

Abstract
Carbon nanotubes (CNTs) are emerging as important occupational and environmental toxicants owing to their increasing prevalence and potential to be inhaled as airborne particles. CNTs are a concern because of their similarities to asbestos, which include fibrous morphology, high aspect ratio, and biopersistence. Limitations in research models have made it difficult to experimentally ascertain the risk of CNT exposures to humans and whether these may lead to lung diseases classically associated with asbestos, such as mesothelioma and fibrosis. In this study we sought to comprehensively compare profiles of lung pathology in mice following repeated exposures to multi-wall CNTs or crocidolite asbestos (CA). We show that both exposures resulted in granulomatous inflammation and increased interstitial collagen, CA exposures caused predominantly bronchoalveolar hyperplasia whereas CNT exposures caused alveolar hyperplasia of type II pneumocytes (T2Ps). T2Ps isolated from CNT-exposed lungs were found to have upregulated proinflammatory genes, including IL-1ß, in contrast to those from CA-exposed. Immunostaining in tissue showed that while both toxicants increased IL-1ß protein expression in lung cells, T2P-specific IL-1ß increases were greater following CNT exposure. These results suggest related but distinct mechanisms of action by CNTs versus asbestos which may lead to different outcomes in the two exposure types.
Keywords: Carbon nanotubes, crocidolite asbestos, fiber toxicology, comparative pathology, lung, mouse
Introduction
Lung diseases in humans and their associated public health impacts are frequently linked to occupational and environmental exposures to inhaled particles (Donaldson et al, 2001, Pope III et al, 1995, Rom et al, 1987). Insoluble fiber particles are noted to be especially injurious due to their aspect ratio and biopersistence (Jones et al, 1997), the most notorious historical example being asbestos. Inhalational exposure to asbestos (particularly amphibole forms such as crocidolite) is known to cause a constellation of pathological features in the lung including granulomatous inflammation, alveolar septal fibrosis, epithelial hyperplasia, and carcinoma. Carbon nanotubes (CNTs), another class of fibrous particles, have extensive utility in developing modern technologies and applications (DeVolder et al, 2013). Due to their unique properties and wide range of applications, it is widely forecasted that CNTs will become ubiquitous in coming years. This has lead to dramatic increases in CNT manufacture and distribution, and the forecasted volume of supply makes occupational and environmental exposures likely. CNTs may become agitated into aerosols during manufacturing and sorting processes similarly to asbestos (Dahm et al, 2012, Han et al, 2008). As these two materials share many properties, including high aspect ratio and biopersistence, it is speculated that they could act similarly as occupational and environmental lung toxicants. Aspect ratio and resistance to breakdown and clearance are known to be determinants in the disease-promoting potential of asbestos (Hesterberg et al, 1996, Miller et al, 1999). Toxicological studies of CNTs have increased in recent years (Bonner, 2013, Porter et al, 2010, Sargent et al, 2014), but knowledge of their toxicology and mechanisms remains underdeveloped and the study of risk assessment of CNTs as lung toxicants comparable to asbestos is far from complete. Isolated studies (Poland et al, 2008, Takagi et al, 2008) have attempted to compare these materials in vivo, but studies of this kind are few and are often focused primarily on the possibility that CNTs may induce pleural mesothelioma similarly to asbestos. In addition, batch-to-batch variation of CNTs which frequently differ significantly in their physiochemical characteristics (i.e., fiber diameter, morphology, manufacturing vestiges such as metals) hampers consensus between independent projects and highlights the need for further work in assessment of CNT toxicology.
First-line pulmonary defenses in response to inhaled particle irritants include clearance and breakdown. Abiotic, insoluble particles which are resistant to breakdown are cleared by mucociliary transport in the conducting airways and by alveolar macrophages (AMs) in the bronchoalveolar regions (Oberdörster, 1988). Globular particulates such as dust and pollen are efficiently cleared by AMs but fiber particles which deposit in the distal airspaces are known to present a challenge to the clearance processes of phagocytic uptake and subsequent AM locomotion to the mucociliary-equipped regions of the airway (Lippman, 1990). It is believed that the shape and aspect ratios of biopersistent fibers such as crocidolite asbestos cause harmful deformations of AMs attempting to take up the particles (Dörger et al, 2000, Nagai & Toyokuni, 2012) while the ends of fibers may puncture or damage cell membranes (frustrated phagocytosis). In addition to inducing mechanical strain, fiber particles have large surface areas which may affect cell and tissue environments with harmful surface reactivity such as the generation of oxidative free radicals (Shukla et al, 2003).
Lung responses to non-biological particles which are not efficiently cleared or which irritate cells and surfaces include inflammation and remodeling of tissue (Oberdörster, 1995). The alveolar epithelium is functional in the role of gas exchange, which is possible because of diffusion through thin, delicate walls lined by type I pneumocytes (T1Ps). The alveolar epithelium is easily damaged due to the fragile nature of gas exchange areas, resulting in acute phase inflammation. Leukocytes (chiefly neutrophils and monocytes) infiltrate airspaces during acute phase inflammation and may remain active beyond the resolution of exudative influx if insulting particles persist in the bronchoalveolar space. Acute inflammation gives way to fibroproliferative regeneration and reinforcing of alveolar structure during which type II pneumocytes (T2Ps) proliferate and differentiate into T1Ps to restore the integrity and functionality of gas exchange areas (Fehrenbach, 2001). In cases of persistent insult such as uncleared fiber particles, infiltrating leukocytes may aggregate into foci of granulomatous inflammation and T2Ps and other epithelial cell types may undergo adaptive changes to the detriment of the gas exchange capacity of alveolar lining (Snyder et al, 1990). In addition, inappropriate deposition of collagen may lead to fibrosis of the septal interstitium and reduced compliance of the alveolus. Septal interstitial fibrosis is one of the key clinical features in the diagnosis of asbestosis caused by chronic exposure to asbestos fibers (Wagner, 1997). Although descriptive in vivo studies have shown that CNT exposures may induce similar changes (Mercer et al, 2011, Ryman-Rasmussen et al, 2009), detailed studies comparing and contrasting subchronic parenchymal lung responses in CNT and asbestos exposures are lacking. In the present study our objective is to compare profiles of lung pathology induced by exposure to either CNTs or crocidolite asbestos in a mouse model of subchronic, repeated-dose exposure. We sought to test the hypothesis that CNT and asbestos exposures induce similar profiles of lung pathology, and that parallels in effector cell recruitment and induction of molecular mediators may account for this similarity. We tested the hypothesis by direct comparison of the histopathology observed in either exposure group, including specific endpoints for granulomatous inflammation, T2P hyperplasia, collagen deposition, and expression of molecular mediators in effector cells and tissues.
Methods
Chemicals and reagents
For routine work, phosphate-buffered saline (PBS), RPMI 1640, formalin, and histological alcohol were obtained through ThermoFisher (Pittsburgh, PA). For humane animal sacrifice, Euthasol was obtained from Butler-Schein (Dublin, OH). Fetal bovine serum (FBS) used was from Atlanta Biologicals (Flowery Branch, GA). Pluronic F127 was from Sigma (St.Louis, MO). Liberase TL and DNaseI were from Roche Diagnostics (Indianapolis, IN). Percoll was from Santa Cruz Biotech (Santa Cruz, CA). All reagents (antibodies and bead-conjugated avidin) for magnet assisted cell sorting (MACS) were purchased from Miltenyi Biotech (Auburn, CA). The following antibodies were obtained from eBioscience (San Diego, CA): anti-CD16/32, anti-CD326-APC. The following antibodies were obtained from BioLegend (San Diego, CA): anti-CD45-Alexa Fluor 700, anti-Ki67, anti-IL-1ß, anti-rabbit IgG-DyLight 488, anti-hamster IgG-DyLight 594, and anti-rat IgG-DyLight 594. Anti-proSPC was obtained from Millipore (Billerica, MA). TRI reagent was from Molecular Research Center (Cincinnati, OH). RNeasy columns were from Qiagen (Valencia, CA). qRT-PCR was carried out using Brilliant III SYBR Green Master Mix (Agilent, Santa Clara, CA) and primers were synthesized by Integrated DNA Technologies (Coralville, IA).
