

Abstract
Polygenetic risk factors and reduced expression of many genes in late-onset Alzheimer’s disease (AD) impedes identification of a target(s) for disease-modifying therapies. We identified a single microRNA, miR-34a that is over expressed in specific brain regions of AD patients as well as in the 3xTg-AD mouse model. Specifically, increased miR-34a expression in the temporal cortex region compared to age matched healthy control correlates with severity of AD pathology. miR-34a over expression in patient’s tissue and forced expression in primary neuronal culture correlates with concurrent repression of its target genes involved in synaptic plasticity, oxidative phosphorylation and glycolysis. The repression of oxidative phosphorylation and glycolysis related proteins correlates with reduced ATP production and glycolytic capacity, respectively. We also found that miR-34a overexpressed neurons secrete miR-34a containing exosomes that are taken up by neighboring neurons. Furthermore, miR-34a targets dozens of genes whose expressions are known to be correlated with synchronous activity in resting state functional networks. Our analysis of human genomic sequences from the tentative promoter of miR-34a gene shows the presence of NFκB, STAT1, c-Fos, CREB and p53 response elements. Together, our results raise the possibilities that pathophysiology-induced activation of specific transcription factor may lead to increased expression of miR-34a gene and miR-34a mediated concurrent repression of its target genes in neural networks may result in dysfunction of synaptic plasticity, energy metabolism, and resting state network activity. Thus, our results provide insights into polygenetic AD mechanisms and disclose miR-34a as a potential therapeutic target for AD.
Keywords: Alzheimer’s disease, microRNAs, miR-34a, oxidative phosphorylation, glycolysis, synaptic plasticity, resting state network
1. Introduction
Late-onset Alzheimer’s disease (AD) accounts for more than 90% of AD cases and is the most common form of dementia (Querfurth and LaFerla, 2010). Currently, there are no effective treatments for AD. Recently genome-wide association studies (GWAS) of AD have identified that nine novel risk factor genes (Seshadri et al., 2010) and their expression may be involved in AD (Allen et al., 2012). Furthermore, genome-wide transcriptome study indicate that many important genes necessary for energy metabolism and synapse activity are downregulated in AD (Liang et al., 2008). fMRI and PET imaging studies of AD patient’s resting state network (RSN) revealed that with progressing AD severity, a decrease in glucose and oxygen metabolism correlates with increased Clinical Dementia Rating Scores (Brier et al., 2012). Although genes that cause loss of functional connections in intra- and inter-network resting state with AD progression remain unknown, many genes are recently reported to be correlated with functional RSN (Richiardi et al., 2015). Thus, in order to develop new therapeutic strategies to treat AD, there is a need to identify novel molecular target(s) that concomitantly dysregulate expression of multiple genes involved in synaptic plasticity, energy metabolism, and RSN. A single 22-nt non-coding microRNA targets many genes and concomitantly down regulates multiple biological pathways by repressing mRNA translation/or degradation (Bartel, 2009). As such, we sought to identify expression of novel microRNAs in AD brains that maximizes the number of target genes possibly involved in cognitive function. Although regulation of memory and synaptic plasticity via a single microRNAs has been previously reported in mice (Gao et al., 2010; Griggs et al., 2013), the causal effects of a single microRNAs involving polygenetic mechanism of AD are not known.
2. Results
2.1 Expression levels of selected miRNAs in human and mouse AD brains
Because a single microRNA targets many genes and concomitantly down regulates multiple biological pathways, we sought to identify expression of novel microRNAs in AD brains that maximizes the number of targets possibly involved in cognitive function. To this end, we selected five miRNAs after data mining of dozens of miRNA target genes from five different miRNA target gene databases (Vlachos and Hatzigeorgiou, 2013). These five miRNAs were hsa-miRNA-34a, 146a, -9, -132, and -15 whose target predicted by all five databases collectively have the potential to deregulate genes associated with synaptic plasticity, neuronal survival, energy metabolism, amyloid precursor protein (APP) metabolism, Aβ elimination, and RSN. We next quantified expression of these miRNAs via qRT-PCR from temporal cortex (TC), frontal cortex (FC), and cerebellum (CB) of subjects with AD and compared them to healthy age-matched control (AMC) (Table 1). Expression analysis showed that in the TC, miR-34a expression was significantly increased compared to AMC (Fig. 1A, left panel), but not in the FC (Fig. 1B), and CB (Fig. 1C). When differentiated by Braak and Braak (B&B) stages, miR-34a expression was significantly increased in the TC of both stage III and stage VI (Fig. 1A, middle panel) but not in the FC or CB. Pearson’s test show significant positive correlation between miR-34a level and B&B stage as well as amyloid angiopathy in the TC (Fig. 1A, right panel), but not in FC or CB. Expression level the other four miRNAs in the same tissues are shown in Table 2. Expression of miR-146a was also increased in the TC and was B&B stage VI specific. There was no significant increase in expression of miR-132, miR-9, and miR-15a in TC region of AD. In case of the FC, changes in expression of miR-146a, -132, -9, and -15a were not significant. But B&B stage specific increased expression of miR-9in frontal cortex was significant. In the CB region, expression changes of miR-146a, -132 and -15a were not significant. miR-9 expression in CB region was significantly reduced.
Table 1.
Human subject list with pathological scores.
| ID | Age | Sex | APOE | PMD | Diagnoses | B&B stage | Atherosclerosis* | Amyloid angiopathy* | Other pathology |
|---|---|---|---|---|---|---|---|---|---|
| 591 | 78 | F | 33 | 5.5 | Normal CERAD 1A | 1 | |||
| 673 | 80 | F | 33 | 1.2 | Normal CERAD 1A | 1 | Mild | ||
| 981 | 74 | F | 24 | 4.7 | Normal CERAD 1B | 3 | Metastatic Carcinoma | ||
| 1035 | 72 | F | 33 | 30 | Normal CERAD 1B | 2 | |||
| 196 | 75 | M | 33 | 18.9 | Normal CERAD 1B | 1 | |||
| 381 | 78 | M | n/a | 9.8 | Normal CERAD 1A | 1 | |||
| 837 | 75 | M | 22 | 29 | Normal CERAD 1A | 1 | Lacunes | ||
| 543 | 72 | F | 34 | 3 | Normal CERAD 1A | 1 | |||
| 707 | 80 | M | 33 | 4.3 | Normal CERAD 1B | 1 | |||
| 795 | 81 | M | 34 | 7.2 | Normal CERAD 1B | 1 | Lacunes | ||
| 357 | 75 | M | 33 | 3 | AD | 3 | Mild | ||
| 347 | 77 | M | 33 | 16.8 | AD | 3 | |||
| 601 | 76 | F | 33 | 10.5 | AD | 3 | |||
| 602 | 78 | M | 34 | 0.8 | AD | 3 | Mild | ||
| 731 | 76 | F | 44 | 5.5 | AD | 3 | Mild | Mild | |
| 1054 | 75 | F | 34 | 20.1 | AD | 3 | Severe | Mild | |
| 1052 | 71 | M | 33 | 25.2 | AD | 6 | Mild | ||
| 1068 | 77 | M | 44 | 25.4 | AD | 6 | Mild | Mild | |
| 1560 | 74 | F | 34 | 20.5 | AD | 6 | |||
| 360 | 75 | F | 33 | 13.3 | AD | 6 | Mild | ||
| 1664 | 79 | F | n/a | 20.8 | AD | 6 | Moderate | Infarcts | |
| 49 | 75 | F | 34 | 24 | AD | 6 | Severe | ||
| 254 | 75 | M | 34 | 2.6 | AD | 6 | Moderate |
Pathological scores were analyzed by a pathologist for atherosclerosis and amyloid angiopathy using CERAD guidelines and NIA-Reagan criteria
Figure 1.
