

Abstract
Binding of peptides to MHC class I (MHC-I) molecules is the most selective event in the processing and presentation of antigens to cytotoxic T lymphocytes (CTL) and insights into the mechanisms that govern peptide-MHC-I binding should facilitate our understanding of CTL biology. Peptide-MHC-I interactions have traditionally been quantified by the strength of the interaction, i.e. the binding affinity, yet it has been show that the stability of the peptide-MHC-I complex is a better correlate of immunogenicity compared to binding affinity. Here, we have experimentally analyzed peptide-MHC-I complex stability of a large panel of human MHC-I allotypes and generated a body of data sufficient to develop neural networks based pan-specific predictor of peptide-MHC-I complex stability. Integrating the neural networks predictors of peptide-MHC-I complex stability with state-of-the-art predictors of peptide-MHC-I binding is shown to significantly improve the prediction of CTL epitopes. The method is publicly available at www.cbs.dtu.dk/services/NetMHCstabpan.
Introduction
Binding and presentation of antigenic peptides by MHC molecules is a central event in the initiation of an adaptive immune response. MHC-I molecules bind peptides derived from the intracellular protein metabolism and present them at the cell surface for scrutiny by effector cells of the immune system. The resulting presentation of peptides of pathogenic origin (e.g. from virus or tumors) can trigger the activation of immune effector cells such as CD8+ cytotoxic T lymphocytes (CTLs). Peptide binding to MHC-I molecules is the single most selective of the many events leading to antigen presentation and considerable efforts have been dedicated to unravel the rules of this event. For a peptide to induce an immune response it must bind with sufficient affinity and stability to the restricting MHC-I element to allow translocation of intact peptide-MHC-I complexes to the cell surface where it should remain associated long enough for circulating CD8+ CTL's of appropriate specificity to arrive and recognize it. Although the Law of Mass Action establishes a very close and direct relationship between affinity and stability, the above logic would suggest that stability might be the better immunogenicity correlate of the two parameters. As previously noted by us (1), several others have suggested that the stability of a pMHC complex correlates with immunogenicity (both for MHC-I (2-8), and for MHC-II (9, 10)); and it has even been suggested that stability correlates better with immunogenicity than affinity does (both for MHC-I (11-14) and MHC-II (15)). Unfortunately, these claims have been supported by limited and to some extend biased experimental data. The former is due to the experimental challenges associated with measuring complex stability, which effectively has meant that affinity has remained the most frequently established correlate of immunogenicity. The latter is due to the frequent use of predicting algorithms to select peptides of interest, which effectively precludes the use of immunogenicity data obtained from such predictions to compare the impact of affinity versus stability.
Using an HLA-A*02:01-transgenic mouse model and examining anti-vaccinia immune responses in an unbiased and comprehensive manner, Assarsson et al. showed that as much as 90% of the peptides that were found to bind with high affinity to HLA-A*02:01, nonetheless, were not recognized after infection (16). Asking whether lack of stability could be part of the explanation of this high rate of false discovery, they examined a small selection of 12 of these peptides spanning dominant, subdominant, cryptic, and non-immunogenic events and concluded that “a suggestive, but not statistically significant, trend was noted for off-rates and dominance”. In a follow-up study (1), we examined the stability of a larger collection of the peptide-HLA-A*02:01 interactions reported by Assarsson et al. and in increasing the sample size we indeed found that stability is a significantly better correlate of immunogenicity than affinity is. We suggested that 30% of the peptides, which are non-immunogenic even though they experimentally can be verified as binders to HLA-A*02:01, lack immunogenicity because they form MHC-I complexes of low stability. Given this we concluded that pMHC-I complex stability is important in the generation of T cell responses.
Over the last decades, accurate and reliable in silico methods capable of predicting the binding affinity of peptide binding to MHC-I have been developed (17-22) and these have been highly successful in narrowing down the search for T cell epitopes. However, the high false discovery rate in terms of immunogenicity (i.e. that only few of the peptides, which are predicted and subsequently shown to bind to MHC-I, are actually recognized by T cells following infection) remains a problem. Recently, we have shown that accurate predictors of pMHC-I complex stability can be developed and that these predict immunogenic peptides to form pMHC-I complexes of higher stability compared to non-immunogenic peptides (1, 23). These studies and corresponding predictors covered a limited set of human MHC-I allotypes. Here, we have generated a large body of pMHC-I complex stability data using a large repository of human MHC-I molecules and synthetic peptides with the aim of developing pan-specific predictors of MHC-I complex stability. Using this data, we report the development of an artificial neural network (ANN) based pan-specific predictor of pMHC-I complex stability and evaluate its accuracy in predicting pMHC-I complex stability. By integrating this novel predictor with state-of-the-art predictors of pMHC-I binding affinity, we find that constructing a model with 80% weight on affinity and 20% weight on stability, the prediction of MHC-I ligands and T-cell epitopes is significantly improved beyond that of any of the two models alone. Current predictors of pMHC-I binding affinity are trained on a significantly larger body of data compared to available pMHC-I complex stability data. To access whether the relative low contribution of stability predictions was influenced by this unbalance in data volume, we evaluate the performance of predictors trained on balanced pMHC-I binding affinity data, and pMHC-I complex stability data. Evaluating such predictors, we find that the integrative model with similar weight on affinity and stability achieves optimal performance, and that this significantly outperforms the two individual models for prediction of both MHC ligands and T cell epitopes.
Materials and Methods
Peptide-MHC-I complex stability
9023 in-house nonameric peptides were predicted for binding to 76 HLA-I molecules using the NetMHCpan 2.8 prediction server (17, 18). Peptides with a predicted high affinity binding (<500nM or 2% rank) to multiple of the available MHC-I molecules were grouped into 12 peptide sets of 383 peptides each. Each peptide set was screened for pMHC-I complex stability on multiple MHC-I allotypes according to table 1 using a scintillation proximity based peptide-MHC-I dissociation assay (24). The reported half-life of pMHC-I complexes is the geometric mean of the half-life from two independent experiments. This data set was combined with an in-house small-scale data set from earlier studies arriving at a data set of 28,939 measurements covering 80 HLA-A and B molecules.