Exposure materials and characterization
High-purity multi-wall CNTs were obtained from Baytubes, C150HP, lot E0006AAD08 (Leverkusen, Germany) as a dry bulk powder form. Crocidolite asbestos (CA) was sourced from IITRI/NIOSH (Chicago, IL). Materials were characterized using transmission electron microscopy (TEM) and zeta potential. Briefly, either CNTs or CA were prepared for delivery by sonication for 30min intervals on ice in PBS with 1% Pluronic F127 until well-dispersed suspensions were obtained as confirmed by brightfield microscopy. CNT suspensions were centrifuged at 3200rcf for 30min to pellet remaining aggregates. CA suspensions were not centrifuged (due to the incomplete solubility of the particles) so weight/volume of CA was matched to the measured concentration of centrifuged CNT stock suspension (as determined by weight and elemental carbon content as measured previously [Birch, 2004a, 2004b]). Zeta potential was measured in as-delivered suspensions using a Malvern Zetasizer Nano (Malvern, UK). CNTs used were further characterized for metal content, surface area, and thermogravimetric analysis as shown previously (Frank et al, 2015).
Animals and Exposure
Animals were approved for use in this study by the University of Cincinnati Institutional Animal Care and Use Committee. Six-week old male Non-swiss Albino CF-1 mice were obtained from Harlan Labs (Haslett, MI) and housed for 1 week prior to exposure. Exposures were administered by oropharyngeal aspiration (Rao et al, 2003) under isoflurane anesthesia. Briefly, anesthetized animals were suspended supine and the tongue was gently restrained. A 50μL aliquot was placed at the back of the tongue and the nostrils were held until two full breaths were taken. Animals were exposed to 33μg/dose of CNT or CA in suspension. Exposures consisted of three doses/week (Mon, Wed, Fri) for three weeks for a total dose of 300μg. The control group received vehicle (PBS with 1% Pluronic F127) using the same dosing schedule. Animals (n=8/treatment group) rested for 5 weeks following exposure, allowing for a total exposure time of 8 weeks.
Sample Preparation
Animals were euthanized by i.p. Euthosol injection and exsanguinated. For animals intended for histology (n=8) the trachea was exposed and cannulated and the left lung was inflated with 10% neutral buffered formalin. The left lungs were then removed, placed into formalin, fixed for 24 hours, and paraffin embedded, sectioned, and stained at the Pathology Research Core at Cincinnati Children’s Hospital Medical Center (CCHMC) in Cincinnati, OH. For animals intended for cell isolation (n=3), the lungs were perfused with 10mL of cold PBS via the right ventricle.
Lung Digestions and Cell Isolation
Digestion medium was formulated as follows: RPMI 1640 medium was supplemented with 100U/I of penicillin/streptomycin (Thermo-Fisher), 500μg/mL DNaseI, and 250μg/mL of Liberase TL. Perfused lungs were inflated with 2mL of digestion medium. Then, 0.5mL of molten 1% low-melting agarose was immediately instilled and the chest cavity was cooled by ice for 2min. The lungs were excised, rinsed in PBS incubated in digestion medium for 80min at 37°C and 5% CO2 and disrupted with 100μm and 40μm mesh. Digestions were centrifuged for 20min at 400rcf through a discontinuous 20%/40%/70% Percoll gradient. The cells at the 20%/40% interphase, consisting of intact, viable T2Ps, were taken for MACS separations. Anti-CD45-biotin and anti-CD326(EpCam)-biotin primary antibodies were used to enrich for and isolate, respectively, T2Ps using anti-biotin microbeads on an autoMACS Pro Separator in the Research Flow Cytometry Core at CCHMC. Cells were 40μm filtered immediately prior to each separation. Purity of T2Ps was monitored by flow cytometry using anti-CD45 (30-F11) and anti-CD326(EpCam, clone G8.8). Isolated cells were immediately pelleted and lysed in TRI reagent (Molecular Research Center, Cincinnati, OH) and stored at −80°C.
RNA Isolation and qRT-PCR
Total RNA from samples in TRI reagent was isolated by phase separation. The yields were cleaned up using RNeasy Plus columns (Qiagen, Valencia, CA). One-step qRT-PCR was carried out using Brilliant III SYBR Green Master Mix with 10ng of isolated RNA/reaction. Primers for IL-1ß, Cxcl1, Cxcl5, Ccl2, RNA polymerase II (Rbp1), hypoxanthine ribosyltransferase (Hprt), and glucuronidase ß (Gusß) were generated using Primer BLAST (www.ncbi.nlm.nih.gov/tools/primer-blast). qRT-PCR was performed on an ABI 7500 real-time thermocycler with the following conditions: RT reaction for 10min at 50°C followed by PCR amplification using 3min at 95°C, 40 × (5s at 95°C, 33s at 60°C). Raw CT values were normalized using the geometric mean of three housekeeping genes (Rbp1, Hprt, Gusß). The ▲▲CT method was used to arrive at the quantification of target gene transcripts in exposed samples relative to the vehicle control. The calculation for the method used the following formula:
Where fold change is a positive number more, less, or equal to 1,TG: Target gene, HKG: Housekeeping gene To represent relative increases or decreases, values of fold change (FC) were linearized by the following formula:
Histology, histochemistry, and immunofluorescent histochemistry (IHC)
Hematoxylin & eosin (H&E) and Masson’s trichrome stains were prepared at CCHMC Pathology Research Core. Further sections for IHC were also prepared by the Core. Slides were examined with a Zeiss Axio Scope.A1 microscope equipped with an AxioCam ICm1 and Zeiss Zen software (Carl Zeiss Microscopy, Thornwood, NY). Slides stained with H&E were scored blind by a board-certified veterinary pathologist. Microscopic findings were scored with a severity grade of 1–4 based upon a characterization of minimal, mild, moderate, or marked, respectively, and present (‘present’ denotes the observation of pigmented macrophages commonly observed in CNT-exposed lungs). Samples received severity scores on specific morphologies including granulomatous inflammation, lymphoid tissue hyperplasia, T2P hyperplasia, and bronchoalveolar (mixed-cell) hyperplasia. Slides stained with Masson’s trichrome were analyzed by quantitative microscopy. Images were taken using 40× objective at 5 regions of interest per lung. Five adjacent, non-overlapping images were captured at each region of interest for a total of 25 images per sample. Areas of high background collagen, such as conducting airways, were avoided. Images were analyzed quantitatively in ImageJ (http://rsbweb.nih.gov/ij/) using color thresholding. Briefly, images were threshold-masked to only display the blue collagen stain. Images were converted to 8-bit form for intensity thresholding and the resulting images (representing high-collagen areas) were measured in terms of pixel intensity. Images were batch-analyzed (identically processed) in this way and the results were analyzed for mean pixel intensity across the whole image. Statistical analysis between groups used a one-way ANOVA with Tukey post-hoc analysis. To obtain representative images from H&E and Masson’s trichrome stained sections, slides were digitally scanned by the CCHMC Pathology Research Core and images were extracted using Aperio ImageScope software.
