (τd = 67 ms) when compared to STN NMDA-R, although the rise times were identical in both neurons. The kinetics of GPe AMPA-R were identical to STN. All GPe neurons received inhibitory axonal collateral from two other GPe neurons (Igege) (Bolam et al., 2000). Each GPe neuron received inhibitory input from all indirect Str MSNs, and these accounted for nearly 80–90% of the total synaptic connections found in GPe (Sims et al., 2008). GPe neurons received a constant bias current Iappgpe (3 μA/cm2) representing the net synaptic input from all sources that were not exclusively modeled.
GPi (or, entopeduncular nucleus, EP) is the primary output nucleus of the BG. The membrane potential, vGPi, of a GPi neuron was calculated using
with ionic currents similar to GPe neurons described above Igegi, Istrgpi and Isngi,ampa are the synaptic inputs from GPe, direct Str MSN, and STN, respectively, all converging onto GPi neurons (Kita, 2001). Each GPi neuron received inhibitory input from two GPe neurons (Igegi) and from all direct Str MSNs (Istrgpi). STN-GPi synaptic connectivity was similar to GPe with a portion of GPi neurons not receiving any synaptic input from STN (Fig. 1(B)), although currently there is no experimental evidence indicating whether or not the finding of two types of neurons in rodent GP(e) also extends to GPi (EP). STN-GPi synaptic dynamics were mediated by only AMPA-R with kinetics identical to those of STN-GPe. GPi neurons also received a constant bias current Iappgpi (3 μA/cm2) similar to GPe. GP neurons in the model were quiescent at resting membrane potential.
2.5 TH Model Neuron
TH neurons were modified from those in a previous model (So, Kent, et al., 2012). The current pulses to TH representing the sensorimotor cortical (SMC) input were replaced by a constant applied current (Iappth = 1.2 μA/cm2) representing the cerebellar input to TH. The membrane potential of a TH neuron, vTh was calculated using
with ionic currents similar to the GPe neurons described above. Each TH neuron received inhibitory input from a single GPi neuron (Igith). Model TH neurons were not spontaneously active at the resting membrane potential.
2.6 Modeling Different States
We modeled three states representing control (normal), 6-OHDA lesioned (PD), and 6-OHDA lesioned plus STN DBS in rats. The PD state, resulting from the loss of striatal dopamine neurons, was implemented by making three changes to the normal state. First, loss of striatal dopamine is accompanied by an increase in acetylcholine levels (Ach) in the Str (Ikarashi et al., 1997). This results in a reduction of M-type potassium current in both the direct and indirect MSNs (Brown, 2010; McCarthy et al., 2011), and was modeled by decreasing the maximal conductance gm from 2.6 to 1.5 mS/cm2. Second, dopamine loss results in reduced sensitivity of direct Str MSN to cortical stimulation (Mallet et al., 2006), which was modeled by decreasing the maximal corticostriatal synaptic conductance gcostr from 0.07 to 0.026 mS/cm2. Finally, striatal dopamine depletion causes an increase in the synaptic strength of intra-GPe axonal collaterals resulting in aberrant GPe firing (Miguelez et al., 2012), and this was modeled by increasing the maximal synaptic conductance ggege from 0.125 to 0.5 mS/cm2. DBS was modeled by applying intracellular current pulses in all STN model neurons so that every pulse evoked one action potential at frequencies in the range of 5–200 Hz (amplitude 300 μA/cm2, duration 0.3 ms).
2.7 Outcome Measures
PD is accompanied by an increase in low-frequency oscillatory activity across the cortex and BG (Mallet, Pogosyan, Márton, et al., 2008; Mallet, Pogosyan, Sharott, et al., 2008; McConnell et al., 2012). Oscillatory power in the beta band correlates with akinesia/bradykinesia (Kühn et al., 2008), while oscillations in the alpha band may be associated with tremor (Shaw & Liao, 2005). Therefore, we quantified the effects of STN DBS at different frequencies on low-frequency oscillatory power in the model GPi in the PD state. Spectral analyses were performed using the Chronux neural signal analysis package (www.chronux.org) (sliding 1 s window, 0.1 s step size and [3 5] tapers [3 is the time-bandwidth product and 5 is the number of tapers]) and MATLAB R2014a. Oscillatory power in the GPi was calculated by integrating the spectral power of GPi spike times in the 7–35 Hz frequency band. Changes in the responses evoked in GPi by CTX activation might be associated with PD motor symptoms (Degos et al., 2005; Kita & Kita, 2011). Therefore, we also quantified the strength and duration of the GPi response (long inhibition) evoked by CTX stimulation and used it as a surrogate to study the frequency dependent effects of STN DBS. The duration of inhibition was the difference between time instances at which the GPi evoked firing rates were just lesser and greater than the mean firing rate. The area corresponding to this duration of long inhibition in the GPi evoked response was quantified as strength.
3 MODEL VALIDATION
Model parameter values were selected based on independent experimental evidence wherever possible, as described above, but the values of several parameters were tuned to match the repsonses evoked in the basal ganglia by cortical stimulation in rats (Kita & Kita, 2011). GPe and GPi applied currents (Iappgpe, Iappgpi) were fixed at (3 μA/cm2) to generate firing rates in STN, GPe and GPi neurons similar those measured in rats. Maximal synaptic and ionic conductances (ggege, gcostr, gm) were either increased or decreased between normal and PD conditions as described under ‘Modeling Different States’ section. We applied supra-threshold stimulus pulses to each cortical neuron (duration 0.3 ms, amplitude 300 μA/cm2, frequency 1 Hz) and analyzed the activity evoked in Str, STN, GPe and GPi using post-stimulus time histograms (PSTH) in the normal and PD states. The PSTH had a bin width of 1 ms and was averaged across 10 neurons for 100 trials. The model PSTHs were compared with experimental results obtained under similar conditions (Kita & Kita, 2011).
3.1 Str Response to CTX Stimulation
CTX stimulation evoked a strong excitatory response in model Str MSNs in both the normal and PD conditions, similar to evoked responses in rats (Kita & Kita, 2011) (Fig. 2). In the PD state, CTX stimulation evoked strong excitation in model Str neurons followed by long-duration GABAergic inhibition due to cortical disfacilitation (Fig. 2(B)). Cortical disfacilitation was due to the inhibition of regular spiking excitatory neurons by fast spiking interneurons. Model Str MSNs exhibited increased firing in the PD state as compared to the normal condition, and this increase in mean firing rate was also observed in rat MSNs following dopamine depletion (Mallet et al., 2006; Pang et al., 2001).
Fig. 2.
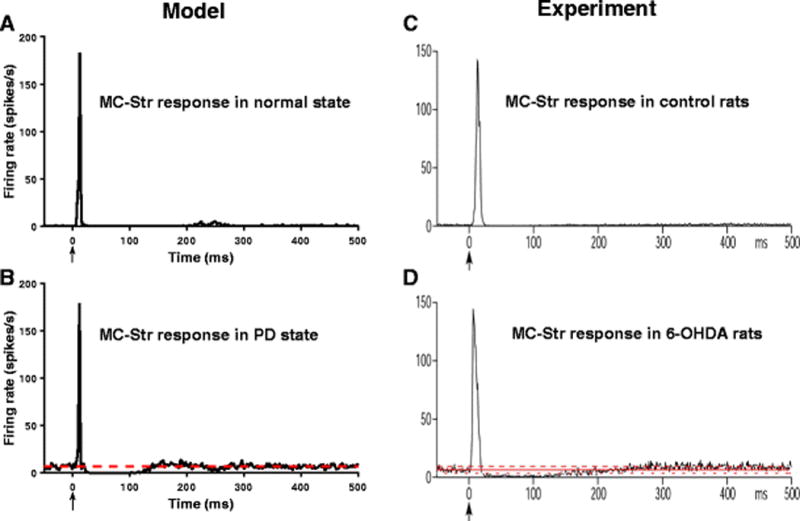
Str responses to CTX stimulation. (A) Model Str PSTH obtained under normal conditions shows strong excitation following CTX stimulation. Str neurons are not spontaneously active under normal conditions. (B) Model Str PSTH obtained during PD state shows strong excitation and long inhibition following CTX stimulation. Str neurons exhibit increased spontaneous firing during PD. (C,D) Experimental PSTHs (Kita & Kita, 2011) match well with model results. The red lines depict the mean firing rate of neurons
3.2 STN Response to Cortical Stimulation
In the normal state, CTX stimulation evoked early excitation followed by late excitation in model STN neurons (Fig. 3(A)) by activation via the hyperdirect pathway of AMPA-R and NMDA-R, respectively. The reduction in firing rate between the early and late excitation was due to the difference in timing between the activation of AMPA-R and NMDA-R rather than synaptic inhibition. Following CTX stimulation in the PD state, model STN neurons exhibited early and late excitation followed by protracted inhibition (Fig. 3(B)) due to the increased late excitation of GPe leading to late inhibition of STN. The model PSTHs were consistent with the experimental PSTHs from rats (Kita & Kita, 2011) (Fig. 3(C),(D)).
Fig. 3.
