

Abstract
Prostaglandin E2 (PGE2), a cyclooxygenase metabolite that generally acts as a systemic vasodepressor, has been shown to have vasopressor effects under certain physiologic conditions. Previous studies have demonstrated that PGE2 receptor signaling modulates angiotensin II (Ang II)-induced hypertension, but the interaction of these two systems in the regulation of vascular reactivity is incompletely characterized. We hypothesized that Ang II, a principal effector of the renin-angiotensin-aldosterone system, potentiates PGE2-mediated vasoconstriction. Here we demonstrate that pre-treatment of arterial rings with 1 nM Ang II potentiated PGE2-evoked constriction in a concentration dependent manner (AUC−Ang II 2.778 ± 2.091, AUC+Ang II 22.830 ± 8.560, ***P<0.001). Using genetic deletion models and pharmacological antagonists, we demonstrate that this potentiation effect is mediated via concurrent signaling between the angiotensin II receptor 1 (AT1) and the PGE2 E-prostanoid receptor 3 (EP3) in the mouse femoral artery. EP3 receptor-mediated vasoconstriction is shown to be dependent on extracellular calcium in combination with proline-rich tyrosine kinase 2 (Pyk2) and Rho-kinase. Thus, our findings reveal a novel mechanism through which Ang II and PGE2 regulate peripheral vascular reactivity.
Keywords: prostaglandin E2, angiotensin II, EP3 receptor, vascular reactivity, Pyk2, AT1 receptor
Graphical Abstract
1. Introduction
High blood pressure is a major risk factor for cardiovascular diseases, including myocardial infarction, stroke and renal failure [1, 2]. The renin-angiotensin-aldosterone-system (RAAS) has long been regarded as a major drug target for treatment of hypertension due to its critical role in maintaining blood pressure through regulation of the vascular, renal, endocrine, and neural systems [3, 4]. Angiotensin II (Ang II), a principal effector of the RAAS system, signals via the G-protein coupled receptors AT1 and AT2. AT1 receptors mediate classical Ang II pressor effects including smooth muscle contraction, increased sympathetic nerve activity, and renal tubular reabsorption of Na+ [5]. The AT2 receptor, although not fully characterized, has been shown to mediate opposing responses [6, 7]. The AT1 receptor induces vasoconstriction by coupling to Gq, thereby leading to activation of phospholipase C and increasing intracellular calcium [7, 8]. Such rises in calcium can then activate kinases that promote an increase in myosin light chain phosphorylation and ultimately to smooth muscle contraction. The AT1 receptor is also known to increase phospholipase A2 activation, resulting in liberation of arachidonic acid and synthesis of prostaglandins [9].
Prostaglandin E2 (PGE2) is an oxygenated metabolite of arachidonic acid, which among many functions modulates blood pressure [10, 11]. PGE2 acts primarily as a vasodilator and vasodepressor; however, under certain circumstances PGE2 can have vasopressor effects [12–17]. These divergent functions are due in part to the ability of PGE2 to activate four distinct E-prostanoid receptors, EP1 – EP4 [10, 18]. EP1 and EP3 receptors facilitate vasoconstriction while the EP2 and EP4 receptors mediate vasodilation [13, 17]. As with the AT1 receptor, activation of the EP1 receptor leads to a rise in intracellular calcium via Gq signaling [19]. The EP3 receptor couples to Gi and G12/13, which inhibits cAMP production, activates Rho GTPase, and elevates intracellular calcium levels through its Gβγ subunit [18]. EP2 and EP4 receptors couple to Gs and upon activation increase cAMP.
We have previously demonstrated that blockade of the EP1 or EP3 receptor has salutary effects in mouse models of Ang II-mediated hypertension. EP1 receptor knockout mice display a reduced rise in mean arterial blood pressure in response to Ang II infusion and are protected against end-organ damage in hypertensive mouse models [20, 21]. Similarly, EP3 receptor knockout mice have a blunted blood pressure response to acute administration of Ang II [22]. Although these results indicate an interaction between Ang II and PGE2 signaling, the molecular mechanisms through which these systems synergize to regulate vascular tone is incompletely characterized.
Previous studies have indicated a unique characteristic of PGE2-mediated vessel reactivity, where pre-stimulation of rat arterial tissue with a contractile agent followed by PGE2 unmasks a significant vasoconstrictive response to PGE2 [23]. Thus, we hypothesized that Ang II, a potent vasoconstrictor, could potentiate PGE2-mediated vasoconstriction. Additionally, we predicted that this enhanced vasoconstriction would be mediated by potentiation of EP3 or EP1 receptor signaling rather than through desensitization of the vasodilator EP receptors. In the present study, we investigate the effects of low dose Ang II “priming” on PGE2-incited contraction in isolated blood vessels to determine whether Ang II can alter PGE2 vessel responses. We demonstrate that Ang II primes vessels to exhibit potentiated vasoconstriction in response to PGE2. This effect is mediated by the AT1 and EP3 receptors and requires calcium-dependent mechanisms involving Rho-kinase (ROCK) and the proline-rich tyrosine kinase 2 (Pyk2).
2. Materials and methods
2.1 Animals
All studies were performed in accordance with the National Institutes of Health animal care standards and approved by the Vanderbilt University Institutional Animal Care and Use Committee. Male mice 9 to 15 weeks old on a C57BL/6 background were used for all studies unless noted otherwise. Generation of EP1 (ptger1), EP2 (ptger2), and EP3 (ptger3) receptor knockout mice has been previously described [16, 20, 24]. Mice deficient for the EP4 receptor (ptger4) have a lethal newborn phenotype when maintained on a C57BL/6 genetic background [25]. We bred C57BL/6 EP4+/− male mice to an outbred strain (CD-1, Charles River laboratories; 10-generation outcross) and observed EP4−/− mice viability into adulthood; thus, these mice were used for the EP4 studies [26, 27].
