

Abstract
Background
Genome-wide association studies have shown a pattern of rare copy number variations (CNVs) and single nucleotide polymorphisms (SNPs) in patients with common variable immunodeficiency disorder (CVID), which was recognizable by a Support Vector Machine (SVM) algorithm. However, rare monogenic causes of CVID may lack such a genetic fingerprint.
Objective
To identify a unique monogenic cause of familial immunodeficiency, and to evaluate the use of SVM to identify patients with possible monogenetic disorders.
Methods
A family with multiple members with a diagnosis of CVID was screened by whole-exome sequencing. The proband and other individuals with mutations associated with CVID-like phenotypes were screened via the SVM algorithm from our recent CVID genome-wide association study. RT-PCR, protein immunoblots, and in vitro plasmablast differentiation assays were performed on patient and control EBV lymphoblastoids cell lines.
Results
Exome sequencing identified a novel heterozygous mutation in IRF2BP2 (c.1652G>A:p.(S551N)) in affected family members. Transduction of the mutant gene into control human B-cells decreased production of plasmablasts in vitro, and IRF2BP2 transcripts and protein expression were increased in proband versus control EBV-lymphoblastoid cell lines. The SVM algorithm categorized the proband and subjects with other immunodeficiency-associated gene variants in TACI, BAFFR, ICOS, CD21, LRBA, and CD27 as genetically dissimilar from polygenic CVID.
Conclusion
A novel IRFBP2 mutation was identified in a family with autosomal dominant CVID. Transduction experiments suggest that the mutant protein has an impact on B-cell differentiation, and is likely a monogenetic cause of the family’s CVID phenotype. Successful grouping by the SVM algorithm suggests that our family and other subjects with rare immunodeficiency disorders cluster separately and lack the genetic pattern that is present in polygenic CVID cases.
Keywords: Common variable immunodeficiency, machine learning, primary antibody deficiency, IRF2BP2, immunoglobulin
Introduction
Common variable immunodeficiency disorder (CVID) is one of the most frequently diagnosed forms of primary immunodeficiency (PID) that requires clinical intervention.1–3 Defined clinically by low quantity of two immunoglobulin classes (including IgG) and poor specific antibody production, it has been thought of as an “umbrella diagnosis” due to the heterogeneity in onset and co-morbidities, including autoimmune disease and risk of malignancy.4 Roughly 5% of cases are familial, and the increasing availability of next generation sequencing technology has permitted identification of an increasing number of causative and associated mutations in individuals with CVID-like disease.5–16
A recent multi-institutional genome-wide array study of CVID showed unique associations with specific single nucleotide polymorphisms (SNP) and copy number variants (CNV), with intraexonic duplications in ORC4L being most highly associated with disease.17 The studied CVID cohort was found to have a unique pattern of SNPs and rare CNV, and a Support Vector Machine (SVM) algorithm was successfully used to identify this pattern in CVID patients versus controls. SVM is a learning algorithm used for non-linear classification and regression analysis.18 It can be trained with a variety of data, and produces a “hyperplane” for subsequent classification. In the original study, the CVID SVM hyperplane successfully classified cases with an accuracy of 91%, positive predictive value of 100%, and negative predictive value of 96%.
Though the SVM results support the polygenic nature of CVID, it was unclear whether patients with monogenic causes of CVID-like disease or risk alleles for CVID (TACI, BAFFR) would lack the genetic fingerprint of the more common polygenic disease. If this were the case, SVM classification of microarray data could be used to separate monogenic disorders from the more common polygenic form of CVID, and could therefore be a useful screening method for identifying cases in which next generation sequencing would be high yield, thereby guiding the proper workup for unknown CVID cases.
Methods & Patients
Human sample collection, DNA extraction, and EBV-lymphoblastoid cell line production
A family with 3 affected individuals with CVID and unaffected family members were enrolled in a research protocol approved by the host institutional review board (Children’s Hospital of Philadelphia). DNA samples from patients with monogenic immunodeficiency disorders with CVID-like phenotypes were provided by collaborating researchers. DNA was extracted from patient peripheral blood using a DNAeasy extraction kit (Qiagen, Hilden, Germany). EBV-immortalized lymphoblastoid cell lines (EBV-LCL) were generated from peripheral blood mononuclear cells (PBMC) of one individual carrying the IRF2BP2 mutation and two samples without the mutation as controls, using a previously published protocol.19
Genetic studies
Whole-exome sequencing using the Agilent SureSelect Human All Exon 50Mb kit was performed on the proband and her family. Variants were matched to disease segregation (which suggested a heterozygous, autosomal dominant pattern), and further narrowed by exclusion of synonymous mutations, in silico analysis of mutation impact, exclusion of variants with minor allele frequency greater than 0.5% in public databases (1000 Genomes, NHLBI 6500 exomes Project) and previously identified in controls by our in-house exome variant database, tissue expression pattern (BioGPS.org), and ties to known immunologic pathways.
High throughput SNP genotyping was performed with the Infinitium HumanHap610 Beadchip, and the PennCNV algorithm was used for CNV calls. A support vector machine algorithm was trained with data from 179 CVID cases and 1917 controls, utilizing the 658 most significantly associated variants from the 2011 study.17 Cytogenetic data from the proband and monogenic cases were subsequently analyzed with the trained algorithm.