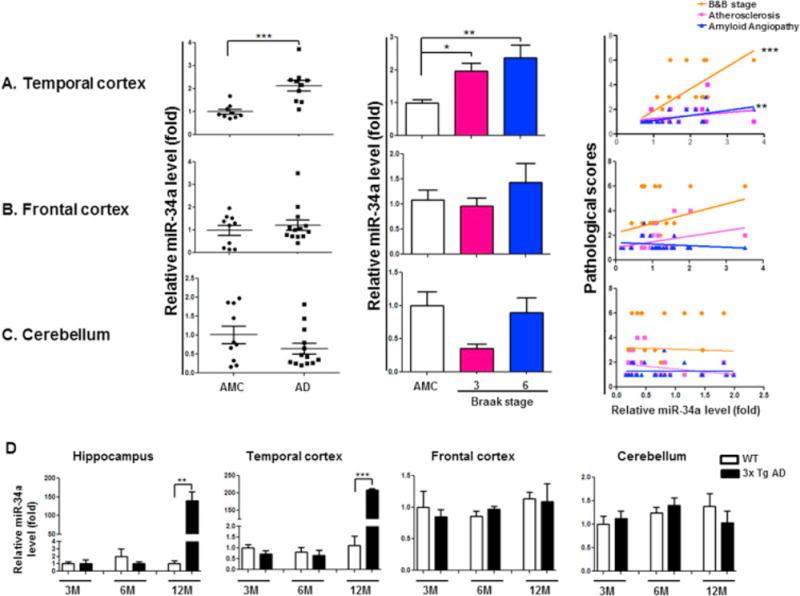
Expression of miR-34a in Human and Mouse Alzheimer’s disease (AD) brain regions. miR-34a is significantly upregulated in AD patients TC and AD mice TC and hippocampus but not in FC and CB region. qRT-PCR analysis was used to measure miR-34a levels from postmortem brain samples from AD patients and age-matched control (AMC) as well as 3xTg AD mice. (A) miR-34a relative fold levels in the TC of human AD patients (n=10) when compared with AMC (n=9). Five AD cases were diagnosed as Braak & Braak (B&B) stage VI and five were diagnosed as stage III. Bar graph shows B&B stage specific increment of miR-34a expression in TC. Pearson’s test analysis on individual AD samples of TC significantly correlated with B&B stage and amyloid angiopathy. (B) miR-34a relative fold levels in the FC of human AD patients (n=13) when compared with AMC (n=10). Seven AD cases were diagnosed as B&B stage III and six were B&B stage VI. No significant increase in miR-34a expression level was found in FC region. Also no significance in B&B stage specific expression and no positive correlation in Pearson’s test found in FC. (C) miR-34a relative fold levels in the CB of human AD patients (n=13) when compared with AMC (n=9). Seven AD cases were diagnosed as B&B stage III and six were B&B stage VI. No significant increase in miR-34a expression level was found in CB region. Also no significance in B&B stage specific expression and no positive correlation with the AD patient’s pathology in Pearson’s test was found in the CB region. (D) miR-34a expression determined by qRT-PCR in different regions of 3x Tg AD mice brain. *p<0.05, **p<0.01, ***p<0.001
Table 2.
miR-146a, -132, -9 and -15a levels in AD human brain.
| Braak stage | |||||
|---|---|---|---|---|---|
|
| |||||
| miRNA | Region | AMC | AD | III | IV |
| TC | 1.00 ± 0.10 | 3.63 ± 1.03* | 1.52 ± 0.21 | 5.75 ± 1.69** | |
| miR-146a | FC | 1.00 ± 0.19 | 5.27 ± 1.86 | 1.44 ± 0.31 | 8.55 ± 2.98** |
| CB | 1.00 ± 0.14 | 3.73 ± 1.63 | 0.16 ± 0.02 | 6.80 ± 2.54** | |
| TC | 1.00 ± 0.23 | 2.76 ± 0.76 | 1.11 ± 0.10 | 4.42 ± 1.21** | |
| miR-132 | FC | 1.00 ± 0.26 | 1.48 ± 0.43 | 0.67 ± 0.12 | 2.18 ± 0.71 |
| CB | 1.00 ± 0.31 | 0.10 ± 0.01** | 0.12 ± 0.01* | 0.10 ± 0.01* | |
| TC | 1.00 ± 0.10 | 1.67 ± 0.32 | 1.02 ± 0.13 | 2.32 ± 0.52** | |
| miR-9 | FC | 1.00 ± 0.17 | 1.22 ± 0.24 | 0.58 ± 0.07 | 1.78 ± 0.32* |
| CB | 1.00 ± 0.11 | 1.51 ± 0.42 | 0.58 ± 0.12 | 2.32 ± 0.63* | |
| TC | 1.00 ± 0.09 | 1.25 ± 0.15 | 1.17 ± 0.14 | 1.32 ± 0.28 | |
| miR-15a | FC | 1.00 ± 0.10 | 1.05 ± 0.12 | 0.71 ± 0.08 | 1.34 ± 0.15 |
| CB | 1.00 ± 0.14 | 1.13 ± 0.18 | 0.74 ± 0.14 | 1.46 ± 0.26 | |
AD patients (n=10) and age-matched controls (AMC, n=9). TC (temporal cortex), FC (frontal cortex) and CB (cerebellum).
Statistical significance:
p<0.05
p<0.01
Although genetics of AD and familial AD (FAD) are different, converging evidences from PIB-PET (Bateman et al., 2012), MRI (Reiman et al., 2012) and the resting state functional connectivity MRI (Thomas et al., 2014) imaging studies suggest that late onset AD and familial AD have similar disease processes. In order to further characterize the similarities between the two AD types, we profiled miRNA expression in brain regions from familial AD model of transgenic AD mice (3xTg AD). Expression of miR-34a was significantly increased in TC of 3xTg AD mice by 12-month of age (Fig. 1D) but not in the FC and CB (Fig 1D) as we have seen in human AD cases. miR-34a expression was also increased in the hippocampus of these mice (Fig. 1D) as reported in human AD (Zovoilis et al., 2011). Expression levels of the other four miRNAs in FC, TC, and CB, and in the hippocampus were not significantly altered compare to the age matched control (Table 3).
Table 3.
miR-146a, -132, -9 and -15a levels in AD mice brain
| miRNA | Group (N) | Brain region (Relative fold Mean ± SEM) | |||
|---|---|---|---|---|---|
| Hip | TC | FC | CB | ||
| miR-146a | 3M C (n=3) | 1.00 ± 0.09 | 1.00 ± 0.14 | 1.00 ± 0.05 | 1.00 ± 0.12 |
| 3M AD (n=4) | 0.96 ± 0.08 | 0.81 ± 0.18 | 0.87 ± 0.08 | 1.09 ± 0.15 | |
| 6M C (n=3) | 0.99 ± 0.03 | 0.81 ± 0.22 | 0.97 ± 0.11 | 1.43 ± 0.05 | |
| 6M AD (n=4) | 0.83 ± 0.04* | 0.73 ± 0.20 | 0.99 ± 0.06 | 1.79 ± 0.17 | |
| 12M WT (n=3) | 1.19 ± 0.01 | 0.78 ± 0.19 | 1.05 ± 0.07 | 2.77 ± 0.66 | |
| 12M AD (n=3) | 0.95 ± 0.10 | 0.79 ± 0.21 | 0.97 ± 0.35 | 1.80 ± 0.57 | |
| miR-132 | 3M WT (n=3) | 1.00 ± 0.16 | 1.00 ± 0.04 | 1.00 ± 0.25 | 1.00 ± 0.02 |
| 3M AD (n=4) | 1.29 ± 0.15 | 1.01 ± 0.04 | 1.09 ± 0.13 | 1.03 ± 0.10 | |
| 6M WT (n=3) | 0.88 ± 0.10 | 0.81 ± 0.11 | 0.98 ± 0.09 | 1.16 ± 0.06 | |
| 6M AD (n=4) | 1.42 ± 0.23 | 0.78 ± 0.11 | 1.06 ± 0.06 | 1.42 ± 0.09 | |
| 12M WT (n=3) | 1.19 ± 0.24 | 0.87 ± 0.15 | 1.04 ± 0.01 | 1.27 ± 0.11 | |
| 12M AD (n=3) | 1.36 ± 0.05 | 0.90 ± 0.16 | 0.99 ± 0.16 | 0.91 ± 0.13 | |
| miR-9 | 3M WT (n=3) | 1.00 ± 0.06 | 1.00 ± 0.14 | 1.00 ± 0.14 | 1.00 ± 0.02 |
| 3M AD (n=4) | 0.80 ± 0.10 | 1.06 ± 0.04 | 0.73 ± 0.10 | 1.08 ± 0.04 | |
| 6M WT (n=3) | 0.75 ± 0.14 | 0.93 ± 0.20 | 0.64 ± 0.03 | 1.80 ± 0.38 | |
| 6M AD (n=4) | 0.76 ± 0.08 | 0.89 ± 0.07 | 0.64 ± 0.05 | 2.27 ± 0.07 | |
| 12M WT (n=3) | 0.91 ± 0.034 | 1.14 ± 0.08 | 0.68 ± 0.08 | 2.18 ± 0.15 | |
| 12M AD (n=3) | 0.60 ± 0.07* | 1.00 ± 0.12 | 0.43 ± 0.10 | 1.62 ± 0.16 | |
| miR-15a | 3M WT (n=3) | 1.00 ± 0.11 | 1.00 ± 0.06 | 1.00 ± 0.22 | 1.00 ± 0.07 |
| 3M AD (n=4) | 0.80 ± 0.03 | 0.98 ± 0.07 | 1.12 ± 0.12 | 1.03 ± 0.06 | |
| 6M WT (n=3) | 0.91 ± 0.01 | 1.16 ± 0.11 | 1.06 ± 0.04 | 1.50 ± 0.20 | |
| 6M AD (n=4) | 0.77 ± 0.06 | 1.02 ± 0.11 | 1.03 ± 0.04 | 1.55 ± 0.09 | |
| 12M WT (n=3) | 0.90 ± 0.02 | 1.22 ± 0.09 | 1.16 ± 0.02 | 1.72 ± 0.12 | |
| 12M AD (n=3) | 0.68 ± 0.03** | 1.01 ± 0.20 | 0.75 ± 0.12* | 1.01 ± 0.13* | |
Statistical significance:
p<0.05
p<0.01 M = month; WT = wildtype mice; AD = 3xTgAD mice
2.2. Functional analysis of the repression of target proteins in increased miR-34a AD brain regions
It has been reported that miR-34c, a family member of miR-34, targets the SIRT1 gene, and elevated level of miR-34c correlates with memory impairment in AD mice (Agostini et al., 2011a). Also, it has been shown that ectopic expression of miR-34a in primary cortical neurons affects dendritic spine morphology and the reduction of amplitude/frequency of miniature excitatory synaptic current (mEPSC) (Agostini et al., 2011b). Scanning all genes targeted by both miR-34 family and miR-146a, we selected miR-34a as a novel candidate based on its potential to target the maximal number of genes involved in brain energy metabolism, synaptic plasticity, and synchronous activity in RSN (Table 4). To assess expression of miR-34a targeted proteins in AD brains, we measured protein levels of selected synaptic plasticity related genes, VAMP2, SYT1, HCN1, NR2A and GLUR1; oxidative phosphorylation related genes, NDUFC2, SDHC, UQCRB, UQCRQ, and COX10; and genes in the glycolytic pathway, H6PD, PFK1, and PFK2 in the TC and CB of the same AD and AMC samples used in our microRNA analysis. Western blot analysis revealed a consistent and significant down regulation of the synaptic proteins, proteins involved in oxidative phosphorylation and glycolysis in the TC, but not in the CB (Fig. 2)
Table 4.
