

Abstract
The parasite Toxoplasma gondii replicates in a specialized intracellular vacuole and causes disease in many species. Protection from toxoplasmosis is mediated by CD8+ T cells, but the T. gondii antigens and host genes required for eliciting protective immunity are poorly defined. Here we identified GRA6, a polymorphic protein secreted in the parasitophorous vacuole, as the source of the immunodominant and protective decapeptide HF10 presented by the H-2Ld major histocompatibility complex class I molecule. Presentation of the HF10–H-2Ld ligand required proteolysis by ERAAP, the endoplasmic reticulum aminopeptidase associated with antigen processing. Consequently, expansion of protective CD8+ T cell populations was impaired in T. gondii–infected ERAAP-deficient mice, which were more susceptible to toxoplasmosis. Thus, endoplasmic reticulum proteolysis is critical for eliciting protective immunity to a vacuolar parasite.
Toxoplasma gondii is an obligate intracellular parasite that infects a wide range of avian and mammalian species and can cause severe disease in humans1. Proinflammatory cytokines produced by lymphocytes are crucial for controlling a variety of intracellular pathogens, including T. gondii2. Cytokines such as tumor necrosis factor3, lymphotoxin-α4 and interferon-γ (IFN-γ)5 mediate protection against the acute and chronic stages of toxoplasmosis. Natural killer cells6,7 as well as CD4+ T cells8 and CD8+ T cells9–11, three lymphocyte subsets that produce these cytokines, have all been suggested to influence T. gondii immunity in mice and humans. Although the relative contribution of each subset probably varies with the stage of disease, the parasite genotype and host genetics, CD8+ T cells have been known for many years to have a critical protective function11,12. Indeed, in H-2d mice, resistance to toxoplasmic encephalitis has been linked to the locus encoding H-2Ld major histocompatibility complex (MHC) class I (ref. 13). Nevertheless, the mechanisms and antigens that elicit the activation and expansion of T. gondii–specific CD8+ T cell populations are not understood.
On the infected cell surface, CD8+ T cells recognize MHC class I molecules presenting peptides derived from intracellular proteins. For endogenously synthesized proteins, antigenic peptides are produced by proteolysis in the cytoplasm, are transported into the endoplasmic reticulum and are further processed by the aminopeptidase ERAAP. In the endoplasmic reticulum, ERAAP trims many precursor peptides to the appropriate length for presentation by MHC class I molecules14,15. Analysis of independent models of ERAAP-deficient mice16–19 has established a key function for ERAAP in shaping a normal repertoire of peptide–MHC class I complexes bearing peptides derived from endogenous and microbial sources. However, whether the modified peptide repertoire noted in the absence of trimming in the endoplasmic reticulum influences development of protective immunity against infectious pathogens remains unknown18. Here we identify the natural antigens required for eliciting protective immunity to T. gondii, which replicates in a specialized parasitophorous vacuole in infected cells. We found that the induction of immunodominant protective CD8+ T cell responses required processing of T. gondii antigens by ERAAP in the endoplasmic reticulum.
RESULTS
ERAAP-deficient mice are susceptible to T. gondii
To assess the requirement for endoplasmic reticulum proteolysis for protection against T. gondii infection, we studied the resistant H-2d mouse strain (B10.D2). In most infected animals, T. gondii can exist as either rapidly growing tachyzoites or bradyzoites that reside in semidormant cysts. We compared the susceptibility of H-2d ERAAP-deficient mice, mice heterozygous for ERAAP deficiency (called ‘ERAAP-heterozygous’ mice here) and wild-type mice to infection by T. gondii tachyzoites and cysts. Survival after challenge with tachyzoites (Fig. 1a) or cysts (data not shown) of a type II strain of T. gondii parasites, Prugniaud (Pru), was significantly impaired in the absence of ERAAP. Consistent with the enhanced susceptibility, parasite growth during acute infection was five- to tenfold higher in splenocytes and peritoneal macrophages from ERAAP-deficient mice than in their ERAAP-heterozygous or wild-type counterparts (Fig. 1b). The parasite burden was also higher in the brains, spleens and livers of ERAAP-deficient mice (Fig. 1c). These results indicate that the ability to control parasite replication is impaired in the absence of ERAAP. Thus, ERAAP determines the ‘genetic susceptibility’ of H-2d mice to T. gondii infection.