RT-PCR and Immunoblotting
RNA was isolated from whole blood obtained from the patient and controls using Trizol (Applied Biosystems, Grand Island, NY) and RNEasy kits (Qiagen). cDNA was produced via high capacity Reverse Transcriptase kit (Applied Biosystems), and custom cDNA primers for IRF2BP2 (both total and isoform 2) and GAPDH were created. Custom primers are detailed in Supplemental S1. RT-PCR was performed using SYBR Green core reagents on a QPCR-7900HT system (Applied Biosystems). Gene dose was calculated via the ΔΔCT method.
For Western blot analysis of IRF2BP2 expression, EBV-LCL were lysed with Nonidet P-40 lysis buffer (Invitrogen). Proteins were separated on 4–12% NuPAGE Bis-Tris gels in MOPS SDS running buffer and transferred overnight onto nitrocellulose membranes (Invitrogen). The membrane was blocked in 3% BSA and cut into two halves. The top half was incubated with rabbit anti-IRF2BP2 polyclonal antibody (Abcam), and the bottom half was incubated with rabbit anti-TATA binding protein monoclonal antibody (Abcam). Subsequently, the membranes were washed, incubated with secondary Ab for 1 h, and washed again; bound Ab was detected with a WesternBright ECL chemiluminescence detection system (Advansta).
In vitro plasmablast differentiation
Plasmablasts were produced from frozen PBMC from the proband and healthy controls as previously described.20 Briefly, PBMCs were plated at 2.5 × 105 cells per well and treated with either CD40L (Axxora, Farmingdale, NY) at 500ng/ml, IL-21 (Peprotech, Rocky Hill, NJ) at 500ng/ml, or CpG ODN (Invivogen, San Diego, CA) at 2.5ug/ml. Cells were harvested after 7 days of incubation and analyzed via flow cytometry utilizing a FACSCalibur (BD Biosciences, San Jose, CA). Cells were stained with CD19-FITC, CD27-APC, and CD38-PE (BD Biosciences). Isotype controls were also utilized. Results were analyzed using FlowJo (Tree Star Inc, Ashland, OR). Plasmablasts were identified as CD19+ CD27+ CD38++ with lymphocyte gating (Supplemental S2).
Constructs
Plasmids containing wild type and mutant (p.S551N) IRF2BP2 with a C-terminal Myc-tag were purchased (Origene, Rockville, MD). Empty pCMV6 plasmid was also obtained for control purposes.
Isolation and transduction of Human B-cells
Primary human B-cells were obtained from sorted PBMC from healthy donors. Purity was confirmed by flow cytometry. Cells were transduced by nucleofection utilizing NuclecofectorR Program U-15 AmaxaR and Human B cell Nucleofector R kit (Lonza, Allendale, NJ). Transduction was confirmed via Western blotting for the Myc tag.
Results
Clinical History
The proband is a 24 year old woman who experienced recurrent sinopulmonary infections beginning in early childhood, which worsened in adolescence. She was evaluated immunologically and found to have gradually worsening hypogammaglobulinemia and poor vaccine responses between ages 17 to 19 years (Table 1) and was diagnosed with CVID. She has been maintained on subcutaneous immunoglobulin with good control of infections. She was diagnosed at age 23 with CVID colitis following onset of chronic diarrhea, and is being treated with Entocort.
Table 1. Humoral Immunophenotype for proband, sibling and father.
Immunoglobulin totals show consistent adolescent onset of CVID in the proband (P), brother (B), and father (F). Values for the father (F) prior to IVIG therapy were not available. Bolded values are abnormal, normal ranges are in parentheses. Values with an asterisk (*) denote levels while on immunoglobulin replacement.
| Patient | F | F | B | B | B | B | P | P | P | P |
|---|---|---|---|---|---|---|---|---|---|---|
| Age at analysis | 46 | 65 | 14 | 15 | 17 | 25 | 17 | 18 | 19 | 23 |
| IgG (mg/dl) | 559* (717–1411) | 1550* (717–1411) | 815 (717–1411) | 712 (600–1600) | 391 (600–1600) | 2030* (717–1411) | 926 (700–1600) | 687 (700–1600) | 620 (700–1600) | 1190* (635–1775) |
| IgG1 | 738 (315–855) | 582 (155–1020) | 313 (155–1020) | 767 (490–1140) | ||||||
| IgG2 | 17 (64–495) | 17 (44–495) | <12 (44–495) | 42 (150–640) | ||||||
| IgG3 | 42 (23–196) | 23 (8–209) | 19 (8–209) | 15 (11–85) | ||||||
| IgA (mg/dl) | <13 (78–391) | <7 (78–391) | <7 (78–391) | <5.9 (70–315) | <5.9 (70–315) | <7 (717–1411) | <7 (70–400) | <7 (70–400) | <6 (70–486) | |
| IgM (mg/dl) | 27 (53–334) | 27 (53–334) | 35 (53–334) | 22.4 (40–250) | 6.8 (40–250) | 12 (53–334) | <10 (40–230) | <10 (40–230) | <4 (71–237) |
Her family history is notable for CVID in both her father as well as her older brother (Figure 1). The proband’s older brother was diagnosed with CVID at age 16 after experiencing chronic sinus infections, and has been treated with IVIG therapy. He also has a history of type I diabetes. The proband’s father was similarly diagnosed with CVID at age 16 in the setting of recurrent sinus infections which have been well controlled on IVIG therapy. He also has a history of psoriasis. The paternal grandparents, aunts, and their families are unaffected, as is the maternal family.
