

Abstract
A series of 1,4-dihydropyridines (DHPs) were investigated as inhibitors of capacitative calcium influx through store-operated calcium (SOC) channels. Such channels activate after ATP-elicited release of inositol trisphosphate (IP3)-sensitive calcium stores in leukemia HL-60 cells. The most potent DHPs were those containing a 4-phenyl group with an electron-withdrawing substituent, such as m- or p-nitro- or m-trifluoromethyl (IC50 values: 3–6 μM). Benzyl esters, corresponding to the usual ethyl/methyl esters of the DHPs developed as L-type calcium channel blockers, retained potency at SOC channels, as did N-substituted DHPs. N-Methylation reduced by orders of magnitude the potency at L-type channels resulting in DHPs nearly equipotent at SOC and L-type channels. DHPs with N-ethyl, N-allyl, and N-propargyl groups also had similar potencies at SOC and L-type channels. Replacement of the usual 6-methyl group of DHPs with larger groups, such as cyclobutyl or phenyl, eliminated activity at the SOC channels; such DHPs instead elicited formation of inositol phosphates and release of IP3-sensitive calcium stores. Other DHPs also caused a release of calcium stores, but usually at significantly higher concentrations than those required for the inhibition of capacitative calcium influx. Certain DHPs appeared to cause an incomplete blockade of SOC channel-dependent elevations of calcium, suggesting the presence of more than one class of such channels in HL-60 cells. N-Methylnitrendipine (IC50 2.6 μM, MRS 1844) and N-propargylnifrendipine (IC50 1.7 μM, MRS 1845) represent possible lead compounds for the development of selective SOC channel inhibitors.
Keywords: Calcium channels, Dihydropyridines, Capacitative calcium influx, Inositol trisphosphate, Store-operated calcium channels
1. Introduction
Cell function is regulated in a wide variety of cells by changes in cytosolic free calcium. In non-excitable cells, changes in intracellular calcium are often associated with receptor-mediated release of calcium from intracellular calcium stores and subsequent capacitative calcium entry due to the activation of SOC channels in the plasma membrane. The activation of SOC channels appears to be linked to the filling state of the calcium stores, but the pathways of activation and inactivation remain unclear [1–3]. A major problem in studying SOC channels has been the lack of selective probes for detection, activation, blockade, or modulation of such channels. A variety of structurally different compounds have been reported to cause some degree of SOC channel blockade [4–9], but there is no clear pattern regarding the structural requirements for the blockade of SOC channels, nor insights into the mechanisms involved. Many of the putative blockers of SOC channels also cause the release of intracellular calcium, rendering them unsatisfactory as specific probes for SOC channels [5,7,9,10].
DHPs are known as a class of potent and selective inhibitors of voltage-dependent L-type calcium channels [11]. Other classes of voltage-sensitive calcium channels, however, can be inhibited by certain DHPs [12]. In addition, certain structurally novel DHPs have proven to be highly selective as antagonists of A3-adenosine receptors [13–15]. DHPs, such as nicardipine, nitrendipine (6), and nifedipine (8), have been reported to cause inhibition of capacitative calcium influx in HL-60 and U937 cells, which lack L-type calcium channels, but high micromolar concentrations are required [10,16]. Certain p-substituted 4-phenyl DHPs inhibit a capacitative calcium current in skeletal muscle cells [17], while nifedipine (8), which is an o-nitro-substituted 4-phenyl DHP, enhances the current in muscle cells [18].
A series of DHPs have now been assayed with HL-60 cells, which lack L-type calcium channels, to provide insight into the structural requirements for inhibition of the SOC channels by DHPs in this cell line. The IP3-sensitive calcium stores were depleted in the HL-60 cells using the receptor agonist ATP to initiate the formation of IP3, the release of intracellular calcium, and the subsequent activation of the SOC channels [10]. The inhibition of SOC channels by the DHPs was determined by monitoring the effects after ATP on intracellular calcium levels using a fluorescent calcium probe. Several DHPs also were tested in GH4C1 cells, labeled with a fluorescent calcium probe, for effects on calcium influx through voltage-sensitive L-type calcium channels to ascertain if there was any correlation between inhibition of L-type calcium channels and SOC channels by the DHPs.