Figure 1.
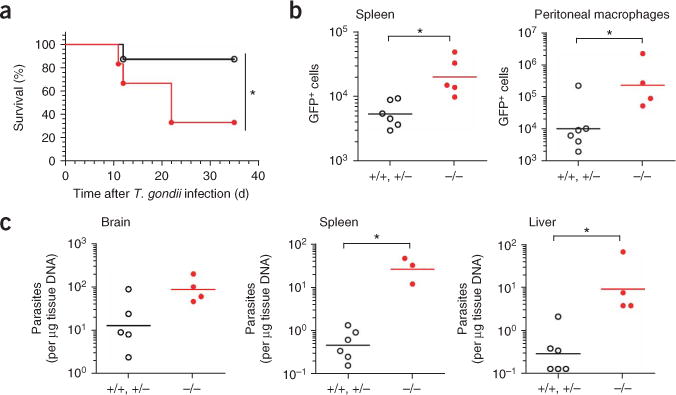
Uncontrolled parasite replication in ERAAP-deficient H-2d mice and greater susceptibility to T. gondii infection. (a) Kaplan-Meier survival curves of B10.D2 (H-2d) mice challenged intraperitoneally with 500 T. gondii Pru tachyzoites. *, P = 0.047 (Mantel-Cox log-rank test). Data are representative of two independent experiments with at least six mice per group. (b,c) Flow cytometry (b) of GFP+ infected splenocytes (*, P = 0.0043) or peritoneal macrophages (*, P = 0.038) and semiquantitative PCR (c) of parasite burden in the brain (*, P > 0.05), spleen (*, P = 0.012) and liver (*, P = 0.013) in mice infected intraperitoneally with 3 × 103 GFP-expressing Pru tachyzoites and analyzed 12 d later. +/+, +/−, pooled wild-type and ERAAP-heterozygous; −/−, ERAAP-deficient. Each symbol represents an individual mouse; small horizontal bars indicate the mean. Data represent two to four independent experiments with at least three mice per group.
Identification of the natural T. gondii antigen GRA6
Studies have shown that ERAAP deficiency alters the repertoire of endogenous or microbial peptides presented by MHC class I molecules on the cell surface as well as CD8+ T cell responses16–20. We hypothesized that T. gondii–derived peptide–MHC class I complexes might also be affected by ERAAP deficiency. Because the natural CD8+ T cell–stimulating T. gondii antigens have remained elusive, we used expression cloning to search for T. gondii proteins presented as peptide–MHC class I complexes to CD8+ T cells21. We immunized resistant BALB/c (H-2d) mice with γ-irradiated tachyzoites, which do not replicate yet do infect cells and elicit immune responses. The immunized mice had both CD8+ T cells and CD4+ T cells that produced IFN-γ in response to T. gondii–infected antigen-presenting cells (APCs) but not in response to APCs loaded with surface antigen 1, a protein from T. gondii proposed to be a T cell–stimulating antigen22,23 (Supplementary Fig. 1 online). We further expanded those T. gondii–specific CD8+ T cell populations by restimulation in vitro (Fig. 2a) and fused them to BWZ.36.CD8α, a T cell receptor-αβ (TCRαβ)-negative fusion partner that contains an inducible reporter gene encoding β-galactosidase (lacZ)24. Occupancy of the TCR in these hybridomas can be assayed by measurement of intracellular lacZ activity. Each of the 31 T. gondii–specific hybridomas obtained from two independent fusions responded to T. gondii–infected APCs expressing H-2Ld but not to those expressing H-2Kd MHC class I molecules (Fig. 2b). Thus, as predicted13, activation of T. gondii–specific hybridomas required H-2Ld and a T. gondii–derived peptide.
Figure 2.