Table 1. Grouping of HLA-I allotypes by predicted binding motif.
| Group | A1 | A2 | A3 | A24 | A26 | B7 | B8 | B27 | B39 | B44 | B58 | B62 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Allotype | HLA-A*01:01 | HLA-A*02:01 | HLA-A*03:01 | HLA-A*23:01 | HLA-A*25:01 | HLA-B*07:02 | HLA-B*08:01 | HLA-B*27:02 | HLA-B*15:09 | HLA-B*18:01 | HLA-B*15:17 | HLA-B*15:01 |
| HLA-A*29:02 | HLA-A*02:03 | HLA-A*11:01 | HLA-A*24:02 | HLA-A*26:01 | HLA-B*35:01 | HLA-B*08:02 | HLA-B*27:03 | HLA-B*15:10 | HLA-B*40:01 | HLA-B*57:01 | HLA-B*15:02 | |
| HLA-A*30:02 | HLA-A*02:10 | HLA-A*30:01 | HLA-A*24:03 | HLA-A*26:02 | HLA-B*35:03 | HLA-B*08:03 | HLA-B*27:05 | HLA-B*38:01 | HLA-B*40:13 | HLA-B*57:02 | HLA-B*15:42 | |
| HLA-A*43:01 | HLA-A*02:11 | HLA-A*31:01 | HLA-A*24:07 | HLA-A*26:03 | HLA-B*39:10 | HLA-B*14:01 | HLA-B*27:20 | HLA-B*39:01 | HLA-B*41:01 | HLA-B*57:03 | HLA-B*46:01 | |
| HLA-A*80:01 | HLA-A*02:12 | HLA-A*33:03 | HLA-A*24:19 | HLA-A*66:01 | HLA-B*42:01 | HLA-B*14:02 | HLA-B*39:02 | HLA-B*44:05 | HLA-B*58:01 | |||
| HLA-A*02:16 | HLA-A*68:01 | HLA-A*32:07 | HLA-A*68:23 | HLA-B*42:02 | HLA-B*39:06 | HLA-B*45:01 | ||||||
| HLA-A*02:19 | HLA-A*74:01 | HLA-B*51:01 | HLA-B*48:01 | |||||||||
| HLA-A*02:50 | HLA-B*54:01 | |||||||||||
| HLA-A*03:19 | HLA-B*55:01 | |||||||||||
| HLA-B*56:01 | ||||||||||||
| HLA-B*81:01 | ||||||||||||
| HLA-B*83:01 |
pMHC-I binding was predicted using the NetMHCpan-2.8 prediction algorithm for 76 MHC-I allotypes, which were subsequently grouped according to overlapping binding motifs resulting in 12 groups. Binding data for the 6 allotypes denoted in italic in table 1 were inconclusive (for details see text).
Data sets
For the training of the stability predictor, only data from HLA molecules characterized by at least one stable binding and two non-stable binding measurements (using a half-life threshold at 37oC of 2 hours) were included. This left a data set of pMHC-I complex half-lifes with 28,166 measurements covering 75 HLA molecules. The data was enriched with 1000 random natural 9mer peptides for each allele. These peptides were selected to have predicted affinities (using NetMHCpan-2.8) weaker than 20.000 nM, and were assigned a stability value of 0 hours. For retraining of the NetMHCpan method, the IEDB data set corresponding to version 2.8 was used. This data set consists of 136,153 peptide affinity measurements covering 152 distinct MHC molecules.
To generate balanced data sets where an equal number of HLA allotypes were represented for both stability and affinity, and for each allotype an equal number of peptide data points and peptide binders were represented, the following procedures were applied: For stability, the peptides were classified into binders and non-binders using a threshold of a stability half-life of 1 hour. For affinity, the same classification was made using an affinity threshold of 500 nM. Next, the subset of 58 allotypes covered by at least one binder and two non-binders for both affinity and stability was identified. For each of these allotypes, the number of binders/non-binders was identified as the lowest number from the stability and affinity data set, respectively, and this number of binders/non-binders was selected from each data set. To ensure the highest degree of peptide diversity, this selection was done prioritizing peptides with measured binding to few HLA alleles. Following this procedure, a balanced data set was constructed for both affinity and stability each containing 17,998 data points covering 58 alleles. As for the complete data set, these data sets were enriched with 1000 random natural 9mer for each allotype. As before, these peptides were selected to have predicted affinities (using NetMHCpan-2.8) weaker than 20.000 nM.
Artificial neural network training
Artificial neural networks (ANN) were trained as a feed-forward ANN method (25) using either Blosum50 encoding with a normalization factor of 5 or sparse encoded with one of the 20 inputs being 0.9 and the remaining 19 being 0.05 with 40, 50 or 60 neurons in the hidden layer. In each case, the pool of unique peptides was split into five sets in a typical fivefold cross-validation scheme with all peptide-HLA-I stability data for a given peptide placed in the same group (26) (in this way, no peptide can belong to more than one group), 4/5 of the data was used for training and the remaining 1/5 was left for testing and early stop. Prior to ANN training, half-life values were transformed to a value between 0 and 1. The transformation used was s=2-t0/th, where s is the transformed value, th is the measured pMHC-I complex half-life (in hours) and t0 is a threshold value that is fitted to obtain a suitable distribution of the data-points for training purpose. From data show in table 2, it is apparent that the HLA-I allotypes display highly variable ability to form stable complexes; by way of example, the 95% percentile (the half-life for the top 5% most stable peptide) for HLA-A*03:01 was 50 times greater than the corresponding value for HLA-B*08:01 (see table 2). This large range in half-life values makes rescaling of data prior to ANN training not trivial, yet essential. Given the data, it is likely that an allotype specific threshold for rescaling would result in the best performing network, as information about the stability range of each specific allotype would be encoded in the network. However, as discussed later, this is not an applicable solution when developing a pan-specific predictor and as an alternative to allotype specific rescaling, we therefore probed different global rescaling thresholds in the range 0.5h to 2h to optimize the predictive performance of the networks.