For quantitative assessment of T2P proliferation, IHC was carried out using standard protocols for deparafinization, rehydration and sodium-citrate antigen retrieval. Sections were blocked with 1% BSA in PBS and stained with anti-proSPC for unambiguous identification of T2Ps and Ki67 to indicate active proliferation. DAPI was used as a counterstain. Appropriate secondary antibodies conjugated to DyLight 488 or DyLight 594 were used to visualize staining. All IHC slides were imaged on a Nikon (Melville, NY) Eclipse 50i fluorescent microscope equipped with an EXFO X-Cite 120 lamp (Lumen Dynamics, Ontario, Canada) and Nikon DS-5M camera system. For fluorescent IHC, three-color overlays were generated using Adobe Photoshop (www.adobe.com) and individual cells were scored as positive or negative for given stains. Cytoplasm of T2Ps exhibited robust staining for proSPC and number of proSPC+ cells was divided by the number of alveoli in the field to measure the average number of T2Ps per alveolus. About 60 alveoli total in regions of interest were measured blind per sample. To assess apoptosis in T2Ps, terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay was performed by the CCHMC Pathology Core. TUNEL staining, along with proSPC staining in serial sections to identify T2P cells, was visualized using DAB.
In further IHC work, sections were co-stained as above with anti-proSPC and anti-IL-1ß with DAPI counterstain. For counting of IL-1ß-positive total cells and T2Ps, positives were normalized to the number of alveoli in the field. Statistical significance of differences between vehicle, CNT-, and CA-exposed groups in these analyses were evaluated using one-way ANOVA with Tukey post-hoc analysis.
Results
Physiochemical Characterization
CNTs prepared for instillation were present as tangled, loose agglomerates (Fig. 1) with occasional free single fibers. The average diameter of CNTs was 10.4nm, with a range of 4.4–21.9nm. Entanglement prevented the accurate measurement of length but most fibers appeared to be longer than 1μm. Agglomerates were roughly in the range of 200–400nm across. CA fibers were present as single fiber particles and splintered fibers. The average CA fiber diameter was 269nm with a range of 116–627nm. CA lengths ranged from 1.87–16.55μm with an average of 6.48μm. Zeta potentials of suspensions were −0.17±0.6 for CNTs and −17.1±1.49 for CA. CNTs in this study exhibited negligible levels of metal contamination. Further characterization for CNTs is available as published previously (Frank et al, 2015).
Fig. 1. Morphology and size distribution of carbon nanotubes (CNTs) and crocidolite asbestos (CA) materials used in this study.

Left side: CNTs presented as loose tangled agglomerates with an average fiber diameter of 10.4 nm. Right side: CA was composed mostly of individual fibers with average diameter of 269.2 nm.
Clinical and Gross Findings
Animals administered either particle exposure did not show any overt clinical symptoms during the exposure period. One animal (from the CA-treated group) was euthanized during the experiment due to injuries sustained from fighting, thus n=8 for CNT- and vehicle-treated groups and n=7 for CA-treated for all histopathological analyses (except where noted). Repeated administrations via oropharyngeal aspirations modestly increased the prevalence of alveolar macrophages (AMs) in the bronchoalveolar space. The distal regions of the lung tissue exhibited more pronounced crowding of AMs in the alveoli, which were characterized by abundant foamy cytoplasm in the vehicle-treated animals as well as particle-exposed lungs. This finding is an expected alteration related to administration of amphiphilic Pluronic surfactant used in the vehicle suspension.
Histopathology
Pathology scores for observed histological features are presented in Fig. 2 and are discussed below. The general extent and distribution of lung pathologies resulting from exposure are shown at low magnification in Figs. 3 and 4. Vehicle treatments did not induce any widespread changes in lung architecture or tissue appearance beyond accumulation of alveolar foamy macrophages (AMs). Limited (scores of ‘1′) T2P hyperplasia (focal) was noted in 3 of 8 vehicle-treated animals. Animals exposed to CNTs or CA both featured mild to moderate multifocal alveolar accumulations of AMs, lymphocytes, and lesser numbers of neutrophils (granulomatous inflammatory foci), with multifocal nodular aggregates of inflammatory cells expanding the alveoli (microgranulomas). AMs in CNT- or CA-treated animals contained variable amounts of coarse pigmented material with the occasional presence of multinucleated AMs.
Fig 2. CNTs and crocidolite cause overlapping but distinct profiles of lung pathology.

Vehicle-treated lungs exhibited little or no microscopic alterations. Both particle-treated groups showed granulomatous inflammation, composed chiefly of macrophages mixed with lymphocytes and low numbers of neutrophils. CNT-exposed lungs predominantly showed lymphoid tissue hyperplasia (interstitial and perivascular) and type II pneumocyte hyperplasia while crocidolite-treated lungs predominantly showed bronchoalveolar hyperplasia.
Fig 3. Extent and distribution of CA- or CNT-induced lung lesions.

Hematoxylin and eosin (H&E) stains. Vehicle-treated lung, (A) 2× and (B) 4×, (C) CA-exposed lung showed granulomatous inflammation and epithelial hyperplasia predominantly at terminal airways and alveolar ducts, (C) 2× and (D) 4×. CNT-exposed lung showed diffuse alveolar hyperplasia in addition to granulomatous inflammation, (E) 2× and (F) 4×.
Fig 4. Regions of interest in CA- or CNT-exposed lung.
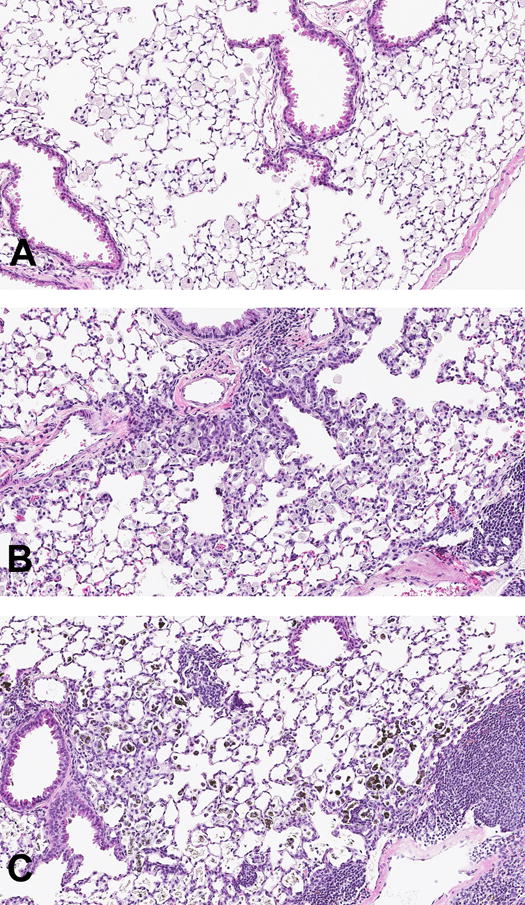
H&E stains, 10×. (A) Vehicle treated lungs showed no change in airway or alveolar structure. (B) CA exposures caused hyperplasia of terminal bronchioles and alveolar ducts. (C) CNT exposures caused hyperplasia of alveolar epithelium and BALT/lymphoid tissue. CNT-laden macrophages are visible as darkly pigmented bodies.
CNT-exposed animals had marked locally extensive T2P hyperplasia, characterized by plump epithelial cells lining the alveolar walls in areas proximal to CNT-laden AMs aggregates (Fig. 5). CNT-exposed lungs also had interstitial and perivascular foci of hyperplastic lymphoid tissue, comprising mostly of small lymphocytes. These irregular lymphoid expansions were observed both adjacent to airways (bronchial/ole associated lymphoid tissue, BALT) and proximal to the more severe areas of epithelial hyperplasia.