miR-34a target genes involved in synaptic plasticity and energy metabolism.
| Gene | miRNA/Target mRNA alignment |
|---|---|
| Synaptic plasticity proteins | |
| VAMP2 (synaptobrevin2) | |
| SYT1 (Synaptotagmin 1) | |
| HCN (Hyperpolarization-activated cyclic nucleotide-sensitive channels) | |
| NR2A (GRIN2)(N-methyl-D-aspartate receptor 2A) | |
| GLUR1 (GRIA1) (Glutamate receptor) | |
| Oxidative phosphorylation proteins | |
| NDUFC2 (NADH dehydrogenase [ubiquinone] 1 subunit C2) | |
| SDHC (Succinate dehydrogenase complex subunit C) | |
| UQCRB (ubiquinol-cytochrome c reductase binding protein) | |
| UQCRQ (ubiquinol-cytochrome c reductase, complex III subunit VII) | |
| COX10 (COX10 heme A:farnesyltransferase cytochrome c oxidase assembly factor) | |
| Glycolysis proteins | |
| H6PD (hexose-6-phosphate dehydrogenase) | |
| PFK1 (PFKM) (Phosphofructokinase-1) | |
| PFK2 (PFKFB3) (Phosphofructokinase-2) | |
Figure 2.
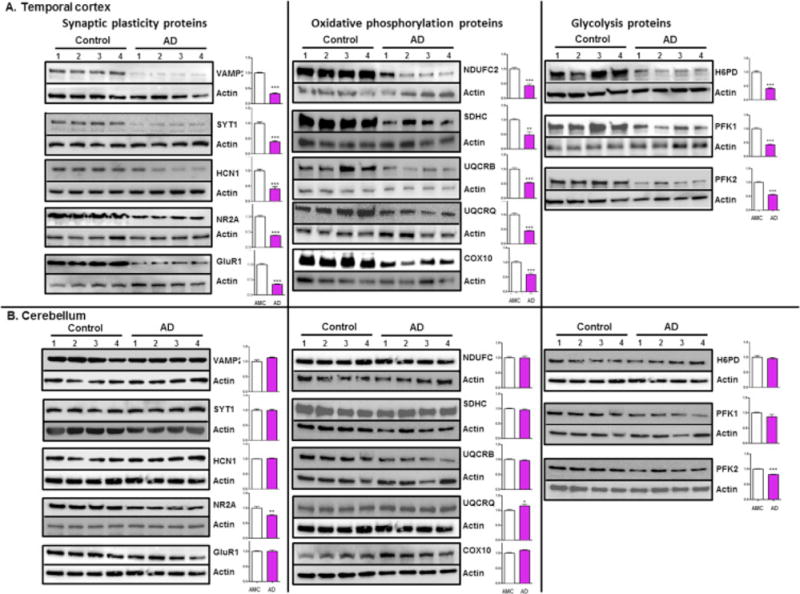
Suppression of miR-34a target proteins in AD patients brain regions. Representative western blot of synaptic plasticity related proteins, oxidative phosphorylation proteins and enzymatic glycolysis proteins probed with respective protein specific antibody in protein samples isolated from four AD patients (B&B stage VI) and AMC are shown in the TC and CB. Representative western blots of β-actin were used as normalization controls. Densitometric quantification of respective protein levels are shown in the adjacent bar graph (fold difference, average of control = 1, data are mean ± SEM, n = 4, *p < 0.05 **p <0.01 ***p<0.001.
2.3. Functional analysis of the repression of target proteins by overexpressing miR-34a in primary neurons
Next, we assessed the functional consequences of miR-34a mediated repression of oxidative phosphorylation and glycolysis related gene transcripts in ectopically expressed miR-34a primary neurons. In order to evaluate the effects of miR-34a on cellular energy metabolisms, we constructed a miR-34a expression vector and measured oxidative phosphorylation by oxygen consumption rate and glycolysis by extracellular acidification rate in miR-34a transfected primary cortical neurons. Our data revealed that oxidative phosphorylation including, ATP production, spare capacity, maximum respiration, and proton leak are severely inhibited in neurons transfected with miR-34a expression vector when compared to vector and mock transfected cells (Fig. 3A). Additionally, glycolytic capacity and glycolytic reserve were significantly reduced in miR-34a transfected neurons (Fig. 3D). Protein analysis of miR-34a target genes revealed that all of the oxidative phosphorylation proteins, NDUFC2, SDHC, UQCRB, UQCRQ, and COX 10 (Fig. 3B and C), and glycolysis proteins, H6PD, PFK1, PFK2, and LDH (Fig. 3E and F) were significantly reduced in miR-34a transfected primary neurons when compared to mock and empty vector transfected controls. Severe reduction of oxidative phosphorylation and glycolysis were not due to cellular death as determined by Calcein AM viability assay of 36-hours post-transfected primary neurons (Fig. 3G). Also, measurement of miR-34a expression in transfected neurons (Fig. 3H) and in exosomes isolated from the growth medium (Fig. 3I) revealed a 10-20-fold increase in miR-34a in neurons and a 50-250-fold increase in media.
Figure 3.