2. Materials and methods
2.1. Materials
Nitrendipine (6), nifedipine (8), diltiazem, and methoxyverapamil (D-600) were obtained from Research Bio-chemicals International (RBI). Dibutyryl cyclic AMP was obtained from the Sigma Chemical Co., and ATP from Fluka. Roswell Park Memorial Institute (RPMI) 1640 medium, F10 nutrient mixture (Ham), fetal bovine serum, horse serum, L-glutamine, trypsin-EDTA, and penicillin/streptomycin (10,000 units/mL of penicillin G sodium and 10,000 μg/mL of streptomycin sulfate) were purchased from Gibco BRL, Life Technologies. Fluo3-AM, fluo4-AM, and fura2-AM were from Molecular Probes, Inc. [3H]myo-Inositol was obtained from NEN Life Science Products. 3-Nitrobenzaldehyde, 3-trifluoromethylbenzaldehyde, ethyl 3-aminocrotonate, ethyl acetoacetate, 3-aminocrotonitrile, 2-methoxyethyl acetoacetate, benzyl acetoacetate, p-nitrobenzyl acetoacetate, ethyl 4,4,4-trifluoroacetoacetate, methyl acetoacetate, ethyl propionylacetate, iodomethane, zinc, ammonium acetate, and sodium hydride were purchased from the Aldrich Chemical Co. All other materials were obtained from standard commercial sources.
2.2. Synthesis
The syntheses of DHPs 1–5, 7, 13–15, and 29 have been reported previously [13,14]. The DHPs 9–12 and 16–28 were prepared in a similar manner using the Hantzsch condensation reaction (Fig. 1, see below). Reduction of the nitro group of DHP 7 to the amine gave DHP 10, which was then acetylated to give DHP 12. N-Methylation with methyl iodide of 7 and 20 gave DHPs 30 and 31, respectively. Alkylation of nitrendipine (6) with methyl iodide, ethyl iodide, allyl bromide, propargyl bromide, or benzyl chloride gave DHPs 32, 33, 34, 35, and 36, respectively (Fig. 1). The yield for 32 was nearly quantitative. For structures, see Table 1.
Fig. 1.

General procedure for synthesis of 1,4-dihydropyridines and N-alkylation.
Table 1.
Inhibition of capacitative calcium influx through SOC channels in HL-60 cells by 1,4-dihydropyridines
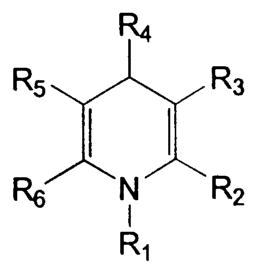
| ||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | R4 | R5 | R6 | IC50a (μM) | Imaxb (%) |
| 1 | H | Me | COOEt | Ph | COOMe | Me | 40 ± 8 | 75 ± 6 |
| 2 | H | Me | COOEt | CH=CH–Ph | COOMe | Me | 15 ± 6 | 50 ± 6 |
| 3 | H | Me | COOEt | C≡C–Ph | COOMe | Me | NIc | NI |
| 4 | H | Me | COOEt | 2-Thienyl | COOEt | Me | NI | NI |
| 5 | H | Me | COOEt | 4-NO2–Ph | COOEt | Me | 6.5 ± 2.8 | 70 ± 10 |
| 6d | H | Me | COOEt | 3-NO2–Ph | COOMe | Me | 3.3 ± 1.2 | 80 ± 6 |
| 7 | H | Me | COOEt | 3-NO2–Ph | COOEt | Me | 5.0 ± 2.1 | 90 ± 3 |
| 8e | H | Me | COOMe | 2-NO2–Ph | COOMe | Me | 16 ± 2 | 75 ± 3 |
| 9 | H | Me | COOEt | 3,5-(NO2)2–Ph | COOEt | Me | 7 | 95 |
| 10 | H | Me | COOEt | 3-NH2–Ph | COOEt | Me | NI | NI |
| 11 | H | Me | COOEt | 3-CF3–Ph | COOEt | Me | 2.6 ± 0.6 | 55 ± 2 |
| 12 | H | Me | COOEt | 3-(NHCOMe)–Ph | COOEt | Me | NI | NI |
| 13 | H | Me | COOEt | 4-OMe–Ph | COOMe | Me | 36 ± 3 | 75 ± 5 |
| 14 | H | Me | COOEt | 2-CF3–Ph | COOMe | Me | 35 ± 4 | 75 ± 4 |
| 15 | H | Me | COOEt | 3-OMe-4-OH–Ph | COOMe | Me | 25 | 75 |
| 16 | H | Me | COOEt | 3-NO2–Ph | COSEt | Me | 8 | 70 |
| 17 | H | Me | COOEt | 3-NO2–Ph | CN | Me | NI | NI |
| 18 | H | Me | COOEt | 3-NO2–Ph | COO(CH2)2OH | Me | 30 | 80 |
| 19 | H | Me | COOEt | 3-NO2–Ph | COO(CH2)2OMe | Me | >100 | NDf |