2.2 Isolated femoral arteries
Following euthanasia, femoral arteries were excised and placed into ice-cold Modified Krebs-Henseleit solution (NaHCO2 25, KH2PO4 1.2, MgSO4 1.2, KCl 4.7, NaCl 118, CaCl2 2.5, Dextrose 10 mM). Vessel rings (~2 mm long) were cut and suspended between 40 μm stainless steel wire in a Danish Myo Technology Model 620M myograph system to record tension. Tissue baths contained 5.0 mL of Krebs-Henseleit at 37 °C aerated with 95% O2, 5% CO2 gas. Vascular preparations were allowed to stabilize for 45 minutes at a resting tension of 2.5 mN, which had been determined to give about 90% of maximal response to 50 mM K+ in mouse femoral arteries. Each preparation was initially challenged with 50 mM K+, followed by 3 washes of Krebs-Henseleit solution before testing experimental reagents. Separate rings were used for each data point to avoid the rapid tachyphylaxis of the AT1 receptors. For Ang II priming, Ang II was added to vessel ring baths for 10 minutes followed by addition of PGE2. At the end of each experiment, endothelium integrity was confirmed by reversal of 1 μM phenylephrine-evoked constriction with 10 μM acetylcholine. Rings were excluded that did not dilate at least 70% of the phenylephrine contraction (tension reduced to < 30% of the phenylephrine response). Finally, rings were washed and retested with 50 mM K+ to assure maintained responsiveness throughout the entire experiment. All drugs were dissolved in water, ethanol (PGE2, nifedipine, ibuprofen, SC-236), or DMSO (DG-041, PF-431396) and then further diluted to ≥ 1:1000 or more in Krebs buffer before being added to the vessel bath. Data were collected and stored using ADInstuments LabChart 7 software.
2.3 Chemicals
Angiotensin II (human), acetylcholine, losartan, ibuprofen, nifedipine, PF-431396 hydrate, salicylic acid, and phenylephrine were purchased from Sigma (St. Louis, MO). Prostaglandin E2 and SC-236 were purchased from Cayman Chemical (Ann Arbor, MI). PD123319 was purchased from Tocris (Bristol, UK). Y-27632 was purchased from Abcam Biochemicals (Cambridge, MA). EGTA was purchased from Thermo Fisher Scientific (Waltham, MA). FAK inhibitor 14 was purchased from Santa Cruz Biotechnology (Dallas, TX). (Sar1,Ile4,8)-Angiotensin II Trifluoroacetate salt was purchased from Bachem (Bubendorf, Switzerland). The Vanderbilt Chemical Synthesis Core synthesized DG-041. Salicylamine was a gift from L. Jackson Roberts (Vanderbilt University).
2.4 Competitive binding of [3H]PGE2
Total kidney membranes (100 μg) from wildtype C57BL/6 mice were incubated with [3H]PGE2 (2 nM) in binding buffer (25mM potassium phosphate, 1mM EDTA, and 10mM MgCl, pH 6.2) at a total volume of 200 μl for 1 hr at 30 °C. Nonspecific binding was determined in the presence of excess unlabeled PGE2 (1 μM). EP1/3 selective agonist sulprostone (1 μM) was used as a positive control. Competitive binding experiments were performed in the presence of 2 nM [3H]PGE2 and 1 μM of competing ligands, either Losartan or PD123319. Binding reactions were terminated by vacuum filtration and radioactivity was quantified [28, 29].
2.5 Data analysis
Vascular responses were quantified as a change in force (maximal contraction-baseline tension) normalized to the first 50 mM K+ response (K-standard) of each ring. All data were graphed as means ± SEM using GraphPad Prism 5 software. Data were fitted with variable slope sigmoidal curves using GraphPad Prism. Area under the curve (AUC) (% contraction/Log[treatment]) values were used to statistically compare concentration response curves (CRC) of PGE2 with or without priming agents. Student’s t test was used to determine significance between AUCs as well as for the [H3]PGE2 binding data. Ang II CRCs were fitted to a variable-slope sigmoidal curve. Comparison of single dose PGE2 to various conditions was performed by one-way ANOVA with Bonferroni post-hoc test. For all studies, P ≤ 0.05 was considered statistically significant. Significance was represented throughout each figure as the following: *= P ≤ 0.05, **= P ≤ 0.01, and ***= P ≤ 0.001.
3. Results
3.1 Enhanced prostaglandin E2-mediated contraction after priming with low-dose Ang II
To investigate the effect of Ang II on PGE2-mediated vasoconstriction, mouse femoral arteries (I.D. ~0.25–0.3 mm) were studied ex vivo using a wire myograph. Femoral arteries were selected for our studies as a model of peripheral vasculature in order to compare to previous studies conducted using rat femoral arteries. Arterial rings were assayed for responsiveness to a range of single Ang II concentrations to determine the concentration response curve (CRC) (Fig. 1A). Arterial rings were then primed with a sub-threshold constrictor concentration of Ang II (1 nM, ~ EC15) followed by a single concentration of PGE2 at varying doses to construct a PGE2 CRC. Exposure of arterial rings to PGE2 alone, even at a high concentration of 1 μM (3.326 ± 0.647 % contraction of K-standard, N = 5) did not cause significant vasoconstriction. However, priming of arterial rings with a submaximal concentration of Ang II (1 nM) resulted in robust PGE2-mediated vessel contraction (Fig. 1B, C). Ang II priming facilitated arterial contraction following exposure to doses of 2 nM PGE2 or greater (AUCuntreated 2.778 ± 2.091, AUCAng II primed 22.830 ± 8.560, t test ***P<0.001) (Fig. 1C).
Figure 1. Ang II priming enhances PGE2-mediated vasoconstriction.

(A) Ang II CRC in wildtype mice. (N = 5–7) Dotted line indicates concentration of Ang II used to prime vessels in B and C. (B) Representative trace of 30 nM PGE2-induced contraction of vessels primed with 1 nM Ang II. (C) Comparison of PGE2 concentration-response curve with (filled circles, N = 5–6) or without (open circles, N = 5) 1 nM Ang II priming. Data for each concentration was collected on separate rings. Results were normalized to each ring’s response to 50 mM KCl. Vertical bars indicate SEM.