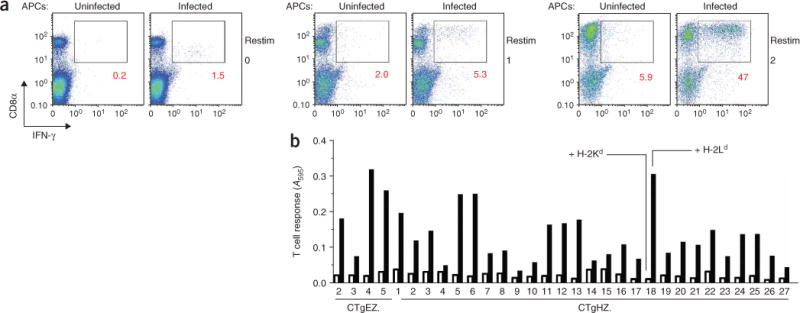
All T. gondii–specific CD8+ T cell hybridomas are stimulated by H-2Ld MHC class I. (a) Flow cytometry of IFN-γ production by CD8+ T cells from the spleens of T. gondii–immunized BALB/c mice, analyzed ex vivo 1 week after challenge (left) or after one restimulation (middle) or two restimulations (right) in vitro with infected J774 macrophages. Numbers under outlined areas indicate percent cells in each. Data are representative of two experiments. (b) The lacZ responses of T cell hybridomas (CTgEZ hybrids from batch E and CTgHZ hybrids from batch H; numbers along axis indicate hybridoma number) after overnight culture together with T. gondii–infected L cells expressing H-2Ld (filled bars) or H-2Kd (open bars), measured with a chromogenic substrate24. The hybridomas were generated by fusion of IFN-γ-producing CD8+ T cells from two independent groups of mice to a lacZ-inducible fusion partner. A595, absorbance at 595 nm. Data are from two experiments.
To identify the antigens from which these peptides were derived, we prepared a plasmid cDNA library with mRNA from T. gondii tachyzoites (Supplementary Fig. 2a,b online). We assessed the ability of L fibroblasts expressing H-2Ld and transfected with small pools of cDNA from the library to stimulate CTgEZ.4, one of the T. gondii–specific hybridomas (Supplementary Fig. 2c). We subdivided the positive pools and rescreened them to obtain individual cDNA clones25. The nucleotide sequences showed that each cDNA encoded either the full-length or an amino-terminally truncated version of the 224–amino acid GRA6 protein26 (Supplementary Fig. 2d). GRA6 is located in the dense granules, the final of three secretory organelles sequentially released by T. gondii after invasion of the host cell. Once secreted into the mature parasitophorous vacuole, GRA6 is believed to become associated with a network of lipid membranes found in the parasitophorous vacuole, but its biological function remains unknown27. We concluded that T. gondii–encoded GRA6 was the source of the antigenic peptide presented by H-2Ld.
Immunodominant protective GRA6 peptide
Next, we identified the T cell–stimulating peptide encoded by the GRA6 cDNA. H-2Ld L fibroblasts transfected with full-length GRA6 cDNA stimulated the CTgEZ.4 hybridoma (Fig. 3a). However, cells transfected with cDNA encoding a GRA6 deletion construct lacking the carboxy-terminal 55 amino acids were inactive, which indicated that the antigenic epitope was located in the deleted residues. By systematically testing all potential peptides predicted to bind H-2Ld MHC class I (data not shown)28, we determined that the decapeptide HPGSVNEFDF (HF10) was recognized by the CTgEZ.4 hybridoma (Fig. 3b). The HF10 peptide was located at the carboxyl terminus of GRA6 and was highly potent in stimulating the T cell hybridoma relative to its amino-terminally truncated or extended analogs. To establish that HF10 was the naturally processed peptide, we fractionated synthetic HF10 analogs (Fig. 3c) as well as extracts of GRA6-transfected H-2Ld L cells and T. gondii–infected bone marrow–derived macrophages (BMDMs) and bone marrow–derived dendritic cells (BMDCs) by high-performance liquid chromatography (HPLC; Fig. 3d). We tested each HPLC fraction for antigenic activity with the CTgEZ.4 hybridoma and H-2Ld L cells as APCs. In all cellular extracts, we found an activity peak corresponding to that of synthetic HF10 (fraction 32). The additional activity peak often present (fractions 34–35) probably represented potential precursors of HF10, as this peak eluted together with that of the longer synthetic 14–amino acid and 15–amino acid peptides29 (Fig. 3d). All the other available hybridomas also responded to GRA6-transfected cells as well as APCs pulsed with the HF10 peptide (Supplementary Fig. 3 online). We concluded that HF10 was the naturally processed product of the GRA6 protein presented by H-2Ld and was recognized by all T cell hybridomas tested.