| 20 | H | Me | COOEt | 3-NO2–Ph | COOCH2Ph | Me | 1.8 ± 0.1 | 95 ± 5 |
| 21 | H | Me | COOCH2Ph | 3-NO2–Ph | COOCH2Ph | Me | 3.0 ± 1.5 | 97 ± 2 |
| 22 | H | Me | COOEt | 3-NO2–Ph | COOCH2-(4-NO2)–Ph | Me | NI | NI |
| 23 | H | Me | COOMe | 3-NO2–Ph | COOMe | Me | 8.0 ± 4.0 | 50 ± 5 |
| 24 | H | Et | COOEt | 3-NO2–Ph | COOEt | Et | 10 | 55 |
| 25 | H | Me | COOEt | 3-NO2–Ph | COSEt | Et | 15 | 75 |
| 26 | H | Me | COOEt | 3-NO2–Ph | COOEt | CF3 | NI | NI |
| 27 | H | Me | COOEt | 3-NO2–Ph | COOCH2Ph | Cyclopropyl | NI | NI |
| 28 | H | Me | COOEt | 3-NO2–Ph | COOCH2Ph | Cyclobutyl | NI | NI |
| 29 | H | Me | COOEt | Me | COOEt | Ph | NI | NI |
| 30 | Me | Me | COOMe | 3-NO2–Ph | COOMe | Me | 2.8 ± 0.3 | 88 ± 4 |
| 31 | Me | Me | COOEt | 3-NO2–Ph | COOEt | Me | 2.0 ± 0.8 | 72 ± 11 |
| 32 | Me | Me | COOEt | 3-NO2–Ph | COOMe | Me | 2.6 ± 1.1 | 84 ± 12 |
| 33 | Et | Me | COOEt | 3-NO2–Ph | COOMe | Me | 6 | 75 |
| 34 | Allyl | Me | COOEt | 3-NO2–Ph | COOMe | Me | 6.8 ± 1.8 | 69 ± 7 |
| 35 | Propargyl | Me | COOEt | 3-NO2–Ph | COOMe | Me | 1.7 ± 1.3 | 78 ± 12 |
| 36 | Benzyl | Me | COOEt | 3-NO2–Ph | COOMe | Me | 28 | 51 |
The IC50 values represent the mean concentration ± SEM (N = 3–6) required for 50% of the maximum inhibition observed for each compound, as shown in Fig. 3A–C. Single values are the average of two determinations.
Imax represents the maximum inhibition ± SEM by the DHP of the sustained calcium levels elicited by 10 μM ATP.
NI: no inhibition observed at concentrations up to 100 μM.
Nitrendipine.
Nifedipine.
ND=not determined.
Equimolar amounts (0.5 to 1.0 mmol) of the appropriate (β-enaminoester, aldehyde, and (β-ketoester were dissolved in 2–5 mL of absolute ethanol. The mixture was sealed in a Pyrex tube and heated with stirring to 90° overnight. After cooling to room temperature, the solvent was evaporated and the product was purified by preparative thin-layer chromatography (silica 60; 1000 mm; Analtech) with petroleum ether–ethyl acetate. The ratio of solvents varied for different compounds from 1:1 to 4:1. The DHPs were characterized by chromatographic analysis, nuclear magnetic resonance spectra, mass spectrometry, and, in most cases, C, H, N analysis. Proton nuclear magnetic resonance spectroscopy was performed on a Varian GEMINI-300 spectrometer, and all spectra were obtained in CDCl3. Chemical-ionization (CI) mass spectrometry was performed with a Finnigan 4600 mass spectrometer, and electron-impact (EI) mass spectrometry with a VG7070F mass spectrometer at 6 kV. Elemental analysis was performed by Galbraith Laboratories, Inc. The yields were as follows: 9, 77%; 10, 83%; 12, 71%; 16, 36%; 17, 76%; 18, 41%; 19, 92%; 20, 55%; 21, 49%; 22, 45%; 23, 20%; 24, 12%; 25, 48%; 26, 89%; 27, 58%; 28, 54%; 30, 22%; 31, 74%; 32, 95%; 33, 21%; 34, 22%; 35, 22%; and 36, 16%.
S-Ethyl 3-oxothiobutyrate, S-ethyl 3-oxothiovalerate, benzyl 3-oxo-3-cyclopropylpropionate, and benzyl 3-oxo-3-cyclobutylpropionate were prepared as described [13]. 2-Hydroxyethyl acetoacetate and p-methoxybenzyl acetoacetate were prepared, respectively, by heating 2,2,6-trimethyl-4H-1,3-dioxin-4-one and either ethylene glycol or p-methoxybenzyl alcohol in toluene for 4 hr. 3,5-Dinitrobenzaldehyde was prepared by pyridinium chlorochromate oxidation of 3,5-dinitrobenzyl alcohol.