3.2 EP3 receptor signaling primed by AT1 receptor activation
To determine which prostaglandin receptor mediates the PGE2-induced vasoconstriction of Ang II-primed vessels, wire myography studies were performed utilizing a combination of pharmacological antagonists and genetic deletion models. Femoral arteries isolated from EP3−/− mice displayed a lack of PGE2-mediated vasoconstriction even with Ang II priming (Fig. 2A, B). PGE2-mediated contraction was then assessed using wildtype femoral arteries treated with the EP3 receptor antagonist, DG-041. Pre-treatment with 1 μM DG-041 had no effect on resting tone or Ang II-mediated constriction (data not shown); however, Ang II failed to potentiate PGE2-mediated vasoconstriction in DG-041 pretreated vessels (Fig. 2B). The apparent PGE2-induced vasodilation of vessels primed with Ang II in the presence of DG-041 (Fig. 2B, far right bar) was not statistically significant compared to zero priming. By contrast, in femoral arteries isolated from EP1−/− mice, Ang II was able to potentiate PGE2-mediated vasoconstriction to a similar level as observed with wildtype mice (Fig. 2B). Although these data indicated that upon Ang II priming the subsequent PGE2-induced contraction was EP3 receptor-mediated, Ang II promoting desensitization of either vasodilator receptor, EP2 or EP4, could also explain the PGE2-induced contraction. Thus, to test the contribution of the EP2 or EP4 receptor, we assessed the effect of PGE2 on EP2−/− and EP4−/− femoral arteries. In the absence of Ang II priming, deletion of either EP2 or EP4 was insufficient to facilitate comparable PGE2-induced vasoconstriction to that observed in wildtype vessels primed with Ang II (Fig. 2B, EP2−/− & EP4−/− white bars). PGE2-mediated contraction of Ang II-primed vessels was notably greater in EP2−/− vessels, presumably from loss of the EP2 receptor vasodilator function (Fig. 2B, EP2−/− black bar, P ≤ 0.01). Arterial rings isolated from EP4−/− mice exhibited responses that were indistinguishable from the responses observed in wildtype vessels.
Figure 2. Effects of EP-receptors on PGE2-induced contraction of Ang II-primed arteries.

(A) Representative wire myography trace of an EP3−/− femoral artery that shows the lack of PGE2 response when primed with Ang II. (B) Comparison of % contraction during 30 nM PGE2-induced vasoconstriction with or without 1 nM Ang II priming in EP1−/−, EP2−/−, EP3−/−, or EP4−/− vessels or in wildtype vessels treated with or without the EP3 antagonist DG-041 (DG) (N = 4–5 for EP1−/−, all other bars N = 5; two EP2−/− mice were one year old). PGE2 alone did not cause significant vasoconstriction in any of the mouse strains (wildtype, EP1−/−, EP2−/−, EP3−/−, EP4−/−). Veh1 indicates Ang II priming of PGE2 solvent (ethanol). Veh2 indicates pre-treatment with DG-041 solvent (DMSO). Wildtype data with or without Ang II are taken from Fig 1C. (C) Ang II concentration-response curves comparing contraction of wildtype (taken from Fig 1A, N = 5–7) and EP3−/− vessels (N = 5–6). EC50 for each was −8.74 and −8.70. Vertical bars indicate SEM.
To investigate whether EP3−/− mice have altered contractile responses to Ang II, concentration-response relationships for wildtype or EP3−/− vessels were compared. The EC50 and Emax for Ang II-induced vasoconstriction were not different between wildtype and EP3−/− mice (Fig. 2C). Interestingly, the CRC to Ang II exhibited a steeper slope in wildtype vessels (Hill coefficient of 2.581) as compared to that observed in vessels isolated from EP3−/− mice (Hill coefficient of 1.198). PGE2 alone did not cause significant vasoconstriction in any of the mice (wildtype, EP1−/−, EP2−/−, EP3−/−, EP4−/−). Taken together, these results indicate the EP3 receptor is necessary for PGE2-induced vasoconstriction potentiated by Ang II.
To identify the Ang II receptor responsible for potentiation of PGE2-mediated vasoconstriction by Ang II, vessel contraction was measured in arterial rings pre-treated (30 minutes) with the AT1 antagonist losartan or the AT2 antagonist PD123319 prior to Ang II priming and PGE2 addition. Losartan blocked the ability of Ang II to potentiate PGE2-induced vasoconstriction, while PD123319 had no significant effect. (Fig. 3A, B, C) To verify that the effect of losartan was not due to off target competitive inhibition of the prostanoid receptors, the ability of either losartan or PD123319 to compete with [3H]PGE2 binding in mouse kidney membrane preparations was assessed. In contrast to the EP1/3 selective agonist, sulprostone, neither losartan nor PD123319 competed with [3H]PGE2 binding (Fig. 3D). Together, these results indicate that the AT1 receptor facilitates the Ang II priming effect on EP3 receptor-mediated contraction.
Figure 3. AT1 antagonist inhibits Ang II priming of PGE2.

(A) Representative wire myography trace of femoral arterial rings primed with 1 nM Ang II, followed by addition of 100 nM PGE2. (B) Representative trace of femoral artery with 30 minutes pretreatment with 1 μM losartan followed by addition of 1 nM Ang II priming of 100 nM PGE2. (C) Comparison between femoral arterial ring responses to 100 nM PGE2 with and without 1 nM Ang II priming, as well as with priming in the presence of an AT1 (losartan) or AT2 antagonist (PD123319) (N = 5). (D) Competition binding in wildtype mouse kidney membranes; displacement of [3H]PGE2 by the EP3 agonist sulprostone, losartan, and PD123319. Graph is normalized to competition binding with cold PGE2. (N = 3). * = P < 0.05
3.3 AT1-mediated β-arrestin pathways are not sufficient to potentiate PGE2-mediated contraction
AT1 has been identified to mediate several physiological responses mainly through activation of Gq; however, some AT1-mediated effects can be attributed to activation of G-protein independent pathways via GRK/β-arrestin/MAPK signaling. To determine whether Ang II-induced potentiation of PGE2-mediated vasoconstriction involved activation of β-arrestin pathways, 30 μM of the biased agonist Sar1,Ile4,8-Angiotensin II (SII) (AT1a KD = 300 nM, [30]) was used in wire myography studies. SII has been previously shown to activate β-arrestin signaling pathways via AT1 while not stimulating the Gq-associated calcium cascade [31]. SII did not evoke vasoconstriction at doses up to 30 μM. SII also did not exhibit a potentiation effect on PGE2-mediated constriction of femoral arteries (Fig. 4A, C). Furthermore, SII blocked Ang II-induced contraction as well as Ang II priming of PGE2-mediated contraction, presumably by acting as a competitive antagonist for Ang II-AT1 priming (Fig. 4B, C).
