

Abstract
The aim of this review is to enlighten the critical roles that the liver plays in cholesterol metabolism. Liver transplantation can serve as gene therapy or a source of gene transmission in certain conditions that affect cholesterol metabolism, such as low-density-lipoprotein (LDL) receptor gene mutations that are associated with familial hypercholesterolemia. On the other hand, cholestatic liver disease often alters cholesterol metabolism. Cholestasis can lead to formation of lipoprotein X (Lp-X), which is frequently mistaken for LDL on routine clinical tests. In contrast to LDL, Lp-X is non-atherogenic, and failure to differentiate between the two can interfere with cardiovascular risk assessment, potentially leading to prescription of futile lipid-lowering therapy. Statins do not effectively lower Lp-X levels, and cholestasis may lead to accumulation of toxic levels of statins. Moreover, severe cholestasis results in poor micellar formation, which reduces cholesterol absorption, potentially impairing the cholesterol-lowering effect of ezetimibe. Apolipoprotein B-100 measurement can help distinguish between atherogenic and non-atherogenic hypercholesterolemia. Furthermore, routine serum cholesterol measurements alone cannot reflect cholesterol absorption and synthesis. Measurements of serum non-cholesterol sterol biomarkers - such as cholesterol precursor sterols, plant sterols, and cholestanol - may help with the comprehensive assessment of cholesterol metabolism. An adequate cholesterol supply is essential for liver-regenerative capacity. Low preoperative and perioperative serum cholesterol levels seem to predict mortality in liver cirrhosis and after liver transplantation. Thus, accurate lipid profile evaluation is highly important in liver disease and after liver transplantation.
Keywords: Cholesterol metabolism, Cholestasis, Liver transplantation, Non-cholesterol sterols, Cholestanol, Donor, Low density lipoprotein receptor mutation, Apolipoprotein B-100, Lipoprotein-X
Core tip: The liver plays key roles in cholesterol metabolism. Cholestatic liver disease leads to alterations of cholesterol metabolism: Cholesterol homeostasis is disturbed and cholesterol synthesis and especially cholesterol absorption are reduced, and lipoprotein X may develop. The latter can interfere with cardiovascular risk assessment. Apolipoprotein B-100 measurement may be useful in such cases. Cholesterol metabolism in cholestasis could be better described using cholesterol precursor sterols, diet-derived plant sterols, and cholestanol (the liver-synthesized derivate of cholesterol). Accurate lipid profile evaluation is particularly important after liver transplantation, when both atherogenic and non-atherogenic hypercholesterolemia may co-exist.
INTRODUCTION
Abundant in the bloodstream and in cell membranes, cholesterol is a critical component of vertebrate cell-membrane structure and function, allowing cells to maintain the permeability and fluidity that is fundamental for all animal life[1,2]. Cholesterol biosynthesis defects, such as Smith-Lemli-Opitz syndrome and lathosterolosis, reveal cholesterol’s importance in normal embryonic development. Lathosterolosis is a defect of postsqualene cholesterol biosynthesis that results in deficient transformation of lathosterol into 7-dehydrocholesterol by sterol-C5-desaturase/dehydrogenase. This disorder is characterized by high serum levels of the cholesterol precursor lathosterol, and low cholesterol levels in cells, plasma, and tissues-which causes multiple congenital anomalies, including microcephaly and progressive cholestasis leading to liver failure[3].
The tightly inter-regulated whole-body cholesterol homeostasis includes the following main components: Intestinal cholesterol absorption, hepatic de-novo cholesterol synthesis, and cholesterol excretion from the body. Recent advances in the field have further clarified the mechanisms of intestinal transporters and regulatory pathways[4-11]. The brain is home to about 23% of total body cholesterol, which is mainly synthesized in situ following blood-brain barrier establishment since dietary cholesterol does not cross this boundary[3]. In contrast to other species, humans exhibit a high cholesterol synthesis rate in the brain only after birth[3].
Under normal circumstances, the liver is the primary site of cholesterol biosynthesis and storage[12]. The liver is also the principal site of cholesterol excretion, converting cholesterol to bile acids and removing free cholesterol as neutral sterols via biliary excretion[4,5,13,14]. Since the liver plays a central role in cholesterol metabolism, liver disease can impact cholesterol metabolism, depending on the type of liver injury (parenchymal, cholestatic, or mixed)[15]. In one case of lathosterolosis, liver transplantation (LT) removed the liver disease, reversing the cholesterol metabolism defect and somewhat improving the postnatal neurological symptoms[3]. Conversely, various changes in cholesterol metabolism can be indicators of hepatic and biliary dysfunction.
In the present review, we aimed to summarize current concepts regarding the regulation of cholesterol metabolism in health and in cholestatic liver disease. We discuss difficulties in assessing cholesterol metabolism, and summarize the cholesterol metabolism disturbances seen in cholestatic liver disease and before and after LT. Cholesterol metabolism in the setting of non-alcoholic steatohepatitis was recently reviewed[16], and is not discussed here.
OVERVIEW OF CHOLESTEROL METABOLISM
The following are the main components involved in liver-related cholesterol metabolism and the control of plasma cholesterol levels: (1) intestinal absorption of dietary and biliary cholesterol; (2) bile acid synthesis; (3) endogenous cholesterol synthesis; (4) biliary excretion of cholesterol; (5) low-density lipoprotein (LDL) receptor activity; (6) very-low-density lipoprotein (VLDL) particle synthesis and transport into circulation; and (7) reverse cholesterol transport from peripheral tissues for biliary or non-biliary excretion [trans-intestinal cholesterol efflux (TICE)], the latter of which has been demonstrated only in animal models[4-11,13,17-19].
ABSORPTION OF DIETARY AND BILIARY CHOLESTEROL IN THE SMALL INTESTINE
Intestine-driven pathways are an important component of cholesterol homeostasis, through which cholesterol is both taken up from and pumped back to the intestinal lumen. Intestinal cholesterol absorption is a selective multistep process that is regulated by multiple sterol-transporter genes at the enterocyte level[17,18]. Uptake of free cholesterol from mixed micelles in the intestinal lumen to enterocytes occurs via the specific transporter protein Niemann-Pick C1 Like 1 (NPC1L1), which is highly expressed in the brush-border membrane of small-intestinal enterocytes[10,11,18,19] (Figure 1). These enterocytes then selectively efflux about half of the free cholesterol and about 90% of plant sterols back to the intestinal lumen via the adenosine triphosphate (ATP)-binding cassette (ABC) G5/G8 transporters[20].