Figure 3.
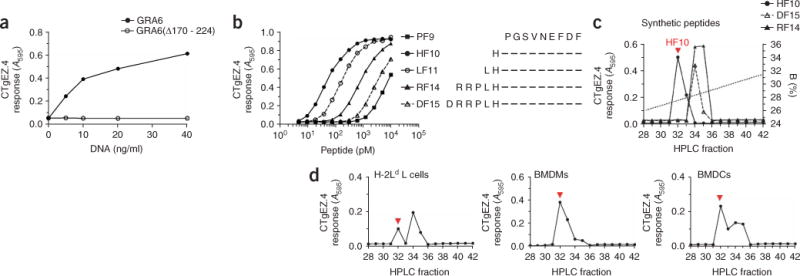
T. gondii–specific CD8+ T cell hybrids recognize the HF10 decapeptide presented by H-2Ld MHC class I. (a) Response of the T. gondii–specific CTgEZ.4 hybridoma to H-2Ld L cells transfected with full-length GRA6 cDNA or cDNA encoding a GRA6 construct lacking the carboxy-terminal residues 170–224 (GRA6Δ170 – 224), assessed as induction of β-galactosidase. (b) Response of CTgEZ.4 to synthetic GRA6 peptides (right) presented exogenously by H-2Ld L cells. (c,d) The CTgEZ.4-stimulatory capacity of synthetic peptides (c) or extracts of GRA6-transfected H-2Ld L cells or infected BMDMs or BMDCs (d), analyzed after fractionation by HPLC. Each fraction was pulsed onto H-2Ld L cells, which served as APCs. B (right margin, c), organic solvent (acetonitrile and 0.1% (vol/vol) trifluoroacetic acid). Red arrowheads indicate the HPLC fraction in which HF10 coelutes. Data in a–d are representative of at least two experiments.
To assess the importance of the HF10–H-2Ld complex in T. gondii–induced immune responses, we monitored CD8+ T cells specific for this complex in mice orally infected with T. gondii cysts. At 4 weeks after infection, approximately 5% of splenic CD8+ T cells and 24% of CD8+ T cells infiltrating the brain were stained by H-2Ld MHC multimers loaded with HF10 but not those loaded with the irrelevant peptide QL9 (QLSPFPFDL; Fig. 4a). As in a published study of T. gondii expressing a model antigen30, antigen-specific CD8+ T cell populations expanded for up to 4 weeks after infection (Fig. 4b). CD8+ T cells specific for HF10–H-2Ld were also readily detected in the spleen (about 2.5%) and were detected at a far higher frequency in the brain (about 12.5%) of mice injected intraperitoneally with tachyzoites (Fig. 4c), and they persisted for over 8 months (data not shown). We concluded that HF10–H-2Ld was a principal naturally processed ligand recognized by CD8+ T cells during T. gondii infection.
Figure 4.