Fig 5. CA- or CNT-induced lung epithelial hyperplasia.
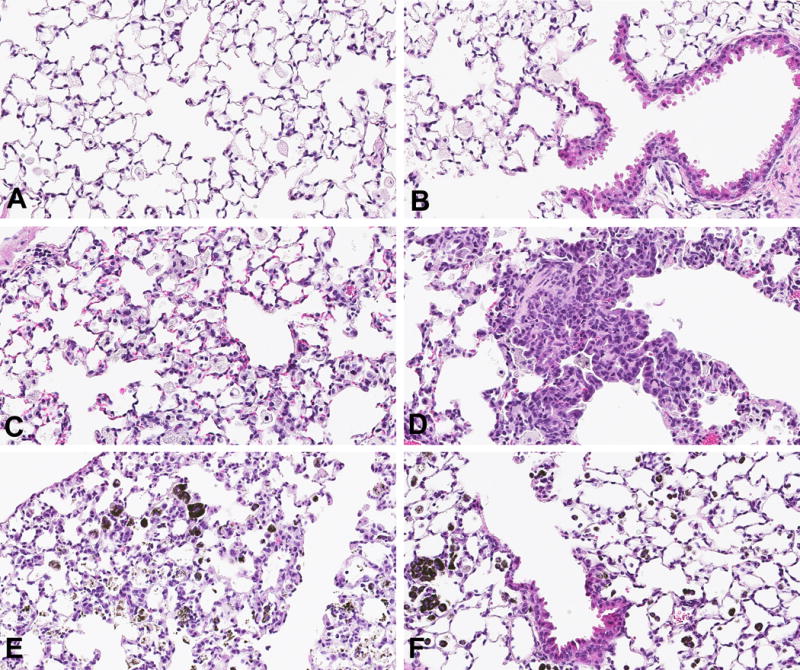
H&E stains, 20×. Vehicle-treated lungs showed little or no type II pneumocyte (T2P) and bronchoalveolar hyperplasia (A & B, respectively). CA-exposed lung showed modest T2P hyperplasia (C) but prominent bronchoalveolar hyperplasia (D). CNT-exposed lung showed more diffuse and severe T2P hyperplasia (E), while showing relatively little bronchoalveolar hyperplasia and peribronchial changes (F) in contrast with what is seen in CA-exposed lung.
In contrast, CA exposures induced significantly less severe T2P hyperplasia and less lymphoid hyperplasia. Individual CA fibers were visible in association with epithelium and AMs. CA-exposed lungs featured bronchoalveolar hyperplasia predominantly at terminal bifurcations and alveolar ducts, which was minimal in CNT-exposed lungs. Neither CNT- nor CA-treated lungs had any appreciable microscopic findings in the pleura.
Masson’s trichrome stain analysis
Increased collagen staining was observed in either particle-exposed lungs and was more predominant in areas of alveolar inflammation and, especially, adjacent to epithelial hyperplastic lesions (Fig. 6). Quantitative microscopy analysis of sections stained with Masson’s trichrome and showed that both CNT and CA exposures increased collagen staining to a similar extent in lung tissue (the increases were statistically non-significant). One sample (CA-treated) was omitted due to faulty staining, thus n=6 for the CA-treated group in this analysis. Additionally, either toxicant induced distinct patterns of interstitial fibrosis, with CA inducing more extensive peribronchial and perivascular foci and CNTs inducing nodular fibrotic foci in alveolar regions.
Fig 6. CA- or CNT-induced interstitial remodeling.
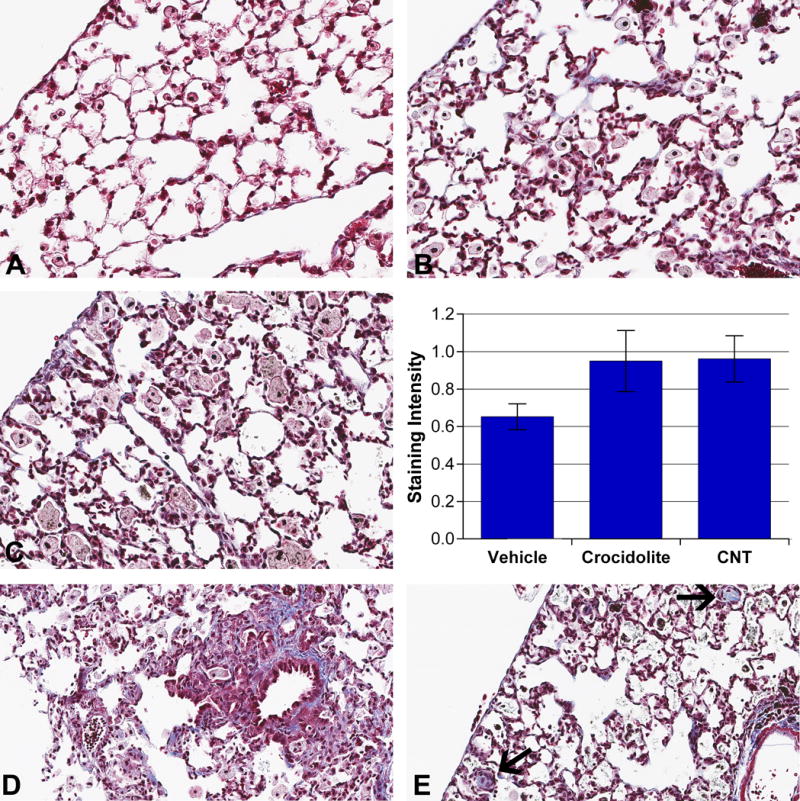
Masson’s trichrome stain, 40×. Vehicle-treated lungs showed no or minimal interstitial collagen staining (A). CA-exposed (B) and CNT-exposed (C) lungs showed increased interstitial collagen staining. Graph: Images of left lung sections stained with Masson’s trichrome were taken in bronchoalveolar regions (25 per slide). Collagen-specific color was isolated using color thresholding in ImageJ and resulting images were measured by mean pixel intensity. Distinct patterns of interstitial fibrosis following exposure were observed where (D) CA induced prominent perivascular and peribronchial fibrotic foci while (E) CNTs induced nodular fibrotic foci (arrows) in alveolar spaces following 8wk exposure to 300μg of either material.
Immunofluorescence Histochemistry for T2Ps
Both CNT and CA exposures significantly increased T2Ps/alveolus compared to vehicle (Fig. 7). CNT-exposed lungs exhibited significantly increased T2Ps compared to CA-exposed. Characteristic hyperplastic epithelial lesions induced by CNT exposures were confirmed to be composed of T2Ps. Hyperplastic lesions associated with CA exposures were shown to be comprised of proSPC+ and proSPC− cells. No trends were observed in the number of alveoli/field. Nuclei of actively proliferating cells within the BALT stained positive for Ki67 and served as internal positive control. No Ki67+ staining was observed in T2Ps in any group. Collectively, the data show that CNT exposures induce T2P hyperplasia by increasing number of T2Ps and do so to a significantly greater extent than CA exposures. The absence of any observed Ki67 staining indicates that proliferation of T2Ps may not be actively occurring at the time of sampling. This suggests that, although active proliferation of T2Ps may have ceased within the 5 weeks following cessation of exposure, hyperplastic T2Ps remain the predominant cell type lining the alveoli at this time point.
Fig. 7. ProSPC+ type II pneumocytes (T2Ps) are increased in hyperplastic lesions induced by CNT exposure.