Figure 4. SII does not potentiate PGE2-mediated contraction.

(A) Representative wire myography trace showing that 10 minute pre-treatment with 30 μM SII did not potentiate 100 nM PGE2-mediated contraction. (B) Representative trace of arterial rings preincubated with 30 μM SII for 10 minutes, followed by priming with 1 nM Ang II and addition of 100 nM PGE2. (C) Quantification of SII results, showing a lack of potentiation by SII on PGE2-induced contraction (3rd bar from left), and SII blockade of PGE2-mediated contraction of Ang II-primed vessels (4th bar from left) (N = 5).
3.4 Extracellular calcium stores and Rho-kinase are required for contraction
Activation of AT1 by Ang II but not SII leads to increased intracellular calcium levels [Ca2+]i in smooth muscle cells to stimulate contraction [31]. The contribution of extracellular stores of calcium to Ang II-induced potentiation of PGE2-mediated vasoconstriction was assessed by analyzing vessels in Ca2+ free buffer. Arterial rings were incubated in calcium free Krebs buffer containing 1 mM EGTA. EGTA was subsequently removed from the bath, and the contractile response of Ang II-primed vessels to PGE2 was tested. In control vessels without Ang II pre-treatment, 100 nM PGE2 did not cause significant contraction in normal Krebs or upon addition of higher calcium concentrations to the bath (3.5 or 4.5 mM), demonstrating that increasing extracellular calcium concentration is not sufficient to evoke potentiated PGE2-mediated contraction (Fig. 5A). In the absence of extracellular Ca2+, femoral arteries exhibited minimal responsiveness to Ang II. Furthermore, Ang II did not potentiate PGE2-induced contraction in Ca2+ free media. However, re-addition of Ca2+ into the bath restored PGE2-mediated contraction (Fig. 5B, C). Pre-treatment (30 minutes) of arteries with the L-type calcium channel blocker, Nifedipine, inhibited the PGE2-mediated contraction (Fig. 5C, far right bar). Taken together, these studies indicate a significant contribution of extracellular sources of calcium, particularly from calcium influx by L-type channels, in mediating PGE2-induced contraction after priming with Ang II. Due to its known role in sensitizing smooth muscle cells to calcium-induced contraction, ROCK was evaluated for its effect on PGE2-induced vasoconstriction of Ang II-primed vessels. Addition of the ROCK inhibitor Y-27638 to PGE2-induced vasoconstriction returned vascular tone to baseline (Fig. 5D & E). These data indicate that ROCK is required for EP3-induced arterial contraction.
Figure 5. Contribution of extracellular calcium and Rho-kinase to PGE2 primed vasoconstriction.

(A) Representative wire myography trace (N = 5) of PGE2-mediated contraction with additional calcium added (normal Krebs 2.5 mM, additional calcium 3.5 or 4.5 mM). (B) Example trace of femoral arterial ring maintained in calcium-free Krebs. Vessels were briefly treated with EGTA, washed, and subsequently administered Ang II, followed by PGE2. Calcium was then added back to the bath to restore contraction. (C) Quantification of PGE2 response with and without Ang II priming, in the presence or absence of calcium, compared to vessels pre-incubated with 300 nM Nifedipine or vehicle control (ethanol) (N = 5–8). (D) Quantification of the effects of the ROCK inhibitor Y-27632 on PGE2-mediated contraction (N = 5). (E) Example trace of Ang II priming of PGE2-induced contraction with ROCK inhibitor, Y-27632, addition during PGE2 response.
3.5 Salicylate diminishes Ang II potentiation of PGE2-mediated contraction
To determine whether Ang II potentiates PGE2-mediated vasoconstriction through its action on cPLA2, resulting in increased prostaglandin biosynthesis, femoral arterial rings were pre-incubated with nonsteroidal anti-inflammatory drugs (NSAID). 100 μM ibuprofen, a potent COX inhibitor (COX-1 IC50 = 7.6 μM, COX-2 IC50 = 7.2 μM) [32], did not affect PGE2-induced constriction of Ang II-primed vessels (Fig. 6A, P>0.05), nor did 100 nM SC-236 (COX-1 IC50 = 17.8 μM, COX-2 IC50 = 10 nM) [33], a COX-2 specific inhibitor. However, 10 mM salicylate, an active metabolite of aspirin and poor COX enzyme inhibitor (COX-1 IC50 = 5 mM, COX-2 IC50 = 0.5–34 mM) [32], significantly attenuated PGE2-mediated vasoconstriction of Ang II-primed vessels (Fig. 6A, P<0.01). Pretreatment with 100 μM salicylamine, a structurally similar compound to salicylate and identified isoketal scavenger, did not inhibit PGE2-mediated vasoconstriction (Fig. 6A, P>0.05) [34]. Inhibition of contraction by salicylate but not ibuprofen or SC-236, suggests that the salicylate effect was a result of a non-COX mechanism.
Figure 6. Pyk2 is necessary for PGE2-mediated contraction of Ang II-primed vessels.