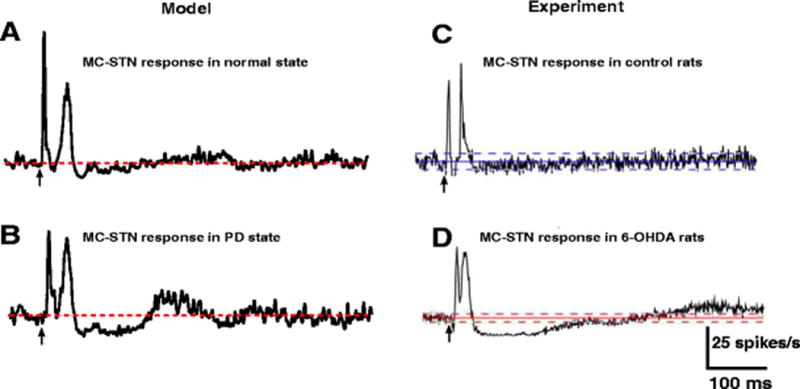
STN responses to CTX stimulation. (A) Model STN PSTH obtained under normal conditions shows early excitation and late excitation following CTX stimulation. (B) Model STN PSTH obtained during PD state shows early excitation, late excitation and long inhibition following CTX stimulation. (C,D) Model PSTHs are comparable with PSTHs obtained from an experimental study (Kita & Kita, 2011). The red and blue lines depict the mean firing rate of neurons
3.3 GPe Response to Cortical Stimulation
In the normal state, model GPe neurons responded to CTX stimulation with early excitation, short inhibition, and weaker late excitation (Fig. 4(A)). The early excitation and short inhibition were mediated by STN and Str, respectively, while the late excitation was mediated by both STN and Str. The GPe response to CTX stimulation in the PD state included early excitation, short inhibition, and large amplitude and long duration late excitation (Fig. 4(B)). The increased late excitation in the PD state was due to the protracted inhibition of Str MSNs, which exhibited higher levels of activity in the PD state that disinhibited the GPe neurons. The model results are similar to the GPe responses in rats (Kita & Kita, 2011) (Fig. 4(C),(D)).
Fig. 4.
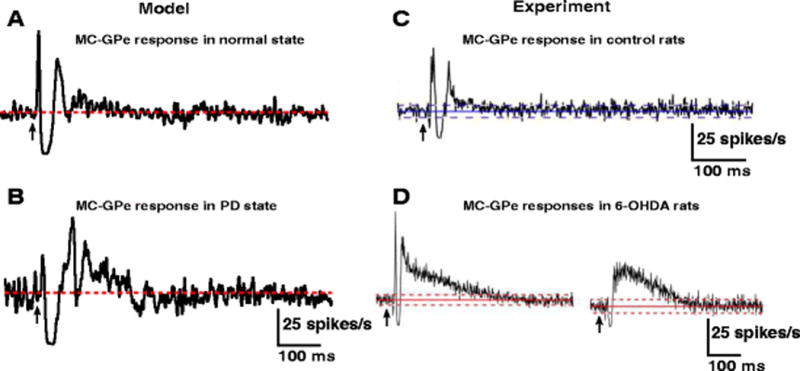
GPe responses to CTX stimulation. (A) Model GPe PSTH obtained under normal condition shows early excitation, short inhibition and weak late excitation following CTX stimulation. (B) Model GPe PSTH obtained during PD state shows weak early excitation, short inhibition and strong late excitation following CTX stimulation. (C,D) Model PSTHs are comparable with PSTHs obtained from an experimental study (Kita & Kita, 2011). The red and blue lines depict the mean firing rate of neurons
3.4 GPi Response to Cortical Stimulation
The responses evoked in Str, GPe, and STN by CTX stimulation converged on GPi neurons, which exhibited two major response patterns. The CTX stimulation generated either early excitation, short inhibition, and late excitation or short inhibition followed by late excitation in model GPi neurons in the normal state (Fig. 5(A),(C)). The early excitation of GPi was due to activation of STN neurons via the hyperdirect pathway, the short inhibition was mediated by the activation of direct MSNs, and the late excitation was mediated by the indirect activation of MSN through GPe and the subsequent disinhibition of GPi. GPi neurons exhibited either one of the two response types depending upon whether or not they received inputs from STN neurons.
Fig. 5.
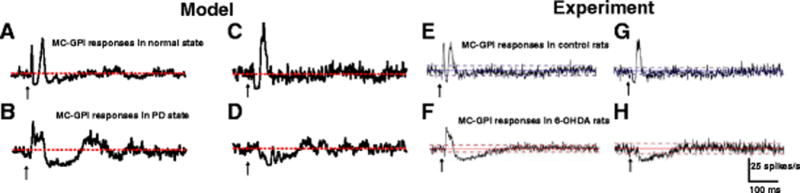
GPi responses to CTX stimulation. (A,C) Model GPi PSTHs obtained under normal conditions show either early excitation, short inhibition and late excitation or short inhibition and late excitation following CTX stimulation. (B,D) Model GPi PSTHs obtained during PD state show either early excitation and long inhibition or only long inhibition following CTX stimulation. (E,F,G,H) Model PSTHs are comparable with PSTHs obtained from an experimental study (Kita & Kita, 2011). The red and blue lines depict the mean firing rate of neurons
The response patterns in GPi in the PD state differed considerably from those under normal conditions. CTX stimulation evoked either early excitation followed by strong, long duration inhibition or only long duration inhibition in model GPi neurons (Fig. 5(B),(D)). The early short inhibition in the normal state was replaced by strong, long duration inhibition. The absence of short inhibition was due to the reduced sensitivity of direct MSNs to CTX stimulation in the parkinsonian (dopamine depleted) condition, while the increased late excitation in GPe and increased long inhibition in STN resulted in strong, long duration inhibition in GPi. The model results are similar to GPi responses measured in rats (Kita & Kita, 2011) (Fig. 5(E),(F),(G),(H)).
3.5 Model Neuron Firing Rates and Patterns
Recordings in 6-OHDA lesioned rats indicate that there is an increase in Str MSN firing rate after administration of 6-OHDA (Kita & Kita, 2011; Pang et al., 2001). Similarly, the firing rates of STN and GPi neurons in 6-OHDA rats are higher than in control, while those of GPe neurons are lower following lesion (Hollerman & Grace, 1992; Mallet, Pogosyan, Márton, et al., 2008). Changes in firing rates of neurons in the model were consistent with these experimental results (Fig. 6): Str, STN and GPi neurons exhibited increased firing rates in the PD condition, while GPe neuron firing rates decreased. In the PD state, the model STN, GPe and GPi neurons exhibited more rhythmic burst-like firing patterns (Fig. 7), and this was consistent with experimental studies (Kita & Kita, 2011; Mallet, Pogosyan, Márton, et al., 2008).
Fig. 6.
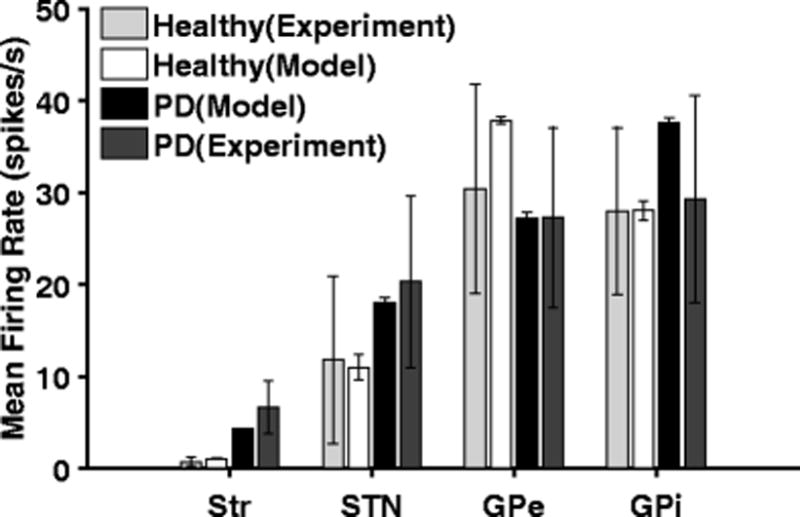
Firing rates of model and experimental (Kita & Kita, 2011) neurons in striatum (Str), subthalamic nucleus (STN), globus pallidus externa (GPe) and globus pallidus interna (GPi) under normal and PD conditions. Standard error bars for model data are shown for 10 ten-second simulations under each condition
Fig. 7.
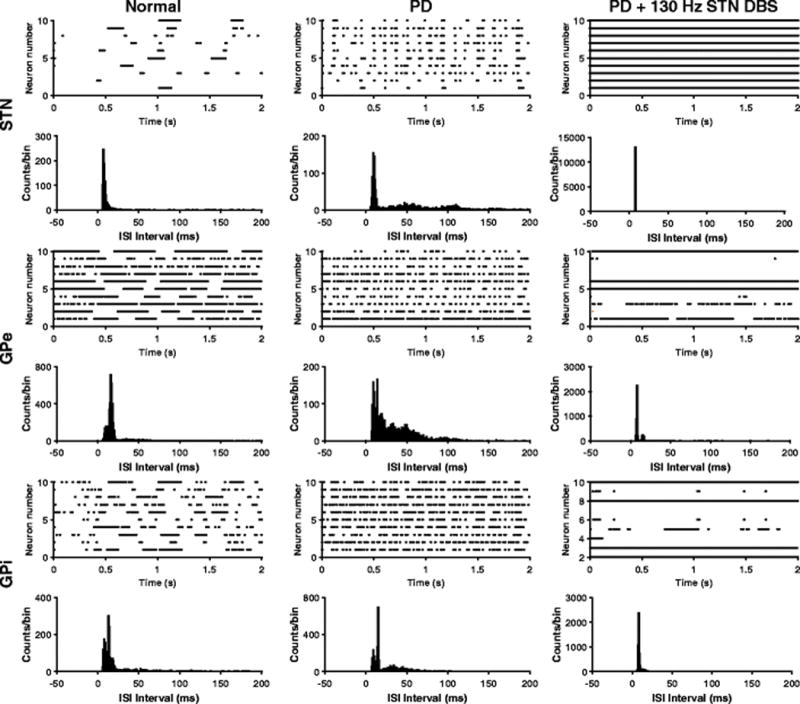
Firing patterns of STN, GPe and GPi neurons. Rastergrams and interspike interval (ISI) plots under normal, PD, and PD condition with 130 Hz STN DBS. During the PD state, neurons fired in a more rhythmic burst fashion, while 130 Hz STN DBS suppressed these bursts by either exciting or inhibiting the firing of GPe/GPi neurons
4 RESULTS
The validated model was used to study the effects of PD and STN DBS on spiking and oscillatory activity of model BG neurons.