2.3. Cell culture
Human leukemic HL-60 cells and rat pituitary GH4C1 cells were from the American Type Culture Collection. The HL-60 cells were grown in suspension in RPMI 1640 medium supplemented with 10% fetal bovine serum, 100 units/mL of penicillin, 100 (μg/mL of streptomycin, and 2 mM L-glutamine. The HL-60 cells were differentiated in supplemented RPMI 1640 medium containing 500 μM dibutyryl cyclic AMP for 48 hr prior to each experiment. The GH4C1 cells were grown in F10 nutrient mixture supplemented with 15% horse serum, 2.5% fetal bovine serum, 100 units/mL of penicillin, and 100 μg/mL of streptomycin. Prior to an experiment, the GH4C1 cells were removed from the cell culture surface with trypsin-EDTA (2 mL, 2 min) and suspended in 20 mL of F10 nutrient mixture.
2.4. Calcium assay
Aliquots (10–20 mL) of suspended cells were subjected to low speed centrifugation (200 g, 25°; HL-60 cells for 10 min, GH4C1 cells for 3 min), and the supernatants were removed. The packed HL-60 cells were resuspended in 10–20 mL of buffered medium containing 1 part supplemented RPMI 1640 medium and 1 part Krebs–Ringer HEPES buffer (KRH: 125 mM NaCl, 1.2 mM KH2PO4, 1.2 mM MgSO4, 2 mM CaCl2, 6 mM glucose, 25 mM HEPES, pH 7.4). The GH4C1 cells were resuspended in buffered saline solution (BSS: 118 mM NaCl; 4.6 mM KCl; 10 mM glucose; 20 mM HEPES; pH 7.4). To the cell suspension was added either 10 μg fluo3-AM for HL-60 cells and 10 μg fura2-AM or 30 μg fluo4-AM for GH4C1 cells in 10 μL dimethyl sulfoxide. In some experiments, fura2-AM was used for HL-60 cells. After 30–40 min in the dark at 25°, the cell suspension was centrifuged as described above and the supernatant removed. The packed cells were resuspended in either KRH or BSS to give a final cell count of approximately 106/mL. A 2-mL aliquot of the dye-loaded cells was transferred into a cuvette and stirred with a magnetic stirrer bar throughout the entire experiment. In most experiments with HL-60 cells, the ATP (10 μM) was added, followed in 2 min by the DHP in dimethyl sulfoxide. For experiments with GH4C1 cells, 40 mM KCl was added followed in 3 min by the DHP in dimethyl sulfoxide. Intracellular calcium levels were monitored by fluorescent spectroscopy using a PTI Deltascan Fluorescence Spectrophotometer (Photon Technology International) set at 506 nm (excitation) and 526 nm (emission) for cells loaded with fluo3 and at 494 nm (excitation) and 516 nm (emission) for cells loaded with fluo4. For cells loaded with fura2, the spectrophotometer was set at 380 and 340 nm for excitation and at 500 nm for emission. Either the ratio is reported or the levels of intracellular calcium were calculated as described [19].
2.5. Inositol phosphate assay
[3H]Inositol (2 μCi/mL) and dibutyryl cyclic AMP were added to the HL-60 cells 48 hr prior to each experiment. The differentiated [3H]inositol-labeled HL-60 cells were then centrifuged (10 min, 200 g, 25°) and resuspended in supplemented RPMI medium to give a final cell concentration of 106/mL. Aliquots (1 mL) of cells were incubated after the addition of LiCl (final concentration, 10 mM) at room temperature for 20 min. The agents were added at the appropriate concentrations, and the cells were incubated further for 10 min at room temperature. The cells were centrifuged (10 min, 200 g, 25°), and the supernatant was removed. The cell pellet was treated with 750 μL of ice-cold formic acid (20 mM), and the suspension was left at 4° for 30 min. The suspensions were neutralized with 60 mM NH4OH and loaded onto anion exchange columns (Bio-Rad AG-1-X8 Resin, 100–200 mesh, formate form, 1 mL vol.). The columns were washed with 10 mL of water, followed by 10 mL of borate/formate buffer (5 mM sodium borate, 60 mM sodium formate), and then the [3H]inositol phosphates were eluted with 5 mL of elution buffer (0.1 M formic acid, 0.2 M ammonium formate). Hydrofluor (2:1, v/v) was added to the eluent, and the radioactivity was measured using a Beckman LS 6500 Multipurpose Scintillation Counter. The anion exchange columns were regenerated with 0.1 M formic acid, 3 M ammonium formate.