Table 2. Summary of pHLA-I stability data.
| t1/2 percentile | t1/2 percentile | ||||||||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| Group | Allotype | Data points | 95 | 5 | Group | Allotype | Data points | 95 | 5 |
| A1 | HLA-A*01:01 | 220 | 40.3 | 0 | B7 | HLA-B*07:02 | 647 | 9.8 | 0 |
| HLA-A*29:02 | 383 | 20.4 | 0.1 | HLA-B*35:01 | 650 | 7.4 | 0 | ||
| HLA-A*30:02 | 367 | 14.1 | 0 | HLA-B*35:03 | 376 | 7 | 0.2 | ||
| HLA-A*43:01 | 359 | 6.7 | 0 | HLA-B*39:10 | 376 | 32.5 | 0 | ||
| HLA-A*80:01 | 351 | 2 | 0 | HLA-B*42:01 | 350 | 34.1 | 0.4 | ||
| HLA-B*42:02 | 376 | 13 | 0 | ||||||
| A2 | HLA-A*02:01 | 1023 | 28.7 | 0.1 | HLA-B*51:01 | 353 | 15 | 0 | |
| HLA-A*02:03 | 379 | 46.7 | 0.3 | HLA-B*54:01 | 357 | 10.8 | 0 | ||
| HLA-A*02:10 | 370 | 31.3 | 0.1 | HLA-B*55:01 | 378 | 9.7 | 0 | ||
| HLA-A*02:11 | 379 | 28.9 | 0.8 | HLA-B*56:01 | 368 | 7.8 | 0 | ||
| HLA-A*02:12 | 372 | 37.4 | 0.4 | HLA-B*81:01 | 367 | 2.7 | 0 | ||
| HLA-A*02:16 | 373 | 36.5 | 0.4 | ||||||
| HLA-A*02:19 | 354 | 39.6 | 0.2 | B8 | HLA-B*08:01 | 465 | 1.2 | 0 | |
| HLA-A*02:50 | 375 | 23.2 | 0.7 | HLA-B*08:03 | 350 | 1.9 | 0 | ||
| HLA-B*14:01 (C67S) | 374 | 0.6 | 0 | ||||||
| A3 | HLA-A*03:01 | 861 | 52.2 | 0 | HLA-B*14:02 (C67S) | 382 | 4.5 | 0 | |
| HLA-A*11:01 | 577 | 71.7 | 0.1 | ||||||
| HLA-A*30:01 | 386 | 37.4 | 0 | B27 | HLA-B*27:02 | 359 | 34.9 | 0 | |
| HLA-A*31:01 | 382 | 77.3 | 0 | HLA-B*27:03 | 368 | 25.1 | 0 | ||
| HLA-A*33:03 | 371 | 56 | 0 | HLA-B*27:05 | 366 | 25.3 | 0 | ||
| HLA-A*68:01 | 387 | 56 | 0 | HLA-B*27:20 | 368 | 8.3 | 0 | ||
| HLA-A*74:01 | 361 | 94.5 | 0 | ||||||
| B39 | HLA-B*15:10 | 363 | 7.7 | 0 | |||||
| A24 | HLA-A*23:01 | 372 | 37.3 | 0.4 | HLA-B*39:01 | 680 | 9.3 | 0 | |
| HLA-A*24:02 | 573 | 30.2 | 0.2 | HLA-B*39:02 | 375 | 14.9 | 0 | ||
| HLA-A*24:03 | 512 | 37.6 | 0.2 | HLA-B*39:06 (C67S) | 379 | 0.5 | 0 | ||
| HLA-A*24:07 | 352 | 24.4 | 0.2 | ||||||
| HLA-A*24:19 | 378 | 2.7 | 0 | B44 | HLA-B*18:01 | 359 | 8 | 0 | |
| HLA-A*32:07 | 364 | 43.5 | 0 | HLA-B*40:01 | 302 | 2.3 | 0 | ||
| HLA-B*41:01 | 368 | 3.5 | 0 | ||||||
| A26 | HLA-A*25:01 | 363 | 21.7 | 0 | HLA-B*44:05 | 352 | 12.1 | 0 | |
| HLA-A*26:01 | 272 | 23.8 | 0 | HLA-B*45:01 | 368 | 5.33 | 0 | ||
| HLA-A*26:02 | 343 | 35.6 | 0 | ||||||
| HLA-A*26:03 | 341 | 15.3 | 0 | B58 | HLA-B*15:17 | 362 | 6.3 | 0 | |
| HLA-A*66:01 | 382 | 3.6 | 0 | HLA-B*57:01 | 349 | 12.4 | 0 | ||
| HLA-A*68:23 | 369 | 9.9 | 0 | HLA-B*57:02 | 363 | 7.2 | 0 | ||
| HLA-B*57:03 | 369 | 7.8 | 0.6 | ||||||
| Others | HLA-A*02:05 | 21 | 26.3 | 0.6 | HLA-B*58:01 | 379 | 21.3 | 0 | |
| HLA-A*32:01 | 32 | 57.2 | 0 | ||||||
| HLA-A*68:02 | 16 | 15.8 | 0.1 | B62 | HLA-B*15:01 | 1070 | 12.5 | 0 | |
| HLA-A*69:01 | 15 | 4.1 | 0 | HLA-B*15:02 | 349 | 5.7 | 0 | ||
| HLA-B*13:02 | 7 | 6 | 0 | HLA-B*46:01 | 361 | 5.3 | 0 | ||
| HLA-B*35:08 | 27 | 8.7 | 0 | ||||||
| HLA-B*40:02 | 19 | 24.4 | 0.5 | ||||||
75 HLA-I allotypes were grouped into 12 “supertype” group by predicted binding motif and screened for pMHC-I complex stability to peptide sets representing each “supertype” binding preference. The extra group “Others” contain molecules with few binding data obtained from an in-house database. For each molecule is indicated the total number of peptide binding measurements, and 95% and 5% percentile half-life values. The 95 percentile value corresponds to the half-time of the top 5% most stable peptide and 5 percentile value to the bottom 5% least stable peptide for each HLA molecule.
For comparison to pan-specific ANNs, the Pearson's correlation coefficient (PCC) was calculated for each allotype included in the training set from the test set predictions. The PCC of each allotype was then compared between networks using a binomial test counting the number of times a network outperformed the other network excluding ties.