Functional effects of ectopically expressed miR-34a on oxidative phosphorylation and glycolysis in primary neurons. (A) ATP synthesis, spare respiratory capacity, maximal respiration, and proton leak was measured in rat primary neurons (E18, 7DIV) 36 hour after transfection with empty vector or with three increasing concentration of miR-34a-expression vector. (B) Representative western blot of oxidative phosphorylation proteins, NDUFC2, SDHC, UQCRB, UQCRQ, and COX10 probed with respective protein specific antibody in protein samples isolated from the transfected primary neurons. β-actin was used as normalization controls. (C) Densitometric quantification of respective protein levels are shown in the adjacent bar graph as fold difference, average of control = 1, data are mean ± SEM, n = 3 independent transfection experiments. (D) Glycolysis rate was measured in rat primary neurons (E18, 7DIV) thirty six hour after transfection with empty vector and with three increasing concentration of miR-34a-expression vector. (E) Representative western blots of enzymatic glycolysis proteins, H6PD, PFK1, PFK2, and LDHA probed with respective protein specific antibody in protein samples isolated from isolated from the transfected primary neurons. β-actin was used as normalization controls. (F) Densitometric quantification of respective protein levels are shown in the adjacent bar graph as fold difference, average of control = 1, data are mean ± SEM, n = 3 independent transfection experiments. (G) Viability of the miR-34a expression plasmid transfected neurons were determined by Calcein AM assay. Level of miR-34a in transfected neurons (H) and in exosomes isolated from the transfected cell culture medium (I) were determined by qRT-PCR. *p < 0.05,**p <0.01 and ***p<0.001
2.4. Interneuronal transfer of miR-34a and spreading of neuron-derived miR-34a-containing GFP-tagged exosomes
Secretion of miR-34a-containing exosomes from neurons to the medium raises the possibility of intercellular transfer of miR-34a leading to dysregulation of many target genes including the genes that cause loss of intra- and inter-network resting state functional connections with AD progression. Indeed published reports provide a mechanism of interneuronal communication involving exosomes released upon synaptic activation specifically bind to other neurons (Mittelbrunn et al., 2011; Squadrito et al., 2014). We tested this possibility in primary neurons by co-microinjection of FITC-labelled miR-34a RNA and Texas Red conjugated dextran, as injected cell marker, in live neurons that were allowed to incubate in neuronal growth medium. The results were striking in that within 2 hour post microinjection incubation, the FITC-labelled miR-34a were transferred from injected neurons (green + red, Fig. 4A) to neighbor cells (green only, Fig. 4A). In order to verify further that miR-34a containing exosomes are secreted and taken up by neighboring neurons, miR-34a expression vector was used for isolating in vitro GFP-labelled exosomes from neurons co-transfected with miR-34a and GFP-CD63 expression vectors, as an exosome specific protein marker. The presence of miR-34a in the purified GFP-labelled exosomes was verified by RT-PCR. These exosomes were transferred first into the primary neurons grown in the upper layer by the exosome mediated transfection and subsequently co-cultured with the bottom layer naive neurons. Presences of GFP in the naive neurons indicate that intraneuronal exosomes from upper layer neurons were taken up by the bottom layer neurons (Fig. 4B). Input exosomes, which had a showed a 4000-fold increase in miR-34a, caused a 300-fold miR-34a increase in the upper layer transfected neurons (Fig. 4C). A 250-fold increase in miR-34a was observed in the exosomes isolated from transwell bottom compartment media and a 150-fold miR-34a increase was seen in the bottom layer naïve neurons during co-culture (Fig. 4C).
Figure. 4.
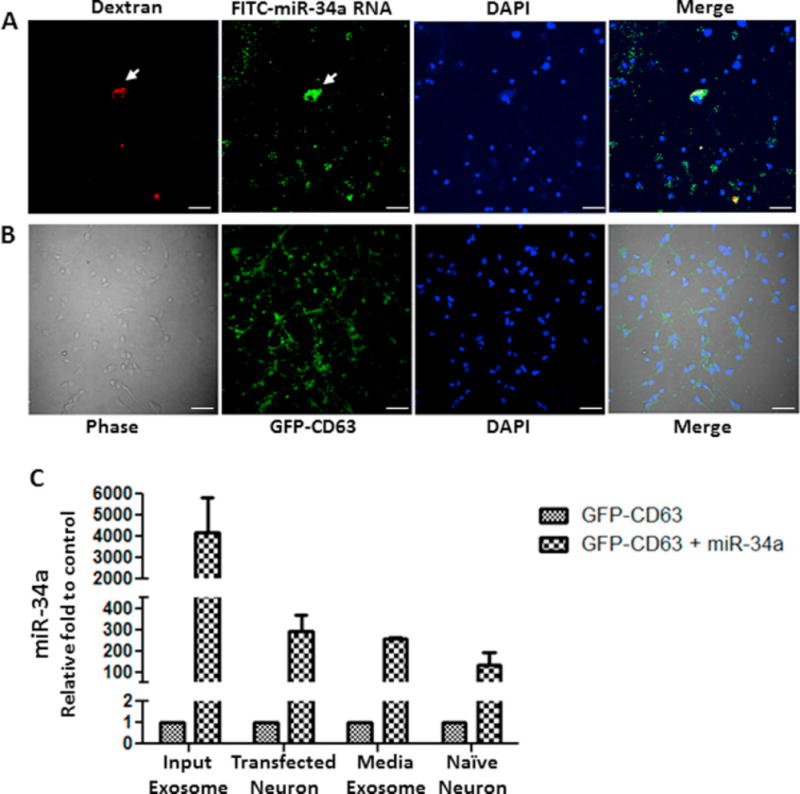
In vitro interneuronal transfer of miR-34a and spreading of neuron-derived miR-34a-containing GFP-tagged exosomes. FITC-labelled miR-34a RNA and Texas Red conjugated dextran were co-microinjected in a primary neuron (arrow) by Fempto tips from Eppendorf attaching with Eppendorf Fempto jet apparatus. After microinjection cells were incubated for 2h in growth medium, fixed and analyzed by confocal microscopy. (A) Representative images of red (dextran), green (FITC-miR-34a RNA), blue (DAPI) and merge. (B) Interneuronal transfer of neuron-derived miR-34a-containing GFP-CD63 tagged exosomes. An in vitro co-culture system (Transwell) was used where primary neuronal cells were seeded in the top compartment which was separated by a porous membrane from bottom compartment containing naive neuronal cells. Top compartmental neurons were transfected with neuronal-derived miR-34a-containing GFP-tagged exosomes. 24 hours after transfection, the neurons on insert membrane were incubated together with naïve neuron in presence of fresh growth medium. 14 hours after co-culture, bottom neuronal cells were fixed and analyzed by confocal microscopy. Representative images of primary neurons for phase contrast, green (GFP), blue (DAPI) and merge. (C) Presence of miR-34a in the input exosomes used for upper layer neurons transfection, in upper layer neurons after input exosome mediated transfection, in exosomes isolated from the bottom layer medium after 14 hours co-culture in Transwell, and in bottom well neurons.
2.5. miR-34a targets genes whose expressions correlate with the synchronous activity in RSN
Our results raise the possibilities that secreted miR-34a containing exosomes from neurons may propagate from pathophysiologically affected brain region to the unaffected neurons in the distal networks, causing dysfunction of whole networks. Recently, it has been shown that secreted exosome containing tau propagates locally from the entorhinal cortex to the dentate gyrus in mice (Asai et al., 2015). TC is a hub of RSN and resting-state functional MRI (rfMRI) studies in human AD patients revealed that loss of functional connections from the TC to hippocampus of AD brain compared to the AMC (Brier et al., 2012; Buckner et al., 2008; Wu et al., 2011). Although genes that cause loss of functional connections in intra- and inter-network resting state with AD progression remain unknown, many genes are recently reported to be correlated with synchronized functional connectivity of RSN (Richiardi et al., 2015). We reasoned that miR-34a may target many of these genes. Our analyses revealed that miR-34a targets dozens of RSN genes (Table 5). Thus there is a possibility that miR-34a could affect RSN activity by targeting the dozens of genes including those that are linked to ion channel activity, synaptic functions, and energy metabolism.
Table 5.
miR-34a target genes involved in RSN

|
miR-34a target genes shown in the above table are those among the set of 136 genes (Richiardi et al., 2015) whose orchestrated expression correlate with the resting state functional networks. The genes listed above are linked to synaptic ion channel activity and synaptic plasticity.
2.6. Analysis of tentative promoter in the miR-34a gene
Cellular sources and molecular mechanisms involved in induction of miR-34a gene transcription in AD brain are not completely understood. Our analysis of 5′-upstream 8.5kb human genomic nucleotide sequences from miR-34a transcription start site shows the presence of NFκB, STAT1, STAT4, c-Fos, CREB and p53 response elements (Fig. 5). Presence of these elements indicates that the brain’s innate immune system, reactive oxygen species, and even over activation of neurons in AD brain may cause activation of these known transcription factors leading to increased miR-34a gene transcription.
Figure 5.
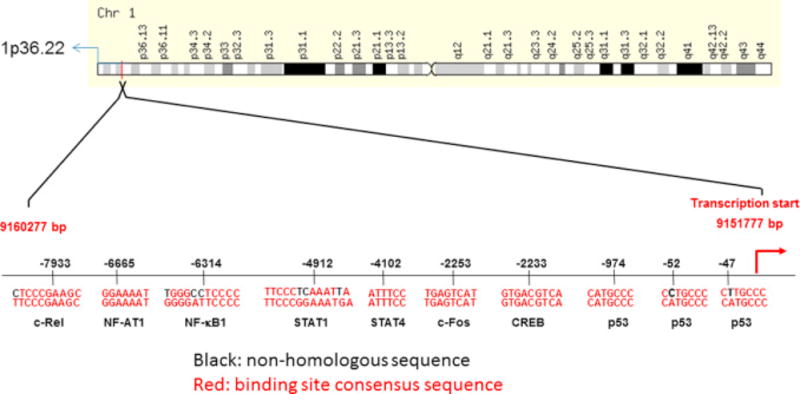
Transcription factor binding sites in the miR-34a promoter. Approximately 8.5 kb of upstream sequences of primary miR-34a transcript site in human genomic nucleotide sequences from chromosome 1 (Assembly GRCh38.p2) was used to search for transcription factor (TF) binding response elements. Using TF-search software, the presence of important transcription factor binding sites were identified and shown in this schematic diagram.