Figure 1.
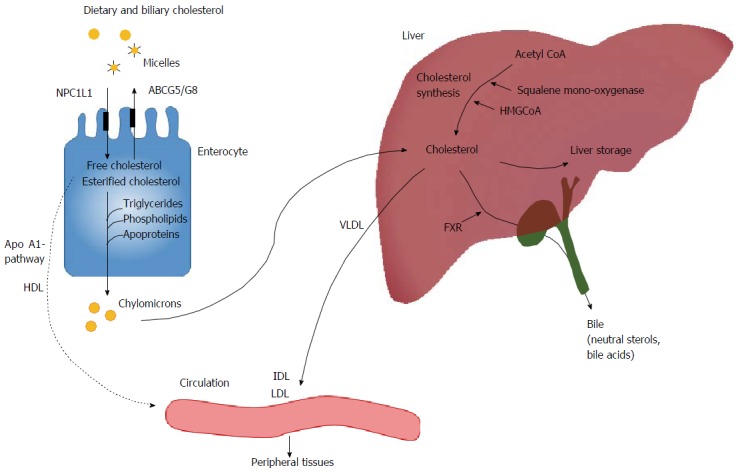
From the gut to the circulation: An overview of the pathways involved in cholesterol absorption and synthesis. IDL: Intermediate density lipoprotein; HDL: High density lipoprotein; LDL: Low density lipoprotein; VLDL: Very low density lipoprotein; NPC1L1: Niemann-Pick C1 like 1; ABCG5: Adenosine triphosphate-binding cassette transporter G5 heterodimer; HMGCoA: 3-hydroxy-3-methyl-glutaryl CoA; FXR: Farnesoid X receptor.
Lipoproteins synthesized in the liver and intestine play central roles in mediating the cholesterol transport to and from tissues through the bloodstream. Within enterocytes, free cholesterol is esterified and assembled - together with triglycerides, phospholipids, and apolipoproteins - to form chylomicrons (lipoproteins). Chylomicrons next enter the lymphatic system and the blood circulation at the thoracic duct, such that chylomicron remnants can be transported to the liver[21] (Figure 1). Some of the cholesterol in enterocytes is generated by endogenous synthesis[4,22]. Cholesterol is also reportedly secreted via an apolipoprotein A1-dependent pathway to form high-density lipoproteins (HDL) in the extracellular milieu, which then enter circulation[21].
CHOLESTEROL SYNTHESIS IN THE LIVER AND PERIPHERAL TISSUE
Cholesterol primarily enters blood circulation from two sources: From intestinal cholesterol absorption, and from the de novo cholesterol synthesis that is ubiquitous in all nucleated cells[1]. The majority of the body’s endogenous cholesterol is produced by the liver[4]. Through a complex 37-step process, cholesterol is synthesized from simpler precursor molecules, starting with acetyl CoA[12]. The rate-limiting factors in the cholesterol synthesis pathway include two enzymes: The target of statins 3-hydroxy-3-methyl-glutaryl CoA reductase and squalene mono-oxygenase, which oxidizes the precursor squalene to lanosterol[1,23] (Figure 1).
The membrane of the endoplasmic reticulum (ER) contains an intracellular feedback system-a tightly controlled protein network that modulates the transcription of genes that mediate cholesterol synthesis and uptake (Figure 2). Sterol regulatory element-binding protein isoform 2 (SREBP-2) is an ER membrane-bound transcription factor that activates genes encoding the enzymes required for cholesterol synthesis[1,6-8,24]. A key event in cholesterol synthesis is the gated movement of SREBP-2 from the ER to the Golgi complex. A crucial ER membrane component, the cleavage-activating protein (SCAP), acts as both an escort for SREBP-2 and a sterol sensor. Immediately after SREBP-2 synthesis in the ER, its COOH-terminal regulatory domain binds to the COOH-terminal domain of SCAP. When cells become cholesterol depleted, SCAP escorts SREBP-2 from the ER to the Golgi apparatus, where SREBP-2 is cleaved by two proteases and then trans-located to the nucleus, where it activates transcription of multiple target genes for cholesterol synthesis. Upon accumulation of excess cellular cholesterol, the SCAP-SREBP complex binds to the resident ER protein INSIG-2, remaining in the ER in a sterol-regulated manner and thereby blocking cholesterol synthesis[25] (Figure 2). Interactions between cholesterol, SCAP, and the SCAP-binding protein INSIG-2 create a sensitive switch that can respond to minor alterations of intracellular cholesterol levels, thus exerting precise control over the cholesterol composition of cell membranes.
Figure 2.
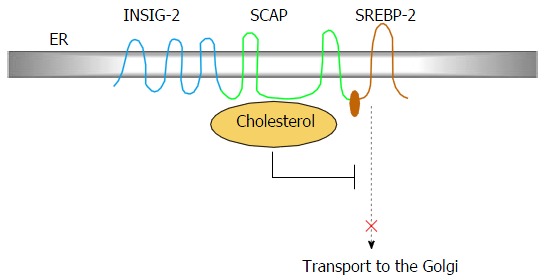
One key event in cholesterol homeostasis is the gated movement of sterol regulatory element-binding protein isoform 2 from the endoplasmic reticulum to the Golgi complex, involving the cleavage-activating protein, and INSIG-2. SREBP-2: Sterol regulatory element-binding protein isoform 2; ER: Endoplasmic reticulum; SCAP: Cleavage-activating protein.
In liver cells, free cholesterol can be excreted as neutral sterols into bile or transformed into bile acids, or it can be esterified and either stored in the liver as cholesterol esters or assembled into VLDL and secreted into circulation. The microsomal transfer protein assembles VLDL from cholesterol esters, triglycerides, phospholipids, free cholesterol, and apolipoprotein B-100 (apo B-100) as its structural protein. Triglycerides in VLDL are subsequently broken down by the enzymes lipoprotein lipase and hepatic lipase, producing intermediate-density lipoproteins (IDLs), followed by LDLs that transport cholesterol to peripheral tissues[21].