3. Results and discussion
Although a variety of SOC channel inhibitors are now known, there has been little investigation into the structural requirements necessary for SOC channel inhibition, and no potent (sub-micromolar) or selective inhibitors are available. Imidazoles, such as SKF 96365 and miconazole [5–7], tricyclics, such as trifluoperazine [7], and DHPs, such as nitrendipine [10], represent distinct structural classes of SOC channel inhibitors. Most imidazoles and tricyclics, in addition to blocking SOC channels, cause the release of intracellular calcium and in some cases influx of calcium [5,7,9,10]. The DHPs in the present study represent a preliminary investigation into the structural requirements for SOC channel inhibition by the DHPs. A typical set of concentration-dependent inhibitions for N-methylnitrendipine (32, MRS 1844) is shown in Fig. 2. Representative composite inhibition curves are shown for several DHPs in panels A–C of Fig. 3. The inhibitory effects of the DHPs with respect to SOC channels are summarized in Table 1.
Fig. 2.

Concentration-dependent inhibition by N-methylnitrendipine (32, MRS 1844) of SOC channels in HL-60 cells. Intracellular calcium levels of calcium were elevated by 10 μM ATP (first arrow) followed by DHP 32 (second arrow). (A) 0.3 μM; (B) 1 μM; (C) 3 μM; (D) 10 μM; (E) 30 μM. A representative experiment is shown. Cells were loaded with fura2-AM. The basal levels of intracellular calcium were calculated to be about 100–150 nM (see Fig. 4).
Fig. 3.

Concentration-dependent inhibition by DHPs of SOC channels in HL-60 cells. Intracellular calcium levels were first elevated with 10 μM ATP, followed by the addition of increasing concentrations of the following agents: (A) 1, 5, nitrendipine (6), and nifedipine (8). (B) 11, 13, and 21. (C) 32 and 35. Values are means ± SEM (N = 3). (D) Structures. Curves fitted with GraphPad Prism (GraphPad Software).
DHP 1 with a phenyl group at the 4-position of the DHP ring showed much greater potency than the 4-phenethynyl DHP 3 or the 4-thienyl DHP 4, both of which were inactive at 100 μM (Table 1). The 4-styryl DHP 2 showed somewhat greater potency than 1, but was less efficacious. The introduction of a variety of substituents on the phenyl ring resulted in a wide range of activities. DHPs 5–7, 9, and 11 with an electron-withdrawing substituent(s), such as nitro or trifluoromethyl in the m- or p-position, were more potent than DHPs 8 and 14 with an o-nitro or o-trifluoromethyl substituent. DHPs 10, 13, and 15 with electron-donating substituents were also less potent. DHP 7 with a m-nitro group was significantly more efficacious than DHP 11 with a m-trifluoromethyl group, but both had similar potencies. Further DHPs, namely 16–28, with a m-nitrophenyl group at the 4-position were synthesized.
DHPs 20 and 21, which are benzyl esters, had inhibitory properties similar to those of the comparable methyl/ethyl esters 6 and 7. Exchanging an ester in 7 for a thioester in 16 had little effect on the potency, but the thioester was less efficacious. DHPs with a nitrile (17) or with polar groups in the ester moiety (18, 19) were much less potent.
DHPs having a methyl group on the ring nitrogen, as in 30–32, were among the most potent inhibitors of SOC channels. A series of N-substituted derivatives of nitrendipine (6) were synthesized. N-Methylnitrendipine (32, MRS 1844) and the N-propargyl (35, MRS 1845) analog were more potent than the N-ethyl (33) or N-allyl (34) derivatives, while the N-benzyl (36) analog was significantly less potent than the other N-substituted DHPs.
DHPs with a cyclopropyl (27), cyclobutyl (28), or phenyl (29) substituent at the 6-position were inactive at SOC channels; all caused an increase in levels of intracellular calcium whether added before or after ATP (Fig. 4A). DHPs 28 and 29 were the most active with half-maximal stimulations at 5 (μM. An increase in calcium levels was also observed with another SOC channel-inactive DHP (3) containing a 4-phenethynyl substituent and a methyl group at the 6-position, but a 10-fold higher concentration was required. The elevation of intracellular calcium elicited by 3 occurred in both the presence and absence of extracellular calcium (Fig. 4B and D). The elevation of intracellular calcium levels by such DHPs would appear to be associated with the generation of inositol phosphates (Fig. 5A), which would cause the release of calcium from IP3-sensitive stores. The DHP-induced generation of inositol phosphates appeared to be additive with the ATP-induced generation of inositol phosphates (Fig. 5B). The DHPs that were the more potent SOC channel inhibitors, such as nitrendipine (6), and compounds 7, 11, 20, 21, 30–32, and 35 did not cause a significant generation of inositol phosphates at the concentrations required for the inhibition of SOC channels (Fig. 5A). At concentrations of 30 μM or greater, most of the DHPs caused an elevation of intracellular calcium levels (data not shown).
Fig. 4.