4.1 Low-Frequency Oscillatory Activity
Model BG neurons exhibited increased oscillatory activity in the beta band (~ 20Hz) in the PD state when compared to normal conditions (Fig. 8(A),(B),(C)), in agreement with experimental observations following unilateral 6-OHDA lesion (Cruz et al., 2012). The model STN and GPi neurons also exhibited low-frequency oscillatory activity in the alpha band (~ 9 Hz) in the PD condition (Fig. 8(A),(B),(C)), similar to oscillatory activity (7–10 Hz) in 6-OHDA lesioned rats (McConnell et al., 2012). We investigated the robustness of oscillatory frequencies first by randomizing the values of the three parameters that were modified to model the PD state (gm, gcostr, ggege), and second by making the connections between model neurons stochastic. Spectral power from ten such trials was calculated and then compared with the spectral power in the PD condition. There were no changes in the peak oscillatory frequencies between the original parameterization or either the new set of randomized parameters or the stochastic connectivity, although the magnitude of spectral power varied (Fig. 8(D),(E),(F),(G),(H),(I)). Spectral analysis of the spike times of model GPi neurons revealed that episodes of beta band oscillatory activity interrupted alpha oscillatory activity in the PD state (Fig. 9(A),(B)), consistent with experimental evidence that episodes of tremor-related oscillations desynchronized beta activity in PD patients (Levy et al., 2002).
Fig. 8.
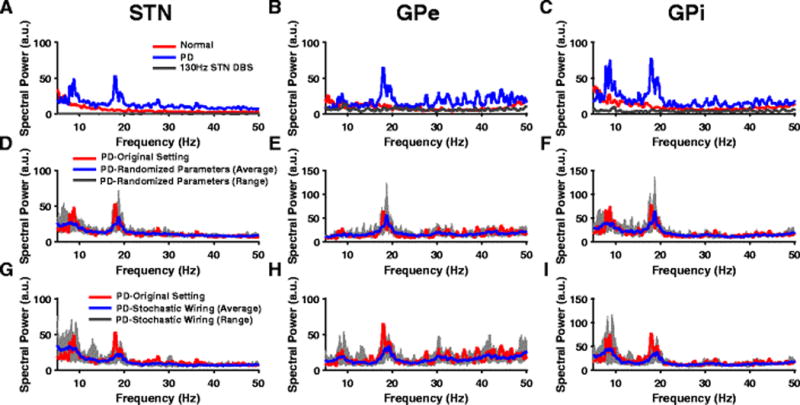
Oscillatory activity across BG nuclei. (A,B,C) Power spectra (PS) of STN, GPe and GPi spike times show exaggerated oscillatory activity in both the alpha and beta band during PD conditions (blue) when compared to the normal state (red). PS show the suppression of these pathological alpha and beta oscillations during HF STN DBS (gray) across all BG nuclei. (D,E,F) PS of STN, GPe and GPi spike times during PD condition show robust oscillatory frequencies across original model parameters (red) and randomized parameters - gm, gcostr, ggege values chosen from an uniform distribution with mean listed in Table 11 (Appendix A). The blue line and gray band represent the average and range respectively across 10 trials. (G,H,I) PS of STN, GPe and GPi spike times during PD condition show robust oscillatory frequencies across original model parameters (red) and a model with stochastic connectivity. Model neurons received synaptic inputs from randomly selected neurons. The blue line and gray band represent the average and range respectively across 10 trials
Fig. 9.
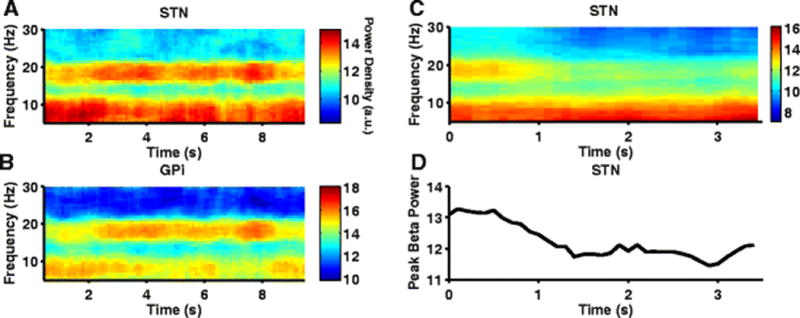
Alpha and beta band oscillatory activity during PD condition in model BG neurons. (A,B) Spectrograms of STN, GPi spike times exhibit prominent oscillations in both the alpha and beta band. GPi oscillatory activity in the alpha band at 9Hz is interrupted by periods of beta frequency oscillation at 20Hz. (C) Spectrogram of STN spike times shows suppression of STN beta band oscillations following reduction of NMDA-R synaptic conductance in STN (mimicking NMDA antagonist infusion) at 1sec. NMDA antagonist did not have any effect on the STN oscillatory activity at 9Hz. (D) Log-transformed STN peak beta band power reduces following simulation of NMDA antagonist infusion at ~1sec
Infusion of an NMDA antagonist (cis-4-[phosphomethyl]-piperidine-2-carboxylic acid) into STN suppressed STN beta band oscillations in 6-OHDA lesioned rats (Pan et al., 2014). Infusion of NMDA antagonist into STN was simulated in the model by reducing the NMDA-R synaptic conductance gcosn,nmda in the hyperdirect pathway to zero. Similar to the experimental observation, reducing the NMDAR synaptic conductance substantially reduced beta oscillatory activity in model STN neurons, but did not alter low-frequency oscillatory activity (Fig. 9(C,D)).
4.2 Model Neuron Firing Rates during STN DBS
The intrinsic activity of STN neurons was masked during HF STN DBS and firing patterns were more regular (Fig. 7). HF STN DBS resulted in both increases and decreases in the firing rate of a greater number of model pallidal neurons than low-frequency STN DBS (Fig. 10(A),(B)), consistent with experimental observations in 6-OHDA lesioned rats (McConnell et al., 2012). Excitation through the STN-GPe pathway resulted in increased firing of some GPe neurons, while inhibition through the axonal collaterals of excited GPe neurons reduced the firing rate of other GPe neurons. The increase in GPi firing rate resulted from activation of the STN-GPi pathway, while reductions in rate were due to excitation of GPe neurons and subsequent inhibition of GPi neurons through the STN-GPe-GPi pathway. Due to this dichotomous response, there was no substantial increase in the mean firing rates of the model GPe or GPi neurons between low and high frequency STN DBS (Fig. 10(A),(B)), matching well the changes in firing rates observed in 6-OHDA lesioned rats during STN DBS (McConnell et al., 2012).
Fig. 10.
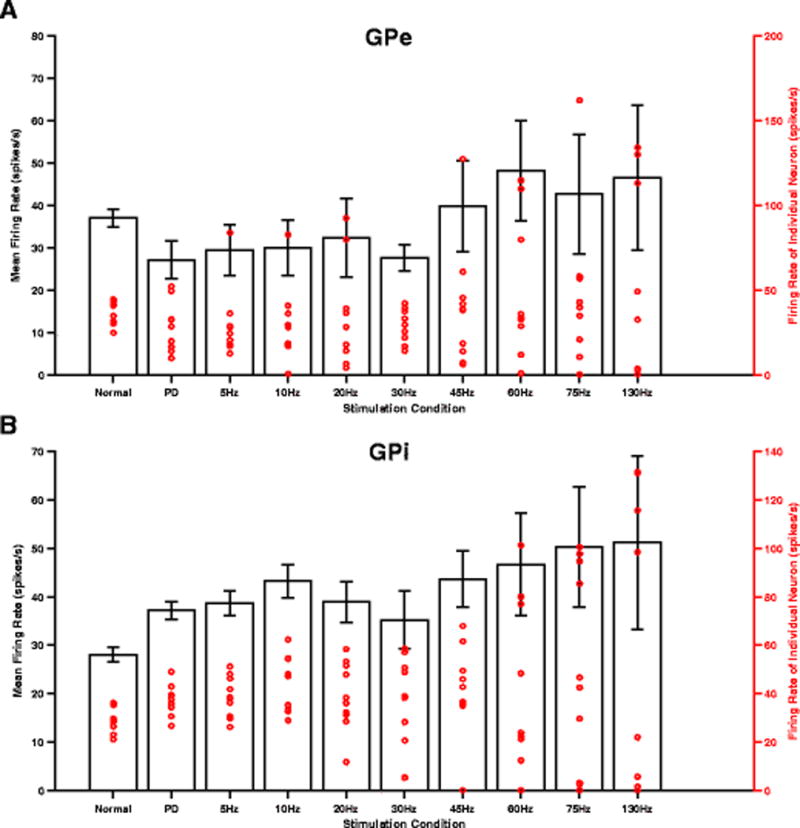
Effects of STN DBS frequency on pallidal neurons firing rate. (A,B) Mean (black bar) and individual (red dot) firing rates of model GPe/GPi neurons. There was no substantial increase in the mean firing rates of model GPe/GPi neurons between low and high frequency STN DBS. Standard error bars are shown for variation in firing rates across 10 neurons for each stimulus frequency. Each red circular marker represents the firing rate of an individual neuron for a total of 10 neurons each in GPe and GPi. Note the activity of a greater number of GPe/GPi neurons being influenced during HF STN DBS than during LF STN DBS
4.3 STN DBS Frequency-Dependent Suppression of Beta Frequency Oscillations
Stimulation frequency is a determinant of the effectiveness of STN DBS in reducing PD symptoms in 6-OHDA lesioned rats (So, McConnell, et al., 2012), and abnormal low-frequency oscillatory activity in the output nuclei of the BG is correlated with PD symptoms in the 6-OHDA rat model of PD (McConnell et al., 2012). We quantified the effectiveness of STN DBS in the model by calculating the total low-frequency (7–35Hz) power of activity in model GPi neurons during DBS normalized to the baseline GPi power in the PD state. STN DBS at frequencies less than 40 Hz did not cause any substantial change in the low-frequency power of model GPi neuron activity. The GPi low-frequency power decreased gradually for DBS frequencies between 50 Hz and 130 Hz, and saturated at DBS frequencies greater than 150 Hz (Fig. 11). The stimulation frequency-dependent suppression of GPi low frequency oscillatory power matched the stimulation frequency-dependent suppression of motor symptoms in 6-OHDA rats (Li et al., 2012; McConnell et al., 2012; Ryu et al., 2013). Further, HF STN DBS suppressed low-frequency oscillations in GPe, STN, and GPi neurons to levels much lower than in the normal and PD states (Fig. 8(A),(B),(C)).