CHOLESTEROL ELIMINATION
Free cholesterol is toxic and mammalian somatic cells cannot catabolize it; thus, the removal of excess intracellular cholesterol by a distinct regulatory system is crucial (Figure 3). Liver X receptors (LXRs) act as whole-body cholesterol sensors. Under physiological conditions, cholesterol pool expansion and high intracellular cholesterol levels raise the intracellular concentration of oxygenated cholesterol metabolites termed oxysterols, which are important intermediate or end products in cholesterol excretion pathways. Oxysterols trigger liver-specific LXR activation, generating a transcriptional response that results in net elimination of cholesterol from the body via mobilization of cholesterol from peripheral tissues and promotion of hepatic excretion[4]. Quantitatively, the most important oxygenation reactions are those involved in the early steps of converting cholesterol into bile acids, a metabolically strictly controlled process. Cholesterol 7α-hydroxylase (CYP7A1) is the initial and rate-limiting enzyme of bile-acid synthesis[7,26]. CYP7A1 gene transcription is inhibited by the farnesoid X nuclear receptor, thereby producing negative feedback that reduces the bile acid synthesis from cholesterol. The farnesoid X receptor-agonist obeticholic acid is currently under investigation for possible use in therapy for primary biliary cholangitis (PBC)[27].
Figure 3.
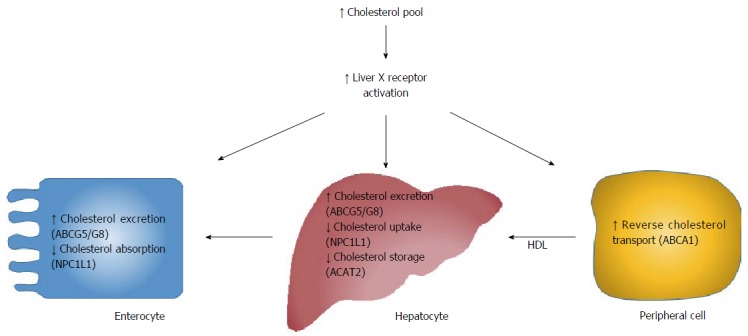
Cholesterol elimination- mechanisms and key transporters. Expansion of the cholesterol pool activates liver X receptor, thereby generating transcriptional responses resulting in cholesterol excretion and catabolism. NPC1L1: Niemann-Pick C1 like 1; ABCA1: ATP-binding cassette transporter sub-family member A 1, also known as the cholesterol efflux regulatory protein; ACAT2: Acetyl-CoA acyltransferase 2; ABCG5: Adenosine triphosphate-binding cassette transporter G5 heterodimer; HDL: High density lipoprotein.
The most important target genes of LXRs include ABCG5/G8, NPC1L1, acetyl-CoA cholesterol acyltransferase 2 (ACAT-2), and ATP-binding cassette transporter sub-family member A 1 (ABCA1; also known as the cholesterol efflux regulatory protein)[4]. Activation of these genes can increase intestinal and hepatic cholesterol excretion (ABCG5/G8), reduce cholesterol absorption (NPC1L1), and reduce cholesterol storage (ACAT-2). ABCA1 is involved in reverse cholesterol transport, in which surplus free cholesterol from peripheral tissues is eliminated from the body via biliary excretion or through the non-biliary TICE pathway[4,5] (Figure 3).
HDL mediates the transfer of cholesterol from peripheral tissues to the liver. Nascent cholesterol-poor pre-β HDL particles take up free cholesterol from peripheral tissues via ABCA1, after which this free cholesterol is esterified by lecithin-cholesterol acyltransferase. The esterified cholesterol is moved to the HDL particle’s hydrophobic core, and progressive lipidation of the HDL particle causes it to mature, enlarge, and become more spherical. The cholesterol esters in mature HDL particles can be removed from circulation by hepatic scavenger receptor B1, or via transfer to apo B-100-containing lipoproteins (VLDL, IDL, and LDL) in a manner mediated by the cholesterol-ester transfer protein. By means of the LDL receptor and the LDL receptor-related protein, the liver can take up the apo B-100-containing lipoprotein particles from circulation[21].
DIFFICULTIES OF ASSESSING CHOLESTEROL METABOLISM IN CHOLESTASIS
Changes in cholesterol metabolism are not mirrored by routine serum cholesterol and lipoprotein measurements[28]. Moreover, the direct methods available to evaluate cholesterol metabolism are complex and laborious, and require labeling techniques, feces collection, and dietary recalls over several days.
In clinical research under steady state conditions, several non-cholesterol sterols that are measurable in serum can serve as valid biomarkers of cholesterol metabolism, especially when expressed as ratios to cholesterol[13,29-31]. Cholesterol precursor sterols, such as desmosterol and lathosterol, are markers of cholesterol synthesis[29]. On the other hand, diet-derived plant sterols (e.g., campesterol and sitosterol) and the liver-synthesized cholesterol metabolite cholestanol are markers of cholesterol absorption efficiency[30]. These markers have been investigated in PBC before and after LT[15,28,32-34]. Compared to that in healthy controls, intestinal cholesterol absorption is reportedly reduced by 2/3 in cases of prolonged severe intrahepatic cholestasis leading to cirrhosis and end-stage liver failure, as seen in PBC[35]. Cholestasis impairs the intestinal absorption of all types of sterols due to poor micellar formation secondary to reduced bile formation and excretion. However, striking increases of serum and hepatic plant sterol and cholestanol levels are also observed, indicating that the serum levels of plant sterols and cholestanol do not correctly mirror cholesterol absorption in cholestasis[15,28]. These changes can be used as biomarkers of the degree of cholestasis, with serum cholestanol/cholesterol being an even more sensitive marker of cholestasis among early-stage PBC patients than serum bilirubin[33]. Moreover, in end-stage cholestasis, serum cholestanol levels increase to levels that are otherwise only seen in the rare genetic disorder cerebrotendinous xanthomatosis[36]. This genetic disorder manifests with extremely high cholesterol deposits in tissues, including nerve tissues, resulting in severe neurologic symptoms[36].