Green: proSPC, Red: Ki-67, with DAPI. Pro-SPC+ cells were minimal in vehicle-treated lungs (A), increased in CA-treated (B), and especially prevalent in CNT-treated (C). Graph: proSPC+ cells were counted manually in lung sections and divided by number of alveoli in the field. CNT-exposed lungs had greatest numbers of type II cells/alveolus, with lesser increase seen with crocidolite exposure. Ki-67+ T2Ps were not observed. **p<.01.
TUNEL assay for apoptosis in T2Ps
Serial sections stained for proSPC and TUNEL, respectively, revealed minimal apoptosis in general (Fig. 8). Enumeration of apoptotic cells in three fields of T2P hyperplasia/sample showed a marginally increased, non-significant prevalence of apoptotic nuclei (not specific to cell type) in CNT- and CA-exposed lungs. Apoptotic cells were also noted to be somewhat more prevalent in regions of marked CA- or CNT-induced granulomatous lesions (not shown). No differences in the number of apoptotic T2Ps were observed, although it was not possible to confirm the identities of some apoptotic cells dependent on the degree of correlation between contiguous serial sections stained for either proSPC or TUNEL. The results here indicate that CNT and CA do not differ considerably in their capacity to induce apoptosis in T2Ps and suggest the differences in T2P hyperplasia are not due to increased cell death in CA-exposed T2Ps.
Fig. 8. Prevalence of apoptotic T2Ps does not differ between CNT- and CA-induced hyperplastic lesions.

Left column: proSPC-stained sections, right column: TUNEL-stained sections, 40× (Hematoxylin counterstain in both sets). Cytoplasm of T2Ps stained dark brown using DAB visualization of proSPC in (A) vehicle-treated, (C) CA-exposed, and (E) CNT-exposed alveolar epithelial lesions. TUNEL staining resulted in purple apoptotic nuclei in serial sections of the lesions (B, E, F, respectively). In general, significant prevalence or increases of apoptotic cells were not observed in any group. Serial sectioning allowed for TUNEL+ nuclei to be co-identified (where possible) as proSPC+ cells (see arrows, C and D, which denote a TUNEL+/proSPC− cell). TUNEL+ T2P cells were uncommon and no trends were observed between exposure groups.
Gene expression in T2Ps
Gene expression of Cxcl1, Cxcl5, Ccl2, and IL-1ß were markedly increased in T2Ps isolated from CNT-exposed lungs compared to vehicle-treated (Fig. 9). CA exposures only modestly increased Cxcl1, Ccl2, and IL-1ß expression and modestly decreased Cxcl5 expression.
Fig. 9. CNT exposures more strongly induced pro-inflammatory mediators in type II pneumocytes (T2Ps) compared to crocidolite.

RNA from isolated T2Ps was subjected to qRT-PCR. Values shown are the relative change from vehicle-treated samples.
Immunofluorescent Histochemistry for IL-1ß
Anti-IL-1ß and anti-proSPC antibodies were used to quantify IL-1ß+ cells and co-identify these with T2Ps in co-stained sections (Fig. 10). The cytoplasm of IL-1ß-expressing cells were stained by anti-IL-1ß and counted manually. Total IL-1ß+ cells were normalized to the number of total cells as enumerated by DAPI-counterstained nuclei and increases were expressed as fold change over vehicle-treated. Three fields were counted blind per sample. Exposures to CNTs or CA both increased total IL-1ß+ cells, with CNT exposures causing a significant difference and CA causing a less significant increase. The number of total IL-1ß+ cells did not differ substantially between treatments. In the same images, cells double-positive for proSPC and IL-1ß were counted and expressed as average number of IL-1ß+ T2Ps per field. No IL-1ß+ T2Ps were observed in the vehicle group, while they appeared modestly increased following CA exposure. Numbers of IL-1ß+ T2Ps were significantly increased in CNT-exposed lung and substantially higher than CA-exposed. This agrees with gene expression data showing increased IL-1ß expression in T2Ps isolated from CNT-exposed lungs. The data collectively show that while both exposures tend to induce the mature IL-1ß protein in lung cells, CNT exposures do so more specifically in T2Ps.
Fig. 10. CNT and CA increase IL-1ß in lung cells, and CNT exposures induce more IL-1ß+ type II cells than CA.

Green: proSPC, Red: IL-1ß, with DAPI. (A) Vehicle-treated lungs showed minimal IL-1ß staining. (B) CA-treated lungs showed IL-1ß increases which were not specific to T2Ps, whereas (C) CNT-treated lungs showed IL-1ß+ T2Ps. Upper graph: IL-1ß+ cells were counted manually and normalized to total number of nuclei. Changes between treatments are expressed as fold change over vehicle. Lower graph: Cells positive for both pro-SPC and IL-1ß were counted and normalized to total cells. Values are average number of double positive cells/field. *p<0.05, **P<0.01.
Discussion
This study compares and contrasts profiles of lung toxicologic pathology following exposures to multi-wall CNTs or CA with results showing that CNTs reproducibly elicit distinct lesions relative to CA particularly with respect to alveolar T2Ps. Although CNT- and asbestos-exposed lung pathology in rodent models have been described individually, to our knowledge, this is the first comparison study to focus on side-by-side assessment of morphological and mechanistic endpoints. Our findings are consistent with and build upon previously published work using mice (Adamson et al, 1987, Bonner et al, 2013, Mercer et al, 2011). It is widely appreciated that the shared characteristics between CNTs and asbestos warrant concern and investigation into their toxicological similarities. Because no history of human exposure to CNTs yet exists, these investigations must use experimental models to gauge the extent to which asbestos and CNTs overlap in their toxicological actions. In vivo comparison studies of CNTs and asbestos often focus on effects in the pleural tissue and mesothelial lining, with the explicit goal of assessing carcinogenic potential as asbestos exposure is notoriously associated with mesothelioma and lung cancer. Two prominent studies (Poland et al, 2008, Takagi et al, 2008) compared the effects of asbestos and CNTs in the pleural lining of the intraperitoneal cavity in mice, agreeing that hyperplastic and neoplastic changes suggest that CNTs may induce preneoplastic lesions. However, such studies frequently exclude comparisons of basic lung pathology caused by either toxicant and appear focused on specific pleural endpoints at the expense of more comprehensive hypothesis-driven investigation of toxicologic pathology. Furthermore, asbestos exposures also cause detrimental effects apart from carcinogenesis, such as interstitial fibrosis and granulomatous inflammation (Wagner, 1997). These effects alone represent a public health burden by directly impacting lung function and complicating coexisting conditions.
Interpretations of experimental results intended to provide information relevant to human health must consider limitations in the ability to accurately model realistic exposure situations. Although many studies take advantage of the lower background variability and specialized phenotypes offered by inbred mouse strains, we opted to use an out-bred mouse model for its more robust health and more realistic modeling of a heterogenous human population. The most relevant route of exposure to CNTs is expected to be inhalation, followed by skin contact. To model inhalation, the current work used oropharyngeal aspiration methods similar to those used in the majority of existing toxicological studies of CNTs. Shvedova et al (2008) showed lung responses to inhaled doses of single-walled CNTs in C57/Bl6 mice were more severe than those following comparable aspirated doses, suggesting the similar trend could be found for the case of multi-wall CNTs. Although inhalation-based studies are more environmentally realistic in nature, they are technically challenging and not feasible in many labs. Future studies should nonetheless consider using inhalational exposures where feasible, particularly where risk assessment and the derivation of safe exposure limits are goals. A further consideration concerns how doses given in animal models approximate actual exposure burdens expected in human occupational scenarios. The repeated doses of CNTs in this study reached a cumulative total of 300 μg/animal. Recent monitoring in modern CNT production facilities reported a mean airborne CNT concentration of 42.6μg/m3 (Kuijpers et al, 2014). Assuming a human light-work breath volume of 1L at 20 breaths a minute, we may predict an inhaled dose of 70μg/8hr workday in a realistic scenario using the Multiple Path Particle Dosimetry model as utilized by Porter et al (2010). Scaling from a mouse alveolar epithelial surface area of .05m2 (Stone et al, 1992) to a human surface area of 102m2, we may predict our model reflects an occupational exposure dose which may be reached in 33.6 years given a 5-day workweek. The CNT exposure dose used here, although high relative to the mouse, therefore approximates exposure burdens humans may feasibly accumulate within a normal occupational lifetime. Nonetheless, as the objective of this study was to compare the relative effects of two toxicants and not to model or assess risk in human exposures, it should be noted that numerous uncertainty factors would need to be considered in extrapolating these results to a realistic occupational health context.