Fig. 11.
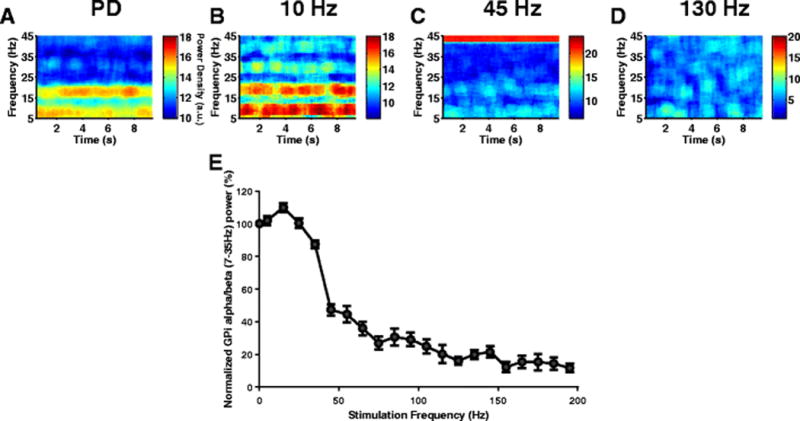
Effect of STN DBS frequency on GPi low-frequency oscillatory activity. (A,B,C,D) Spectrograms of GPi spike times during PD and three different STN DBS stimulus frequencies (10 Hz, 45 Hz and 130 Hz). During PD, GPi neurons exhibited synchronized oscillatory activity in both the alpha and beta band. 10 Hz STN DBS slightly increased this oscillatory activity. Although 45 Hz STN DBS reduced the GPi oscillatory activity, it did not completely suppress the oscillations. 130 Hz STN DBS completely suppressed the GPi oscillations and reversed PD symptoms. (E) Effect of STN DBS frequency on model GPi neurons 7–35Hz power. Standard error bars are shown for 10 ten-second simulations for each stimulus frequency
4.4 STN-DBS Frequency-Dependent Normalization of Abnormal GPi Activity Evoked by CTX Stimulation
Changes in the activity evoked in the BG by CTX might be causative of motor deficits observed in PD (Degos et al., 2005). In the model during PD conditions, CTX stimulation evoked abnormal responses in GPi characterized by the absence of early short inhibition present in the healthy condition and the presence of strong, long-lasting late inhibition not present in the healthy condition. We quantified the effects of STN DBS on the responses evoked in GPi by CTX stimulation in the model. STN DBS at frequencies less than 30 Hz did not cause any substantial change in the GPi response evoked by CTX stimulation (Fig. 12(A)). Both the strength and duration of late inhibition decreased gradually for stimulus frequencies between 45 Hz and 130 Hz (Fig. 12(A)). During 130Hz STN DBS, the strength and duration of late inhibition in the GPi response evoked by CTX stimulation in the PD state was greatly normalized (Fig. 12(B),(C),(D)). However, HF STN DBS did not restore the early short inhibition in the GPi response evoked by CTX stimulation under normal conditions (Fig. 12(B)).
Fig. 12.
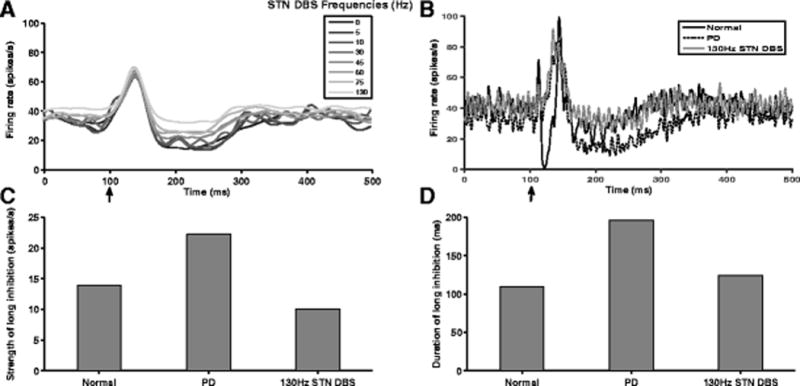
Frequency-dependent effects of STN DBS on GPi responses evoked by CTX stimulation. (A) PSTHs showing CTX stimulation evoked GPi activity at various STN DBS frequencies. (B) PSTHs showing CTX stimulation induced GPi responses under normal, PD and 130 Hz STN DBS conditions. 130 Hz STN DBS effectively normalized the enhanced late inhibition observed in GPi response relative to PD. However, 130 Hz STN DBS failed to restore the early short inhibition seen in the GPi response under normal conditions. Arrow indicates the time (100 ms) at which the CTX was stimulated by a single pulse. (C,D) 130 Hz STN DBS reduced both the strength and duration of late inhibition which were exaggerated during PD to values similar to those seen under normal conditions
5 DISCUSSION
We developed a computational model of the cortical-basal ganglia-thalamus circuit in the 6-OHDA lesioned rat model of PD, including a closed-loop connection between thalamus and cortex. Following extensive validation, demonstrating that the model replicated a wealth of experimental data, we used the model to quantify the effects of STN DBS on low-frequency oscillatory activity. The model was validated by comparing responses evoked by CTX stimulation in Str, STN, GPe, and GPi model neurons with experimental PSTHs. The model accounted for the key differences observed in the response patterns between the normal and PD states. Second, the firing rates and patterns observed in the normal and PD states were consistent with those in experimental studies. Finally, the two key emergent properties of the model – oscillatory activity across different nuclei and stimulation frequency-dependent suppression of this oscillatory activity – also matched well with experimental studies. Finally, the frequency-dependent effects of STN DBS in suppressing pathological low-frequency oscillatory activity paralleled the frequency-dependent normalization of abnormal responses evoked in the output nucleus of the BG by CTX stimulation.
5.1 Importance of CTX Induced Responses in GPi
The timing of the GPi response components evoked by CTX stimulation reflects the importance of the direct, indirect and hyperdirect pathways in the normal functioning of the BG. According to the “center-surround model”, the sequence of activation of the three BG pathways is functionally significant (Nambu et al., 2002). When a movement is initiated, the cortex exerts a rapid and strong excitatory influence on the output nucleus of the BG (GPi) via the hyperdirect pathway. Excitation of GPi, which results in inhibition of TH, is thought to negate all competing motor programs. Next, the cortical activation of the direct pathway results in strong inhibition of GPi, which likely disinhibits the TH. This allows the CTX to transmit the selected motor program efficiently through the TH. Finally, the activation of the indirect pathway again causes excitation of the GPi and subsequent inhibition of the TH. The functional implication is believed to be that unwanted motor programs are suppressed, which further aids in the transmission of only the selected motor program. Nambu and colleagues conducted their study in non-human primates, but the hypotheses they put forward regarding the center-surround model may also apply in rats since the GPi response patterns to CTX stimulation are similar in both animals (Kita & Kita, 2011).
However, in 6-OHDA lesioned rats, CTX stimulation evokes abnormal responses in GPi with notable differences being the insensitivity of the direct pathway to CTX stimulation and the increased firing of indirect pathway neurons (Kita & Kita, 2011; Mallet et al., 2006). These alterations in the normal functioning of the CTX-BG pathways resulted in abnormal CTX stimulation induced GPi responses in the model. HF STN DBS partially restored the normal functioning of BG pathways in the model by normalizing the abnormal CTX stimulation induced GPi response that was caused due to the increased firing of indirect pathway neurons. However, HF STN DBS failed to restore the component that was lost as a result of the reduced sensitivity of the direct pathway neurons to CTX stimulation. Hence, we predict in 6-OHDA lesioned rats that increased firing of indirect pathway neurons and transmission of this activity to the BG output nuclei might contribute to PD motor symptoms.