Prediction of T cell epitopes and MHC ligands
The predictive performance of the different prediction methods was evaluated on a large set of 9mer T cell epitopes and MHC-I ligands. T-cell epitopes were extracted from both the IEDB (27) and SYFPEITHI (28) databases, while MHC-I ligands where obtained from the SYFPEITHI database only. The evaluation data sets were filtered for predicted binding affinity to the reported MHC restriction element, peptide length and presence in the training data set as described by Jørgensen et al. (23). Briefly, all T cell epitopes and ligands were filtered for predicted binding affinity to the restricting MHC using NetMHCpan-2.8 with a rank threshold for binding of <=10% rank to exclude erroneously annotated epitopes/ligands. The evaluation set was further restricted to exclude HLA-peptide pairs present in the training data set. This filtered data set consisted of 1268 ligands representing 35 allotypes and 1230 epitopes representing 28 allotypes respectively. As the data were unevenly distributed among the allotypes, a balanced data set was extracted limiting the number of ligands/epitope per allotype to 100 and filtering out those allotypes with less than 10 data points. This final evaluation set consisted of 1058 ligands and 598 epitopes, covering 31 and 23 different allotypes, respectively. For evaluation of methods trained on balanced binding data sets, the epitope and ligand data set consisted, after filtering and size restriction, of 1085 MHC ligands and 856 T cell epitopes, covering 31 and 23 different allotypes, respectively. The increase in size of the evaluation data is explained by the dramatic reduction in the size of affinity data in balanced training data set (from 136,153 to 17,998 data points.
The performance of the ANN methods was evaluated as described earlier (17, 29) using the area under the receiver operating characteristic curve (AUC) performance measure: the source protein of each ligand/epitope was fragmented into 9mers, the reported ligand/epitope was annotated as positive and every other 9mer as negative. In this way, one AUC value is calculated for each HLA-ligand/epitope pair, and the performance of a method is reported as the average of the AUC over all HLA-ligand/epitope pairs.
Results
Large scale analysis of peptide-HLA-I stability
We have recently reported the generation of a predictor trained on pMHC-I stability data and shown that such a tool can accurately predict immunogenic MHC-I ligands and that combining this tool with state-of-the-art predictors of pMHC-I binding (NetMHCpan) can improve the predictive performance of T cell epitopes and ligands (1, 23). These studies were conducted on a representative set of human MHC-I allotypes and the resulting predictive tool has limited allotype coverage. Here, we have sought to reinforce these findings and generate sufficient pMHC-I stability data to develop pan-specific predictors of pMHC-I stability. Having 76 HLA-I allotypes and >9,000 nonameric peptides at our disposal, we devised a strategy to allow rapid screening of pHLA-I stability. Using NetMHCpan-2.8 all nonameric peptides were predicted for binding to each HLA-I allotype. Next, peptides predicted to bind to multiple HLA-I allotypes were grouped resulting in twelve peptide sets that could be used to screen for stability on 4 to 12 different allotypes (see table 1). This allowed us to screen the same peptide set on multiple HLA-I allotypes, increasing the throughput of stability assay. To fit the experimental layout, each peptide group was limited to contain 383 peptides. Having compiled the twelve peptide sets, each HLA-I allotype was screened for pMHC-I stability to the representative peptide set using a scintillation proximity assay based pMHC-I dissociation assay (24). For 70 of the 76 allotypes, we were able to obtain conclusive data (data for 6 allotypes denoted in italic in table 1 were inconclusive). Combining this data set with a small-scale in-house data set resulted in a data set of 28,939 individual pMHC-I half-life data points covering 80 HLA-I allotypes. Peptides forming stable pMHC-I complexes (t½>1h) were found for most HLA-I allotypes. Of these, only data from allotypes covered by at least one binding and two non-binding peptides (defined using a half-life threshold of 2 hours) were included in the training data for the pan-specific predictor, arriving at the data set of 28,166 9-mer measurements covering 75 HLA molecules (see table 2).
Development of Artificial Neural Networks predictors of pHLA-I complex stability
Artificial neural networks (ANN) constitute a powerful machine-learning framework for data mining and ANNs have successfully been applied to develop highly efficient predictors of peptide-MHC-I binding (17, 18). To obtain a pan-specific predictor, the NetMHCpan approach earlier developed for pan-specific prediction of peptide-MHC binding affinity was applied. Here, an MHC-I pseudo-sequence was extracted from the polymorphic MHC-I residues in potential contact with the bound peptide (17), and this pseudo sequence was encoded together with each peptide in the training. The data were partitioned and the pan-specific stability predictor was trained as described in Materials and Methods. Prior to training, the half-life values were rescaled as described in materials and methods. Both allotype specific and global rescaling thresholds ranging from 0.5h to 2h (here denoted t0), were used to identify the optimal rescaling. The highest performance was obtained using allotype specific rescaling values (PCC=0.693) (see figure 1). However, using allotype specific rescaling is not an viable option since our current aim is to develop a pan-specific prediction method, which by its very nature should be applicable also to allotypes without experimental data i.e. such a method cannot be made dependent on the availability of specific allotype data. Moving forward, we therefore selected the global rescaling threshold with the highest performance (t0=1h, PCC=0.676). This performance is slightly lower than the performance of the network using allotype specific rescaling, yet statistically comparable (p=0.901, binomial test excluding ties).
Figure 1. Predictive performance of ANN predictors of pMHC-I complex stability.
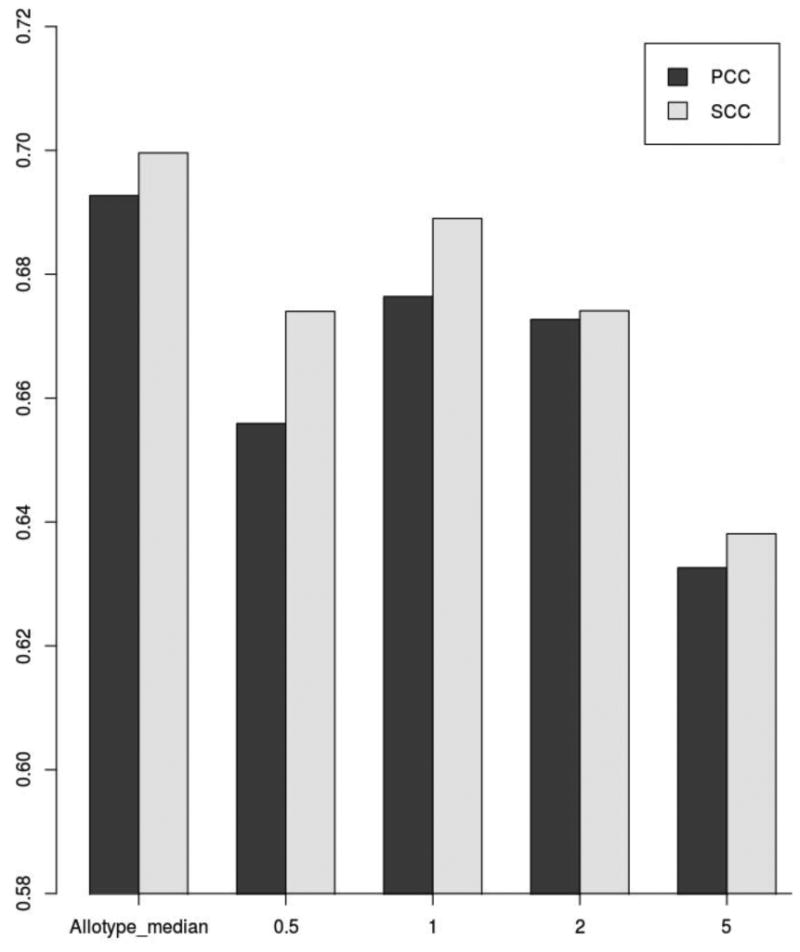
The graphic shows the average PCC,and SCC values for the predictors trained with different t0 thresholds.