Figure 1.
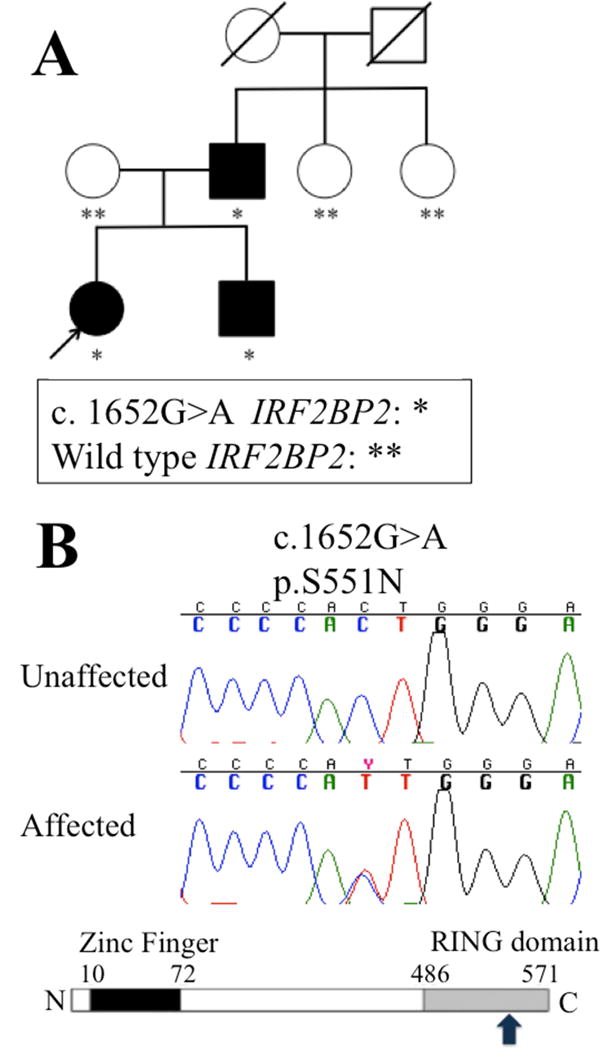
(A) IRF2BP2 c. 1652G>A segregated only in family members diagnosed with CVID. (B) The resulting p.S551N mutation occurs in the RING domain of IRF2BP2 (denoted by the arrow).
Immunologic phenotype of the proband and family
Evaluation of the proband at age 18 years showed hypogammaglobulinemia with undetectable IgM and IgA (Table 1) and poor specific antibody production following vaccination to S. pneumoniae (1 of 14 serotypes >1.3mcg/ml) and N. meningitidis (0 of 4 strains protective). Of note, her father and brother also had absent IgA and low IgM, and her brother had undetectable IgG2. Lymphocyte flow cytometry showed normal total B-cells in all subjects, but was notable for a relative decrease in switched memory B-cells in the proband and her father (Table 2).
Table 2. Cellular Immunophenotype for proband (P), sibling (S) and father (F).
Assessments of T, B and NK cells in each patient across multiple timepoints. Bolded values are abnormal, normal ranges are in parentheses.
| Patient | F | F | F | B | B | B | P | P |
|---|---|---|---|---|---|---|---|---|
| Age at analysis (years) | 45 | 49 | 62 | 17 | 22 | 25 | 18 | 23 |
| Leukocytes (thousands of cells/ul) | 8.32 (4–11) | 7.93 (5.8–9.3) | ||||||
| CD3+ T cells (cells/ul) | 1364 (794–2508) | 1282 (794–2503) | 1336 (617–2254) | 2560 (958–2388) | 1642 (677–2383) | 2318 (677–2383) | 1118 (1000–2600) | 1359 (700–2100) |
| CD4+ T cells (cells/ul) | 1048 (501–1608) | 980 (490–1546) | 1118 (430–1513) | 1544 (533–1674) | 1000 (424–1509) | 1339 (424–1509) | 590 (530–1500) | 620 (300–1400) |
| CD8+ T cells (cells/ul) | 266 (256–900) | 287 (265–1509) | 197 (101–839) | 945 (284–958) | 598 (169–955) | 889 (169–955) | 428 (330–1100) | 569 (200–900) |
| CD4:CD8 ratio | 3.94 (1.2–3.65) | 3.4 (0.5–3.3) | 5.67 (>1) | 1.63 (1.10–3.25) | 1.67 (>1) | 1.51 (>1) | 1.38 (0.9–3.7) | 1.1(1–3.6) |
| CD19+ B cells (cells/ul) | 116 (98–434) | 33 (60–597) | 90 (31–409) | 558 (75–660) | 476 (99–527) | 502.1 (90–539) | 388 (110–570) | 215 (100–500) |
| Percent Switched memory B cells (CD27+IgM-/CD27- % of CD19 cells for F, B and CD19+ CD27+IgD- for P) | 0% (2.3–26.5%) | 2.1% (2.3–26.5) | 0.4% (no nl range quoted) | 0.6%, (4.6–33%) (CD27+Ig M-CD19+ 0.3% (3.9–30.7%)) | ||||
| CD16/CD56+ NK cells (cells/ul) | 200 (113–461) | 171 (110–857) | 92 (102–565) | 122 (101–678) | 163 (101–678) | 44 (90–600) |
As decreased B-cell maturation in vitro has been described in many monogenic immunodeficiency disorders,20 we sought to test B-cell maturation in the proband. Stimulation of the patient’s PBMC with IL-21, CD40L, and CPG for 7 days showed decreased formation of B-cell plasmablasts (CD19+CD27+CD38high) in comparison with control PBMC in response to all stimulants (Figure 2).