5.2 Neural Activity in 6-OHDA Lesioned Rats and Parkinsonian Primates
In both the 6-OHDA-lesioned rat and the MPTP-treated non-human primate models of PD, striatal dopamine depletion results in an increase in the firing rate of indirect Str MSNs (DeLong, 1990; Mallet et al., 2006; Pang et al., 2001). This is consistent with the classical model of PD that hypothesizes that SNc dopaminergic neurons exert an inhibitory effect on the indirect Str MSN and the loss of this inhibition results in PD symptoms. Also common to both animal models is the presence of exaggerated, synchronized pathological low-frequency oscillatory activity across BG nuclei and CTX in the parkinsonian state (Mallet, Pogosyan, Márton, et al., 2008; Raz et al., 2000), and suppression of such activity by effective STN DBS (Hammond et al., 2007; McConnell et al., 2012). The responses evoked in different BG nuclei by CTX stimulation are also similar in rats and non-human primates (Kita & Kita, 2011; Nambu et al., 2000; Tremblay & Filion, 1989). The downstream effects of STN DBS are also similar across the two species: behaviorally effective STN DBS evokes both excitation and inhibition in both rat and non-human primate GPi/SNr neurons (Bosch et al., 2011; Dorval et al., 2008; Hahn & McIntyre, 2010; McConnell et al., 2012). One of the model predictions is the existence of a population of GPi neurons that does not receive any input from STN. Our decision to extend the experimental observation of two populations of GPe neurons in rodents – one that receives STN input and one that does not (Mallet, Pogosyan, Márton, et al., 2008) – to GPi, enabled the model GPi neurons to exhibit two different response types to CTX stimulation, and these response types matched well with experimental responses (Kita & Kita, 2011). The GPi neurons that did not receive STN input, had a greater probability of being inhibited during HF STN DBS through the STN-GPe-GPi pathway. Hence, the presence of a diverse population of GPi neurons, with varying levels of synaptic input from STN, might be one of sources of the heterogeneous GPi responses observed during STN DBS. Other potential sources include the activation of inhibitory Str-GPi (direct pathway) and GPe-GPi fibers of passage (Bosch et al., 2011).
One major difference between rats and non-human primates is the firing rate of BG neurons. The firing rates of STN, GPe, and GPi neurons in rats are much lower when compared to non-human primates in both normal and parkinsonian conditions (Wichmann & Soares, 2006). Our model adequately accounts for this difference and the mean firing rate of all model BG neurons was< 40 spikes/s in both normal and PD states. The differences in firing rates likely underlie the variations in the frequency-dependent effects of DBS between the animal models. While low frequency stimulation (~50 Hz) was sufficient to mask and regularize the intrinsic activity of a model neuron firing at a low rate, higher frequency stimulation (>100 Hz) was necessary to achieve similar effects in a neuron that fired at a higher rate (Grill et al., 2004). In primates, STN DBS frequencies above 100 Hz relieve symptoms, while frequencies below 50 Hz are usually ineffective (Fogelson et al., 2005; Timmermann et al., 2004). However, in rats, the therapeutic window of STN-DBS starts as low as 50 Hz and reaches peak effectiveness at around 130 Hz (Li et al., 2012; Ryu et al., 2013). The STN DBS frequency tuning profile in the model was similar to these experimental studies in rats. Another major difference between the two animal models is the response of GPe neurons to HF STN DBS. Behaviorally effective STN DBS evokes both excitation-inhibition patterns in rat GPe neurons (McConnell et al., 2012), whereas a majority of non-human primate GPe neurons are excited during effective HF STN DBS (Dorval et al., 2008; Hahn & McIntyre, 2010). The model GPe neurons exhibited this dichotomous response during HF STN DBS similar to the experimental study in rats.
5.3 Prior Computational Models of the BG Circuit
Efforts continue to understand better the therapeutic mechanism of STN DBS using computational models of the BG. Initial attempts to explain the therapeutic mechanism of HF STN DBS used the classical rate model of PD (Albin et al., 1989). According to this model, dopamine depletion results in an imbalance characterized by decreased activation of the direct pathway and an increased activation of the indirect pathway. Increased activation of the indirect pathway leads to a decrease in GPe firing rate and a subsequent increase in the STN and GPi firing rates. The firing rate of GPi is further increased by the decreased activation of the direct pathway. Therefore, a hyperactive GPi during PD increases inhibition of the TH, which results in bradykinesia/akinesia. Single unit recordings across BG nuclei in 6-OHDA rat and MPTP-treated monkey support the classical rate model (Bergman et al., 1994; Hollerman & Grace, 1992; Mallet, Pogosyan, Márton, et al., 2008; Wichmann & Soares, 2006). However, the classical rate model failed to explain the therapeutic mechanism of STN DBS. Results from experimental studies suggest that DBS activates the efferent axons of the stimulated nucleus (Anderson et al., 2003; Hashimoto et al., 2003). Hence, HF STN DBS should increase the firing rate of GPi neurons. However, according to the classical rate model, a hyperactive GPi during HF STN DBS should lead to a more bradykinetic state than those observed during PD. This prediction of the rate model is in contrast with the clinical outcome observed during HF STN DBS in PD patients. Therefore, the classical rate model does not convincingly explain the therapeutic effects of HF STN DBS and, collectively, these observations suggested that it is not just the firing rate, but also the pattern of neural firing that needs to be considered to explain the therapeutic mechanism of HF STN DBS.
Rubin and Terman (RT) (Rubin & Terman, 2004) developed a biophysical computational model of the BG network. In the PD state, the BG neurons exhibit more burst-like firing, and this pattern of activity was reflected in the RT model. However, despite representation of the activity patterns observed during PD, the RT model did not reproduce the frequency dependent effects of STN DBS on PD symptoms (So, Kent, et al., 2012), as frequencies as low as 20 Hz were effective in suppressing a model proxy for symptom, which is inconsistent with clinical observations (Birdno & Grill, 2008). So et al. (So, Kent, et al., 2012) revised the properties of the RT model to account for the frequency-dependent effects of STN DBS. However, the firing rates and patterns of activity observed in model BG neurons during PD in the revised model are not consistent with those seen in the 6-OHDA lesioned rat.
Kang and Lowery developed a biophysical model of the cortico-BG-thalamic circuit that included the hyperdirect pathway but not the striatum (Kang & Lowery, 2013). The direct and indirect pathways representing the striatal inputs to GPi and GPe respectively were modeled using constant applied currents. The key prediction of the model was the emergence of oscillatory activity in STN depending upon the synaptic strength of hyperdirect pathway. Although the model accounted for the pathological oscillatory activity similar to those seen in PD, the model was not validated against any experimental data, and the oscillations were achieved as a result of parameter tuning rather than being an emergent property of a validated model. The firing rate of GPi neurons (110 ± 15 spikes/s) in the Kang and Lowery model was comparable to those seen in primates rather than rats. The cortico-basal ganglia-thalamic model that we developed was validated both at the cellular and network levels and reproduced key features of experimental data from the 6-OHDA lesioned rat model of PD.
5.4 Mechanism of STN DBS
At least three sites are possible sources of pathological low-frequency oscillatory activity in PD. Firstly, cortical neurons exhibit synchronous beta oscillations in PD, as seen in CTX local field potentials in 6-OHDA lesioned rats (Mallet, Pogosyan, Sharott, et al., 2008), and there is evidence for generation of the beta rhythm in CTX (Yamawaki et al., 2008). Hence, the CTX is a potential source of low-frequency oscillations in the PD state independent of its synaptic inputs. A second possible source of pathological low-frequency oscillations is the Str (McCarthy et al., 2011), as an increase in Str ACh as a result of dopamine loss is sufficient for Str neurons to generate oscillations in the 8–30Hz band. Finally, the reciprocally connected STN-GPe network is capable of generating oscillations without any synaptic inputs from the CTX or Str (Plenz & Kital, 1999). In the model, BG beta band oscillatory actvity was suppressed when the NMDA synaptic conductance of the hyperdirect pathway was reduced. Hence, the model supports the hypothesis that beta oscillatory activity generated in the CTX enters the BG through the STN, which receive strong excitatory projections from the CTX, and oscillatory input from STN drives GPe and GPi to oscillate in the beta band as observed in the 6-OHDA lesioned rat (Moran et al., 2011).
Regardless of the source, propagation of pathological low-frequency oscillatory activity to the GPi occurs through the STN, and this might explain why surgical interventions involving the STN are effective for relieving PD motor symptoms. STN lesion silences its efferents to GPi and GPe, such that pathological low-frequency oscillatory activity cannot reach the output of the BG. In the model, HF STN DBS suppressed pathological low-frequency oscillations by exciting some GPi neurons through the STN-GPi pathway and inhibiting other GPi neurons through the STN-GPe-GPi pathway. Excited GPi neurons showed a decrease in pathological burst activity and exhibited a more regularized firing, while inhibited GPi neurons simply did not transmit the pathological activity to the TH. A greater proportion of neurons were inhibited and excited during effective HF STN DBS when compared to ineffective LF STN DBS. Therefore, the therapeutic effects of HF STN DBS might arise from the ability to both excite and inhibit greater numbers of neurons in the output nucleus of the BG through the STN-GPe and STN-GPe-GPi pathway when compared to LF STN DBS. The STN is strategically located and able to influence GPi neurons both directly and indirectly. GPi neurons in a computational model of the BG circuit representative of non-human primates exhibited hetergenous responses during STN DBS similar to our study (Humphries & Gurney, 2012). The authors suggested that an optimum distribution of GPi excitatory/inhibitory responses was necessary for STN DBS to be effective, and concluded that HF STN DBS was better equipped to produce those effects than LF STN DBS. Behaviorally effective HF STN DBS in 6-OHDA-lesioned rats resulted in both excitation and inhibition of SNr neurons similar to those observed in the model (Bosch et al., 2011). The excitation and inhibition of SNr neurons during STN DBS was due to the activation of STN efferents to SNr and GPe efferents to SNr passing through STN respectively. The same study also showed an increase in the number of SNr neurons being inhibited and excited during HF STN DBS than during LF STN DBS.
Acknowledgments
This work was supported by grants from the US National Institutes of Health (NIH R37 NS040894 and NIH R01 NS079312). The authors would like to thank the Duke Shared Cluster Resource team for computational support.
All transmembrane potentials (v) are expressed in mV, intrinsic and synaptic conductances in mS/cm2, currents in μA/cm2, and time constants in msec. For all cell models the membrane capacitance is 1 μA/cm2
Thalamic Neuron Model
Table 2.