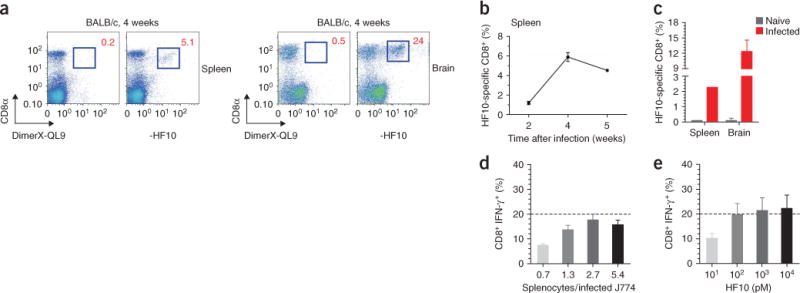
The HF10–H-2Ld complex is the only detectable ligand recognized by CD8+ T cells in T. gondii–infected H-2d mice. (a) Frequency of HF10-specific CD8+ cells after oral infection of BALB/c mice with T. gondii cysts. Numbers adjacent to outlined areas indicate percent CD8+ cells stained by an H-2Ld multimer (DimerX) loaded with HF10 or QL9 (irrelevant H-2Ld peptide). Data are representative of two experiments with two mice (brain) or three mice (spleen). (b) Expansion of HF10–H-2Ld–specific splenic CD8+ T cell populations over time in mice infected orally (n = 6 mice total, with only one mouse analyzed at the 5-week time point). Data are presented as mean ± s.e.m., corrected for background with QL9, and are representative of two experiments. (c) Frequency of HF10–H-2Ld–specific CD8+ cells in BALB/c mice infected intraperitoneally with 3 × 103 T. gondii tachyzoites and analyzed 7 weeks after infection. Data (mean and s.e.m.) are representative of two experiments with brains from four mice and spleen cells pooled from four mice. (d,e) Flow cytometry of IFN-γ production by CD8+ splenic T cells activated by T. gondii–infected J774 APCs (d) or HF10-loaded J774 APCs (e) 4 weeks after B10.D2 mice were infected intraperitoneally with 5 × 103 tachyzoites. Splenocyte/J774 cell ratio in e is 2.7. Dashed lines indicate the response elicited by 100 pM HF10 peptide. Data (mean and s.e.m.) are representative of two experiments with three mice each.
The dominance of CD8+ T cells specific for HF10–H-2Ld was unexpected because the T. gondii genome contains over 8,000 protein-encoding genes31. We therefore assessed the relative frequency of CD8+ T cells specific for HF10 versus other potential antigens among all IFN-γ-producing CD8+ T cells elicited by T. gondii (Fig. 4d,e). Approximately 18% of CD8+ T cells from T. gondii–infected mice produced IFN-γ in response to stimulation with T. gondii–infected J774 mouse macrophages. The frequency of IFN-γ-producing cells elicited by stimulation with the same J774 cells loaded with the HF10 peptide alone was approximately 20%. Thus, despite the potential antigenic complexity of the T. gondii, HF10 was apparently responsible for the entire CD8+ T cell response to this parasite. Yet this restricted antigen specificity was not due to the proliferation of a single T cell clone because at least two different TCR variable β-chains (Vβ2 and Vβ8.3) were present among the HF10-specific T cells (Supplementary Fig. 4 online). Together with the analysis of the panel of T cell hybridomas that all recognized the same antigen, these results suggest that the GRA6-derived HF10 is an immunodominant T. gondii antigen in H-2d mice.
The dominance of the HF10-specific CD8+ T cell populations suggested that immunization with this peptide might protect mice against toxoplasmosis. To test that hypothesis, we immunized B10.D2 mice with BMDCs pulsed with HF10 or another irrelevant H-2Ld-binding peptide (YL9 (YPHFMPTNL)). When challenged with live Pru tachyzoites, all control mice succumbed to infection within 12 d. In contrast, all HF10-immunized mice survived acute as well as chronic infection (Fig. 5a). As carboxy-terminal peptides from GRA6 have been reported to be B cell antigens32, we assessed whether protection was due to CD8+ T cells specific for HF10–H-2Ld by immunizing C57BL/6 H-2b mice with HF10 and YL9 (Fig. 5b). None of these mice were protected from T. gondii infection, which demonstrated that protection from disease was MHC restricted and thus was unlikely to be due to an antibody response. Furthermore, prior depletion of CD8+ cells from H-2d mice abolished the protective effect of HF10 immunization (Fig. 5c,d), which directly demonstrated that CD8+ cells were critical for protection. We concluded that in H-2d mice, HF10 not only was an immunodominant epitope but was also able to elicit a protective CD8+ T cell response during T. gondii infection.
Figure 5.