3. Discussion
In the present study, we have identified a novel microRNA, miR-34a that is over expressed in affected brain regions of AD patients as well as in transgenic AD mice. Our assessment of bioinformatics network and pathway analyses of the miR-34a target genes indicated the ability of miR-34a to affect molecular processes that are intrinsically linked to the regulation of pre- and post-synaptic neuronal excitability, mitochondria oxidative phosphorylation, glycolysis, and resting state functional connectivity.
In particular, we have demonstrated that miR-34a overexpression correlates with the repression of pre-synaptic plasticity related proteins, and VAMP2 (Synaptobrevin 2) and SYT1 (Synaptotagmin 1) proteins in TC region of AD brains. Our pathway analysis of VAMP2 revealed that VAMP2 is the hub of interacting protein networks whose constituents include SYT1, SNAP25, STX1A, SNAP25, and UNC 13B and these all are also the targets of miR-34a, as identified by our miR-34a target scan analysis. Thus miR-34a mediated repression of these pre-synaptic proteins may affect the process known to be involved in both fast synaptic-vesicle endocytosis and fast Ca2+-triggered synaptic vesicle exocytosis (Schoch et al., 2001; Tang et al., 2006).
We have also found that miR-34a over expression correlated with the repression of post-synaptic ion channel proteins including HCN, NR2A, and GluR1 in the TC regions of AD brains. Function of HCN is known to be involved in synaptic inputs integration by temporal synchrony in the synaptic buttons of pyramidal neurons (Vaidya and Johnston, 2013). NR2A ion channel opening at the post synaptic spines generates long-term potentiation (LTP), a form of plasticity involved in learning and memory (Bast et al., 2005), and GluR1 channel activity is essential for LTP, LTD, spine enlargement during LTP, and the retention of memories (Lee et al., 2003; Matsuzaki et al., 2004). Thus our results indicate the ability of miR-34a to affect both pre- and post-synaptic activity. Although it remains to be seen whether repression of synaptic plasticity-related gene by miR-34a relate to the impairment of cognitive function in AD, a published report have demonstrated that miR-34c, a family member of miR-34, targets SIRT1 gene, and elevated level of miR-34c correlates with memory impairment in AD mice (Agostini et al., 2011a). Also, it has been reported that ectopic expression of miR-34a in primary cortical neurons affects dendritic spine morphology and the reduction of amplitude/frequency of miniature excitatory synaptic current (mEPSC) (Agostini et al., 2011b). It has also been shown that ectopic expression of miR-34a down regulates synaptic plasticity-related gene, Arc in primary hippocampal neurons (Wibrand et al., 2012).
Memory formation, storage, and retrieval by pre- and post-synaptic activity requires large amounts of metabolic energy. AD is associated with characteristic and progressive reductions in regional glucose metabolism (CMRg) (Silverman et al., 2001). Also, it has been shown that the 3xTg AD mice has significant reductions in fluorodeoxyglucose (FDG) uptake into cortical and subcortical gray matter (Nicholson et al., 2010). These reductions in PET may be associated with reduced expression of nuclear genes encoding subunits of the mitochondrial electron transport chain. We have demonstrated that miR-34a targets genes encoding NDUFC2, SDHC, UQCRB, UQCRQ, and COX10 that are components of electron transport chain, and their expression is reduced in the miR-34a over expressed TC regions of AD patient cases. Also, we found that the reduced expression of these genes in ectopically expressed miR-34a in primary cortical neuronal culture correlates with severely reduced in mitochondria function. This reduction is not due to cellular death as indicated by our calcein AM assay. Although it has been shown that ectopic expression of miR-34a induces apoptosis in various tumor cell lines including colon cancer (Chang et al., 2007), neuroblastoma (Welch et al., 2007) and lung cancer (Raver-Shapira et al., 2007), it does not in induce apoptosis not in primary diploid cells (He et al., 2007).
It remains to be seen whether the reduced expression of complex I, complex II, complex III, or complex IV proteins in AD relate to cognitive dysfunction, published reports demonstrated that the gamma oscillations in the hippocampus require high complex I gene expression and strong functional performance of mitochondria (Kann et al., 2011) and impaired mitochondrial function abolishes gamma oscillations in the hippocampal networks (Whittaker et al., 2011). In a transgenic mouse model of AD, it has been shown that theta-gamma coupling is defective in the principal output region of the hippocampus, the subiculum (Goutagny et al., 2013). Optogenetic inhibition of synchronized high-frequency gamma oscillation in the entorhinal-hippocampal circuits of mouse brain causes reduction of correct execution of working-memory guided behavior (Yamamoto et al., 2014). These observations suggests that miR-34a provides a potential polygenetic mechanism causing energy metabolism dysfunction that could relate to working memory dysfunction characteristic symptoms found in human AD as well as in transgenic AD mice.
Glucose is not only a substrate for oxidative phosphorylation but also a substrate for glycolysis in the pentose phosphate pathway (PPP). It has been reported that for the long-term survival, neurons manage their redox state via the PPP (Herrero-Mendez et al., 2009). We have reported here miR-34a target genes for the glycolytic pathway, including H6PD, PFK1, and PFK2 and their protein levels were reduced in the TC regions of AD cases where miR-34a expression was high. Also, we found that the reduced expression of these genes in ectopically expressed miR-34a in primary cortical neuronal culture correlates with reduced glycolysis. Glucose-6 phosphate dehydrogenase is the gatekeeper of the PPP and it has an important role in protecting brain against reactive oxygen species (Brand and Hermfisse, 1997; Mejias et al., 2006). Phosphofructokinase 1 & 2 (PFK1 and 2) are key regulators of glycolysis and control PPP in neurons (Herrero-Mendez et al., 2009). Thus miR-34a, by inhibiting PPP could increases oxidative stress, contributing to neurodegeneration.
Besides the miR-34a target genes that link synaptic plasticity and energy metabolism many other genes relevant to cognitive functions are also the targets of miR-34a. Synchronous activity in different organizational levels of resting state networks (RSN) facilitates cognitive performances in a wide range of cognitive domains including performances in working and episodic memory (Sala-Llonch et al., 2012; Vincent et al., 2006). Connectivity of functional RSN has been shown to be disrupted in AD (Brier et al., 2012; Thomas et al., 2014). Our demonstration that secretion of miR-34a-containing exosomes from neurons to the medium and subsequent interneuronal transfer, raises the possibility that miR-34a leads to dysregulation of many target genes, including the genes that cause loss of intra- and inter-network resting state functional connectivity during AD progression. Although genes that cause loss of functional connections in intra- and inter-network resting state with AD progression remain unknown, many genes are recently reported to be correlated with synchronized functional connectivity of RSN (Richiardi et al., 2015). Our miR-34a target gene analysis revealed that it targets dozens of genes linked to ion channel and synaptic function and raises the possibilities that miR-34a is capable of disrupting many functions of RSN including episodic and working memory. It remains to be seen whether miR-34a spreading in vivo relate to the repression of these genes and affects cognitive functions. Molecular mechanisms that cause induction of miR-34a gene expression in AD brain regions are not completely understood. Our miR-34a promoter analysis by transcription factor search revealed the presence of p53, NFkB, fos, c-rel, STAT1, and CREB transcription factor binding response elements. These transcription factors for example NFkB, c-rel and STAT1, are known to be activated in response to inflammation (Jin et al., 2013; Lee, 2013; Sarnico et al., 2009); CREB, and fos are activated by neuronal excitation (Impey et al., 1998; Morgan and Curran, 1986); and p53 by oxidative stress (Gambino et al., 2013). It has been shown that SIRT1, p53 and miR-34a are involved in a positive feedback loop for miR-34a gene transcription in a fashion whereby increased miR-34a suppresses its target SIRT1 expression, thereby resulted in increased p53 transcriptional activity leading to continued up-regulation of miR-34a (Yamakuchi et al., 2008). Thus miR-34a may up regulate its own gene transcription through this positive feedback loop. There is also a possible involvement of this SIRT1, P53 and miR-34a feedback loop in β-amyloid production. Our search identified that miR-34a targets the α-secretase gene, ADAM10. It has been reported that SIRT1 prevents Aβ peptide generation in neurons (Donmez et al., 2010). miR-34a, by suppressing its target SIRT1, could lead ultimately to an increase in β-amyloid production. Also, suppression of both SIRT1 and ADAM10 by miR-34a, could lead to an increased p53 transcriptional activity, resulting in a continued increase in Aβ production.