Figure 2.
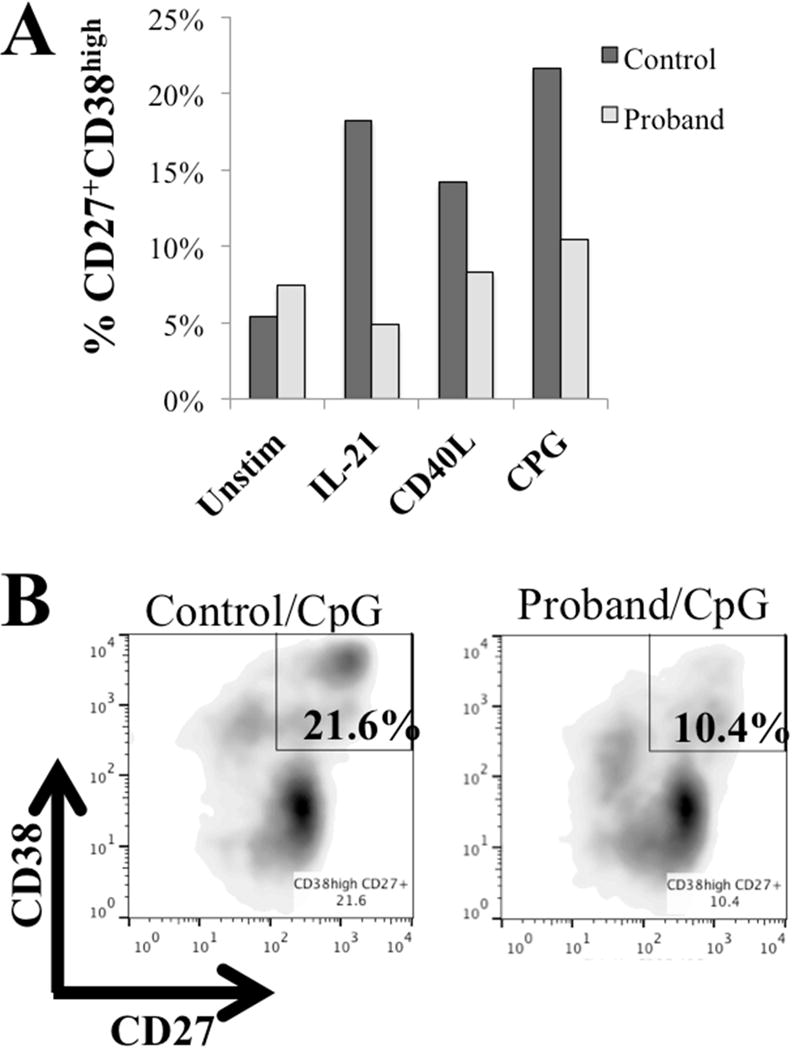
(A) Development of B-cell plasmablasts was decreased in the proband versus healthy control 7 days after stimulation with IL21, CD40L, or CPG. (B) Plasmablasts (CD19+CD27+CD38++) at day 7 were decreased in the proband at day 7 versus a healthy control.
IRB2BP2 mutation co-segregates with disease in the proband and family
Whole exome sequencing of the proband yielded 12 non-synonymous variants that matched an autosomal dominant pattern of disease. Of these, 5 were predicted to be damaging via in silico analysis utilizing SIFT and Polyphen2 (Supplemental S3). Three of the variant genes (AMBP, IRF2BP2, and PIK3C2G) have been tied to immunologic pathways (www.genecards.org). The AMBP and PIK3C2G variants were both previously repeated described in existing public databases, and were deemed unlikely to be causative. Sanger sequencing confirmed a heterozygous c. 1652G>A:p.(S551N) mutation in IRF2BP2 (Mendelian Inheritance in Man (MIM) 615332; NM_182972.2), which was confirmed to match the extended familial pattern of disease (Figure 1A, B). The serine residue at this position is highly conserved across species (Supplemental S4),21 and the p.S551N variant has not been described in public genomic databases (1000 Genomes, NHLBI 6500 exomes Project, EXAC v0.3) nor the Baylor Center for Mendelian Genomics Database of over 6,000 exomes.22
The biological disease relevance of the autosomal dominant inheritance pattern for this variant is supported by the fact that the IRF2BP2 gene is damage-intolerant. No viable IRF2BP2−/− mice are known to exist; conditional knockout of the gene is proposed to be necessary for studying its function.23 Only 3 loss of function variants are reported in the ExAC database: 2 are heterozygous splice donor changes of unknown significance, and 1 is a heterozygous frameshift base insertion at amino acid position 586 (downstream from p.S551N). In the Baylor Center for Mendelian Genomics Database, no frameshift, stopgain, or splice site variants have been observed in IRF2BP2. In addition, no individuals have been identified in the database who carry homozygous or compound heterozygous missense variants in the gene.