Elevation of intracellular calcium levels by certain DHPs in HL-60 cells. (A) Effect of the addition of 10 μM DHP 29 after the elevation of intracellular calcium levels by 10 μM ATP in the presence of external calcium. (B) Effect of the addition of 10 μM DHP 29 before the elevation of intracellular calcium levels by ATP in the presence of external calcium. (C) Effect of the addition of 10 μM DHP 29 after the elevation of intracellular calcium levels by ATP in the absence of external calcium. (D) Effect of 10 μM DHP 29 before the elevation of intracellular calcium levels by ATP in the absence of external calcium. External calcium was removed by the addition of 2 mM EGTA to the cell suspensions 2 min prior to the addition of the agents. Similar effects were obtained for 28, while higher concentrations of 3 and 27 were required for similar effects.
Fig. 5.

Effect of DHPs on the generation of [3H]inositol phosphates in HL-60 cells. (A) [3H]Inositol-labeled HL-60 cells were incubated with 10 μM DHP, and the accumulation of [3H]inositol phosphates was determined as described in “Section 2.” (B) Effect of 10 μM DHP 3 or 29 in combination with 10 μM ATP on the accumulation of [3H]inositol phosphates. The generation of [3H]inositol phosphates by 10 μM ATP alone is set arbitrarily at 100%. Values are means ± SEM (N = 3).
Certain generalities as to structure-activity relationships for DHPs as blockers of SOC channels in HL-60 cells are possible. With regard to the aryl R4-substituent (see Table 1), an electron-withdrawing group at the m-position of the phenyl ring (5, 9, 11, 20, 21) or the p-position (5) enhanced activity relative to that of the DHP with an unsubstituted phenyl ring (1). The nature of the ester moieties (COOMe, COOEt, COSEt, COOCH2Ph) at R3 and R5 (6, 7, 16, 20, 21, 23) had no or only modest effects on activity. However, certain larger ester moieties (18,19, 22) or a nitrile moiety (17) were not tolerated. Virtually all the DHPs had methyl groups at R2 and R6. Replacement of such methyl groups with ethyl groups (24) had little effect, but replacement of one methyl with CF3, cycloalkyl, or phenyl (26–29) eliminated activity. N-Methylation at the R1-position of DNPs (6 → 32, 7 → 31) slightly increased potency. Nitrendipines with N-ethyl, N-allyl, or N-propargyl substituents (33–35) all were potent, while an N-benzyl (36) was poorly tolerated. Most of the DHPs caused at least a 70% apparent inhibition of the SOC channel-dependent elevation of intracellular calcium (Table 1). However, a few DHPs (2, 11, 23, 24, 36) caused only a maximal apparent inhibition of 50–65%. It is possible that more than one class of SOC channel exists in HL-60 cells and that one subclass is poorly inhibited by certain DHPs. Some of the DHPs alone caused significant elevation of intracellular calcium at concentrations greater than 10 μM (data not shown), and this might result in an apparently less than complete inhibition of SOC channels by such DHPs.
It has been suggested that the inhibitory effect of the DHPs at SOC channels might reflect structural similarities between the voltage-dependent L-type calcium channels and the voltage-independent SOC channels [16]. Thirteen structurally diverse DHPs were tested for inhibition of L-type calcium channels in GH4C1 cells (Table 2). All of the DHPs caused inhibition of L-type calcium channels. In most instances, inhibition was observed at much lower concentrations (1100- to 3000-fold) than what was required for inhibition of SOC channels (Table 2). There were some striking exceptions, namely the styryl DHP 2, which appeared only 15-fold more potent at the L-type channels, and the p-methoxyphenyl DHP 13, which was only 12-fold more potent, and the N-substituted DHPs (30, 32–35), which had greatly reduced potencies as inhibitors of L-type calcium channels. Such N-substituted DHPs each had similar potencies at L-type channels and SOC channels. The present results indicate that DHP sites on the two channels are quite different with respect to tolerance of certain substituents at R4 and to N-substitution. The L-type calcium channels are also inhibited by benzothiazepines, such as diltiazem, and by phenylalkylamines, such as methoxyverapamil [11], neither of which caused inhibition of SOC channels in HL-60 cells at concentrations up to 100 μM ([5], and data not shown). Certain of the DHPs caused a rapid but only a partial reduction of the intracellular calcium influx through L-type calcium channels, suggestive of more than one subtype of such channels in GH4C1 cells. There has been a report of both “fast” and “slow” effects of DHPs on calcium channels in such pituitary tumor cells [20].
Table 2.