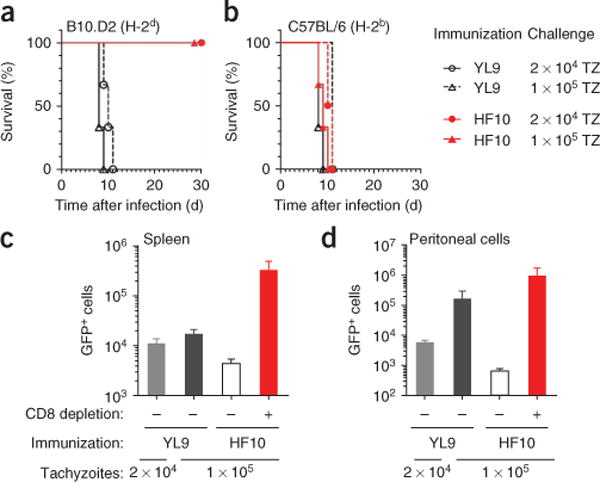
Immunization with HF10 protects mice from toxoplasmosis. (a,b) Survival of B10.D2 mice (a) or C57BL/6 mice (b) immunized with BMDCs pulsed with HF10 or YL9 (irrelevant H-2Ld peptide) and challenged with either of two doses of T. gondii tachyzoites (TZ). Data are representative of two independent experiments with three mice per group. (c,d) Infected (GFP+) splenocytes (c) and peritoneal cells (d) 10 d after infection of B10.D2 mice previously immunized with BMDCs pulsed with HF10 or YL9; mice were left undepleted (−) or were depleted of CD8+ cells 36 h before immunization by one intraperitoneal injection of depleting antibody (+). Data (mean and s.e.m.) are representative of two experiments with two to three mice per condition.
Processing and generation of HF10–H-2Ld complexes
To understand why ERAAP-deficient mice were more susceptible to T. gondii infection we analyzed the processing mechanisms by which infected cells generated HF10–H-2Ld complexes. Treatment of infected BMDMs with the proteasome inhibitors epoxomicin or lactacystin abrogated activation of the CTgEZ.4 hybridoma (Fig. 6a,b), which demonstrated that proteasomes were required for the generation of HF10–H-2Ld complexes. Studies using ovalbumin as a model antigen secreted by tachyzoites have reported that the peptide transporter associated with antigen processing (TAP) is required for presentation of ovalbumin peptide to CD8+ T cells33,34. We therefore assessed the requirement for TAP in presentation of the GRA6 epitope by transducing H-2Ld into TAP-deficient or wild-type C57BL/6 BMDMs. In contrast to wild-type cells, TAP-deficient BMDMs failed to stimulate the CTgEZ.4 hybdroma (Fig. 6c). Thus, TAP transport was essential for presentation of the HF10–H-2Ld complex on the cell surface.
Figure 6.
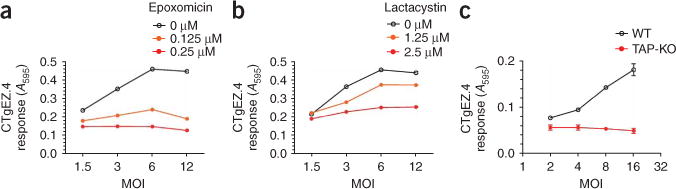
Presentation of the HF10–H-2Ld complex requires proteasomal activity and TAP transport. (a,b) Induction of β-galactosidase by the CTgEZ.4 hybridoma in response to BMDMs pretreated for 2 h with the proteasome inhibitors epoxomicin (a) or lactacystin (b) and infected for 8 h with T. gondii. Data are representative of two independent experiments. (c) Induction of β-galactosidase by CTgEZ.4 in response to H-2b BMDMs from C57BL/6J mice (WT) or TAP-deficient mice (TAP-KO) transduced with H-2Ld before T. gondii infection. Measurement of GFP expression at a multiplicity of infection (MOI) of 8 showed that 77% of C57BL/6J cells and 73% of TAP-deficient cells were infected. Data are representative of two independent experiments.