The liver is almost solely responsible for the secretion of sterols (e.g., cholesterol, plant sterols, and cholestanol) from the human body via bile, which is regulated by hepatic proteins, including ABCG5/G8, NPC1L1, and LXRs[4-11]. ABCG5/G8 is active in cholesterol and sterol excretion across the canalicular membrane into bile. To our knowledge, no human studies have been performed to clarify how these sterol transporters function on the biliary canalicular level in intrahepatic cholestasis. Mutations in the genes of these transporters cause phytosterolemia, characterized by increased intestinal absorption and reduced biliary secretion of plant sterols, cholesterol, and cholestanol. Interestingly, Miettinen et al[37] reported the case of a patient with phytosterolemia who presented with cholestatic liver disease necessitating LT. Following LT, the grossly elevated pre-transplant serum levels of plant sterols decreased to values only slightly above normal. This case highlights that the liver apparently plays a predominant role in maintaining sterol balance, since the intestinal ABCG5/G8 defect was not altered by LT[37].
LIPOPROTEIN X
Despite reduced cholesterol synthesis, high serum total and LDL cholesterol concentrations and even xanthomata are common features in PBC and other forms of cholestatic liver disease[35,38-47]. In PBC, serum total cholesterol varies widely, ranging from 2.9 to 46.1 mmol/L (112-1779 mg/dL), and even up to 83 mmol/L (3204 mg/dL)[43].
High serum LDL cholesterol concentration is associated with atherosclerosis. Apo B-100 is present in all liver-derived atherogenic lipoproteins-including VLDL, IDL, LDL, and lipoprotein (a). However, in chronic cholestasis, LDL cholesterol measured using standard hospital laboratory methods is frequently elevated due to abnormal lipoprotein X (Lp-X), which is distinct from apo B-100-containing lipoproteins. Lp-X is characterized by a vesicular structure comprising a 30- to 70-nm lipid bilayer enclosing an aqueous compartment. Lp-X possesses strikingly high contents of unesterified cholesterol and phospholipids; low contents of cholesterol esters and triglycerides; small amounts of albumin and apolipoproteins C, E, and A-1; and no or a low concentration of apo B-100. Lp-X and LDL have the same density and are thus indistinguishable by standard lipoprotein ultracentrifugation. On the other hand, the physical size of Lp-X is in the range of VLDL or larger. Routine clinical laboratory methods currently used to measure LDL cholesterol are markedly affected by the presence of Lp-X, leading to false interpretations of elevated LDL cholesterol levels. Nuclear magnetic resonance spectroscopy measurements of lipoproteins reveal that Lp-X exist in PBC patients more commonly than currently recognized[48]. This phenomenon explains why high LDL cholesterol concentrations within the context of PBC, when actually caused by Lp-X, show no association with atherosclerotic events[38-41].
Lp-X formation is typically associated with a low apo B-100 concentration together with a high total cholesterol concentration[44]. The usual target level of apo B-100 is below 90 mg/dL, corresponding to a true LDL cholesterol concentration of below 3.0 mmol/L (116 mg/dL)[49-51]. The ratio of apo B-100 to total cholesterol is normally around 1:2, but may be 1:10 in cases of severe Lp-X formation[45]. Since an elevated apo B-100 concentration is a risk factor for atherosclerosis[49-51], apo B-100 concentrations should be measured when considering lipid-lowering treatment in PBC and other cholestatic conditions (Figure 4). Even when LDL cholesterol levels are high, cholesterol-lowering medication is unnecessary in cases where apo B-100 is below 90 mg/dL, since this suggests prevalence of non-atherogenic Lp-X. Lp-X resolves after successful cholestasis treatment[52].
Figure 4.
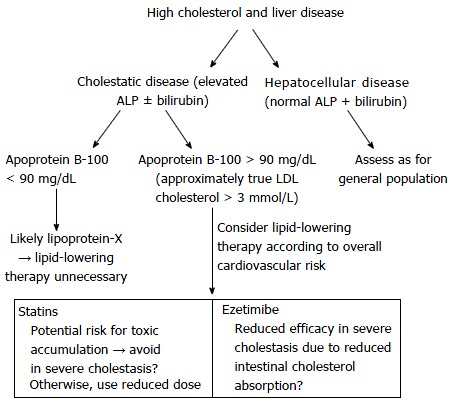
Algorithm for assessing hyperlipidemia in the setting of chronic liver disease. LDL: Low density lipoprotein; ALP: Alkaline phosphatase.
Importantly, most statins are excreted into bile and, thus, cholestatic liver disease may lead to toxic levels of drug accumulation[41]. Furthermore, in Lp-X-related hypercholesterolemia, statin therapy does not effectively lower cholesterol levels because Lp-X does not undergo LDL receptor-mediated hepatic clearance[43,48]. Therefore, statins must be used cautiously in cholestatic conditions. Moreover, cholesterol absorption is low in severe cholestasis due to poor micellar formation, potentially diminishing the effect of ezetimibe, which lowers cholesterol levels by decreasing intestinal cholesterol absorption. In severe cholestasis, a lipid phenotype suggesting high cardiovascular risk necessitates accurate evaluation with consultation of a lipidologist. An additional caveat is that elevated Lp-X may affect various laboratory tests - for instance, potentially leading to pseudohyponatremia[45]. Although hypercholesterolemia is well-acknowledged in PBC, Lp-X formation is often neglected[53].
CHOLESTEROL AND LIVER REGENERATION
An ample cholesterol supply is critical for liver regeneration and for hepatocyte, stellate cell, and Kupffer cell function[54]. The importance of a circulating cholesterol supply for liver regeneration is exemplified following liver resection, where declining serum cholesterol coincides with intrahepatic cholesterol accumulation. In parallel, a serum total cholesterol concentration of below 2.8 mmol/L (108 mg/dL) in decompensated liver cirrhosis is associated with reduced transplant-free survival[55]. Additionally, among patients with non-cholestatic cirrhosis who underwent LT, a recipient serum total cholesterol level of below 1.8 mmol/L (69 mg/dL) at LT was associated with reduced post-LT graft outcome, independent of relevant donor, graft, and pre-operative recipient variables[56]. Both recipient cholesterol levels and the expressions of cholesterol metabolism genes in the liver graft could conceivably influence liver graft cholesterol availability and graft regeneration[56].
DONOR-DERIVED HYPERCHOLESTEROLEMIA
The LDL receptor is critical in mediating the catabolism of cholesterol-enriched particles and is abundant in the liver, with hepatocytes expressing up to 70%-80% of all LDL receptors in humans[57]. Pathogenic mutations in the LDL receptor gene cause familial hypercholesterolemia (FH) characterized by markedly elevated serum total and LDL cholesterol levels, tendon xanthomas, and early atherosclerosis. LT presents an effective therapy for homozygous FH.