Together, our experimental data along with our analysis of predicted miR-34a target genes raise the possibilities that locally, AD pathology-induced activation of specific transcription factor may lead to increased expression of miR-34a gene in specific brain region(s) and initiates synaptic dysfunction by concurrently repressing its polygenetic targets. Continued accumulation of miR-34a along with the interneuronal transfer of miR-34a-loaded exosomes may affect neural networks dedicated to memory circuits, including RSN. fcMRI studies in a large cohort of human subjects demonstrated a decline in oxygen use even in very mild [clinical dementia rating (CDR 0.5)] and mild (CDR 1) cases of AD (Brier et al., 2012). The CDR stages in the AD subjects used in our studies are not known. The exact relationship of miR-34a expression with CDR stages of AD remains to be determined. Thus, our results provide insights into polygenetic AD mechanisms and disclose miR-34a as the potential therapeutic targets for AD.
4. Experimental procedure
4.1. Human Brain Tissues
Brain tissue from the temporal cortex, frontal cortex and cerebellum of AD patients and age-matched control (AMC) subjects were obtained from Kathleen Price Bryan Brain Bank, Bryan Alzheimer’s Disease Research Center, Duke University Medical Center, Durham, NC. Brain tissues were used in accordance with the institutional review board/ethical guidelines of the donor institution. The characteristics of the study population are summarized in Table 1.
4.2. AD mouse Brain Tissues
Both male and female 3xTg-AD mouse model (B6;129-Psen1tm1MpmTg(APPSwe,tauP301L) 1Lfa/Mmjax) (Oddo et al., 2003) and control (B6129SF2/J) were purchased from the Jackson laboratory and were housed with food and water ad libitum in sterile microisolator cages with sterile bedding at the West Virginia University Health Science Center animal facility. At the age of 3, 6, 12 months, mouse brains were perfused with DEPC treated PBS via cardiac puncture to remove residual blood. Brains were dissected into hippocampus, temporal cortex, frontal cortex and cerebellum. Tissues were flash-frozen in liquid nitrogen and stored at −80 °C until used for biochemical analysis. All animals were treated by experimental protocols approved by the WVU HSC Institutional Animal Care and Use Committee.
4.3. In Silico Search and miRNA Expression Profiling
The target mRNAs that have potential binding sites for individual microRNAs were identified by searching them on public databases endowed with prediction algorithms, such as TargetScan (http://targetscan.org), PicTar (http://pictar.mdc-berlin.de), miRBase (http://www.mirbase.org), microRNA.org (http://www.microRNA.org), and PITA miRNA prediction tool.
Total RNA enriched in microRNAs was isolated from approximately 5 mg of individual frozen brain tissue samples by using miRNeasy Micro Kit isolation kit (Qiagen). RNA concentration was determined using Nano drop 2000 spectrophotometer (Thermo Scientific). 2μg of total RNA containing microRNAs was reverse transcribed in a total volume of 10μl reaction mix to make cDNA by using miScript II RT Kit (Qiagen). Expression of five microRNAs namely hsa-miR-34a, -146a, -132,- 9, and -15a that collectively has potential to deregulate genes known to be involved in synaptic function, innate immunity, neuronal survival, and energy metabolism were determined by quantitative RT-PCR. Quantitative RT-PCR was performed using target specific miScript primer assays and the miScript SYBR® Green PCR kits (Qiagen). The RT-PCR reaction was in triplicate for each sample using a StepOne Plus PCR system (Applied Biosystems) for 40 cycles as follows: 10 sec at 95 °C, 30 sec at 55 °C, 30 sec at 70 °C. Negative control reactions were included as wells containing only master mix and nuclease-free water without any template cDNA. All of microRNA specific primers were from Qiagen and they were miScript Primer Assay Hs_miR-34a (MS00003318), Hs_miR-146a (MS00003535), Hs_miR-132 (MS00003458), Hs_miR-15a_1 (MS00003178), Hs_miR-9 (MS00010752) and Hs_RNU6 (MS00033740). The expression levels of target genes were standardized against those of the RNU6 gene detected in identical cDNA samples. Quantification of PCR amplified microRNA specific cDNA was done by comparative cycle threshold CT method (ΔΔCT). With the comparative CT method, the StepOne software measures amplification of the target and of the endogenous control in samples and in a reference sample.
4.4. Western Blot Analysis
To prepare total protein extract, frozen brain tissues were homogenized in RIPA lysis buffer with addition of protease inhibitors cocktail (Sigma, St. Louis, MO, USA), followed by centrifugation at 13,400 g for 5 min at room temperature (RT). The supernatant was collected and protein samples were separated by electrophoresis run on a precast SDS-PAGE Mini-PROTEAN TGX Gells 4–20% (Bio-Rad Laboratories). The protein concentration was determined by a Bradford assay kit (Bio-Rad). After gel electrophoresis, the protein was transferred onto a PVDF membranes Immobilion (EMD Millipore) which were immune-labelled at 4 °C overnight with the following antibodies: a mouse monoclonal VAMP-1/2 (SP10, Santa Cruz Biotechnologies), a mouse monoclonal Synaptotagmin I (15, Santa Cruz Biotechnologies), an affinity purified goat polyclonal HCN 1 (v-17, Santa Cruz Biotechnologies), a rabbit monoclonal NMDAR2A (EPR2465-2, Abcam), a rabbit monoclonal Glutamate Receptor 1, AMPA subtype (EPR5479, Abcam), a rabbit monoclonal NDUFC2 (EPR16499, Abcam), a rabbit monoclonal SDHC (EPR 11035b, Abcam), a rabbit monoclonal UQCRB (EPR15591, Abcam), an affinity purified goat polyclonal UQCRQ (C-12, Santa Cruz Biotechnologies), an affinity purified rabbit polyclonal COX 10 (ab135468, Abcam), a mouse monoclonal H6PD (C-10, Santa Cruz Biotechnologies), a mouse monoclonal PFK1 (ab 77159, Abcam), and a rabbit monoclonal PFK2 (EPR12594, Abcam). After overnight incubation with primary antibodies the membranes were washed and followed by incubation at room temperature for 60 min with appropriate anti-rabbit, anti-mouse, secondary antibody conjugated with horseradish peroxidase (Bio-Rad Laboratories). The specific reaction was visualized by using a chemiluminescent substrate (Pierce, Rockford, IL, USA) followed by imaging by UVP Chemi Doc-It Imager (UVP, USA). After the antibodies were stripped by incubating the membranes at 50°C for 30 min in stripping buffer composed of 62.5 mM Tris-HCl, pH 6.7, 2% SDS and 100 mM 2-mercaptoethanol, they were processed for relabeling with a mouse monoclonal antibody against actin protein (C2, Santa Cruz Biotechnologies), an internal control for protein loading. Immunoblotting bands were quantified by densitometry using UVP software (UVP, USA).
4.5. Construction of miR-34a Expression Vector
The precursor of hsa-miR-34a (GeneBank Accession No. EF570048.1) was amplified by PCR with PfuTurbo DNA polymerase (Stratagene, USA) using human chromosomal DNA isolated from HEK 293 cells and a set of primers (Forward 5′-AACTAggatccCCCACATTTCCTT and Reverse 5′-AATCCaagcttGCAGAAGAGCTTC). Then, it was cloned in the expression vector PrnaU6.1/hygro (GenScript, USA) at the BamHI/HindIII cloning site.
4.6. Preparation of Primary Rat Neuronal Cultures
At embryonic day 18 (E18), pregnant rats were anesthetized and cervically dislocated. The brains of pups were removed and placed into magnesium (Mg2+) free Hank’s balance salt solution (HBSS). Cortices were dissected under a microscope, washed, and placed into neurobasal culture media supplemented with B27 and pen-strep (all from Gibco). The cortices were triturated using a graded series of fine polished Pasteur pipettes, and then filtered through a 40 μm nylon cell strainer (Becton Dickinson Labware). The neurons were plated on poly-L-lysine coated dishes, and cultured in vitro in 95% humidity and 5% CO2 atmosphere for required days. At day 2 cells were treated with 5 μM 1-beta-D-arabinofuranosylcytosine (AraC, Sigma-Aldrich, USA) to inhibit glial cell growth.