Inhibition of calcium influx through voltage-dependent L-type calcium channels in GH4C1 cells by DHPs: comparison to inhibition of SOC channels in HL-60 cells
|
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | R4 | R5 | R6 | L-type channel | SOC channel IC50c (μM) | Ratio SOC/L-type | |
| Imaxa (%) | IC50b (μM) | |||||||||
| 1 | H | Me | COOEt | Ph | COOMe | Me | 54 | 0.01 | 40 ± 8 | 4000 |
| 2 | H | Me | COOEt | CH=CH–Ph | COOMe | Me | 40 | 1 | 15 ± 6 | 15 |
| 6d | H | Me | COOEt | 3-NO2–Ph | COOMe | Me | 92 ± 4 | 0.003 ± 0.001 | 3.3 ± 1.2 | 1100 |
| 7 | H | Me | COOEt | 3-NO2–Ph | COOEt | Me | 77 | 0.003 | 5.0 ± 2.1 | 1600 |
| 8e | H | Me | COOMe | 2-NO2–Ph | COOMe | Me | 85 ± 3 | 0.01 ± 0.005 | 16 ± 2 | 1600 |
| 13 | H | Me | COOEt | 4-OMe–Ph | COOMe | Me | 60 | 3 | 36 ± 3 | 12 |
| 20 | H | Me | COOEt | 3-NO2–Ph | COOCH2Ph | Me | 55 | 0.001 | 1.8 ± 0.1 | 1800 |
| 21 | H | Me | COOCH2Ph | 3-NO2–Ph | COOCH2Ph | Me | 60 | 0.001 | 3.0 ± 1.5 | 3000 |
| 30 | Me | Me | COOMe | 3-NO2–Ph | COOMe | Me | 70 | 2 | 2.8 ± 0.3 | 1.4 |
| 32 | Me | Me | COOEt | 3-NO2–Ph | COOMe | Me | 86 ± 7 | 1.3 ± 0.3 | 2.6 ± 1.1 | 2.0 |
| 33 | Et | Me | COOEt | 3-NO2–Ph | COOMe | Me | 82 ± 4 | 2.3 ± 0.4 | 6 | 2.6 |
| 34 | Allyl | Me | COOEt | 3-NO2–Ph | COOMe | Me | 72 ± 5 | 3.8 ± 1.8 | 6.8 ± 1.8 | 1.8 |
| 35 | Propargyl | Me | COOEt | 3-NO2–Ph | COOMe | Me | 86 ± 7 | 2.1 ± 0.2 | 1.7 ± 1.3 | 0.8 |
Imax represents the maximum mean inhibition ± SEM (N = 3) by the DHP of the elevation of calcium levels elicited by depolarization of the cell membrane with 40 mM KCl. Single values are the average of two determinations.
The IC50 values represent the mean concentration ± SEM (N = 3) required for 50% of the maximum inhibition observed for each compound. Single values are the average of two determinations.
The IC50 values are from Table 1.
Nitrendipine.
Nifedipine.
In conclusion, the classical DHPs, such as nitrendipine (6) and nifedipine (8), while being SOC channel blockers, are many fold more potent as blockers of L-type calcium channels. The N-substituted DHPs, exemplified by N-methylnitrendipine (32, MRS 1844), represent lead compounds for the development of selective SOC channel blockers. However, the present N-substituted DHPs (31–35) are not selective for SOC channels, since, although potency at L-type channels is reduced, it is not abolished, and the DHPs (31–35) now have comparable potencies at the two classes of calcium channels. The N-substituted DHPs do not elicit increases in intracellular calcium at concentrations required to block SOC channels in HL-60 cells. Release of calcium by the classical, widely used imidazole blockers of SOC channels, such as SKF 96365, miconazole, and clotrimazole, has represented a major disadvantage to their use [5,7,9,10]. Thus, the N-substituted DHPs appear to represent at present the only class of SOC channel blockers without the undesirable side-effect of releasing intracellular calcium. Further structural modification of N-substituted DHPs hopefully will result in even more potent and selective SOC channel blockers.