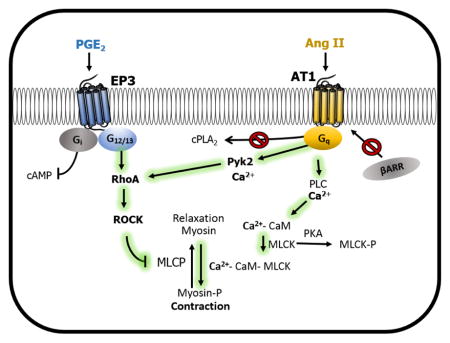
(A) PGE2-induced contraction of femoral arterial rings pretreated for 30 minutes with the indicated concentrations of ibuprofen, SC-236, salicylate, salicylamine, or control solvent for ibuprofen/SC-236 (EtOH) followed by priming with Ang II and subsequent addition of PGE2 (N = 7–10). Salicylate attenuated PGE2-induced constriction of vessels primed with Ang II (P<0.05 1way ANOVA). (B) Preincubation with the Pyk2 inhibitor PF-431396 blocked PGE2-induced contraction of Ang II-primed vessels, while inhibiting FAK with FAK inhibitor 14 did not affect contraction (N = 5). Vehicle represents control solvent for PF-431396 (N = 5). Vertical bars indicate SEM. (C) Schematic of key proteins involved in PGE2-induced contractile responses. Prior activation of the AT1a receptor by Ang II facilitates vasoconstriction via a PGE2 – EP3 receptor pathway. PGE2-induced vasoconstriction is dependent upon signaling through a Ca2+-Pyk2-ROCK cascade, as opposed to other defined pathways such as activation of phospholipase A2 or signaling via β-arrestin. EP3 and AT1 signaling converges through Pyk2-ROCK to sensitize smooth muscle cells to calcium and thus trigger a significant contractile response.
3.6 Effects of proline-rich tyrosine kinase 2 inhibitors
Salicylate has been shown to inhibit the non-receptor tyrosine kinase Pyk2 [35, 36]. To test whether salicylate exerted its effect by inhibition of Pyk2, the Pyk2 inhibitor PF-431396 was tested. PF-431396 abolished PGE2-induced contraction of Ang II-primed vessels (Fig 6B). Since Pyk2 is structurally similar to focal adhesion kinase (FAK), arteries were pre-treated with a FAK specific inhibitor, 10 μM FAK inhibitor 14, to test the contribution of FAK (Fig 6B). The FAK inhibitor did not affect Ang II priming of PGE2, indicating that Pyk2 underlies PGE2-mediated contraction. Collectively, these findings demonstrate that Ang II potentiates vasoconstriction induced by PGE2 via convergent signaling between the AT1 and EP3 receptors. These convergent events occur through a Ca2+-Pyk2-ROCK mechanism and are independent of β-arrestin signaling and AT1-mediated activation of PLA2 (Fig 6, C).
4. Discussion
In the present study, we demonstrate that PGE2 functions as a vasoconstrictor in femoral arteries pretreated with low-dose Ang II. In contrast, PGE2 has no detectable effect on vascular tone in the absence of priming by Ang II. This finding reveals a novel interaction between Ang II and PGE2 in regulating peripheral vascular reactivity. Although other constrictors may be able to potentiate EP3 receptor-induced contraction, indicated by published results with rat femoral arteries primed with thromboxane or phenylephrine [23], Ang II is of particular interest due to its central role in the development of cardiovascular disease. One contributing factor of high blood pressure is increased peripheral vascular resistance [37]. Increased formation of vasoconstrictors and altered effectiveness of vasodilators may underlie such impairments. PGE2 has been demonstrated to function primarily as a vasodilator and vasodepressor when intravenously infused into many species, including humans, dogs, rabbits, mice and rats; however, in certain physiologic settings PGE2 acts as a vasoconstrictor [12, 16, 17, 38–43]. Our studies indicate that in the presence of Ang II, PGE2 exhibits vasoconstrictor effects in mouse femoral arteries. Because Ang II and PGE2 are well known to regulate peripheral vasculature, contractile responses mediated by concurrent signaling by Ang II and PGE2 may contribute to vascular dysfunction associated with hypertension.
The Ang II concentration of 1 nM used for priming femoral arteries in our studies is greater than typical reported plasma concentrations of Ang II, which have been measured to be in the pM range [44, 45]. However, evidence indicates that tissue sources of Ang II may contribute more to vascular contractions than circulating Ang II, and interstitial levels of Ang II have been detected in the nM range in femoral arteries and kidneys [46, 47]. Therefore, our priming concentration of Ang II, which produced only a modest contractile response prior to PGE2 administration (Figure 1), may approximate levels observed by AT1 receptors in vivo and could thus be relevant to vascular responsiveness.
Studies presented here demonstrate that the EP3 receptor is required for PGE2-mediated vasoconstriction. We found that PGE2-induced vasoconstriction following Ang II priming was lost upon knock-out of the EP3 receptor. Previous studies indicate that EP3−/− mice have lower mean arterial pressure (MAP) in response to acute or chronic Ang II infusions [22]. The observed changes in vascular reactivity described in the present studies may contribute to the reduction in MAP in response to Ang II observed in EP3−/− mice [22]. Of note, femoral arteries from EP3−/− mice exhibited Ang II concentration response curves similar to wildtype femoral arteries, suggesting the alterations in vascular reactivity to PGE2 observed in EP3−/− femoral vessels are not due to a loss of Ang II responsiveness. In contrast, a previously reported study investigating the effect of EP3 receptor signaling on mouse mesenteric vasculature indicated that Ang II-mediated contraction was altered by knockout of the EP3 receptor [22]. Considering these findings, our data suggest differences in the interactions between Ang II and PGE2 signaling that are specific to particular vascular beds.
Our findings support a model, diagrammed in Figure 6C, in which activation of the AT1 receptor by Ang II sensitizes smooth muscles cells to calcium, thereby facilitating a contractile response to PGE2. This vasoconstriction does not require AT1-mediated production of endogenous prostaglandins or β-arrestin scaffolding. Importantly, our studies identified Pyk2 as a tyrosine kinase crucial for PGE2-induced contraction. This finding aligns well with previous investigations of Pyk2 function. Pyk2 is a calcium-dependent tyrosine kinase that acts upstream of the RhoA/ROCK pathway [35, 36]. Pyk2 can be activated by Ang II and plays a role in the ability of Ang II to mediate many cellular functions, including vasoconstriction [35, 48]. The EP3 receptor has been identified to mediate contractile responses through activation of ROCK [22, 23, 49]. Therefore, Ang II signaling may activate Pyk2, thereby sensitizing the ROCK pathway to EP3 signaling (depicted in Figure 6C); alternatively, EP3 and AT1 signaling may concurrently converge through a Pyk2-RhoA-ROCK pathway. Activation of ROCK would ultimately lead to inhibition of myosin phosphatase and enhance Ca2+ sensitivity of myosin filaments. The inhibition of Ang II priming of PGE2-mediated contraction observed with salicylate and the specific Pyk2 inhibitor (PF-431396) may also explain some of the beneficial cardiovascular effects of salicylate.