4.7. Measurement of Mitochondrial Respiration (OCR, Oxygen Consumption Rate)
Approximately 20,000 primary cortical neurons (E18) were seeded in poly L-lysine coated XF 96 cell culture microplate (Seahorse Bioscience, North Billerica, MA) in neurobasal medium supplemented with B27 growth factors. 3 days after growth neurons were transfected with miR-34a expression plasmid DNA by Lipofectamine 2000 (Invitrogen, Life technologies, USA) according to the supplier’s protocol. Thirty six hours after transfection, OCR was measured by XFe 96 Extracellular Flux Analyzer using a Mito Stress Kit (Seahorse Biosciences) according to the supplier’s protocol. This analyzer monitored in real time indices of mitochondrial function and was measured as follows. Oligomycin, FCCP, and antimycin A plus rotenone were added separately in a sensor cartridge injected sequentially through ports in the XF Assay cartridges to final concentrations of 1 μg/ml, 1 μM and 10 μM, respectively. This allowed us to determine the basal level of oxygen consumption, the amount of oxygen consumption linked to ATP production, the level of non-ATP-linked oxygen consumption (proton leak), the maximal respiration capacity and the non-mitochondrial oxygen consumption. For each experimental condition six replicate wells were used.
4.8. Measurement of the Extracellular Acidification Rate (ECAR)
Similar to OCR, primary cortical neurons were cultured and transfected with miR-34a expression plasmid DNA. Thirty six hours after transfection, ECAR was measured by XFe 96 Extracellular Flux Analyzer using glycolysis stress kit (Seahorse Biosciences) which measures three key parameters of glycolytic function: glycolysis, glycolytic capacity, and glycolytic reserve were determined as follows. The first injection is a saturating concentration of glucose (10 mM). Glucose is taken up by the neurons and catabolized through the glycolytic pathway to lactate, producing ATP and protons. The extrusion of protons into the surrounding medium produces a rapid increase in ECAR. This glucose-induced response is reported as the rate of glycolysis (or glycolytic flux) under a basal condition. The second injection is oligomycin, an ATP synthase inhibitor. Oligomycin inhibits mitochondrial ATP production and thus shifts the energy production to glycolysis, with the subsequent increase in ECAR revealing the maximum glycolytic capacity of the cells. The final injection is 2-DG, a glucose analog, which inhibits glycolysis through competitive binding to glucose hexokinase, the first enzyme in the glycolytic pathway. The resulting decrease in ECAR further confirms that the ECAR produced in the experiment is due to glycolysis. The difference between glycolytic capacity and glycolysis rate defines glycolytic reserve.
4.9 Microinjection of FITC-labelled miR-34a into Primary Rat Cortical Neurons
hsa-miR-34a mimic (Qiagen) was labelled with FITC using Label It kits (Mirus) according to the supplier’s protocol. Briefly, 10 nm of mimic was used in a 100μl reaction volume with the supplied reagents. After incubation at 37 °C for 1h, the reaction was stopped by adding stop buffer. Unreacted label was separated by column purification and bound labelled miR-34a mimic was eluted in 30 μl elution buffer. Purified FITC-labelled miR-34a mimic and Texas Red conjugated dextran were co-microinjected into a primary neuron grown in a poly-lysine coated glass coverslips by Fempto tips attached with a Fempto jet apparatus (Eppendorf). Immediately after microinjection, cells were incubated for 2h in neurobasal growth medium supplemented with B27, fixed with paraformaldehyde and analyzed by confocal microscopy.
4.10. Exosome Purification
Rat cortical primary neuronal cultures were grown in poly-L-lysine coated 150mm dishes for 7 days in vitro (7DIV) in neurobasal medium supplemented with B27. Six of each 150mm dishes with 75% confluence were transfected either GFP-tagged-human CD63 expression vector (Origene) alone or co-transfected with miR-34a expression vector by lipofectamine 2000. Thirty six hours after transfection, exosomes were isolated from the cell culture media by filtration and ultracentrifugation steps according to the method previously described (Thery et al., 2006). Briefly, cells were centrifuged (320 g for 5 min) and the combined media were filtered through steriflip (0.22 μm pore size) (Millipore). Exosomes were pelleted by ultracentrifugation at 100,000 g for 60 min at 4 °C using Beckman SW 28 rotor running in Beckman Coulter Optima L-100 XP ultracentrifuge (Beckman Coulter). Exosomes were washed once with sterile phosphate buffered saline and washed exosomes were collected by ultracentrifugation. Washed pellets of exosomes were re-suspended in 1ml of sterile phosphate buffer. Presence or absence of miR-34a were determined by isolating RNAs using miRNeasy kit (Qiagen) followed by qRT-PCR as described in the above method section.
4.11. Transwell Co-Cultures
Polyester (PET) membrane transwell inserts for 6-well plates with a 0.2μm pore-sized filter (Corning) were used following the manufacturer’s instructions. For imaging the GFP-labelled exosome transfer, 100,000 cortical primary neurons were seeded into each of six separate well inserts and cultured in neurobasal medium supplemented with B27. After two days of growth, neurons were transfected with neuronal-derived miR-34a–containing GFP-tagged exosomes. Transfected neurons were incubated in presence of growth medium and twenty four hours after transfection, cells in the inserts were washed with neurobasal medium and the inserts were placed on the top of primary neurons grown in poly-L-lysine coated glass coverslips placed in 6 well plates containing growth medium. Both the neuronal cells in the top inserts and the coverslips in the bottom of the 6 well plates were cultured together for fourteen hours in growth medium. After the incubation, neuronal cells in the glass coverslips were fixed with paraformaldehyde and analyzed by Zeiss LSM 710 confocal microscopy.
For the quantitation of miR-34a transfer through exosome, a total of 36 inserts were used. 18 each were transfected with either GFP-CD63-labelled exosomes or GFP-CD63 labelled exosome containing miR-34a by the same method as described above. Twenty four hours after transfection, the transfected neurons on the insert were washed with growth media and placed on the naïve primary neurons grown in 6 well plates. Fourteen hours after the co-culture, RNAs were isolated from top insert and bottom well neurons, and from exosomes isolated from bottom layer culture medium as described above. Quantitative RT-PCR was done to assess the amount of miR-34a in input (transfected amount), the transfected top insert neurons, in co-cultured bottom naive neurons and the bottom co-cultured naïve cell medium.
4.12. Statistical Analyses
Results from the experiments are reported as means ± SEM. All quantitative data are assessed for significance using either a Student’s t-test, a one-way ANOVA with Tukey’s post-hoc test or Pearson’s correlation analysis by GraphPad Prism software. A p value < 0.05 is used to establish significance.
Highlights.
Cognitive impairment in sporadic Alzheimer’s disease (AD) arises due to polygenic dysfunction.
The potential for a single miRNA linking synaptic dysfunction and altered bioenergetics is explored.
Five miRNAs were selected by bioinformatics that maximizes their polygenic targets involved in AD.
miR-34a in AD brains and primary neurons repressed proteins related to synaptic plasticity and energy metabolism.
Identifying polygenic targets discloses miR-34a as a potential therapeutic target for AD.