Integrating stability and affinity based predictions of pMHC-I interaction
We have previously reported that combining predictors of pMHC-I affinity with predictors of pMHC-I stability improve the prediction of T cell epitopes and MHC-I ligands (23). However, this analysis was limited to a small set of allotypes. Here, we have expanded this observation to a much larger set of MHC molecules. For this purpose, we extracted T cell epitopes and MHC-I ligands from the IEDB (27) and SYFPEITHI (28) to use as benchmark data sets for evaluation of the combined predictors. We applied a simple weighted average between affinity and stability-based predictions to identify the optimal contribution of each to the prediction of T cell epitopes and ligands. In a 5-fold cross validation, a ratio of affinity-based and stability-based predictions of 0.9:0.1 and 0.8:0.2 was consistently found to be optimal with respect to the prediction of MHC-I ligands and MHC-I epitopes, respectively (see figure 2). These combinations significantly outperformed each of the methods in isolation (p=0.0013, binomial test excluding ties for MHC-I ligands, and p=0.0005, binomial test excluding ties for MHC-I epitopes) thus consolidating our earlier findings (23).
Figure 2. Performance of epitope/ligand predictions in terms of AUC.
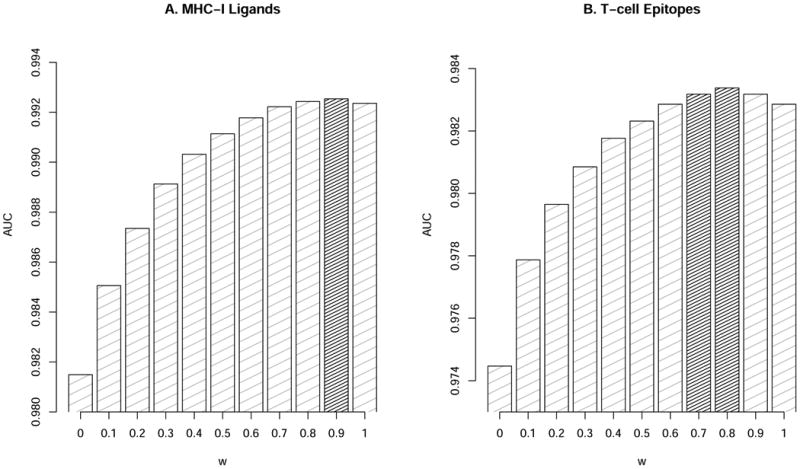
Predictive performance for the stability (w=0) and affinity (w=1) predictors and their combinations for (A) MHC-I Ligands and (B) T-cell Epitopes. The grey columns show combinations with significantly improve (p<0.05) performance compared to both affinity alone (w=1), and stability alone (w=0) and in-significant difference (p>0.05) compared to the optimal model (the value of w with highest AUC value). Statistical differences were evaluated using binomial test excluding ties.
Analysis of the predictive performance using balanced networks
The above analysis shows that the affinity predictions currently contribute more than the stability predictions to the predictive power of the optimal method. However, the differences in the amounts of data available for the training of the affinity and stability based predictors are substantial with ∼140,000 affinity measurements covering > 140 MHC-I allotypes compared to a “mere” ∼28,000 stability measurements covering 75 HLA alleles. To investigate the impact of this difference, we extracted balanced data sets from the pMHC-I binding affinity and stability data sets containing the same number of “positives” and “negatives” data point (for details see Materials and Methods). Next, predictors of pMHC-I binding affinity and stability were trained as described above using a rescaling factor t0=1 hours for the stability network. Using the afore mentioned weighted average scheme and the large set of IEDB and SYFPEITHI T cell epitopes and MHC ligands, we find that the combination of the two methods also here significantly improved the prediction beyond each of the individual methods with an optimal ratio of 0.7:0.3 for MHC-I ligands and 0.6:0.4 for MHC-I epitopes (p-value < 0.05 binomial test excluding ties) (see figure 3). Thus, using methods trained on size balanced data sets, the relative importance of stability predictions compared to that of affinity predictions is increased. Also, we consistently in both benchmarks find that the relative importance of stability is higher for the prediction of T cell epitopes than for the prediction of MHC ligands.
Figure 3. Performance of epitope/ligand predictions in terms of AUC for methods trained using balanced data sets.
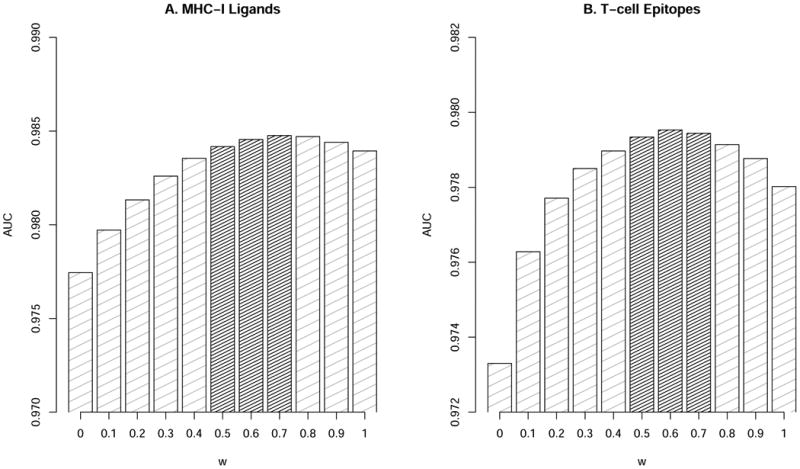
Prediction performance for the stability (w=0) and affinity (w=1) methods training on balanced data sets and their combinations for (A) MHC-I Ligands and (B) T-cell epitopes. The grey columns show combinations with significantly improve (p<0.05) performance compared to both affinity alone (w=1), and stability alone (w=0) and in-significant difference (p>0.05) compared to the optimal model (the value of w with highest AUC value). Statistical differences were evaluated using binomial test excluding ties.