TH neuron model equations
| Current | Equation | Gating variables | Parameters | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Il | gl * (vTh − El) |
gl
= 0.05 El = −70 |
|||||||
| INa |
|
|
|
gNa =
3 ENa = 50 |
|||||
|
|
|
|
|||||||
| IK | gK * (0.75 * (1−h))4 * (vTh−Ek) | Same h as in INa |
gK
= 5 EK = −75 |
||||||
| It |
|
|
|
gt =
5 Et = 0 |
|||||
|
| |||||||||
| Igith | ggith * (vTh−Esyn) * S |
|
ggith
= 0.112 Esyn = −85 τ = 5 td = 5 1 GPi → 1Th |
||||||
| Iappth | 1.2 | ||||||||
External Globus Pallidus Neuron Model
Table 3.
GPe neuron model equations
| Current | Equation | Gating variables | Parameters | ||||
|---|---|---|---|---|---|---|---|
| Il | gl * (vGPe − El) |
gl
= 0.1 El = −65 |
|||||
| INa |
|
|
|
gNa =
120 ENa = 55 |
|||
|
| |||||||
| IK | gK * n4 * (vGPe−EK) |
|
gK =
30 EK = −80 |
||||
|
| |||||||
| It |
|
|
|
gt =
0.5 Et = 0 |
|||
|
| |||||||
| ICa |
|
|
gCa
= 0.15 ECa = 120 |
||||
| Iahp |
|
gahp
= 10 Eahp = −80 |
|||||
| Isnge,ampa | gsnge,ampa * (vGPe−Esyn) * S |
|
gsnge,ampa is
uniformly distributed with mean =
0.15 Esyn = 0 τr = 0.4 τd = 2.5 td = 2 2 STN → 1 GPe |
||||
| Isnge,nmda | gsnge,nmda * (vGPe−Esyn) * S |
|
gsnge,nmda is
uniformly distributed with mean =
0.001 Esyn = 0 τr = 2 τd = 67 td = 2 2 STN → 1 GPe |
||||
| Igege | ggege * (vGPe−Esyn) * S |
|
Esyn
= −85 τ = 5 td = 1 2 GPe → 1 GPe |
||||
| Istrgpe | gstrgpe * (vGPe−Esyn) * S |
|
gstrgpe =
0.5 Esyn = −85 τ = 5 td = 5 10 Str → 1 GPe |
||||
| Iappgpe | 3 | ||||||
Internal Globus Pallidus Neuron Model
Table 4.
GPi neuron model equations
| Current | Equation | Gating variables | Parameters | ||||
|---|---|---|---|---|---|---|---|
| Il | gl * (vGPi−El) |
gl
= 0.1 El = −65 |
|||||
| INa |
|
|
|
gNa =
120 ENa = 55 |
|||
|
| |||||||
| IK | gK * n4 * (vGPi−EK) |
|
gK =
30 EK = −80 |
||||
|
| |||||||
| It |
|
|
|
gt =
0.5 Et = 0 |
|||
|
| |||||||
| ICa |
|
|
gCa
= 0.15 ECa = 120 |
||||
| Iahp |
|
gahp
= 10 Eahp = −80 |
|||||
| Isngi | gsngi * (vGPi−Esyn) * S |
|
gsngi is
uniformly distributed with mean =
0.15 Esyn = 0 τ = 5 τd = 1.5 2 STN → 1 GPi |
||||
| Igegi | ggegi * (vGPi−Esyn) * S |
|
ggegi =
0.5 Esyn = −85 τ = 5 td = 3 2 GPe → 1 GPi |
||||
| Istrgpi | gstrgpi * (vGPi−Esyn) * S |
|
gstrgpi =
0.5 Esyn = −85 τ = 5 td = 5 10 Str → 1 GPi |
||||
| Iappgpe | 3 | ||||||
Subthalamic Nucleus Neuron Model
Table 5.
STN neuron model equations
| Current | Equation | Gating variables | Parameters | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Il | gl * (vSTN−El) |
gl
= 0.35 El = −60 |
|||||||
| INa |
|
|
|
gNa =
49 ENa = 60 |
|||||
|
|
|
||||||||
| IK | gK * n4 * (vSTN−EK) |
|
gK =
57 EK = −90 |
||||||
|
| |||||||||
| It |
|
|
|
gt = 5 [Ca]o = 2000 [Ca]i,initial = 0.005 |
|||||
|
|
|
||||||||
| ICak | gCak * r2 * (vSTN−Ek) |
|
gCa =
1 Ek = −90 |
||||||
| τr = 2 | |||||||||
| Ia | ga * a2 * b * (vSTN−Ek) |
|
|
ga =
5 Ek = −90 |
|||||
|
|
|
||||||||
| IL | gL * c2 * d1 * d2 * (vSTN − ECa) |
|
|
|
gL = 15 [Ca]o = 2000 [Ca]i,initial = 0.005 |
||||
|
|
|
τd2 = 130 | |||||||
| Igesn | ggesn * (vSTN−Esyn) * S |
|
ggesn
= 0.5 Esyn = −85 τr = 0.4 τd = 7.7 td = 4 2 GPe → 1 STN |
||||||
| Icosn,ampa | gcosn,ampa * (vSTN−Esyn) * S |
|
gcosn,ampa =
0.15 Esyn = 0 τr = 0.5 τd = 2.49 td = 5.9 2 CTX → 1 STN |
||||||
| Icosn,nmda | gcosn,nmda * (vSTN−Esyn) * S |
|
gcosn,nmda =
0.003 Esyn = 0 τr = 2 τd = 90 td = 5.9 10 CTX → 1 XTN |
||||||
Striatum Medium Spiny Neuron Model
Table 6.
MSN neuron model equations
| Current | Equation | Gating variables | Parameters | |||
|---|---|---|---|---|---|---|
| Il | gl * (vStr−El) |
gl =
0.1 El = −67 |
||||
| INa | gNa * m3 * h * (vStr−ENa) |
|
|
gNa =
100 ENa = 50 |
||
|
|
|
|||||
| IK | gK * n4 * (vStr−EK) |
|
gK =
80 EK = −100 |
|||
|
| ||||||
| Im | gm * p * (vStr−Em) |
|
Em = −100 | |||
|
| ||||||
| Igaba | ggaba * (vStr−Esyn) * S |
|
Esyn
=
−80 ggaba = 0.1/N 4 Str → 1 Str (Indir) 3 Str → 1 Str (dir) |
|||
| Icostr | gcostr * (vStr−Esyn) * S |
|
gcostr
= 0.07 Esyn = 0 τ = 5 td = 5.1 1 CTX → 1 Str |
|||
Cortical Regular Spiking Projection Neuron Model
Table 7.
CTX regular spiking neuron model parameters
| Parameter | Value |
|---|---|
| ars | 0.02 |
| brs | 0.2 |
| crs | −65 |
| drs | 8 |
Table 8.
CTX Regular Spiking neuron model equations
| Current | Equation | Gating variables | Parameters | |
|---|---|---|---|---|
| Iie | gie * (vrs−Esyn) * S |
|
gie =
0.2 Esyn = −85 τ = 5 td = 1 4 FSI → 1 RS |
|
| Ithco | gthco * (vrs−Esyn) * S |
|
gthco
= 0.15 Esyn = 0 τ = 5 td = 5.6 1 Th → 1 RS |
Cortical Fast Spiking Interneuron Model
Table 9.
CTX fast spiking interneuron model parameters
| Parameter | Value |
|---|---|
| afsi | 0.1 |
| bfsi | 0.2 |
| cfsi | −65 |
| dfsi | 2 |
Table 10.
CTX fast spiking interneuron model equations
| Current | Equation | Gating variables | Parameters | |
|---|---|---|---|---|
| Iei | gei * (vfsi−Esyn) * S |
|
gei
= 0.1 Esyn = 0 τ = 5 td = 1 4 RS → 1 FSI |
Table 11.