Acknowledgments
This work was supported by NIH PO1 AG022550 and NIH P01 AG027956, NIH/NIGMS Award Number U54GM104942 and NIH grant P20 GM109098.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agostini M, et al. Neuronal differentiation by TAp73 is mediated by microRNA-34a regulation of synaptic protein targets. Proc Natl Acad Sci U S A. 2011a;108:21093–8. doi: 10.1073/pnas.1112061109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agostini M, et al. microRNA-34a regulates neurite outgrowth, spinal morphology, and function. Proc Natl Acad Sci U S A. 2011b;108:21099–104. doi: 10.1073/pnas.1112063108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen M, et al. Novel late-onset Alzheimer disease loci variants associate with brain gene expression. Neurology. 2012;79:221–8. doi: 10.1212/WNL.0b013e3182605801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asai H, et al. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci. 2015;18:1584–93. doi: 10.1038/nn.4132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–33. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bast T, da Silva BM, Morris RG. Distinct contributions of hippocampal NMDA and AMPA receptors to encoding and retrieval of one-trial place memory. J Neurosci. 2005;25:5845–56. doi: 10.1523/JNEUROSCI.0698-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman RJ, et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med. 2012;367:795–804. doi: 10.1056/NEJMoa1202753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand KA, Hermfisse U. Aerobic glycolysis by proliferating cells: a protective strategy against reactive oxygen species. FASEB J. 1997;11:388–95. doi: 10.1096/fasebj.11.5.9141507. [DOI] [PubMed] [Google Scholar]
- Brier MR, et al. Loss of intranetwork and internetwork resting state functional connections with Alzheimer’s disease progression. J Neurosci. 2012;32:8890–9. doi: 10.1523/JNEUROSCI.5698-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckner RL, Andrews-Hanna JR, Schacter DL. The brain’s default network: anatomy, function, and relevance to disease. Ann N Y Acad Sci. 2008;1124:1–38. doi: 10.1196/annals.1440.011. [DOI] [PubMed] [Google Scholar]
- Chang TC, et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell. 2007;26:745–52. doi: 10.1016/j.molcel.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donmez G, et al. SIRT1 suppresses beta-amyloid production by activating the alpha-secretase gene ADAM10. Cell. 2010;142:320–32. doi: 10.1016/j.cell.2010.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Gambino V, et al. Oxidative stress activates a specific p53 transcriptional response that regulates cellular senescence and aging. Aging Cell. 2013;12:435–45. doi: 10.1111/acel.12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, et al. A novel pathway regulates memory and plasticity via SIRT1 and miR-134. Nature. 2010;466:1105–9. doi: 10.1038/nature09271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goutagny R, et al. Alterations in hippocampal network oscillations and theta-gamma coupling arise before Abeta overproduction in a mouse model of Alzheimer’s disease. Eur J Neurosci. 2013;37:1896–902. doi: 10.1111/ejn.12233. [DOI] [PubMed] [Google Scholar]
- Griggs EM, et al. MicroRNA-182 regulates amygdala-dependent memory formation. J Neurosci. 2013;33:1734–40. doi: 10.1523/JNEUROSCI.2873-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, et al. A microRNA component of the p53 tumour suppressor network. Nature. 2007;447:1130–4. doi: 10.1038/nature05939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrero-Mendez A, et al. The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat Cell Biol. 2009;11:747–52. doi: 10.1038/ncb1881. [DOI] [PubMed] [Google Scholar]
- Impey S, et al. Cross talk between ERK and PKA is required for Ca2+ stimulation of CREB-dependent transcription and ERK nuclear translocation. Neuron. 1998;21:869–83. doi: 10.1016/s0896-6273(00)80602-9. [DOI] [PubMed] [Google Scholar]
- Jin P, et al. Anti-inflammatory and anti-amyloidogenic effects of a small molecule, 2,4-bis(p-hydroxyphenyl)-2-butenal in Tg2576 Alzheimer’s disease mice model. J Neuroinflammation. 2013;10:2. doi: 10.1186/1742-2094-10-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kann O, et al. Gamma oscillations in the hippocampus require high complex I gene expression and strong functional performance of mitochondria. Brain. 2011;134:345–58. doi: 10.1093/brain/awq333. [DOI] [PubMed] [Google Scholar]
- Lee HK, et al. Phosphorylation of the AMPA receptor GluR1 subunit is required for synaptic plasticity and retention of spatial memory. Cell. 2003;112:631–43. doi: 10.1016/s0092-8674(03)00122-3. [DOI] [PubMed] [Google Scholar]
- Lee M. Neurotransmitters and microglial-mediated neuroinflammation. Curr Protein Pept Sci. 2013;14:21–32. doi: 10.2174/1389203711314010005. [DOI] [PubMed] [Google Scholar]
- Liang WS, et al. Alzheimer’s disease is associated with reduced expression of energy metabolism genes in posterior cingulate neurons. Proc Natl Acad Sci U S A. 2008;105:4441–6. doi: 10.1073/pnas.0709259105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki M, et al. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004;429:761–6. doi: 10.1038/nature02617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mejias R, et al. Neuroprotection by transgenic expression of glucose-6-phosphate dehydrogenase in dopaminergic nigrostriatal neurons of mice. J Neurosci. 2006;26:4500–8. doi: 10.1523/JNEUROSCI.0122-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittelbrunn M, et al. Unidirectional transfer of microRNA-loaded exosomes from T cells to antigen-presenting cells. Nat Commun. 2011;2:282. doi: 10.1038/ncomms1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan JI, Curran T. Role of ion flux in the control of c-fos expression. Nature. 1986;322:552–5. doi: 10.1038/322552a0. [DOI] [PubMed] [Google Scholar]
- Nicholson RM, et al. Regional cerebral glucose uptake in the 3xTG model of Alzheimer’s disease highlights common regional vulnerability across AD mouse models. Brain Res. 2010;1347:179–85. doi: 10.1016/j.brainres.2010.05.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, et al. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–21. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Querfurth HW, LaFerla FM. Alzheimer’s disease. N Engl J Med. 2010;362:329–44. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- Raver-Shapira N, et al. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol Cell. 2007;26:731–43. doi: 10.1016/j.molcel.2007.05.017. [DOI] [PubMed] [Google Scholar]
- Reiman EM, et al. Brain imaging and fluid biomarker analysis in young adults at genetic risk for autosomal dominant Alzheimer’s disease in the presenilin 1 E280A kindred: a case-control study. Lancet Neurol. 2012;11:1048–56. doi: 10.1016/S1474-4422(12)70228-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richiardi J, et al. BRAIN NETWORKS. Correlated gene expression supports synchronous activity in brain networks. Science. 2015;348:1241–4. doi: 10.1126/science.1255905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala-Llonch R, et al. Brain connectivity during resting state and subsequent working memory task predicts behavioural performance. Cortex. 2012;48:1187–96. doi: 10.1016/j.cortex.2011.07.006. [DOI] [PubMed] [Google Scholar]
- Sarnico I, et al. NF-kappaB p50/RelA and c-Rel-containing dimers: opposite regulators of neuron vulnerability to ischaemia. J Neurochem. 2009;108:475–85. doi: 10.1111/j.1471-4159.2008.05783.x. [DOI] [PubMed] [Google Scholar]
- Schoch S, et al. SNARE function analyzed in synaptobrevin/VAMP knockout mice. Science. 2001;294:1117–22. doi: 10.1126/science.1064335. [DOI] [PubMed] [Google Scholar]
- Seshadri S, et al. Genome-wide analysis of genetic loci associated with Alzheimer disease. Jama. 2010;303:1832–40. doi: 10.1001/jama.2010.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman DH, et al. Positron emission tomography in evaluation of dementia: Regional brain metabolism and long-term outcome. JAMA. 2001;286:2120–7. doi: 10.1001/jama.286.17.2120. [DOI] [PubMed] [Google Scholar]
- Squadrito ML, et al. Endogenous RNAs modulate microRNA sorting to exosomes and transfer to acceptor cells. Cell Rep. 2014;8:1432–46. doi: 10.1016/j.celrep.2014.07.035. [DOI] [PubMed] [Google Scholar]
- Tang J, et al. A complexin/synaptotagmin 1 switch controls fast synaptic vesicle exocytosis. Cell. 2006;126:1175–87. doi: 10.1016/j.cell.2006.08.030. [DOI] [PubMed] [Google Scholar]
- Thery C, et al. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol. 2006:22. doi: 10.1002/0471143030.cb0322s30. Chapter 3, Unit 3. [DOI] [PubMed] [Google Scholar]
- Thomas JB, et al. Functional connectivity in autosomal dominant and late-onset Alzheimer disease. JAMA Neurol. 2014;71:1111–22. doi: 10.1001/jamaneurol.2014.1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaidya SP, Johnston D. Temporal synchrony and gamma-to-theta power conversion in the dendrites of CA1 pyramidal neurons. Nat Neurosci. 2013;16:1812–20. doi: 10.1038/nn.3562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent JL, et al. Coherent spontaneous activity identifies a hippocampal-parietal memory network. J Neurophysiol. 2006;96:3517–31. doi: 10.1152/jn.00048.2006. [DOI] [PubMed] [Google Scholar]
- Vlachos IS, Hatzigeorgiou AG. Online resources for miRNA analysis. Clin Biochem. 2013;46:879–900. doi: 10.1016/j.clinbiochem.2013.03.006. [DOI] [PubMed] [Google Scholar]
- Welch C, Chen Y, Stallings RL. MicroRNA-34a functions as a potential tumor suppressor by inducing apoptosis in neuroblastoma cells. Oncogene. 2007;26:5017–22. doi: 10.1038/sj.onc.1210293. [DOI] [PubMed] [Google Scholar]
- Whittaker RG, et al. Impaired mitochondrial function abolishes gamma oscillations in the hippocampus through an effect on fast-spiking interneurons. Brain. 2011;134:e180. doi: 10.1093/brain/awr018. author reply e181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wibrand K, et al. MicroRNA regulation of the synaptic plasticity-related gene Arc. PLoS One. 2012;7:e41688. doi: 10.1371/journal.pone.0041688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, et al. Altered default mode network connectivity in Alzheimer’s disease–a resting functional MRI and Bayesian network study. Hum Brain Mapp. 2011;32:1868–81. doi: 10.1002/hbm.21153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamakuchi M, Ferlito M, Lowenstein CJ. miR-34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci U S A. 2008;105:13421–6. doi: 10.1073/pnas.0801613105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto J, et al. Successful execution of working memory linked to synchronized high-frequency gamma oscillations. Cell. 2014;157:845–57. doi: 10.1016/j.cell.2014.04.009. [DOI] [PubMed] [Google Scholar]
- Zovoilis A, et al. microRNA-34c is a novel target to treat dementias. Embo j. 2011;30:4299–308. doi: 10.1038/emboj.2011.327. [DOI] [PMC free article] [PubMed] [Google Scholar]