In this study, CA induced bronchoalveolar hyperplasia which was most prominent in alveolar ducts and terminal bifurcations. This is consistent with published reports that amphibole asbestos preferentially induces lesions in distal airways and alveolar ducts in rodents (Manning et al, 2002, Shannahan et al, 2012). Other exposures known to cause bronchoalveolar hyperplasia include silicon carbide whiskers (Akiyama et al, 2007) and repeated ozone (Hassett et al, 1985). In contrast, CNT-induced hyperplastic changes were prominent throughout the alveolar spaces and characterized by T2P hyperplasia. T2Ps isolated from CNT-exposed lungs displayed a proinflammatory phenotype compared to CA and expressed IL-1ß. Several factors may contribute to the differences in toxicological pathology observed here, including scale and fiber morphology differences. CNTs used in this study were thin (mean diameter: 10.4nm), non-rigid fibers forming 200–400nm agglomerates, whereas CA fibers were rigid fibers 269.2nm in average diameter and up to 16μm long. Published work (Hesterberg et al, 1996, Lippmann, 1990, Nagai et al, 2011) robustly supports the role of size and morphology as possible pathogenic determinants in fiber-induced injury, thus different paradigms of fiber toxicology may be induced by thin, agglomerated CNT fibers versus thick, rigid CA fibers. It has been observed (Hirano et al, 2008) that multi-wall CNTs in the diameter range of 60–80nm are rigid and exist as fibers comparable to CA as opposed to the fibrous particulates formed by the thinner CNTs used in the current study. T2Ps or T1Ps may be sensitive to CNTs based on these differences in dimension. Furthermore, CNTs may preferentially come to rest in alveoli in proximity to alveolar pneumocytes as a result of their morphology and/or size whereas CA fibers may accumulate in distal airways and ducts without readily contacting T1Ps and T2Ps. It was not possible to accurately monitor the deposition of CNTs or CA with regard to localization in this study.
Apart from size and morphology differences, CNTs and CA differed in their metal content. CNTs had negligible metal contamination and no detectable iron, whereas CA is known to contain iron oxides (Gulumian et al, 1993). We expect the presence of iron may play a role in the differences seen in lung pathology. On further note, CNTs in this study present a large surface area of 193.6 m2/g (Frank et al, 2015), compared to the ~8 m2/g normally observed in UICC crocidolite (Goodglick et al, 1990, Gulumian et al, 1993). The high surface area seen in CNTs may underpin qualitative differences in their toxicity versus CA (Hubbs et al, 2011).
We show here that T2Ps are particularly more affected by CNT exposures relative to CA. This was most evident in histopathologically distinct T2P hyperplasia, increased T2P number which cannot be explained by differences in apoptosis, and expression of IL-1ß specific to T2Ps. T2P hyperplasia occurs as a reparative response of the alveolar epithelium in an attempt to reestablish alveolar wall integrity and function. In a classic model of experimental lung fibrosis, bleomycin induces hyperplasia of and apoptosis in T2Ps (Wang et al, 2000) as well as altered IL-1ß signaling and it is substantiated that these effects underlie bleomycin-driven fibrosis in the lung (Gasse et al, 2007). Both toxicant exposures increased collagen staining in the lung interstitium as assessed by Masson’s trichrome. Although CA is a classical pro-fibrotic agent, we did not observe extensive fibrosis following exposure to either toxicant despite using relatively high cumulative doses. We suggest that extensive fibrotic foci were not seen due firstly to the duration of exposure, as fibrotic responses may not be extensive at this time of sampling and are expected to continue to develop based on previous studies in rodents (Adamson et al, 1987, Cyphert et al, 2012, Davis et al, 1978). Second, we note that extensive fibrotic responses are commonly shown in inbred strains of mice known to be susceptible to fibrosis (e.g., C57Bl/6), whereas the outbred animals in this study may be relatively resistant to pulmonary fibrosis (Schrier et al, 1983). In addition, inhalation routes may be more conducive to inducing fibrotic reactions (Shvedova et al, 2008). The present findings appear consistent with previous studies (Davis et al, 1978, Kamp et al, 1993, Wagner et al, 1974) of amphibole asbestos which used outbred rodent models of exposure.
Conclusion
In conclusion, we show that CNT toxicity appears to directly affect alveolar pneumocytes and T2Ps to produce features of lung pathology both common and distinct from asbestos. These differences may precede or be associated with different long-term outcomes in cases of CNT or asbestos exposure. Future work should address the extent to which this observation is true using CNTs of different size and morphology and what role this may have in the mechanism of action of CNTs versus asbestos in their respective exposure-related outcomes. Although research interests in the mechanisms of asbestos toxicity have historically been somewhat limited despite the notoriety of this toxicant, the rise of CNT materials underscores a critical need to conduct comprehensive hypothesis-based research into these materials both separately and in comparison.
Acknowledgments
This work was supported by the National Institutes of Health/National Institute for Environmental Health Sciences Center for Environmental Genetics (CEG) funds [2P30ES006096-16A1 to JSY], Gene-Environment Interactions Training Program (GEITP) fellowship [T32ES016646 to EAF], and National Institute of Occupational Safety and Health/Education and Research Center (ERC) funding [T42OH008432-07 to EAF], and the University of Cincinnati funds (to JSY).