Although the present studies focused on peripheral vascular reactivity, the observed interaction between the EP3 and AT1 receptors may be important in other tissue types and physiologic events associated with hypertension. For example, studies using rats have indicated that intracerebroventricular infusion of PGE2 results in a rise in blood pressure and heart rate [50]. Interestingly, these responses were exacerbated in spontaneously hypertensive rats (SHRs), a hypertension model known to have elevated brain levels of Ang II [51]. These findings suggest a potential interaction of Ang II and PGE2 in the central nervous system that might affect blood pressure. Future studies investigating additional mechanisms through which the EP3 and AT1 receptors synergize and how this interaction relates to the pathology of hypertension may reveal novel therapeutic strategies.
Acknowledgments
This work was supported by a Merit Award from the Department of Veterans Affairs (BX000616 to R. Breyer), National Institutes of Health grants (DK37097 to R. Breyer; HL096967 and HL109199 to J. Reese), and by proceeds from the Dr. Aida Nureddin fund. M. Kraemer was supported by an American Heart Association Pre-doctoral Fellowship (13PRE14190017). There are no disclosures to report in relation to this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Coffman TM. Under pressure: the search for the essential mechanisms of hypertension. Nat Med. 2011;17(11):1402–9. doi: 10.1038/nm.2541. nm.2541 [pii] [DOI] [PubMed] [Google Scholar]
- 2.Calhoun DA, Jones D, Textor S, Goff DC, Murphy TP, Toto RD, et al. Resistant hypertension: diagnosis, evaluation, and treatment. A scientific statement from the American Heart Association Professional Education Committee of the Council for High Blood Pressure Research. Hypertension. 2008;51(6):1403–19. doi: 10.1161/HYPERTENSIONAHA.108.189141. HYPERTENSIONAHA.108.189141 [pii] [DOI] [PubMed] [Google Scholar]
- 3.Stegbauer J, Coffman TM. New insights into angiotensin receptor actions: from blood pressure to aging. Curr Opin Nephrol Hypertens. 2011;20(1):84–8. doi: 10.1097/MNH.0b013e3283414d40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Paul M, Poyan Mehr A, Kreutz R. Physiology of local renin-angiotensin systems. Physiol Rev. 2006;86(3):747–803. doi: 10.1152/physrev.00036.2005. 86/3/747 [pii] [DOI] [PubMed] [Google Scholar]
- 5.Weir MR, Dzau VJ. The renin-angiotensin-aldosterone system: a specific target for hypertension management. Am J Hypertens. 1999;12(12 Pt 3):205S–13S. doi: 10.1016/s0895-7061(99)00103-x. [DOI] [PubMed] [Google Scholar]
- 6.Inagami T. Molecular biology and signaling of angiotensin receptors: an overview. J Am Soc Nephrol. 1999;10(Suppl 11):S2–7. [PubMed] [Google Scholar]
- 7.Wynne BM, Chiao CW, Webb RC. Vascular Smooth Muscle Cell Signaling Mechanisms for Contraction to Angiotensin II and Endothelin-1. J Am Soc Hypertens. 2009;3(2):84–95. doi: 10.1016/j.jash.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nguyen Dinh Cat A, Touyz RM. Cell signaling of angiotensin II on vascular tone: novel mechanisms. Curr Hypertens Rep. 2011;13(2):122–8. doi: 10.1007/s11906-011-0187-x. [DOI] [PubMed] [Google Scholar]
- 9.Cat A, Touyz R. Cell signaling of angiotensin II on vascular tone: novel mechanism. 2011 doi: 10.1007/s11906-011-0187-x. [DOI] [PubMed] [Google Scholar]
- 10.Narumiya S, Sugimoto Y, Ushikubi F. Prostanoid receptors: structures, properties, and functions. Physiol Rev. 1999;79(4):1193–226. doi: 10.1152/physrev.1999.79.4.1193. [DOI] [PubMed] [Google Scholar]
- 11.Yuhki K, Kojima F, Kashiwagi H, Kawabe J, Fujino T, Narumiya S, et al. Roles of prostanoids in the pathogenesis of cardiovascular diseases: Novel insights from knockout mouse studies. Pharmacol Ther. 2011;129(2):195–205. doi: 10.1016/j.pharmthera.2010.09.004. S0163-7258(10)00188-9 [pii] [DOI] [PubMed] [Google Scholar]
- 12.Hockel GM, Cowley AW. Prostaglandin E2-induced hypertension in conscious dogs. Am J Physiol. 1979;237(4):H449–54. doi: 10.1152/ajpheart.1979.237.4.H449. [DOI] [PubMed] [Google Scholar]
- 13.Audoly LP, Tilley SL, Goulet J, Key M, Nguyen M, Stock JL, et al. Identification of specific EP receptors responsible for the hemodynamic effects of PGE2. Am J Physiol. 1999;277(3 Pt 2):H924–30. doi: 10.1152/ajpheart.1999.277.3.H924. [DOI] [PubMed] [Google Scholar]
- 14.Ariumi H, Takano Y, Masumi A, Takahashi S, Hirabara Y, Honda K, et al. Roles of the central prostaglandin EP3 receptors in cardiovascular regulation in rats. Neurosci Lett. 2002;324(1):61–4. doi: 10.1016/s0304-3940(02)00174-x. S030439400200174X [pii] [DOI] [PubMed] [Google Scholar]
- 15.Lawrence RA, Jones RL. Investigation of the prostaglandin E (EP-) receptor subtype mediating relaxation of the rabbit jugular vein. Br J Pharmacol. 1992;105(4):817–24. doi: 10.1111/j.1476-5381.1992.tb09063.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kennedy CR, Zhang Y, Brandon S, Guan Y, Coffee K, Funk CD, et al. Salt-sensitive hypertension and reduced fertility in mice lacking the prostaglandin EP2 receptor. Nat Med. 1999;5(2):217–20. doi: 10.1038/5583. [DOI] [PubMed] [Google Scholar]