The source of the performance gain
Having demonstrated that inclusion stability data led to an improved predictive performance, we next asked ourselves what was the source of this gain in performance. The two predictors for binding affinity and stability are highly correlated (supplementary figure S1), and one could ask if the gain in performance demonstrated above results from the different nature of data (stability versus affinity) or simply because more data were included in the method development. We addressed this question by identifying the list peptides for which both binding affinity and stability measurements existed. This set contained 7600 peptides covering 58 allotypes. Given this data set, we retrained three new pan-specific predictors; a pan-specific affinity predictor trained on affinity peptide data set excluding the peptide data set with both affinity and stability measurements (termed A); a pan-specific affinity predictor trained on affinity data from the peptides with both affinity and stability measurements (termed B); and a pan-stability predictor trained on stability data from the peptides with both affinity and stability measurements (termed C). Both B and C were trained as described above using the same peptide data, identical data partitioning and same number of added artificial random negatives. A was trained using the NetMHCpan protocol described in (18). Next, linear weighted combinations of A+B and A+C were constructed (the optimal weights were identified separately for each method), and the performance of the optimal combined methods was compared on the epitope benchmark data set. Doing this, we find that the method combining affinity and stability (A+C) significantly outperformed the method combining affinity with affinity (A+B) (p-values < 0.001, binomial test excluding ties). This result thus demonstrates that the observed increased performance is due to the different nature of the data, and not due to the simple fact that more data was included.
The NetMHCstabpan server
The final pan-specific MHC class I peptide stability prediction method trained on the complete data set of 28,166 9-mer measurements covering 75 HLA alleles was implemented as a web-server which is publicly available at http://www.cbs.dtu.dk/services/NetMHCstabpan. The server allows for prediction of pMHC-I binding stability to any HLA molecule of known sequence. Predictions for non-9-mer peptides are made using the approximation method described in (30). Submissions are accepted in two formats, as a list of peptides, or as protein sequences in FASTA format. If the submission is in FASTA format, the predictions are made for all overlapping peptides of the user selected length(s) in the sequence. MHC molecules can be selected from a predefined list, or the user can upload a full length MHC protein sequence in FASTA format. The output is given as a table where each row contains the peptide, the predicted stability score, converted half-life in hours and a percentile rank score. The predictor also allows the user to include affinity predictions (calculated by NetMHCpan-2.8). Selecting this adds the predicted affinity score, the converted binding affinity in nM, a combination of affinity and stability scores using the previously described ratio of 0.8:0.2 (this ratio can be altered by the user), and a percentile rank score of the combined prediction score estimated from the harmonic mean of the stability and affinity rank values, respectively to the output.
Discussion
Binding of peptides to MHC class I molecules is the single most selective step in antigen presentation to CD8+ T cells, and substantial experimental and computational work has been dedicated to characterize this event. For a peptide to be selected for antigen presentation and trigger translocation to the cell surface in complex with a restricting MHC-I molecule, it must bind with sufficient affinity and stability. Moreover once presented, it must remain in the complex with the MHC on the cell surface for sufficient time to allow recognition of a circulating CTL. Given this, it has been argued that both binding stability and affinity are essential correlates to peptide immunogenicity and immune dominance, and furthermore we have in a set of recent publications suggested that of the two, stability is the better predictor of CTL immunogenicity (1, 23).
Here, we have extended these previous studies, and generated a large set of almost 30,000 peptide stability binding measurements covering more than 75 HLA class I molecules. The data was created using a cost-effective screening strategy, where peptides were first screening for predicted binding affinity using NetMHCpan v2.8, and second partitioned into 12 groups with predicted shared binding characteristics, allowing screening against multiple HLA allotypes leading to efficient identification of stable binders. From this data, we have constructed a pan-specific predictor of HLA-I peptide binding stability. To the best of our knowledge, this is the first pan-specific predictor of HLA-I stability.
This pan-specific predictor was constructed following the pipeline defined earlier for the pan-specific predictor of HLA-I affinity, NetMHCpan (17). The predictive performance was evaluated using data representing T-cell epitopes or ligands, which were downloaded from the SYFPEITHI and IEDB databases. Comparing the predictive performance of the stability predictor to that of the NetMHCpan-2.8 affinity predictor, demonstrated that the affinity-based predictor significantly outperformed the stability prediction. However, integrating the two predictors with a relative weight on affinity of 80%, showed a significantly improved prediction of CTL epitopes compared to any of the two methods alone.
At first glance the emphasis on the affinity predictor may appears at odds with the notion that stability is the better correlate of CTL immunogenicity. However, comparing the relative importance of stability and affinity-based predictors is not a straightforward task since there are two main biases that lean in favor of generating an accurate affinity based method; a greater quantity and diversity of data available for training of the method. The difference in the amount of data available for the training of affinity and stability based predictors is substantial with ∼140,000 affinity measurements covering > 140 MHC-I allotypes compared to ∼28,000 stability measurements covering 70 HLA alleles. To level this difference, we constructed a balanced set of binding data with the same number of allotypes and data points (i.e. binders and non-binders) for the affinity and stability data sets, and used these balanced data to retrain pan-specific binding affinity and stability predictors. When integrating the predictions of these two methods, and evaluating the performance on the large set of HLA ligands and T cell epitopes, we find that the relative weight on binding affinity was decreased to ∼60%. While the balanced data set has leveled the difference in size and allele coverage, one important aspect remains very different between the two data sets. From the ∼18,000 data points in the balanced data set, the stability data has 5,581 unique peptides whereas the affinity data set has 9,161 unique peptides. This difference in peptide diversity is a consequence of the strategy used to select the peptides for the stability experiments. In retrospect, we made a suboptimal strategy decision, which unfortunately cannot be resolved with the existing stability data set. As more, and more diverse, stability data will be generated in the future, we speculate that the relative importance between the two methods will be shifted further in favor of emphasizing binding stability.