Abbreviations
- CNT
carbon nanotubes
- CA
crocidolite asbestos
- T2P
type II pneumocyte
- T1P
type I pneumocyte
- AM
alveolar macrophage
- IL-1ß
interleukin 1ß
- proSPC
pro-surfactant protein C
- TEM
transmission electron microscopy
- MACS
magnetically-assisted cell sorting
- qRT-PCR
quantitative real-time polymerase chain reaction
- FBS
fetal bovine serum
References
- Adamson IY, Bowden DH. Response of mouse lung to crocidolite asbestos. 2. Pulmonary fibrosis after long fibres. The Journal of pathology. 1987;152(2):109–117. doi: 10.1002/path.1711520207. [DOI] [PubMed] [Google Scholar]
- Akiyama I, Ogami A, Oyabu T, Yamato H, Morimoto Y, Tanaka I. Pulmonary effects and biopersistence of deposited silicon carbide whisker after 1-year inhalation in rats. Inhalation toxicology. 2007;19(2):141–147. doi: 10.1080/08958370601051784. [DOI] [PubMed] [Google Scholar]
- Birch ME. Monitoring of diesel particulate exhaust in the workplace. In: Schlecht PC, O’Connor PF, editors. NIOSH Manual of Analytical Methods (NMAM) 4th. Department of Health and Human Services, Public Health Service, Center for Disease Control and Prevention, National Institute for Occupational Safety and Health. DHHS(NIOSH); 154, Cincinnati, Ohio: 2004a. Third Supplement to NMAM. [Google Scholar]
- Birch ME. NIOSH Method 5040 update. In: Schlecht PC, O’Connor PF, editors. NIOSH Manual of Analytical Methods (NMAM) 4th. Department of Health and Human Services, Public Health Service, Center for Disease Control and Prevention, National Institute for Occupational Safety and Health. DHHS(NIOSH); 154, Cincinnati, Ohio: 2004b. Third Supplement to NMAM. [Google Scholar]
- Bonner JC, Silva RM, Taylor AJ, Brown JM, Hilderbrand SC, Castranova V, Porter D, Elder A, Oberdörster G, Harkema JR, Bramble LA, Kavanagh TJ, Botta D, Nei A, Pinkerton KE. Interlaboratory evaluation of rodent pulmonary responses to engineered nanomaterials: the NIEHS Nano GO Consortium. Environmental health perspectives. 2013;121(6):676–682. doi: 10.1289/ehp.1205693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyphert JM, Padilla-Carlin DJ, Schladweiler MC, Shannahan JH, Nyska A, Kodavanti UP, Gavett SH. Long-term response of rats to single intratracheal exposure of Libby amphibole or amosite. Journal of Toxicology and Environmental Health, Part A. 2012;75(3):183–200. doi: 10.1080/15287394.2012.641203. [DOI] [PubMed] [Google Scholar]
- Dahm MM, Evans DE, Schubauer-Berigan MK, Birch ME, Fernback JE. Occupational exposure assessment in carbon nanotube and nanofiber primary and secondary manufacturers. Ann Occup Hyg. 2012;56(5):542–556. doi: 10.1093/annhyg/mer110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JM, Beckett ST, Bolton RE, Collings P, Middleton AP. Mass and number of fibres in the pathogenesis of asbestos-related lung disease in rats. British journal of cancer. 1978;37(5):673. doi: 10.1038/bjc.1978.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Volder MF, Tawfick SH, Baughman RH, Hart AJ. Carbon nanotubes: present and future commercial applications. Science. 2013;339(6119):535–539. doi: 10.1126/science.1222453. [DOI] [PubMed] [Google Scholar]
- Donaldson K, Stone V, Clouter A, Renwick L, MacNee W. Ultrafine particles. Occupational and environmental medicine. 2001;58(3):211–216. doi: 10.1136/oem.58.3.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dörger M, Münzing S, Allmeling AM, Krombach F. Comparison of the phagocytic response of rat and hamster alveolar macrophages to man-made vitreous fibers in vitro. Human & experimental toxicology. 2000;19(11):635–640. doi: 10.1191/096032700669330178. [DOI] [PubMed] [Google Scholar]
- Fehrenbach H. Alveolar epithelial type II cell: defender of the alveolus revisited. Respir Res. 2001;2(1):33–46. doi: 10.1186/rr36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasse P, Mary C, Guenon I, Noulin N, Charron S, Schnyder-Candrian S, Schnyder B, Akira S, Quesniaux VFJ, Ryffel B, Couillin I. IL-1R1/MyD88 signaling and the inflammasome are essential in pulmonary inflammation and fibrosis in mice. The Journal of clinical investigation. 2007;117(12):3786. doi: 10.1172/JCI32285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodglick LA, Kane AB. Cytotoxicity of long and short crocidolite asbestos fibers in vitro and in vivo. Cancer research. 1990;50(16):5153–5163. [PubMed] [Google Scholar]
- Gulumian M, Bhoolia DJ, Dutoit RSJ, Rendall REG, Pollak H, Vanwyk JA, Rhempula M. Activation of UICC crocidolite: the effect of conversion of some ferric ions to ferrous ions. Environmental research. 1993;60(2):193–206. doi: 10.1006/enrs.1993.1027. [DOI] [PubMed] [Google Scholar]
- Han JH, Lee EJ, Lee JH, So KP, Lee YH, Bae GN, Lee S, Ji JH, Cho MH, Yu IJ. Monitoring multiwalled carbon nanotube exposure in carbon nanotube research facility. Inhalation Toxicol. 2008;20(8):741–749. doi: 10.1080/08958370801942238. [DOI] [PubMed] [Google Scholar]
- Hassett C, Mustafa MG, Elashoff RM. Murine lung carcinogenesis following exposure to ambient ozone concentrations. Journal of the National Cancer Institute. 1985;75(4):771–777. [PubMed] [Google Scholar]
- Hesterberg TW, Miiller WC, Musselman RP, Kamstrup O, Hamilton RD, Thevenaz P. Biopersistence of man-made vitreous fibers and crocidolite asbestos in the rat lung following inhalation. Toxicological Sciences. 1996;29(2):267–279. doi: 10.1006/faat.1996.0031. [DOI] [PubMed] [Google Scholar]
- Hirano S, Kanno S, Furuyama A. Multi-walled carbon nanotubes injure the plasma membrane of macrophages. Toxicology and applied pharmacology. 2008;232(2):244–251. doi: 10.1016/j.taap.2008.06.016. [DOI] [PubMed] [Google Scholar]
- Hubbs AF, Mercer RR, Benkovic SA, Harkema J, Sriram K, Schwegler-Berry D, Goravanahally MP, Nurkiewicz TR, Castranova V, Sargent LM. Nanotoxicology—A pathologist’s perspective. Toxicologic pathology. 2011;39(2):301–324. doi: 10.1177/0192623310390705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones AD, Miller BG, Cullen RT, Searl A, Davis JMG, Buchanan D, Donaldson K, Soutar CA, Bolton RE. The colt fibre research programme: aspects of toxicological risk assessment. The Annals of Occupational Hygiene. 1997;41:244–250. [Google Scholar]
- Kamp DW, Israbian VA, Yeldandi AV, Panos RJ, Graceffa P, Weitzman SA. Phytic acid, an iron chelator, attenuates pulmonary inflammation and fibrosis in rats after intratracheal instillation of asbestos. Toxicologic pathology. 1995;23(6):689–695. doi: 10.1177/019262339502300606. [DOI] [PubMed] [Google Scholar]
- Kuijpers E, Vermeulen R, Tromp P, Fransman W, Brouwer D, Godderis L, Vlaanderen J, Bekker C, Pronk A. 0195 Carbon nanotube exposure assessment for a study on early biological effects; the CANTES study. Occup Environ Med. 2014;71(Suppl 1):A25–A25. [Google Scholar]
- Lippmann M. Effects of fiber characteristics on lung deposition, retention, and disease. Environmental Health Perspectives. 1990;88:311. doi: 10.1289/ehp.9088311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning CB, Cummins AB, Jung MW, Berlanger I, Timblin CR, Palmer C, Taatjes DJ, Hemenway D, Vacek P, Mossman BT. A mutant epidermal growth factor receptor targeted to lung epithelium inhibits asbestos-induced proliferation and proto-oncogene expression. Cancer research. 2002;62(15):4169–4175. [PubMed] [Google Scholar]