On the other hand, we recently reported a case in which an LDL receptor mutation was unintentionally transmitted from a donor to an LT recipient, causing severe hypercholesterolemia in the recipient[58]. Prior to LT, the patient had hepatic epithelioid hemangioendothelioma without cirrhosis or cholestasis and exhibited no dyslipidemia. Following LT, the recipient’s lipid levels were similar to those observed in FH, but her genomic DNA was normal in this regard. DNA was extracted from biopsy specimens of the liver allograft, and subjected to sequencing of the LDL receptor coding region, revealing a heterozygous splicing mutation in intron 9 that was previously reported as an FH-associated pathogenic mutation[58]. This finding essentially represents a transgenic model, consistent with previous evidence suggesting that most LDL cholesterol uptake in the body occurs in the liver and is mediated by LDL receptors. Since heterozygous FH is not extremely rare (prevalence 1/200 to 1/500[59]), our report raises concern of LT recipients acquiring unidentified FH from LT donors, especially since FH manifestations are extrahepatic and thus easily overseen during donor evaluation[60].
POST-TRANSPLANT FOLLOW-UP
Hyperlipidemia reportedly occurs in 40%-66% of patients following LT[61]. Many mechanisms contribute to post-LT hypercholesterolemia and hypertriglyceridemia, including genetic susceptibility, diet, obesity, metabolic syndrome, diabetes, cholestatic problems, and immunosuppressive medication. The immunosuppressive drug cyclosporine induces hypercholesterolemia by inhibiting sterol 27-hydroxylase, a key enzyme in the bile synthesis pathway. Corticosteroids are usually tapered in the early post-LT period, and thus have minimal long-term influence on serum lipids[61]. Post-transplant cholestasis is also relatively common, and often secondary to anastomotic or non-anastomotic biliary stricturing[62]. Prolonged cholestasis may lead to Lp-X formation, but very few post-LT cases are reported[52,63,64].
Compared to the general population, LT recipients more commonly experience cardiovascular events, especially LT recipients with metabolic syndrome and/or diabetes[65]. The overall lipoprotein profile in LT recipients is generally proatherogenic, but variation exists[66], warranting an individualized detailed assessment of cardiovascular risk. Hepatic steatosis is considered a manifestation of metabolic syndrome and/or diabetes and is associated with a proatherogenic profile. Importantly, liver graft steatosis is increasingly detected. Thus, lipid profile assessment should include apo B-100 quantification in addition to the routine measurements of total, LDL, and HDL cholesterol, and total triglycerides. It is assumed that reducing intrahepatic lipids reduces the risks of hepatic and cardiovascular complications. Recent data suggest that treatment with a combination of dietary intervention, weight loss, and ezetimibe (which is well tolerated and can be combined with a statin) can reduce LDL cholesterol and apo B-100 concentrations in these patients[22,67-70].
CONCLUSION
Various liver disorders, particularly cholestasis, affect cholesterol metabolism and can cause variable hypercholesterolemia, including Lp-X appearance. Mistaking Lp-X for LDL cholesterol may interfere with cardiovascular risk assessment, leading to the prescription of futile lipid-lowering therapy. Lipid panel assessment should be regularly performed in all LT recipients, and at LT evaluation. Apo B-100 measurement can help in distinguishing between atherogenic and non-atherogenic hypercholesterolemia. Therefore, the measurement of apo B-100 can help in evaluating overall cardiovascular risk, as well as the effects of therapy during follow-up. This is particularly important after LT, when cholestasis and Lp-X may coexist with true atherogenic hypercholesterolemia and increased cardiovascular risk.
ACKNOWLEDGMENTS
I recently changed my name from Katriina Nikkilä to Katriina Nemes. My previous publication history has been released by using the name Katriina Nikkilä (e.g., Nikkilä K) partly shown also in the actual references.
Footnotes
Conflict-of-interest statement: The authors declare no conflict of interests for this article.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Manuscript source: Invited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: Finland
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B
Grade C (Good): C
Grade D (Fair): D
Grade E (Poor): 0
Peer-review started: April 6, 2016
First decision: May 17, 2016
Article in press: July 13, 2016
P- Reviewer: Stieger B, Tiao MM, Zhao YL S- Editor: Gong ZM L- Editor: A E- Editor: Li D
References
- 1.Ikonen E. Cellular cholesterol trafficking and compartmentalization. Nat Rev Mol Cell Biol. 2008;9:125–138. doi: 10.1038/nrm2336. [DOI] [PubMed] [Google Scholar]
- 2.Maxfield FR, van Meer G. Cholesterol, the central lipid of mammalian cells. Curr Opin Cell Biol. 2010;22:422–429. doi: 10.1016/j.ceb.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Calvo PL, Brunati A, Spada M, Romagnoli R, Corso G, Parenti G, Rossi M, Baldi M, Carbonaro G, David E, et al. Liver transplantation in defects of cholesterol biosynthesis: the case of lathosterolosis. Am J Transplant. 2014:14; 960–965. doi: 10.1111/ajt.12645. [DOI] [PubMed] [Google Scholar]
- 4.Bonamassa B, Moschetta A. Arteriosclerosis: lessons from LXR and the intestine. Trends Endocrinol Metab. 2013;24:120–128. doi: 10.1016/j.tem.2012.10.004. [DOI] [PubMed] [Google Scholar]