Even though the current tool is trained on the hitherto largest set of peptide MHC-I stability data, the volume of the data set remains small compared to the data available for peptide MHC-I affinity. Furthermore, the current tool has been trained on nonamer peptide data only limited to the HLA-A and HLA-B isotypes. Given this and experiences from earlier works (18, 21, 31, 32), we strongly expect the performance of the tools will increase as more data covering different peptide length, HLA isotypes/allotypes (potentially also MHC allotypes covering other species) become available.
It should be noted that even though we here have demonstrated a high performance for prediction of T cell epitopes driven by binding affinity and stability, other factors including self versus non-self peptides similarity (33, 34), and T cell propensity (35) are essential in order to characterize and predict peptide immunogenicity.
In conclusion, we have here developed the first pan-specific predictor for binding stability of peptide HLA-I complexes, and have demonstrated how this tool can significantly enhance the ability to predict T cell peptide immunogenicity. A webserver implementing the method is available at publicly www.cbs.dtu.dk/services/NetMHCstabpan
Supplementary Material
Footnotes
This work was supported by Federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under Contract No. HHSN272201200010C, and HHSN27200900045C and from the Agencia Nacional de Promoción Científica y Tecnológica, Argentina (PICT-2012-0115).
Reference List
- 1.Harndahl M, Rasmussen M, Roder G, Dalgaard Pedersen I, Sorensen M, Nielsen M, Buus S. Peptide-MHC class I stability is a better predictor than peptide affinity of CTL immunogenicity. European journal of immunology. 2012;42:1405–1416. doi: 10.1002/eji.201141774. [DOI] [PubMed] [Google Scholar]
- 2.van der Burg SH, Visseren MJ, Brandt RM, Kast WM, Melief CJ. Immunogenicity of peptides bound to MHC class I molecules depends on the MHC-peptide complex stability. J Immunol. 1996;156:3308–3314. [PubMed] [Google Scholar]
- 3.Micheletti F, Guerrini R, Formentin A, Canella A, Marastoni M, Bazzaro M, Tomatis R, Traniello S, Gavioli R. Selective amino acid substitutions of a subdominant Epstein-Barr virus LMP2-derived epitope increase HLA/peptide complex stability and immunogenicity: implications for immunotherapy of Epstein-Barr virus-associated malignancies. European journal of immunology. 1999;29:2579–2589. doi: 10.1002/(SICI)1521-4141(199908)29:08<2579::AID-IMMU2579>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 4.Burrows JM, Wynn KK, Tynan FE, Archbold J, Miles JJ, Bell MJ, Brennan RM, Walker S, McCluskey J, Rossjohn J, Khanna R, Burrows SR. The impact of HLA-B micropolymorphism outside primary peptide anchor pockets on the CTL response to CMV. European journal of immunology. 2007;37:946–953. doi: 10.1002/eji.200636588. [DOI] [PubMed] [Google Scholar]
- 5.Nicholls S, Piper KP, Mohammed F, Dafforn TR, Tenzer S, Salim M, Mahendra P, Craddock C, van Endert P, Schild H, Cobbold M, Engelhard VH, Moss PA, Willcox BE. Secondary anchor polymorphism in the HA-1 minor histocompatibility antigen critically affects MHC stability and TCR recognition. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:3889–3894. doi: 10.1073/pnas.0900411106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spierings E, Gras S, Reiser JB, Mommaas B, Almekinders M, Kester MG, Chouquet A, Le Gorrec M, Drijfhout JW, Ossendorp F, Housset D, Goulmy E. Steric hindrance and fast dissociation explain the lack of immunogenicity of the minor histocompatibility HA-1Arg Null allele. J Immunol. 2009;182:4809–4816. doi: 10.4049/jimmunol.0803911. [DOI] [PubMed] [Google Scholar]
- 7.Lipford GB, Bauer S, Wagner H, Heeg K. In vivo CTL induction with point-substituted ovalbumin peptides: immunogenicity correlates with peptide-induced MHC class I stability. Vaccine. 1995;13:313–320. doi: 10.1016/0264-410x(95)93320-9. [DOI] [PubMed] [Google Scholar]
- 8.van Stipdonk MJ, Badia-Martinez D, Sluijter M, Offringa R, van Hall T, Achour A. Design of agonistic altered peptides for the robust induction of CTL directed towards H-2Db in complex with the melanoma-associated epitope gp100. Cancer research. 2009;69:7784–7792. doi: 10.1158/0008-5472.CAN-09-1724. [DOI] [PubMed] [Google Scholar]
- 9.Lazarski CA, Chaves FA, Jenks SA, Wu S, Richards KA, Weaver JM, Sant AJ. The kinetic stability of MHC class II:peptide complexes is a key parameter that dictates immunodominance. Immunity. 2005;23:29–40. doi: 10.1016/j.immuni.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 10.Hall FC, Rabinowitz JD, Busch R, Visconti KC, Belmares M, Patil NS, Cope AP, Patel S, McConnell HM, Mellins ED, Sonderstrup G. Relationship between kinetic stability and immunogenicity of HLA-DR4/peptide complexes. European journal of immunology. 2002;32:662–670. doi: 10.1002/1521-4141(200203)32:3<662::AID-IMMU662>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 11.Pogue RR, Eron J, Frelinger JA, Matsui M. Amino-terminal alteration of the HLA-A*0201-restricted human immunodeficiency virus pol peptide increases complex stability and in vitro immunogenicity. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:8166–8170. doi: 10.1073/pnas.92.18.8166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brooks JM, Colbert RA, Mear JP, Leese AM, Rickinson AB. HLA-B27 subtype polymorphism and CTL epitope choice: studies with EBV peptides link immunogenicity with stability of the B27:peptide complex. J Immunol. 1998;161:5252–5259. [PubMed] [Google Scholar]
- 13.Abdel-Motal UM, Friedline R, Poligone B, Pogue-Caley RR, Frelinger JA, Tisch R. Dendritic cell vaccination induces cross-reactive cytotoxic T lymphocytes specific for wild-type and natural variant human immunodeficiency virus type 1 epitopes in HLA-A*0201/Kb transgenic mice. Clin Immunol. 2001;101:51–58. doi: 10.1006/clim.2001.5095. [DOI] [PubMed] [Google Scholar]