IRF2BP2 transcripts and protein level are increased in the proband
Quantitative PCR of IRF2BP2 from proband and control EBV lymphoblastoid cell lines showed a higher expression level in the proband versus normal control (Figure 3A). Comparative evaluation of transcript levels from isoform 1 versus isoform 2 (which differ in the length of exon 1) showed a uniform increase in the IRF2BP2 expression level in the proband.
Figure 3.
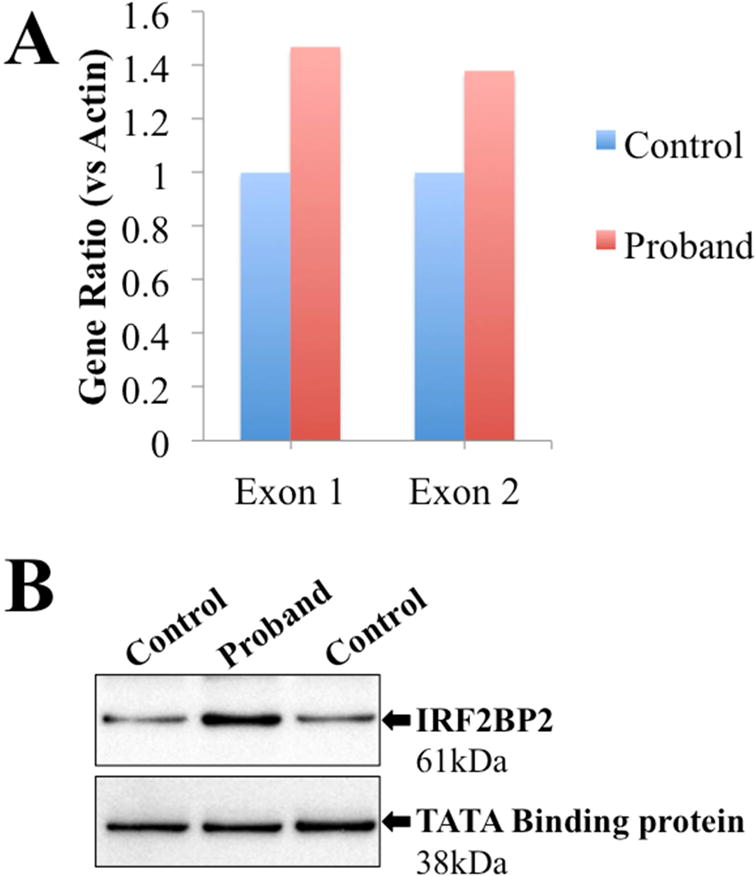
(A) RT-PCR of IRF2BP2 showed increased transcripts in the proband versus healthy control (versus actin) with agreement between primers specific for exon 1 (present in isoform 1 of IRF2BP2) and exon 2 (present in both isoforms 1 and 2). (B) Western blot showed increased protein expression of IRF2BP2 in lysates of EBV-LCL from the proband versus control. TATA Binding protein is shown as loading control.
Protein immunoblotting for IRF2BP2 in control and proband-derived EBV-LCL showed a band at the predicted size (61kD) in both samples, with a more prominent band in the patient versus the control (Figure 3B).
Transfection of mutant IRF2BP2 impacts differentiation of B-cell plasmablasts
Given the decrease in B-cell plasmablast formation in vitro, we sought to determine whether mutant IRF2BP2 impacted B-cell maturation. Nucleofection of healthy control B-cells with the wild type IRF2BP2 led to an increase in development of B-cell plasmablasts after 7 days particularly with stimulation with CPG (Figure 4A). However, nucleofection with IRF2BP2 p.S551N led to decreased plasmablast development, which was similar to that seen with nucleofection of empty vector. This suggests that the mutation impairs but does not ablate plasmablast differentiation.
Figure 4.