- 5.Degirolamo C, Sabbà C, Moschetta A. Intestinal nuclear receptors in HDL cholesterol metabolism. J. Lipid Res. 2015;56:1262–1270. doi: 10.1194/jlr.R052704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Horton JD, Goldstein JL, Brown MS. SREBs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weber LW, Boll M, Stampfl A. Maintaining cholesterol homeostasis: Sterol regulatory element-binding proteins. World J Gastroenterol. 2004;10:3081–3087. doi: 10.3748/wjg.v10.i21.3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eberlé D, Hegarty B, Bossard P, Ferré P, Foufelle F. SREBP transcription factors: master regulators of lipid homeostasis. Biochimie. 2004;86:839–848. doi: 10.1016/j.biochi.2004.09.018. [DOI] [PubMed] [Google Scholar]
- 9.Moschetta A. Nuclear receptors and cholesterol metabolism in the intestine. Atheroscler Suppl. 2015;17:9–11. doi: 10.1016/S1567-5688(15)50003-2. [DOI] [PubMed] [Google Scholar]
- 10.Davis HR Jr, Altmann SW. Niemann-Pick C1 Like 1 (NPC1L1) an intestinal sterol transporter. Biochim Biophys Acta. 2009;1791:679–683. doi: 10.1016/j.bbalip.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 11.Betters JL, Yu L. NPC1L1 and cholesterol transport. FEBS Lett. 2010;584:2740–2747. doi: 10.1016/j.febslet.2010.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343:425–430. doi: 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- 13.Gylling H. Clinical utility of serum markers of cholesterol absorption and synthesis. Curr Opin Lipidol. 2014;25:207–212. doi: 10.1097/MOL.0000000000000069. [DOI] [PubMed] [Google Scholar]
- 14.Turner S, Voogt J, Davidson M, Glass A, Killion S, Decaris J, Mohammed H, Minehira K, Boban D, Murphy E, et al. Measurement of reverse cholesterol transport pathways in humans: in vivo rates of free cholesterol efflux, esterification, and excretion. J Am Heart Assoc. 2012;1:e001826. doi: 10.1161/JAHA.112.001826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nikkilä K, Höckerstedt K, Miettinen TA. High cholestanol and low campesterol-to-sitosterol ratio in serum of patients with primary biliary cirrhosis before liver transplantation. Hepatology. 1991;13:663–669. [PubMed] [Google Scholar]
- 16.Arguello G, Balboa E, Arrese M, Zanlungo S. Recent insights on the role of cholesterol in non-alcoholic fatty liver disease. Biochim Biophys Acta. 2015;1852:1765–1778. doi: 10.1016/j.bbadis.2015.05.015. [DOI] [PubMed] [Google Scholar]
- 17.Sudhop T, Lütjohann D, von Bergmann K. Sterol transporters: target of natural sterols and new lipid lowering drugs. Pharmacol Ther. 2005;105:333–341. doi: 10.1016/j.pharmthera.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 18.Hui DY, Labonté ED, Howles PN. Development and physiological regulation of intestinal lipid absorption. III. Intestinal transporters and cholesterol absorption. Am J Physiol Gastrointest Liver Physiol. 2008;294:G839–G843. doi: 10.1152/ajpgi.00061.2008. [DOI] [PubMed] [Google Scholar]
- 19.Phan BA, Dayspring TD, Toth PP. Ezetimibe therapy: mechanism of action and clinical update. Vasc Health Risk Manag. 2012;8:415–427. doi: 10.2147/VHRM.S33664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ostlund RE Jr, McGill JB, Zeng CM, Covey DF, Stearns J, Stenson WF, Spilburg CA. Gastrointestinal absorption and plasma kinetics of soy Delta(5)-phytostanols in humans. Am J Physiol Endocrinol Metabol. 2002;282:E911–E 916. doi: 10.1152/ajpendo.00328.2001. [DOI] [PubMed] [Google Scholar]
- 21.Hussain MM. Intestinal lipid absorption and lipoprotein formation. Curr Opin Lipidol. 2014;25:200–206. doi: 10.1097/MOL.0000000000000084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arca M. Alterations of intestinal lipoprotein metabolism in diabetes mellitus and metabolic syndrome. Atheroscler Supp. 2015;17:12–16. doi: 10.1016/S1567-5688(15)50004-4. [DOI] [PubMed] [Google Scholar]
- 23.Buhaescu I, Izzedine H. Mevalonate pathway: a review of clinical and therapeutical implications. Clin Biochem. 2007;40:575–584. doi: 10.1016/j.clinbiochem.2007.03.016. [DOI] [PubMed] [Google Scholar]
- 24.Radhakrishnan A, Goldstein JL, McDonald JG, Brown MS. Switch-like control of SREBP-2 transport triggered by small changes in ER cholesterol: a delicate balance. Cell Metab. 2008;8:512–521. doi: 10.1016/j.cmet.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yabe D, Brown MS, Goldstein JL. Insig-2, a second endoplasmic reticulum protein that binds SCAP and blocks export of sterol regulatory element-binding proteins. Proc Natl Acad Sci USA. 2002;99:12753–12758. doi: 10.1073/pnas.162488899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Repa JJ, Berge KE, Pomajzl C, Richardson JA, Hobbs H, Mangelsdorf DJ. Regulation of ATP-binding cassette sterol transporters ABCG5 and ABCG8 by liver X receptors alfa and beeta. J. Biol Chem. 2002;277:18793–18800. doi: 10.1074/jbc.M109927200. [DOI] [PubMed] [Google Scholar]
- 27.Corpechot C. Primary biliary cirrhosis beyond ursodeoxycholic acid. Semin Liver Dis. 2016;36:15–26. doi: 10.1055/s-0035-1571273. [DOI] [PubMed] [Google Scholar]
- 28.Nikkilä K, Höckerstedt K, Miettinen TA. Serum and hepatic cholestanol, squalene and noncholesterol sterols in man: a study on liver transplantation. Hepatology. 1992;15:863–870. doi: 10.1002/hep.1840150519. [DOI] [PubMed] [Google Scholar]