- 14.Vertuani S, Sette A, Sidney J, Southwood S, Fikes J, Keogh E, Lindencrona JA, Ishioka G, Levitskaya J, Kiessling R. Improved immunogenicity of an immunodominant epitope of the HER-2/neu protooncogene by alterations of MHC contact residues. J Immunol. 2004;172:3501–3508. doi: 10.4049/jimmunol.172.6.3501. [DOI] [PubMed] [Google Scholar]
- 15.Grohmann U, Belladonna ML, Bianchi R, Orabona C, Silla S, Squillacioti G, Fioretti MC, Puccetti P. Immunogenicity of tumor peptides: importance of peptide length and stability of peptide/MHC class II complex. Cancer immunology, immunotherapy : CII. 1999;48:195–203. doi: 10.1007/s002620050565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Assarsson E, Sidney J, Oseroff C, Pasquetto V, Bui HH, Frahm N, Brander C, Peters B, Grey H, Sette A. A quantitative analysis of the variables affecting the repertoire of T cell specificities recognized after vaccinia virus infection. J Immunol. 2007;178:7890–7901. doi: 10.4049/jimmunol.178.12.7890. [DOI] [PubMed] [Google Scholar]
- 17.Nielsen M, Lundegaard C, Blicher T, Lamberth K, Harndahl M, Justesen S, Roder G, Peters B, Sette A, Lund O, Buus S. NetMHCpan, a method for quantitative predictions of peptide binding to any HLA-A and -B locus protein of known sequence. PLoS ONE. 2007;2:e796. doi: 10.1371/journal.pone.0000796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoof I, Peters B, Sidney J, Pedersen LE, Sette A, Lund O, Buus S, Nielsen M. NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics. 2009;61:1–13. doi: 10.1007/s00251-008-0341-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karosiene E, Lundegaard C, Lund O, Nielsen M. NetMHCcons: a consensus method for the major histocompatibility complex class I predictions. Immunogenetics. 2011 doi: 10.1007/s00251-011-0579-8. [DOI] [PubMed] [Google Scholar]
- 20.Peters B, Sette A. Generating quantitative models describing the sequence specificity of biological processes with the stabilized matrix method. BMC bioinformatics. 2005;6:132. doi: 10.1186/1471-2105-6-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Andreatta M, Nielsen M. Gapped sequence alignment using artificial neural networks: application to the MHC class I system. Bioinformatics. 2015 doi: 10.1093/bioinformatics/btv639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lundegaard C, Lamberth K, Harndahl M, Buus S, Lund O, Nielsen M. NetMHC-3.0: accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8-11. Nucleic acids research. 2008 doi: 10.1093/nar/gkn202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jorgensen KW, Rasmussen M, Buus S, Nielsen M. NetMHCstab - predicting stability of peptide:MHC-I complexes; impacts for CTL epitope discovery. Immunology. 2013 doi: 10.1111/imm.12160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harndahl M, Rasmussen M, Roder G, Buus S. Real-time, high-throughput measurements of peptide-MHC-I dissociation using a scintillation proximity assay. Journal of immunological methods. 2011;374:5–12. doi: 10.1016/j.jim.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nielsen M, Lundegaard C, Worning P, Lauemoller SL, Lamberth K, Buus S, Brunak S, Lund O. Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci. 2003;12:1007–1017. doi: 10.1110/ps.0239403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nielsen M, Lundegaard C, Lund O. Prediction of MHC class II binding affinity using SMM-align, a novel stabilization matrix alignment method. BMC bioinformatics. 2007;8:238. doi: 10.1186/1471-2105-8-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim Y, Ponomarenko J, Zhu Z, Tamang D, Wang P, Greenbaum J, Lundegaard C, Sette A, Lund O, Bourne PE, Nielsen M, Peters B. Immune epitope database analysis resource. Nucleic acids research. 2012;40:W525–530. doi: 10.1093/nar/gks438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rammensee HG, Friede T, Stevanoviic S. MHC ligands and peptide motifs: first listing. Immunogenetics. 1995;41:178–228. doi: 10.1007/BF00172063. [DOI] [PubMed] [Google Scholar]
- 29.Stranzl T, Larsen MV, Lundegaard C, Nielsen M. NetCTLpan: pan-specific MHC class I pathway epitope predictions. Immunogenetics. 2010;62:357–368. doi: 10.1007/s00251-010-0441-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lundegaard C, Lund O, Nielsen M. Accurate approximation method for prediction of class I MHC affinities for peptides of length 8, 10 and 11 using prediction tools trained on 9mers. Bioinformatics. 2008 doi: 10.1093/bioinformatics/btn128. [DOI] [PubMed] [Google Scholar]
- 31.Rasmussen M, Harndahl M, Stryhn A, Boucherma R, Nielsen LL, Lemonnier FA, Nielsen M, Buus S. Uncovering the peptide-binding specificities of HLA-C: a general strategy to determine the specificity of any MHC class I molecule. J Immunol. 2014;193:4790–4802. doi: 10.4049/jimmunol.1401689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nielsen M, Andreatta M. NetMHCpan-3.0; improved prediction of binding to MHC class I molecules integrating information from multiple receptor and peptide length datasets. Genome Medicine. 2016;8:1–9. doi: 10.1186/s13073-016-0288-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frankild S, de Boer RJ, Lund O, Nielsen M, Kesmir C. Amino acid similarity accounts for T cell cross-reactivity and for “holes” in the T cell repertoire. PLoS ONE. 2008;3:e1831. doi: 10.1371/journal.pone.0001831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bresciani A, Paul S, Schommer N, Dillon MB, Bancroft T, Greenbaum J, Sette A, Nielsen M, Peters B. T-cell recognition is shaped by epitope sequence conservation in the host proteome and microbiome. Immunology. 2016;148:34–39. doi: 10.1111/imm.12585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Calis JJ, Maybeno M, Greenbaum JA, Weiskopf D, De Silva AD, Sette A, Kesmir C, Peters B. Properties of MHC class I presented peptides that enhance immunogenicity. PLoS Comput Biol. 2013;9:e1003266. doi: 10.1371/journal.pcbi.1003266. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.