Abbreviations
- DHPs
1,4-dihydropyridines
- IP3
inositol trisphosphate
- and SOC
store-operated calcium
References
- 1.Berridge MJ. Capacitative calcium entry. Biochem J. 1995;312:1–11. doi: 10.1042/bj3120001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Putney JW., Jr . Capacitative calcium entry. Austin, TX: R.G. Landes Co; 1997. [Google Scholar]
- 3.Putney JW., Jr TRP, inositol 1,4,5-trisphosphate receptors, and capacitative calcium entry. Proc Natl Acad Sci USA. 1999;96:14669–71. doi: 10.1073/pnas.96.26.14669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krautwurst D, Hescheler J, Arndts D, Losel W, Hammer R, Shultz G. Novel potent inhibitors of receptor-activated nonselective calcium currents in HL60 cells. Mol Pharmacol. 1993;43:655–9. [PubMed] [Google Scholar]
- 5.Daly JW, Lueders J, Padgett WL, Shin Y, Gusovsky F. Maitotoxin-elicited calcium influx in cultured cells. Effect of calcium-channel blockers. Biochem Pharmacol. 1995;50:1187–97. doi: 10.1016/0006-2952(95)00257-z. [DOI] [PubMed] [Google Scholar]
- 6.Clementi E, Meldolesi J. Pharmacological and functional properties of voltage-independent Ca2+ channels. Cell Calcium. 1996;19:269–79. doi: 10.1016/s0143-4160(96)90068-8. [DOI] [PubMed] [Google Scholar]
- 7.Harper JL, Daly JW. Inhibitors of store-operated calcium channels: imidazoles, phenothiazines and other tricyclics. Drug Dev Res. 1999;47:107–17. [Google Scholar]
- 8.Guse AH, de Wit C, Klokow T, Schweitzer K, Mayr GW. Unique properties of the capacitative Ca2+-entry antagonist LU 52396: its inhibitory activity depends on the activation state of the cells. Cell Calcium. 1997;22:91–7. doi: 10.1016/s0143-4160(97)90109-3. [DOI] [PubMed] [Google Scholar]
- 9.Harper JL, Daly JW. Effect of calmidazolium analogs on calcium influx in HL-60 cells. Biochem Pharmacol. 2000;60:317–24. doi: 10.1016/s0006-2952(00)00349-x. [DOI] [PubMed] [Google Scholar]
- 10.Harper JL, Shin Y, Daly JW. Loperamide: a positive modulator for store-operated calcium channels? Proc Natl Acad Sci USA. 1997;94:14912–7. doi: 10.1073/pnas.94.26.14912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Triggle DJ. Calcium channel drugs: structure-function relationships and selectivity of action. J Cardiovasc Pharmacol. 1991;18(Suppl 10):S1–6. [PubMed] [Google Scholar]
- 12.Furukawa T, Yamakawa T, Midera T, Sagawa T, Mori Y, Nukada T. Selectivities of dihydropyridine derivatives in blocking Ca2+ channel subtypes expressed in Xenopus oocytes. J Pharmacol Exp Ther. 1999;291:464–73. [PubMed] [Google Scholar]
- 13.van Rhee AM, Jiang JL, Melman N, Olah ME, Styles GL, Jacobson KA. Interaction of 1,4-dihydropyridine and pyridine derivatives with adenosine receptors: selectivity for A3 receptors. J Med Chem. 1996;39:2980–9. doi: 10.1021/jm9600205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang J, van Rhee AM, Chang L, Patchornik A, Ji XD, Evans P, Melman N, Jacobson KA. Structure-activity relationships of 4-(phenylethynyl)-6-phenyl-1,4-dihydropyridines as highly selective A3 adenosine receptor antagonists. J Med Chem. 1997;40:2596–608. doi: 10.1021/jm970091j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li AH, Moro S, Melman N, Ji XD, Jacobson KA. Structure-activity relationships and molecular modeling of 3,5-diacyl-2,4-dialkylpyridine derivatives as selective A3 adenosine receptor antagonists. J Med Chem. 1998;41:3186–201. doi: 10.1021/jm980093j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Willmott N, Choudhury Q, Flower RJ. Functional importance of the dihydropyridine-sensitive, yet voltage-insensitive store-operated Ca2+ influx of U937 cells. FEBS Lett. 1996;394:159–64. doi: 10.1016/0014-5793(96)00939-8. [DOI] [PubMed] [Google Scholar]
- 17.Hopf FW, Reddy P, Hong J, Steinhardt RA. A capacitative current in cultured skeletal muscle cells is mediated by the calcium-specific leak channel and inhibited by dihydropyridine compounds. J Biol Chem. 1996;271:22358–67. doi: 10.1074/jbc.271.37.22358. [DOI] [PubMed] [Google Scholar]
- 18.Fong PY, Turner PR, Denetclaw WF, Steinhardt RA. Increased activity of calcium leak channels in myotubes of Duchenne human and mdx mouse origin. Science. 1990;250:673–6. doi: 10.1126/science.2173137. [DOI] [PubMed] [Google Scholar]
- 19.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–50. [PubMed] [Google Scholar]
- 20.Cataldi M, Taglialatela M, Palagiano F, Secondo A, di Caprariis P, Amoroso S, di Renzo G, Annunziato L. Effects of monidipine and nitrendipine enantiomers on the plateau phase of K+-induced intracellular increase in GH3 cells. Eur J Pharmacol. 1999;376:169–78. doi: 10.1016/s0014-2999(99)00149-1. [DOI] [PubMed] [Google Scholar]