- 29.Miettinen TA, Tilvis RS, Kesäniemi YA. Serum plant sterols and cholesterol precursors reflect cholesterol absorption and synthesis in volunteers of a randomly selected male population. Am J Epidemiol. 1990;131:20–31. doi: 10.1093/oxfordjournals.aje.a115479. [DOI] [PubMed] [Google Scholar]
- 30.Miettinen TA, Tilvis RS, Kesäniemi YA. Serum cholestanol and plant sterol levels in relation to cholesterol metabolism in middle-aged men. Metabolism. 1989;38:136–140. doi: 10.1016/0026-0495(89)90252-7. [DOI] [PubMed] [Google Scholar]
- 31.Miettinen TA, Gylling H, Nissinen MJ. The role of serum non-cholesterol sterols as surrogate markers of absolute cholesterol synthesis and absorption. Nutr Metab Cardiovasc Dis. 2011;21:765–769. doi: 10.1016/j.numecd.2011.05.005. [DOI] [PubMed] [Google Scholar]
- 32.Nikkilä K, Miettinen TA, Höckerstedt KV, Isoniemi H. Sterol parameters as markers of liver function in primary biliary cirrhosis before and after liver transplantation. Transpl Int. 2005;18:221–225. doi: 10.1111/j.1432-2277.2004.00002.x. [DOI] [PubMed] [Google Scholar]
- 33.Nikkilä K, Nissinen MJ, Gylling H, Isoniemi H, Miettinen TA. Serum sterols in patients with primary biliary cirrhosis and acute liver failure before and after liver transplantation. J Hepatol. 2008;49:936–945. doi: 10.1016/j.jhep.2008.07.026. [DOI] [PubMed] [Google Scholar]
- 34.Nikkilä K, Höckerstedt K, Miettinen TA. Liver transplantation modifies serum cholestanol, cholesterol precursor and plant sterol levels. Clin Chim Acta. 1992;208:205–218. doi: 10.1016/0009-8981(92)90077-4. [DOI] [PubMed] [Google Scholar]
- 35.Gylling H, Färkkilä M, Vuoristo M, Miettinen TA. Metabolism of cholesterol and low- and high-density lipoproteins in primary biliary cirrhosis: cholesterol absorption and synthesis related to lipoproteins levels and their kinetics. Hepatology. 1995;21:89–95. [PubMed] [Google Scholar]
- 36.Bhattacharyya AK, Lin DS, Connor WE. Cholestanol metabolism in patients with cerebrotendinous xanthomatosis: absorption, turnover, and tissue deposition. J Lipid Res. 2007;48:185–192. doi: 10.1194/jlr.M600113-JLR200. [DOI] [PubMed] [Google Scholar]
- 37.Miettinen TA, Klett EL, Gylling H, Isoniemi H, Patel SB. Liver transplantation in a patient with sitosterolemia and cirrhosis. Gastroenterology. 2006;130:542–547. doi: 10.1053/j.gastro.2005.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Crippin JS, Lindor KD, Jorgensen R, Kottke BA, Harrison JM, Murtaugh PA, Dickson ER. Hypercholesterolemia and atherosclerosis in primary biliary cirrhosis: what is the risk? Hepatology. 1992;15:858–862. doi: 10.1002/hep.1840150518. [DOI] [PubMed] [Google Scholar]
- 39.Longo M, Crosignani A, Battezzati PM, Squarcia Giussani C, Invernizzi P, Zuin M, Podda M. Hyperlipidaemic state and cardiovascular risk in primary biliary cirrhosis. Gut. 2002;51:265–269. doi: 10.1136/gut.51.2.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chang PY, Lu SC, Su TC, Chou SF, Huang WH, Morrisett JD, Chen CH, Liau CS, Lee YT. Lipoprotein-X reduces LDL atherogenicity in primary biliary cirrhosis by preventing LDL oxidation. J Lipid Res. 2004;45:2116–2122. doi: 10.1194/jlr.M400229-JLR200. [DOI] [PubMed] [Google Scholar]
- 41.Sorokin A, Brown JL, Thompson PD. Primary biliary cirrhosis, hyperlipidemia, and atherosclerotic risk: a systematic review. Atherosclerosis. 2007;194:293–299. doi: 10.1016/j.atherosclerosis.2006.11.036. [DOI] [PubMed] [Google Scholar]
- 42.Citkowitz E. Was the low-density lipoprotein cholesterol level really elevated? Liver Transpl. 2011;17:1234, author reply 1235. doi: 10.1002/lt.22317. [DOI] [PubMed] [Google Scholar]
- 43.Wong ML, Raghavan RP, Hedger NA, Ellis RD, Meeking DR, Albon L. The use of plasmapheresis in managing primary biliary cirrhosis presenting with profound hypercholesterolaemia. The British Journal of Diabetes and Vascular Diseases. 2012;12:156–158. [Google Scholar]
- 44.Ooi YK, Mietus-Snyder M, Torres C, Mohan P, Harahsheh A. Lipoprotein-X-accumulation: a mimic of familial hypercholesterolemia. Consult Pediatricians. 2013;12:63–65. [Google Scholar]
- 45.Hussain I, Ahmad Z, Garg A. Extreme hypercholesterolemia presenting with pseudohyponatremia - a case report and review of the literature. J Clin Lipidol. 2015;9:260–264. doi: 10.1016/j.jacl.2014.11.007. [DOI] [PubMed] [Google Scholar]
- 46.Joukhadar R, Chiu K. Severe hypercholesterolemia in patients with graft-vs-host disease affecting the liver after stem cell transplantation. Endocr Pract. 2012;18:90–97. doi: 10.4158/Ep11212.RA. [DOI] [PubMed] [Google Scholar]
- 47.Chow A, Rifici VA, Schneider SH. Lipoprotein-X in a patient with lymphoplasmacytic sclerosing cholangitis: an unusual cause of secundary hypercholesterolemia. AACE Clinical Case Reports. 2016;2:e20–e 24. [Google Scholar]
- 48.Foley KF, Silveira MG, Hornseth JM, Lindor KD, McConnell JP. A patient with primary biliary cirrhosis and elevated LDL cholesterol. Clin Chem. 2009;55:187–191, discussion 191-192. doi: 10.1373/clinchem.2008.108720. [DOI] [PubMed] [Google Scholar]
- 49.Assessment by the AACC Lipoproteins and Vascular Diseases Division Working Group on Best Practices. Cole TG, Contois JH, Csako G, McConnell JP, Remaley AT, Devaraj S, Hoefner DM, Mallory T, Sethi AA, Warnick GR. Association of apolipoprotein B and nuclear magnetic resonance spectroscopy-derived LDL particle number with outcomes in 25 clinical studies. Clin Chem. 2013;59:752–770. doi: 10.1373/clinchem.2012.196733. [DOI] [PubMed] [Google Scholar]