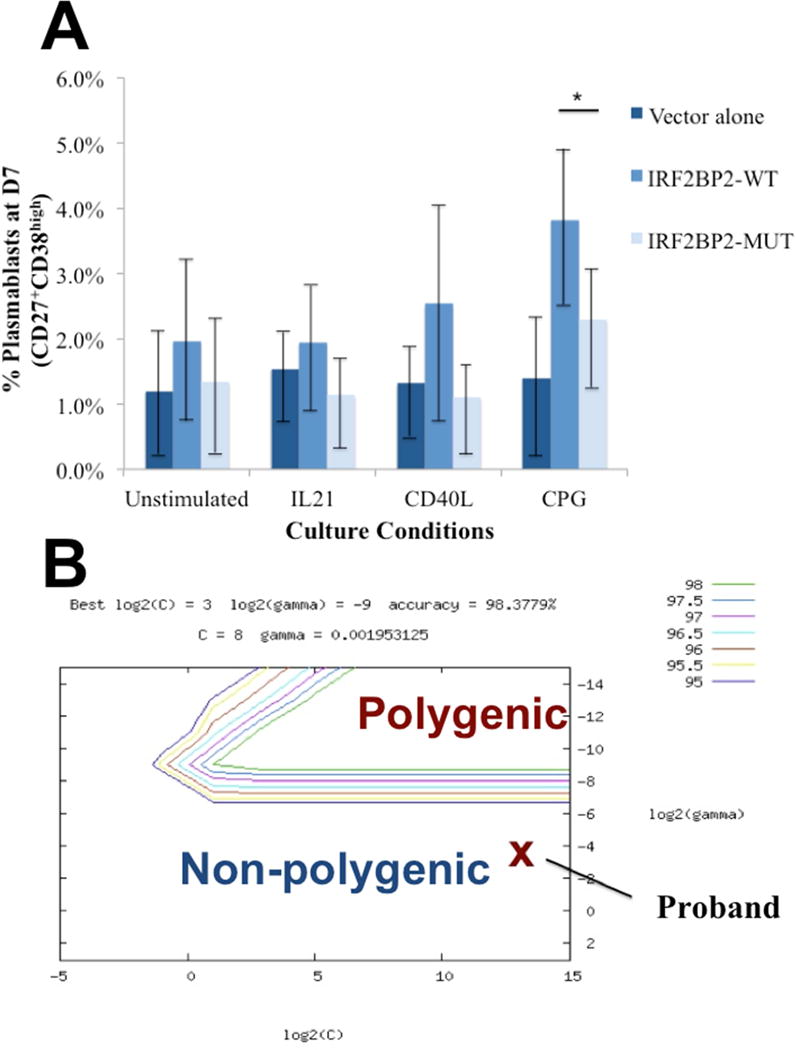
(A) Nucleofection of human B-cells with mutant IRF2BP2 construct (p.S551N) leads to reduced plasmablast formation 7 days after CPG stimulation versus wild type IRF2BP2 nucleofection, which reached statistical significance (p<0.02, denoted by *). Data shown reflects mean percentage of plasmablast formation after 7 days of culture, and represents five duplicate runs. (B) On the SVM hyperplane, the proband was predicted to fall in the non-polygenic region.
Proband and other monogenic CVID-like subjects cluster separately from the polygenic form of CVID by SVM classification
With the finding of a novel variant that segregates with affected individuals in this family, we next sought to determine if their cytogenetic pattern differed from polygenic CVID using the pretrained CVID SVM algorithm. Analysis of the top 658 SNP/CNVs by the CVID SVM algorithm predicted that the proband in family 1 fell outside the region of polygenic CVID cases on the hyperplane (Figure 4B). Similar analysis was performed for 6 subjects with several forms of monogenic immunodeficiency resulting in CVID-like disease (LRBA, IL21, BAFFR, ICOS, and CD27 deficiency) as well as 3 subjects with CVID and mutations in TACI, and all were similarly predicted to be genetically unique from polygenic CVID subjects (Table 3).
Table 3. Support Vector Algorithm predictions for monogenic CVID-like immunodeficiency disorders.
Of 9 additional subjects evaluated, all were uniformly grouped as different from polygenic CVID by the SVM algorithm.
| Subject # | Immune disorder | Mutation | CVID algorithm prediction |
|---|---|---|---|
| 1 | CD27 deficiency | p.Trp8X | Non-polygenic |
| 2 | LRBA | p.Arg1683X | Non-polygenic |
| 3 | LRBA | p.Iso2657Ser | Non-polygenic |
| 4 | BAFFR | c.189-96del | Non-polygenic |
| 5 | ICOS | c.126-568 del | Non-polygenic |
| 6 | CD21 | c.1225+1G>C | Non-polygenic |
| 7 | TACI mutation | p.Cys104Arg (heterozygous) | Non-polygenic |
| 8 | TACI mutation | p.Cys104Arg (heterozygous) | Non-polygenic |
| 9 | TACI mutation | p.Cys104Arg (heterozygous) | Non-polygenic |
Discussion
Here we describe a novel variant in IRF2BP2, which was present in 3 family members with an autosomal dominant pattern of CVID. The impact of the IRF2BP2 p.S551N mutation on differentiation of plasmablasts in vitro suggests that the mutation impacts B-cell maturation in a dominant fashion, which suggests a monogenic cause of CVID-like immunodeficiency. We postulate that this is likely a rare variant within the CVID population, as linkage studies in other cases of familial CVID have not shown associations with the 1q42 region (B. Grimbacher, unpublished data).
IRF2BP2 encodes an 87kD protein, which has been described to act as a transcriptional co-pressor of the NFAT family, and increased expression has been shown to downregulate production of cytokines including IL-2 and IL-4.24 It contains an N-terminal zinc finger, and a C-terminal RING domain, both of which are essential for repression activity. Position 551 is in the RING domain, which has been shown to be the region of interaction with NFAT. Mutations in IRF2BP2 have been described in the setting of malignancies including CNS lymphoma and chondrosarcoma.25,26 The mechanistic biology of the IRF2BP2 p.S551N mutation is unclear, though its impact on plasmablast formation particularly following CPG stimulation suggests that IRF2BP2 is downstream of TLR9. The low percentage of switched memory B-cells in the proband and father suggests that IRF2BP2 may have a role in the development or survival of memory B-cells. Our studies have confirmed that the introduction of the mutant protein in primary B-cells from healthy individuals interrupts the normal biology of this cell type (Figure 4A). Further studies will be essential to determine the exact mechanism and how it might be affecting the normal IRF2BP2 interactome.