- 17.Zhang Y, Guan Y, Schneider A, Brandon S, Breyer RM, Breyer MD. Characterization of murine vasopressor and vasodepressor prostaglandin E(2) receptors. Hypertension. 2000;35(5):1129–34. doi: 10.1161/01.hyp.35.5.1129. [DOI] [PubMed] [Google Scholar]
- 18.Hata AN, Breyer RM. Pharmacology and signaling of prostaglandin receptors: multiple roles in inflammation and immune modulation. Pharmacol Ther. 2004;103(2):147–66. doi: 10.1016/j.pharmthera.2004.06.003. S0163-7258(04)00096-8 [pii] [DOI] [PubMed] [Google Scholar]
- 19.Wright DH, Abran D, Bhattacharya M, Hou X, Bernier SG, Bouayad A, et al. Prostanoid receptors: ontogeny and implications in vascular physiology. Am J Physiol Regul Integr Comp Physiol. 2001;281(5):R1343–60. doi: 10.1152/ajpregu.2001.281.5.R1343. [DOI] [PubMed] [Google Scholar]
- 20.Guan Y, Zhang Y, Wu J, Qi Z, Yang G, Dou D, et al. Antihypertensive effects of selective prostaglandin E2 receptor subtype 1 targeting. J Clin Invest. 2007;117(9):2496–505. doi: 10.1172/JCI29838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bartlett CS, Boyd KL, Harris RC, Zent R, Breyer RM. EP1 disruption attenuates end-organ damage in a mouse model of hypertension. Hypertension. 2012;60(5):1184–91. doi: 10.1161/HYPERTENSIONAHA.112.199026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen L, Miao Y, Zhang Y, Dou D, Liu L, Tian X, et al. Inactivation of the E-prostanoid 3 receptor attenuates the angiotensin II pressor response via decreasing arterial contractility. Arterioscler Thromb Vasc Biol. 2012;32(12):3024–32. doi: 10.1161/ATVBAHA.112.254052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hung GH, Jones RL, Lam FF, Chan KM, Hidaka H, Suzuki M, et al. Investigation of the pronounced synergism between prostaglandin E2 and other constrictor agents on rat femoral artery. Prostaglandins Leukot Essent Fatty Acids. 2006;74(6):401–15. doi: 10.1016/j.plefa.2006.04.002. S0952-3278(06)00052-4 [pii] [DOI] [PubMed] [Google Scholar]
- 24.Ceddia RP, Lee D, Maulis MF, Carboneau BA, Threadgil DW, Poffenberger G, et al. The PGE2 EP3 receptor regulates diet-induced adiposity in male mice. Endrocrinology. 2015 doi: 10.1210/en.2015-1693. Epub accepted Oct. 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schneider A, Guan Y, Zhang Y, Magnuson MA, Pettepher C, Loftin CD, et al. Generation of a conditional allele of the mouse prostaglandin EP4 receptor. Genesis. 2004;40(1):7–14. doi: 10.1002/gene.20048. [DOI] [PubMed] [Google Scholar]
- 26.Gao Q, Zhan P, Alander CB, Kream BE, Hao C, Breyer MD, et al. Effects of global or targeted deletion of the EP4 receptor on the response of osteoblasts to prostaglandin in vitro and on bone histomorphometry in aged mice. Bone. 2009;45(1):98–103. doi: 10.1016/j.bone.2009.03.667. [DOI] [PubMed] [Google Scholar]
- 27.Nguyen M, Camenisch T, Snouwaert JN, Hicks E, Coffman TM, Anderson PA, et al. The prostaglandin receptor EP4 triggers remodelling of the cardiovascular system at birth. Nature. 1997;390(6655):78–81. doi: 10.1038/36342. [DOI] [PubMed] [Google Scholar]
- 28.Hata AN, Zent R, Breyer MD, Breyer RM. Expression and molecular pharmacology of the mouse CRTH2 receptor. J Pharmacol Exp Ther. 2003;306(2):463–70. doi: 10.1124/jpet.103.050955. [DOI] [PubMed] [Google Scholar]
- 29.Natarajan C, Hata AN, Hamm HE, Zent R, Breyer RM. Extracellular loop II modulates GTP sensitivity of the prostaglandin EP3 receptor. Mol Pharmacol. 2013;83(1):206–16. doi: 10.1124/mol.112.080473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holloway AC, Qian H, Pipolo L, Ziogas J, Miura S, Karnik S, et al. Side-chain substitutions within angiotensin II reveal different requirements for signaling, internalization, and phosphorylation of type 1A angiotensin receptors. Mol Pharmacol. 2002;61(4):768–77. doi: 10.1124/mol.61.4.768. [DOI] [PubMed] [Google Scholar]
- 31.Kendall RT, Strungs EG, Rachidi SM, Lee MH, El-Shewy HM, Luttrell DK, et al. The beta-arrestin pathway-selective type 1A angiotensin receptor (AT1A) agonist [Sar1,Ile4,Ile8]angiotensin II regulates a robust G protein-independent signaling network. J Biol Chem. 2011;286(22):19880–91. doi: 10.1074/jbc.M111.233080. M111.233080 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Warner TD, Giuliano F, Vojnovic I, Bukasa A, Mitchell JA, Vane JR. Nonsteroid drug selectivities for cyclo-oxygenase-1 rather than cyclo-oxygenase-2 are associated with human gastrointestinal toxicity: a full in vitro analysis. Proc Natl Acad Sci U S A. 1999;96(13):7563–8. doi: 10.1073/pnas.96.13.7563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Penning TD, Talley JJ, Bertenshaw SR, Carter JS, Collins PW, Docter S, et al. Synthesis and biological evaluation of the 1,5-diarylpyrazole class of cyclooxygenase-2 inhibitors: identification of 4-[5-(4-methylphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]benze nesulfonamide (SC-58635, celecoxib) J Med Chem. 1997;40(9):1347–65. doi: 10.1021/jm960803q. [DOI] [PubMed] [Google Scholar]