- 50.Sniderman AD, Williams K, Contois JH, Monroe HM, McQueen MJ, de Graaf J, Furberg CD. A meta-analysis of low-density lipoprotein cholesterol, non-high-density lipoprotein cholesterol, and apolipoprotein B as markers of cardiovascular risk. Circ Cardiovasc Qual Outcomes. 2011;4:337–345. doi: 10.1161/CIRCOUTCOMES.110.959247. [DOI] [PubMed] [Google Scholar]
- 51.Varvel SA, Dayspring TD, Edmons Y, Thiselton DL, Ghaedi L, Voros S, McConnel JP, Sasinowski M, Dall T, Warnick GR. Discordance between apolipoprotein B and low-density lipoprotein particle number is associated with insulin resistance in clinical practice. J Clin Lipidol. 2015;9:247–255. doi: 10.1016/j.jacl.2014.11.005. [DOI] [PubMed] [Google Scholar]
- 52.Jankowski K, Wyzgal A, Wierzbicka A, Tronina O, Durlik M, Pruszczyk P. Rapid normalization of severe hypercholesterolemia mediated by lipoprotein X after liver transplantation in a patient with cholestasis - a case report. Acta Biochim Pol. 2015;62:621–623. doi: 10.18388/abp.2015_971. [DOI] [PubMed] [Google Scholar]
- 53.Purohit T, Cappell MS. Primary biliary cirrhosis: Pathophysiology, clinical presentation and therapy. World J Hepatol. 2015;7:926–941. doi: 10.4254/wjh.v7.i7.926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Delgado-Coello B, Briones-Orta MA, Macias-Silva M, Mas-Oliva J. Cholesterol: recapitulation of its active role during liver regeneration. Liver Int. 2011;31:1271–1284. doi: 10.1111/j.1478-3231.2011.02542.x. [DOI] [PubMed] [Google Scholar]
- 55.Jiang M, Liu F, Xiong WJ, Zhong L, Xu W, Xu F, Liu YB. Combined MELD and blood lipid level in evaluating the prognosis of decompensated cirrhosis. World J Gastroenterol. 2010;16:1397–1401. doi: 10.3748/wjg.v16.i11.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ginanni Corradini S, Siciliano M, Parlati L, Molinaro A, Cantafora A, Poli E, Mennini G, Melandro F, Vestri AR, Merli M, et al. Recipient perioperative cholesterolaemia and graft cholesterol metabolism gene expression predict liver transplant outcome. Liver Int. 2014;34:e290–e301. doi: 10.1111/liv.12351. [DOI] [PubMed] [Google Scholar]
- 57.Goldstein JL, Brown MS. The LDL receptor. Arterioscler Thromb Vasc Biol. 2009;29:431–438. doi: 10.1161/ATVBAHA.108.179564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nikkilä K, Åberg F, Isoniemi H. Transmission of LDLR mutation from donor through liver transplantation resulting in hypercholesterolemia in the recipient. Am J Transplant. 2014;14:2898–2902. doi: 10.1111/ajt.12961. [DOI] [PubMed] [Google Scholar]
- 59.Nordestgaard BG, Chapman MJ, Humphries SE, Ginsberg HN, Masana L, Descamps OS, Wiklund O, Hegele RA, Raal FJ, Defesche JC, et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J. 2013;34:3478–3490a. doi: 10.1093/eurheartj/eht273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Najam O, Ray KK. Familial hypercholesterolemia: a review of the natural history, diagnosis, and management. Cardiol Ther. 2015;4:25–38. doi: 10.1007/s40119-015-0037-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sethi A, Stravitz RT. Review article: medical management of the liver transplant recipient - a primer for non-transplant doctors. Aliment Pharmacol Ther. 2007;25:229–245. doi: 10.1111/j.1365-2036.2006.03166.x. [DOI] [PubMed] [Google Scholar]
- 62.Ben-Ari Z, Pappo O, Mor E. Intrahepatic cholestasis after liver transplantation. Liver Transpl. 2003;9:1005–1018. doi: 10.1053/jlts.2003.50212. [DOI] [PubMed] [Google Scholar]
- 63.Tejera P, Karalis D, Xiao G, Simon B, Amori R. Severe hyper?cholesterolemia and cholestatic liver disease after liver transplant: a case of lipoprotein X. The ICE/ENDO 2014 International Congress of Endocrinology and the 96th annual meeting of Endocrine Society in Chicago, June 21-24, 2014 Chicago, SUN-0851 [Google Scholar]
- 64.Yeh H, Kitchens WH, Elias N, Kelsey PB, Markmann JF, Hertl M. Hyperlipidemia due to biliary stricture after living-donor liver transplantation. Transplantation. 2011;92:e29–e30. doi: 10.1097/TP.0b013e31822d095d. [DOI] [PubMed] [Google Scholar]
- 65.Madhwal S, Atreja A, Albeldawi M, Lopez R, Post A, Costa MA. Is liver transplantation a risk for cardiovascular disease? A meta-analysis of observational studies. Liver Transpl. 2012;18:1140–1146. doi: 10.1002/lt.23508. [DOI] [PubMed] [Google Scholar]
- 66.Chhatrala R, Siddiqui MB, Stravitz RT, Driscoll C, Sanyal A, Sargeant C, Luketic V, Sharma A, Sterling R, Matherly S, et al. Evolution of serum atherogenic risk in liver transplant recipients: Role of lipoproteins and metabolic and inflammatory markers. Liver Transpl. 2015;21:623–630. doi: 10.1002/lt.24100. [DOI] [PubMed] [Google Scholar]
- 67.Chan DC, Watts GF, Gan SK, Ooi EM, Barrett PH. Effect of ezetimibe on hepatic fat, inflammatory markers, and apolipoprotein B-100 kinetics in insulin-resistant obese subjects on a weight loss diet. Diabetes Care. 2010;33:1134–1139. doi: 10.2337/dc09-1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Åberg F, Koljonen V, Nikkilä K, Boyd S, Arola J, Isoniemi H. Thiazolidinedione therapy versus lifestyle recommendation in the treatment of post-liver transplant graft steatosis. Ann Transplant. 2014;19:389–396. doi: 10.12659/AOT.890664. [DOI] [PubMed] [Google Scholar]
- 69.Farnier M. Ezetimibe/statin combination therapy to treat patients with type 2 diabetes. Atheroscler Suppl. 2015;17:2–8. doi: 10.1016/S1567-5688(15)50002-0. [DOI] [PubMed] [Google Scholar]
- 70.Averna M. The effect of ezetimibe on NALFD. Atheroscler Suppl. 2015;17:27–34. doi: 10.1016/S1567-5688(15)50007-X. [DOI] [PubMed] [Google Scholar]