Learning algorithms such as SVM have been used in multiple facets of medical research, and have been increasingly used in genetics to predict disease associations with genomic findings.27–29 SVM may also be a valuable method of screening for monogenic diseases within polygenic populations by searching for differences in the genetic pattern of SNP and rare CNV. We previously described the use of this approach in the CVID population.17 In familial clusters with a high penetrance of disease, SVM may be particularly useful for screening for monogenic disorders. In this study, the proband and 6 additional subjects with monogenic immunodeficiencies were found to have a dissimilar pattern of CNV and SNPs from polygenic CVID by the SVM algorithm. Interestingly, 3 individuals with C104R mutations in TACI, which imparts a high risk of development of CVID, were also grouped with the individuals with monogenic disorders. Though these individuals were not screened for other monogenic immunodeficiency disorders, the fact that all of them were predicted to be genetically dissimilar from the majority of CVID patients suggests that the presence of this particular TACI mutation contributes strongly to a unique and limited genetic load (distinct from polygenic CVID) in the individuals with these risk alleles. Though the SVM algorithm is unable to separate all cases with high accuracy, testing of the algorithm with the original discovery cohort suggested a high negative predictive value (96%), suggesting that the ten predictions are unlikely to be spurious. Further testing will determine the utility of this method in separating monogenic disorders from polygenic CVID. The same approach may also be useful for screening for monogenic disorders within populations with early-onset autoimmune disease such as type I diabetes or inflammatory bowel disease.
In conclusion, we have described a novel mutation in IRF2BP2, which appears to impact B-cell maturation, and may represent an autosomal dominant cause of CVID-like immunodeficiency. Additional study of IRF2BP2 may yield further understanding of its importance in B-cell development.
Supplementary Material
Key Messages.
A mutation in IRF2BP2 was identified as a novel cause of autosomal dominant CVID.
Machine learning algorithms may be useful in separating patients with monogenic immunodeficiency from those with CVID based on pattern of genetic polymorphisms and copy number variants.
Acknowledgments
We would like to thank the patient and her family for their participation, Dr. Bodo Grimbacher for his generous sample contributions and critical manuscript review, the staff of the Center for Applied Genomics, the CHOP Division of Allergy & Immunology, and Drs. Luigi Notarangelo, Mike Recher, Kiki Tesselaar, and James Lupski for their support of this study.
Funding: This study was supported by an Institute Development Fund from CHOP, U01HG006830, a donation to CAG from the Kubert Estate Foundation, and by the Jeffrey Modell Foundation.
Abbrevations
- CVID
Common variable immunodeficiency disorder
- CNV
copy number variation
- SNP
single nucleotide polymorphism
- EBV-LCL
Epstein-Barr virus immortalized lymphoblastoid cell line
- PBMC
peripheral blood mononuclear cells
- SVM
Support vector machine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
COI: The authors have no relevant conflicts of interest to disclose.
References
- 1.Lucas M, Lee M, Lortan J, Lopez-Granados E, Misbah S, Chapel H. Infection outcomes in patients with common variable immunodeficiency disorders: relationship to immunoglobulin therapy over 22 years. J Allergy Clin Immunol. 2010;125(6):1354–60. e4. doi: 10.1016/j.jaci.2010.02.040. [DOI] [PubMed] [Google Scholar]
- 2.Gathmann B, Mahlaoui N, Ceredih, et al. Clinical picture and treatment of 2212 patients with common variable immunodeficiency. J Allergy Clin Immunol. 2014;134(1):116–26. doi: 10.1016/j.jaci.2013.12.1077. [DOI] [PubMed] [Google Scholar]
- 3.Yong PF, Thaventhiran JE, Grimbacher B. “A rose is a rose is a rose,” but CVID is Not CVID common variable immune deficiency (CVID), what do we know in 2011? Adv Immunol. 2011;111:47–107. doi: 10.1016/B978-0-12-385991-4.00002-7. [DOI] [PubMed] [Google Scholar]
- 4.Resnick ES, Moshier EL, Godbold JH, Cunningham-Rundles C. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood. 2012;119(7):1650–7. doi: 10.1182/blood-2011-09-377945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Zelm MC, Smet J, Adams B, et al. CD81 gene defect in humans disrupts CD19 complex formation and leads to antibody deficiency. J Clin Invest. 2010;120(4):1265–74. doi: 10.1172/JCI39748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van Zelm MC, Reisli I, van der Burg M, et al. An antibody-deficiency syndrome due to mutations in the CD19 gene. N Engl J Med. 2006;354(18):1901–12. doi: 10.1056/NEJMoa051568. [DOI] [PubMed] [Google Scholar]
- 7.Romberg N, Chamberlain N, Saadoun D, et al. CVID-associated TACI mutations affect autoreactive B cell selection and activation. J Clin Invest. 2013;123(10):4283–93. doi: 10.1172/JCI69854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Losi CG, Silini A, Fiorini C, et al. Mutational analysis of human BAFF receptor TNFRSF13C (BAFF-R) in patients with common variable immunodeficiency. J Clin Immunol. 2005;25(5):496–502. doi: 10.1007/s10875-005-5637-2. [DOI] [PubMed] [Google Scholar]