- 34.Zagol-Ikapitte IA, Matafonova E, Amarnath V, Bodine CL, Boutaud O, Tirona RG, et al. Determination of the Pharmacokinetics and Oral Bioavailability of Salicylamine, a Potent γ-Ketoaldehyde Scavenger, by LC/MS/MS. Pharmaceutics. 2010;2(1):18–29. doi: 10.3390/pharmaceutics2010018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ying Z, Giachini FR, Tostes RC, Webb RC. Salicylates dilate blood vessels through inhibiting PYK2-mediated RhoA/Rho-kinase activation. Cardiovasc Res. 2009;83(1):155–62. doi: 10.1093/cvr/cvp084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mills RD, Mita M, Nakagawa J, Shoji M, Sutherland C, Walsh MP. A role for the tyrosine kinase Pyk2 in depolarization-induced contraction of vascular smooth muscle. J Biol Chem. 2015;290(14):8677–92. doi: 10.1074/jbc.M114.633107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nyberg M, Gliemann L, Hellsten Y. Vascular function in health, hypertension, and diabetes: effect of physical activity on skeletal muscle microcirculation. Scand J Med Sci Sports. 2015;25(Suppl 4):60–73. doi: 10.1111/sms.12591. [DOI] [PubMed] [Google Scholar]
- 38.Lydford SJ, McKechnie KC, Dougall IG. Pharmacological studies on prostanoid receptors in the rabbit isolated saphenous vein: a comparison with the rabbit isolated ear artery. Br J Pharmacol. 1996;117(1):13–20. doi: 10.1111/j.1476-5381.1996.tb15148.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.BERGSTROM S, DUNER H, von EULER U, PERNOW B, SJOVALL J. Observations on the effects of infusion of prostaglandin E in man. Acta Physiol Scand. 1959;45:145–51. doi: 10.1111/j.1748-1716.1959.tb01686.x. [DOI] [PubMed] [Google Scholar]
- 40.Breyer RM, Kennedy CR, Zhang Y, Guan Y, Breyer MD. Targeted gene disruption of the prostaglandin E2 EP2 receptor. Adv Exp Med Biol. 2002;507:321–6. doi: 10.1007/978-1-4615-0193-0_49. [DOI] [PubMed] [Google Scholar]
- 41.Eklund B, Carlson LA. Central and peripheral circulatory effects and metabolic effects of different prostaglandins given I.V. to man. Prostaglandins. 1980;20(2):333–47. doi: 10.1016/s0090-6980(80)80051-7. [DOI] [PubMed] [Google Scholar]
- 42.Armstrong JM, Boura AL, Hamberg M, Samuelsson B. A comparison of the vasodepressor effects of the cyclic effects of the cyclic endoperoxides PGG, and PGH2 with those of PGD2 and PGE2 in hypertensive and normotensive rats. Eur J Pharmacol. 1976;39(2):251–8. doi: 10.1016/0014-2999(76)90133-3. [DOI] [PubMed] [Google Scholar]
- 43.Ishida K, Matsumoto T, Taguchi K, Kamata K, Kobayashi T. Protein kinase C delta contributes to increase in EP3 agonist-induced contraction in mesenteric arteries from type 2 diabetic Goto-Kakizaki rats. Pflugers Arch. 2012;463(4):593–602. doi: 10.1007/s00424-012-1088-9. [DOI] [PubMed] [Google Scholar]
- 44.Schulz A, Jankowski J, Zidek W, Jankowski V. Absolute quantification of endogenous angiotensin II levels in human plasma using ESI-LC-MS/MS. Clin Proteomics. 2014;11(1):37. doi: 10.1186/1559-0275-11-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Catt KJ, Cain MD, Zimmet PZ, Cran E. Blood angiotensin II levels of normal and hypertensive subjects. Br Med J. 1969;1(5647):819–21. doi: 10.1136/bmj.1.5647.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schuijt MP, de Vries R, Saxena PR, Schalekamp MA, Danser AH. Vasoconstriction is determined by interstitial rather than circulating angiotensin II. Br J Pharmacol. 2002;135(1):275–83. doi: 10.1038/sj.bjp.0704452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nishiyama A, Seth DM, Navar LG. Angiotensin II type 1 receptor-mediated augmentation of renal interstitial fluid angiotensin II in angiotensin II-induced hypertension. J Hypertens. 2003;21(10):1897–903. doi: 10.1097/01.hjh.0000084755.37215.c7. [DOI] [PubMed] [Google Scholar]
- 48.Sabri A, Govindarajan G, Griffin TM, Byron KL, Samarel AM, Lucchesi PA. Calcium- and protein kinase C-dependent activation of the tyrosine kinase PYK2 by angiotensin II in vascular smooth muscle. Circ Res. 1998;83(8):841–51. doi: 10.1161/01.res.83.8.841. [DOI] [PubMed] [Google Scholar]
- 49.Kobayashi K, Murata T, Hori M, Ozaki H. Prostaglandin E2-prostanoid EP3 signal induces vascular contraction via nPKC and ROCK activation in rat mesenteric artery. Eur J Pharmacol. 2011;660(2–3):375–80. doi: 10.1016/j.ejphar.2011.03.032. S0014-2999(11)00344-X [pii] [DOI] [PubMed] [Google Scholar]
- 50.Takahashi H, Buñag RD. Pressor responses to centrally-administered prostaglandin E2 in spontaneously hypertensive rats. Hypertension. 1981;3(4):426–32. doi: 10.1161/01.hyp.3.4.426. [DOI] [PubMed] [Google Scholar]
- 51.Campbell DJ, Duncan AM, Kladis A, Harrap SB. Angiotensin peptides in spontaneously hypertensive and normotensive Donryu rats. Hypertension. 1995;25(5):928–34. doi: 10.1161/01.hyp.25.5.928. [DOI] [PubMed] [Google Scholar]