- Mercer RR, Hubbs AF, Scabilloni JF, Wang L, Battelli LA, Friend S, Castranova V, Porter DW. Pulmonary fibrotic response to aspiration of multi-walled carbon nanotubes. Part Fibre Toxicol. 2011;8(1):21. doi: 10.1186/1743-8977-8-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BG, Searl A, Davis JM, Donaldson K, Cullen RT, Bolton RE, Buchanan D, Soutar CA. Influence of fibre length, dissolution and biopersistence on the production of mesothelioma in the rat peritoneal cavity. Annals of Occupational Hygiene. 1999;43(3):155–166. [PubMed] [Google Scholar]
- Nagai H, Okazaki Y, Chew SH, Misawa N, Yamashita Y, Akatsuka S, Ishihara T, Yamashita K, Yoshikawa Y, Yasui H, Jiang L, Ohara H, Takahashi T, Ichihara G, Kostarelos K, Miyata Y, Shinohara H, Toyokuni S. Diameter and rigidity of multiwalled carbon nanotubes are critical factors in mesothelial injury and carcinogenesis. Proceedings of the National Academy of Sciences. 2011;108(49):E1330–E1338. doi: 10.1073/pnas.1110013108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai H, Toyokuni S. Differences and similarities between carbon nanotubes and asbestos fibers during mesothelial carcinogenesis: shedding light on fiber entry mechanism. Cancer science. 2012;103(8):1378–1390. doi: 10.1111/j.1349-7006.2012.02326.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberdörster G. Lung clearance of inhaled insoluble and soluble particles. Journal of Aerosol Medicine. 1988;1(4):289–330. [Google Scholar]
- Oberdörster G. Lung particle overload: implications for occupational exposures to particles. Regulatory Toxicology and Pharmacology. 1995;21(1):123–135. doi: 10.1006/rtph.1995.1017. [DOI] [PubMed] [Google Scholar]
- Poland CA, Duffin R, Kinloch I, Maynard A, Wallace WA, Seaton A, Stone V, Brown S, MacNee W, Donaldson K. Carbon nanotubes introduced into the abdominal cavity of mice show asbestos-like pathogenicity in a pilot study. Nature nanotechnology. 2008;3(7):423–428. doi: 10.1038/nnano.2008.111. [DOI] [PubMed] [Google Scholar]
- Pope CA, III, Thun MJ, Namboodiri MM, Dockery DW, Evans JS, Speizer FE, Heath CW., Jr Particulate air pollution as a predictor of mortality in a prospective study of US adults. American journal of respiratory and critical care medicine. 1995;151(3_pt_1):669–674. doi: 10.1164/ajrccm/151.3_Pt_1.669. [DOI] [PubMed] [Google Scholar]
- Porter DW, Hubbs AF, Mercer RR, Wu N, Wolfarth MG, Sriram K, Leonard S, Battelli L, Schwegler-Berry D, Friend S, Andrew M, Chen BT, Tsuruoka S, Endo M, Castranova V. Mouse pulmonary dose-and time course-responses induced by exposure to multi-walled carbon nanotubes. Toxicology. 2010;269(2):136–147. doi: 10.1016/j.tox.2009.10.017. [DOI] [PubMed] [Google Scholar]
- Rao GVS, Tinkle S, Weissman D, Antonini J, Kashon M, Salmen R, Battelli L, Willard P, Hubbs A, Hoover M. Efficacy of a technique for exposing the mouse lung to particles aspirated from the pharynx. Journal of Toxicology and Environmental Health Part A. 2003;66(15–16):1441–1452. doi: 10.1080/15287390306417. [DOI] [PubMed] [Google Scholar]
- Rom WN, Bitterman PB, Rennard SI, Cantin A, Crystal RG. Characterization of the lower respiratory tract inflammation of nonsmoking individuals with interstitial lung disease associated with chronic inhalation of inorganic dusts. American Review of Respiratory Disease. 1987;136(6):1429–1434. doi: 10.1164/ajrccm/136.6.1429. [DOI] [PubMed] [Google Scholar]
- Ryman-Rasmussen JP, Tewksbury EW, Moss OR, Cesta MF, Wong BA, Bonner JC. Inhaled multiwalled carbon nanotubes potentiate airway fibrosis in murine allergic asthma. American journal of respiratory cell and molecular biology. 2009;40(3):349–358. doi: 10.1165/rcmb.2008-0276OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sargent LM, Porter DW, Staska LM, Hubbs AF, Lowry DT, Battelli L, Siegrist KJ, Kashon ML, Mercer RR, Bauer KB, Chen BT, Salisbury JL, Frazer D, McKinney W, Andrew M, Tsuruoka S, Endo M, Fluharty KL, Castranova V, Reynolds SH. Promotion of lung adenocarcinoma following inhalation exposure to multi-walled carbon nanotubes. Part Fibre Toxicol. 2014;11(1):3. doi: 10.1186/1743-8977-11-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannahan JH, Nyska A, Cesta M, Schladweiler MC, Vallant BD, Ward WO, Ghio AJ, Gavett SH, Kodavanti UP. Subchronic pulmonary pathology, iron overload, and transcriptional activity after Libby amphibole exposure in rat models of cardiovascular disease. Environmental health perspectives. 2012;120(1):85. doi: 10.1289/ehp.1103990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shvedova AA, Kisin E, Murray AR, Johnson VJ, Gorelik O, Arepalli S, Hubbs AF, Mercer RR, Keohavong P, Sussman N, Jin J, Yin J, Stone S, Chen BT, Deye G, Maynard A, Castranova V, Baron PA, Kagan VE. Inhalation vs. aspiration of single-walled carbon nanotubes in C57BL/6 mice: inflammation, fibrosis, oxidative stress, and mutagenesis. American Journal of Physiology-Lung Cellular and Molecular Physiology. 2008;295(4):L552–L565. doi: 10.1152/ajplung.90287.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrier DJ, Kunkel RG, Phan SH. The Role of Strain Variation in Murine Bleomycin-lnduced Pulmonary Fibrosis 1, 2. American Review of Respiratory Disease. 1983;127(1):63–66. doi: 10.1164/arrd.1983.127.1.63. [DOI] [PubMed] [Google Scholar]
- Shukla A, Gulumian M, Hei TK, Kamp D, Rahman Q, Mossman BT. Multiple roles of oxidants in the pathogenesis of asbestos-induced diseases. Free Radical Biology and Medicine. 2003;34(9):1117–1129. doi: 10.1016/s0891-5849(03)00060-1. [DOI] [PubMed] [Google Scholar]
- Snyder LS, Hertz MI, Harmon KR, Bitterman PB. Failure of lung repair following acute lung injury. Regulation of the fibroproliferative response (Part 1) CHEST Journal. 1990;98(3):733–738. doi: 10.1378/chest.98.3.733. [DOI] [PubMed] [Google Scholar]
- Stone KC, Mercer RR, Gehr P, Stockstill B, Crapo JD. Allometric relationships of cell numbers and size in the mammalian lung. Am J Respir Cell Mol Biol. 1992;6(2):235–243. doi: 10.1165/ajrcmb/6.2.235. [DOI] [PubMed] [Google Scholar]
- Takagi A, Hirose A, Nishimura T, Fukumori N, Ogata A, Ohashi N, Kitajima S, Kanno J. Induction of mesothelioma in p53+/− mouse by intraperitoneal application of multi-wall carbon nanotube. The Journal of toxicological sciences. 2008;33(1):105–116. doi: 10.2131/jts.33.105. [DOI] [PubMed] [Google Scholar]
- Wagner GR. Asbestosis and silicosis. The Lancet. 1997;349(9061):1311–1315. doi: 10.1016/S0140-6736(96)07336-9. [DOI] [PubMed] [Google Scholar]
- Wagner JC, Berry G, Skidmore JW, Timbrell V. The effects of the inhalation of asbestos in rats. British journal of cancer. 1974;29(3):252. doi: 10.1038/bjc.1974.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Ibarra-Sunga O, Verlinski L, Pick R, Uhal BD. Abrogation of bleomycin-induced epithelial apoptosis and lung fibrosis by captopril or by a caspase inhibitor. American Journal of Physiology-Lung Cellular and Molecular Physiology. 2000;279(1):L143–L151. doi: 10.1152/ajplung.2000.279.1.L143. [DOI] [PubMed] [Google Scholar]