Healthy and PD state parameters
| Conditions | gcostr of direct pathway | gm | ggege |
|---|---|---|---|
| Healthy | 0.07 | 2.6 | 0.125 |
| PD | 0.026 | 1.5 | 0.5 |
References
- Agid Y, Javoy-Agid F, Ruberg M. Biochemistry of neurotransmitters in Parkinson’s disease. Movement disorders. 1987;2(7):166–230. [Google Scholar]
- Albin RL, Young AB, Penney JB. The functional anatomy of basal ganglia disorders. Trends in neurosciences. 1989;12(10):366–375. doi: 10.1016/0166-2236(89)90074-x. [DOI] [PubMed] [Google Scholar]
- Anderson ME, Postupna N, Ruffo M. Effects of high-frequency stimulation in the internal globus pallidus on the activity of thalamic neurons in the awake monkey. Journal of neurophysiology. 2003;89(2):1150–1160. doi: 10.1152/jn.00475.2002. [DOI] [PubMed] [Google Scholar]
- Baufreton J, Kirkham E, Atherton JF, Menard A, Magill PJ, Bolam JP, Bevan MD. Sparse but selective and potent synaptic transmission from the globus pallidus to the subthalamic nucleus. Journal of neurophysiology. 2009;102(1):532–545. doi: 10.1152/jn.00305.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergman H, Wichmann T, Karmon B, DeLong M. The primate subthalamic nucleus. II. Neuronal activity in the MPTP model of parkinsonism. Journal of neurophysiology. 1994;72(2):507–520. doi: 10.1152/jn.1994.72.2.507. [DOI] [PubMed] [Google Scholar]
- Birdno MJ, Grill WM. Mechanisms of deep brain stimulation in movement disorders as revealed by changes in stimulus frequency. Neurotherapeutics. 2008;5(1):14–25. doi: 10.1016/j.nurt.2007.10.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blesa J, Przedborski S. Parkinson’s disease: animal models and dopaminergic cell vulnerability. Frontiers in neuroanatomy. 2014;8 doi: 10.3389/fnana.2014.00155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolam J, Hanley J, Booth P, Bevan M. Synaptic organisation of the basal ganglia. Journal of anatomy. 2000;196(04):527–542. doi: 10.1046/j.1469-7580.2000.19640527.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch C, Degos B, Deniau JM, Venance L. Subthalamic nucleus high-frequency stimulation generates a concomitant synaptic excitation–inhibition in substantia nigra pars reticulata. The Journal of physiology. 2011;589(17):4189–4207. doi: 10.1113/jphysiol.2011.211367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brocker DT, Swan BD, Turner DA, Gross RE, Tatter SB, Miller Koop M, Grill WM. Improved efficacy of temporally non-regular deep brain stimulation in Parkinson’s disease. Experimental neurology. 2013;239:60–67. doi: 10.1016/j.expneurol.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DA. Muscarinic acetylcholine receptors (mAChRs) in the nervous system: some functions and mechanisms. Journal of molecular neuroscience. 2010;41(3):340–346. doi: 10.1007/s12031-010-9377-2. [DOI] [PubMed] [Google Scholar]
- Chang H, Kitai S. Projection neurons of the nucleus accumbens: an intracellular labeling study. Brain research. 1985;347(1):112–116. doi: 10.1016/0006-8993(85)90894-7. [DOI] [PubMed] [Google Scholar]
- Chang H, Wilson C, Kitai S. A Golgi study of rat neostriatal neurons: light microscopic analysis. Journal of Comparative Neurology. 1982;208(2):107–126. doi: 10.1002/cne.902080202. [DOI] [PubMed] [Google Scholar]
- Cruz AV, Mallet N, Magill PJ, Brown P, Averbeck BB. Effects of dopamine depletion on information flow. PNAS. 2012;109(44):18126–18131. doi: 10.1152/jn.00094.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degos B, Deniau JM, Thierry AM, Glowinski J, Pezard L, Maurice N. Neuroleptic-induced catalepsy: electrophysiological mechanisms of functional recovery induced by high-frequency stimulation of the subthalamic nucleus. The Journal of neuroscience. 2005;25(33):7687–7696. doi: 10.1523/JNEUROSCI.1056-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLong MR. Primate models of movement disorders of basal ganglia origin. Trends in neurosciences. 1990;13(7):281–285. doi: 10.1016/0166-2236(90)90110-v. [DOI] [PubMed] [Google Scholar]
- Dorval AD, Russo GS, Hashimoto T, Xu W, Grill WM, Vitek JL. Deep brain stimulation reduces neuronal entropy in the MPTP-primate model of Parkinson’s disease. Journal of neurophysiology. 2008;100(5):2807–2818. doi: 10.1152/jn.90763.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farries MA, Kita H, Wilson CJ. Dynamic spike threshold and zero membrane slope conductance shape the response of subthalamic neurons to cortical input. The Journal of neuroscience. 2010;30(39):13180–13191. doi: 10.1523/JNEUROSCI.1909-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogelson N, Kühn AA, Silberstein P, Limousin PD, Hariz M, Trottenberg T, Brown P. Frequency dependent effects of subthalamic nucleus stimulation in Parkinson’s disease. Neuroscience letters. 2005;382(1):5–9. doi: 10.1016/j.neulet.2005.02.050. [DOI] [PubMed] [Google Scholar]
- Fujimoto K, Kita H. Response characteristics of subthalamic neurons to the stimulation of the sensorimotor cortex in the rat. Brain research. 1993;609(1):185–192. doi: 10.1016/0006-8993(93)90872-k. [DOI] [PubMed] [Google Scholar]
- Götz T, Kraushaar U, Geiger J, Lübke J, Berger T, Jonas P. Functional properties of AMPA and NMDA receptors expressed in identified types of basal ganglia neurons. The Journal of neuroscience. 1997;17(1):204–215. doi: 10.1523/JNEUROSCI.17-01-00204.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grill WM, Snyder AN, Miocinovic S. Deep brain stimulation creates an informational lesion of the stimulated nucleus. Neuroreport. 2004;15(7):1137–1140. doi: 10.1097/00001756-200405190-00011. [DOI] [PubMed] [Google Scholar]
- Hahn PJ, McIntyre CC. Modeling shifts in the rate and pattern of subthalamopallidal network activity during deep brain stimulation. Journal of computational neuroscience. 2010;28(3):425–441. doi: 10.1007/s10827-010-0225-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond C, Bergman H, Brown P. Pathological synchronization in Parkinson’s disease: networks, models and treatments. Trends in neurosciences. 2007;30(7):357–364. doi: 10.1016/j.tins.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Hashimoto T, Elder CM, Okun MS, Patrick SK, Vitek JL. Stimulation of the subthalamic nucleus changes the firing pattern of pallidal neurons. The Journal of neuroscience. 2003;23(5):1916–1923. doi: 10.1523/JNEUROSCI.23-05-01916.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollerman JR, Grace AA. Subthalamic nucleus cell firing in the 6-OHDA-treated rat: basal activity and response to haloperidol. Brain research. 1992;590(1):291–299. doi: 10.1016/0006-8993(92)91108-q. [DOI] [PubMed] [Google Scholar]
- Hornykiewicz O. Biochemical aspects of Parkinson’s disease. Neurology. 1998;51(2 Suppl 2):S2–S9. doi: 10.1212/wnl.51.2_suppl_2.s2. [DOI] [PubMed] [Google Scholar]
- Humphries MD, Gurney K. Network effects of subthalamic deep brain stimulation drive a unique mixture of responses in basal ganglia output. European journal of neuroscience. 2012;36(2):2240–2251. doi: 10.1111/j.1460-9568.2012.08085.x. [DOI] [PubMed] [Google Scholar]
- Ikarashi Y, Takahashi A, Ishimaru H, Arai T, Maruyama Y. Regulation of Dopamine D< sub> 1</sub> and D< sub> 2</sub> Receptors on Striatal Acetylcholine Release in Rats. Brain research bulletin. 1997;43(1):107–115. doi: 10.1016/s0361-9230(96)00351-6. [DOI] [PubMed] [Google Scholar]
- Izhikevich EM. Simple model of spiking neurons. IEEE Transactions on neural networks. 2003;14(6):1569–1572. doi: 10.1109/TNN.2003.820440. [DOI] [PubMed] [Google Scholar]
- Jankovic J, Rajput AH, McDermott MP, Perl DP. The evolution of diagnosis in early Parkinson disease. Archives of neurology. 2000;57(3):369–372. doi: 10.1001/archneur.57.3.369. [DOI] [PubMed] [Google Scholar]
- Kang G, Lowery MM. Interaction of oscillations, and their suppression via deep brain stimulation, in a model of the cortico-basal ganglia network. Neural Systems and Rehabilitation Engineering, IEEE Transactions on. 2013;21(2):244–253. doi: 10.1109/TNSRE.2013.2241791. [DOI] [PubMed] [Google Scholar]
- Kita H. Neostriatal and globus pallidus stimulation induced inhibitory postsynaptic potentials in entopeduncular neurons in rat brain slice preparations. Neuroscience. 2001;105(4):871–879. doi: 10.1016/s0306-4522(01)00231-7. [DOI] [PubMed] [Google Scholar]
- Kita H, Kita T. Cortical stimulation evokes abnormal responses in the dopamine- depleted rat basal ganglia. The Journal of neuroscience. 2011;31(28):10311–10322. doi: 10.1523/JNEUROSCI.0915-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kita H, Kitai S. Intracellular study of rat globus pallidus neurons: membrane properties and responses to neostriatal, subthalamic and nigral stimulation. Brain research. 1991;564(2):296–305. doi: 10.1016/0006-8993(91)91466-e. [DOI] [PubMed] [Google Scholar]
- Kühn AA, Kempf F, Brücke C, Doyle LG, Martinez-Torres I, Pogosyan A, Hariz MI. High-frequency stimulation of the subthalamic nucleus suppresses oscillatory β activity in patients with Parkinson’s disease in parallel with improvement in motor performance. The Journal of neuroscience. 2008;28(24):6165–6173. doi: 10.1523/JNEUROSCI.0282-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy R, Ashby P, Hutchison WD, Lang AE, Lozano AM, Dostrovsky JO. Dependence of subthalamic nucleus oscillations on movement and dopamine in Parkinson’s disease. Brain. 2002;125(6):1196–1209. doi: 10.1093/brain/awf128. [DOI] [PubMed] [Google Scholar]
- Li Q, Ke Y, Chan DC, Qian ZM, Yung KK, Ko H, Yung WH. Therapeutic deep brain stimulation in Parkinsonian rats directly influences motor cortex. Neuron. 2012;76(5):1030–1041. doi: 10.1016/j.neuron.2012.09.032. [DOI] [PubMed] [Google Scholar]