- 9.Grimbacher B, Hutloff A, Schlesier M, et al. Homozygous loss of ICOS is associated with adult-onset common variable immunodeficiency. Nat Immunol. 2003;4(3):261–8. doi: 10.1038/ni902. [DOI] [PubMed] [Google Scholar]
- 10.van Montfrans JM, Hoepelman AI, Otto S, et al. CD27 deficiency is associated with combined immunodeficiency and persistent symptomatic EBV viremia. J Allergy Clin Immunol. 2012;129(3):787–93. e6. doi: 10.1016/j.jaci.2011.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuijpers TW, Bende RJ, Baars PA, et al. CD20 deficiency in humans results in impaired T cell-independent antibody responses. J Clin Invest. 2010;120(1):214–22. doi: 10.1172/JCI40231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sekine H, Ferreira RC, Pan-Hammarstrom Q, et al. Role for Msh5 in the regulation of Ig class switch recombination. Proc Natl Acad Sci U S A. 2007;104(17):7193–8. doi: 10.1073/pnas.0700815104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lopez-Herrera G, Tampella G, Pan-Hammarstrom Q, et al. Deleterious mutations in LRBA are associated with a syndrome of immune deficiency and autoimmunity. Am J Hum Genet. 2012;90(6):986–1001. doi: 10.1016/j.ajhg.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee CE, Fulcher DA, Whittle B, et al. Autosomal-dominant B-cell deficiency with alopecia due to a mutation in NFKB2 that results in nonprocessable p100. Blood. 2014;124(19):2964–72. doi: 10.1182/blood-2014-06-578542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li J, Jorgensen SF, Maggadottir SM, et al. Association of CLEC16A with human common variable immunodeficiency disorder and role in murine B cells. Nature communications. 2015;6:6804. doi: 10.1038/ncomms7804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maggadottir SM, Li J, Glessner JT, et al. Rare variants at 16p11.2 are associated with common variable immunodeficiency. J Allergy Clin Immunol. 2015;135(6):1569–77. doi: 10.1016/j.jaci.2014.12.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Orange JS, Glessner JT, Resnick E, et al. Genome-wide association identifies diverse causes of common variable immunodeficiency. J Allergy Clin Immunol. 2011;127(6):1360–7. e6. doi: 10.1016/j.jaci.2011.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee Y. Support vector machines for classification: a statistical portrait. Methods in molecular biology. 2010;620:347–68. doi: 10.1007/978-1-60761-580-4_11. [DOI] [PubMed] [Google Scholar]
- 19.Tosato G, Cohen JI. Generation of Epstein-Barr Virus (EBV)-immortalized B cell lines. In: Coligan John E, et al., editors. Current protocols in immunology. 2007. Chapter 7: Unit 722. [DOI] [PubMed] [Google Scholar]
- 20.Recher M, Berglund LJ, Avery DT, et al. IL-21 is the primary common gamma chain-binding cytokine required for human B-cell differentiation in vivo. Blood. 2011;118(26):6824–35. doi: 10.1182/blood-2011-06-362533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kent WJ, Sugnet CW, Furey TS, et al. The human genome browser at UCSC. Genome Res. 2002;12(6):996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lupski JR, Gonzaga-Jauregui C, Yang Y, et al. Exome sequencing resolves apparent incidental findings and reveals further complexity of SH3TC2 variant alleles causing Charcot-Marie-Tooth neuropathy. Genome medicine. 2013;5(6):57. doi: 10.1186/gm461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Teng AC, Kuraitis D, Deeke SA, et al. IRF2BP2 is a skeletal and cardiac muscle-enriched ischemia-inducible activator of VEGFA expression. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2010;24(12):4825–34. doi: 10.1096/fj.10-167049. [DOI] [PubMed] [Google Scholar]
- 24.Carneiro FR, Ramalho-Oliveira R, Mognol GP, Viola JP. Interferon regulatory factor 2 binding protein 2 is a new NFAT1 partner and represses its transcriptional activity. Molecular and cellular biology. 2011;31(14):2889–901. doi: 10.1128/MCB.00974-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bruno A, Boisselier B, Labreche K, et al. Mutational analysis of primary central nervous system lymphoma. Oncotarget. 2014;5(13):5065–75. doi: 10.18632/oncotarget.2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Panagopoulos I, Gorunova L, Bjerkehagen B, Boye K, Heim S. Chromosome aberrations and HEY1-NCOA2 fusion gene in a mesenchymal chondrosarcoma. Oncology reports. 2014;32(1):40–4. doi: 10.3892/or.2014.3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li L, Gao Q, Mao X, Cao Y. New support vector machine-based method for microRNA target prediction. Genetics and molecular research: GMR. 2014;13(2):4165–76. doi: 10.4238/2014.June.9.3. [DOI] [PubMed] [Google Scholar]
- 28.Jiang Q, Wang G, Jin S, Li Y, Wang Y. Predicting human microRNA-disease associations based on support vector machine. International journal of data mining and bioinformatics. 2013;8(3):282–93. doi: 10.1504/ijdmb.2013.056078. [DOI] [PubMed] [Google Scholar]
- 29.Orru G, Pettersson-Yeo W, Marquand AF, Sartori G, Mechelli A. Using Support Vector Machine to identify imaging biomarkers of neurological and psychiatric disease: a critical review. Neurosci Biobehav Rev. 2012;36(4):1140–52. doi: 10.1016/j.neubiorev.2012.01.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.