- Mallet N, Ballion B, Le Moine C, Gonon F. Cortical inputs and GABA interneurons imbalance projection neurons in the striatum of parkinsonian rats. The Journal of neuroscience. 2006;26(14):3875–3884. doi: 10.1523/JNEUROSCI.4439-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallet N, Pogosyan A, Márton LF, Bolam JP, Brown P, Magill PJ. Parkinsonian beta oscillations in the external globus pallidus and their relationship with subthalamic nucleus activity. The Journal of neuroscience. 2008;28(52):14245–14258. doi: 10.1523/JNEUROSCI.4199-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallet N, Pogosyan A, Sharott A, Csicsvari J, Bolam JP, Brown P, Magill PJ. Disrupted dopamine transmission and the emergence of exaggerated beta oscillations in subthalamic nucleus and cerebral cortex. The Journal of neuroscience. 2008;28(18):4795–4806. doi: 10.1523/JNEUROSCI.0123-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsden C, Parkes J, Quinn N. Movement disorders. 1. Vol. 198. London: Butterworth; 1982. Fluctuations of disability in Parkinson’s disease: clinical aspects; pp. 96–122. [Google Scholar]
- McCarthy M, Moore-Kochlacs C, Gu X, Boyden E, Han X, Kopell N. Striatal origin of the pathologic beta oscillations in Parkinson’s disease. Proceedings of the National Academy of Sciences. 2011;108(28):11620–11625. doi: 10.1073/pnas.1107748108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell GC, So RQ, Hilliard JD, Lopomo P, Grill WM. Effective deep brain stimulation suppresses low-frequency network oscillations in the basal ganglia by regularizing neural firing patterns. The Journal of neuroscience. 2012;32(45):15657–15668. doi: 10.1523/JNEUROSCI.2824-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miguelez C, Morin S, Martinez A, Goillandeau M, Bezard E, Bioulac B, Baufreton J. Altered pallido-pallidal synaptic transmission leads to aberrant firing of globus pallidus neurons in a rat model of Parkinson’s disease. The Journal of physiology. 2012;590(22):5861–5875. doi: 10.1113/jphysiol.2012.241331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran RJ, Mallet N, Litvak V, Dolan RJ, Magill PJ, Friston KJ, Brown P. Alterations in brain connectivity underlying beta oscillations in Parkinsonism. PLoS computational biology. 2011;7(8):e1002124. doi: 10.1371/journal.pcbi.1002124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moro E, Lozano AM, Pollak P, Agid Y, Rehncrona S, Volkmann J, Hariz MI. Long-term results of a multicenter study on subthalamic and pallidal stimulation in Parkinson’s disease. Movement disorders. 2010;25(5):578–586. doi: 10.1002/mds.22735. [DOI] [PubMed] [Google Scholar]
- Nakanishi H, Kita H, Kitai S. Intracellular study of rat substantia nigra pars reticulata neurons in an in vitro slice preparation: electrical membrane properties and response characteristics to subthalamic stimulation. Brain research. 1987;437(1):45–55. doi: 10.1016/0006-8993(87)91525-3. [DOI] [PubMed] [Google Scholar]
- Nakanishi H, Kita H, Kitai S. Intracellular study of rat entopeduncular nucleus neurons in an in vitro slice preparation: response to subthalamic stimulation. Brain research. 1991;549(2):285–291. doi: 10.1016/0006-8993(91)90469-c. [DOI] [PubMed] [Google Scholar]
- Nambu A, Tokuno H, Hamada I, Kita H, Imanishi M, Akazawa T, Hasegawa N. Excitatory cortical inputs to pallidal neurons via the subthalamic nucleus in the monkey. Journal of neurophysiology. 2000;84(1):289–300. doi: 10.1152/jn.2000.84.1.289. [DOI] [PubMed] [Google Scholar]
- Nambu A, Tokuno H, Takada M. Functional significance of the cortico– subthalamo–pallidal ‘hyperdirect’pathway. Neuroscience research. 2002;43(2):111–117. doi: 10.1016/s0168-0102(02)00027-5. [DOI] [PubMed] [Google Scholar]
- Nicola SM, Surmeier DJ, Malenka RC. Dopaminergic modulation of neuronal excitability in the striatum and nucleus accumbens. Annual review of neuroscience. 2000;23(1):185–215. doi: 10.1146/annurev.neuro.23.1.185. [DOI] [PubMed] [Google Scholar]
- Otsuka T, Abe T, Tsukagawa T, Song WJ. Conductance-based model of the voltage-dependent generation of a plateau potential in subthalamic neurons. Journal of neurophysiology. 2004;92(1):255–264. doi: 10.1152/jn.00508.2003. [DOI] [PubMed] [Google Scholar]
- Pan MK, Tai CH, Liu WC, Pei JC, Lai WS, Kuo CC. Deranged NMDAergic cortico-subthalamic transmission underlies parkinsonian motor deficits. The Journal of clinical investigation. 2014;124(10):4629. doi: 10.1172/JCI75587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang Z, Ling GY, Gajendiran M, Xu ZC. Enhanced excitatory synaptic transmission in spiny neurons of rat striatum after unilateral dopamine denervation. Neuroscience letters. 2001;308(3):201–205. doi: 10.1016/s0304-3940(01)02031-6. [DOI] [PubMed] [Google Scholar]
- Plenz D, Kital ST. A basal ganglia pacemaker formed by the subthalamic nucleus and external globus pallidus. Nature. 1999;400(6745):677–682. doi: 10.1038/23281. [DOI] [PubMed] [Google Scholar]
- Quinn N, Luthert P, Honavar M, Marsden C. Pure akinesia due to Lewy body Parkinson’s disease: a case with pathology. Movement disorders. 1989;4(1):85–89. doi: 10.1002/mds.870040112. [DOI] [PubMed] [Google Scholar]
- Rajput A, Sitte H, Rajput A, Fenton M, Pifl C, Hornykiewicz O. Globus pallidus dopamine and Parkinson motor subtypes Clinical and brain biochemical correlation. Neurology. 2008;70(16 Part 2):1403–1410. doi: 10.1212/01.wnl.0000285082.18969.3a. [DOI] [PubMed] [Google Scholar]
- Raz A, Vaadia E, Bergman H. Firing patterns and correlations of spontaneous discharge of pallidal neurons in the normal and the tremulous 1-methyl-4-phenyl-1, 2, 3, 6- tetrahydropyridine vervet model of parkinsonism. The Journal of neuroscience. 2000;20(22):8559–8571. doi: 10.1523/JNEUROSCI.20-22-08559.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin JE, Terman D. High frequency stimulation of the subthalamic nucleus eliminates pathological thalamic rhythmicity in a computational model. Journal of computational neuroscience. 2004;16(3):211–235. doi: 10.1023/B:JCNS.0000025686.47117.67. [DOI] [PubMed] [Google Scholar]
- Ryu SB, Bae EK, Kim J, Hwang YS, Im C, Chang JW, Kim KH. Neuronal Responses in the Globus Pallidus during Subthalamic Nucleus Electrical Stimulation in Normal and Parkinson’s Disease Model Rats. The Korean Journal of Physiology & Pharmacology. 2013;17(4):299–306. doi: 10.4196/kjpp.2013.17.4.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw FZ, Liao YF. Relation between activities of the cortex and vibrissae muscles during high-voltage rhythmic spike discharges in rats. Journal of neurophysiology. 2005;93(5):2435–2448. doi: 10.1152/jn.00999.2004. [DOI] [PubMed] [Google Scholar]
- Sims RE, Woodhall GL, Wilson CL, Stanford IM. Functional characterization of GABAergic pallidopallidal and striatopallidal synapses in the rat globus pallidus in vitro. European journal of neuroscience. 2008;28(12):2401–2408. doi: 10.1111/j.1460-9568.2008.06546.x. [DOI] [PubMed] [Google Scholar]
- So RQ, Kent AR, Grill WM. Relative contributions of local cell and passing fiber activation and silencing to changes in thalamic fidelity during deep brain stimulation and lesioning: a computational modeling study. Journal of computational neuroscience. 2012;32(3):499–519. doi: 10.1007/s10827-011-0366-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- So RQ, McConnell GC, August AT, Grill WM. Characterizing effects of subthalamic nucleus deep brain stimulation on methamphetamine-induced circling behavior in hemi-Parkinsonian rats. Neural Systems and Rehabilitation Engineering, IEEE Transactions on. 2012;20(5):626–635. doi: 10.1109/TNSRE.2012.2197761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taverna S, Ilijic E, Surmeier DJ. Recurrent collateral connections of striatal medium spiny neurons are disrupted in models of Parkinson’s disease. The Journal of neuroscience. 2008;28(21):5504–5512. doi: 10.1523/JNEUROSCI.5493-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmermann L, Wojtecki L, Gross J, Lehrke R, Voges J, Maarouf M, Schnitzler A. Ten-Hertz stimulation of subthalamic nucleus deteriorates motor symptoms in Parkinson’s disease. Movement disorders. 2004;19(11):1328–1333. doi: 10.1002/mds.20198. [DOI] [PubMed] [Google Scholar]
- Tremblay L, Filion M. Responses of pallidal neurons to striatal stimulation in monkeys with MPTP-induced parkinsonism. Brain research. 1989;498(1):17–33. doi: 10.1016/0006-8993(89)90395-8. [DOI] [PubMed] [Google Scholar]
- Walker HC, Huang H, Gonzalez CL, Bryant JE, Killen J, Knowlton RC, Guthrie BL. Short latency activation of cortex by clinically effective thalamic brain stimulation for tremor. Movement disorders. 2012;27(11):1404–1412. doi: 10.1002/mds.25137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver FM, Follett K, Stern M, Hur K, Harris C, Marks WJ, Moy CS. Bilateral deep brain stimulation vs best medical therapy for patients with advanced Parkinson disease: a randomized controlled trial. Jama. 2009;301(1):63–73. doi: 10.1001/jama.2008.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichmann T, Soares J. Neuronal firing before and after burst discharges in the monkey basal ganglia is predictably patterned in the normal state and altered in parkinsonism. Journal of neurophysiology. 2006;95(4):2120–2133. doi: 10.1152/jn.01013.2005. [DOI] [PubMed] [Google Scholar]
- Xu W, Russo GS, Hashimoto T, Zhang J, Vitek JL. Subthalamic nucleus stimulation modulates thalamic neuronal activity. The Journal of neuroscience. 2008;28(46):11916–11924. doi: 10.1523/JNEUROSCI.2027-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamawaki N, Stanford IM, Hall SD, Woodhall GL. Pharmacologically induced and stimulus evoked rhythmic neuronal oscillatory activity in the primary motor cortex< i> in vitro</i>. Neuroscience. 2008;151(2):386–395. doi: 10.1016/j.neuroscience.2007.10.021. [DOI] [PubMed] [Google Scholar]