Next, we assessed the ability of ERAAP-deficient cells to present the GRA6 peptide to T. gondii–specific CD8+ T cells. Compared with wild-type counterparts, BMDMs, BMDCs and peritoneal macrophages from ERAAP-deficient mice were considerably compromised in their ability to activate the CTgEZ.4 hybridoma (Fig. 7a,b and Supplementary Fig. 5 online). However, we detected no difference in the frequency or maturation status of wild-type and ERAAP-deficient cells infected with T. gondii or in the capacity of these cells to present exogenous HF10 peptide to the same CTgEZ.4 hybridoma (Fig. 7c,d and Supplementary Fig. 6 online).
Figure 7.
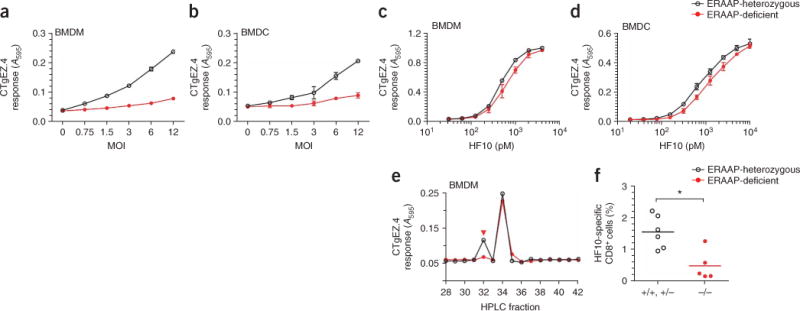
Presentation of HF10–H-2Ld complexes requires proteolysis in the endoplasmic reticulum. (a,b) Induction of β-galactosidase by the CTgEZ.4 hybridoma in response to ERAAP-heterozygous or ERAAP-deficient BMDMs (a) or BMDCs (b) infected for 8 h with T. gondii at a multiplicity of infection of 8. Approximately 90% of ERAAP-heterozygous BMDMs, 93% of ERAAP-deficient BMDMs, 69% of ERAAP-heterozygous BMDCs and 70% of ERAAP-deficient BMDCs were infected, as determined by GFP expression. (c,d) Presentation of synthetic HF10 peptide at various concentrations (horizontal axis) by ERAAP-heterozygous or ERAAP-deficient BMDMs (c) or BMDCs (d) to CTgEZ.4. Data (mean ± s.e.m.) are representative of three (b,d) or six (a,c) experiments. (e) Induction of β-galactosidase by CTgEZ.4 in response to peptide extracts fractionated by HPLC from ERAAP-deficient or ERAAP-heterozygous BMDMs infected as described in a. Data are representative of four experiments. (f) Frequency of HF10-Ld–multimer–binding CD8+ cells in mouse spleens at 12 d after infection with 3 × 103 Pru tachyzoites. Each symbol represents one mouse; small horizontal lines indicate the mean. *, P = 0.0173. Data are representative of three independent experiments.
To directly establish the function of ERAAP in generating HF10, we extracted naturally processed peptides from ERAAP-deficient and ERAAP-heterozygous infected BMDMs (Fig. 7e). Analysis of HPLC fractions showed two peaks of antigenic activity. In both ERAAP-containing and ERAAP-deficient extracts, antigenic activity was similar in fraction 34, which eluted together with the synthetic peptides RF14 (RRPLHPGSVNEFDF) and DF15 (DRRPLHPGSVNEFDF), which could serve as precursors of HF10 (Fig. 3c). Notably, however, although it was recovered from extracts of infected ERAAP-heterozygous macrophages, the HF10 peptide (fraction 32) was barely detected in extracts of ERAAP-deficient cells. To definitively demonstrate the requirement for endoplasmic reticulum proteolysis in generating the HF10–H-2Ld ligand in vivo, we measured the T. gondii–induced CD8+ T cell responses of ERAAP-deficient mice (Fig. 7f). Indeed, the frequency of HF10-specific CD8+ T cells was significantly lower in ERAAP-deficient mice than in wild-type or ERAAP-heterozygous mice. Our data collectively suggest that the impaired ability of ERAAP-deficient APCs to generate the HF10–H-2Ld complexes is directly responsible for the diminished expansion of HF10-specific CD8 T cell populations.
DISCUSSION
We have shown that despite the existence of thousands of potential T. gondii antigens, the immune response to T. gondii infection was dominated by CD8+ T cells specific for a single peptide processed from the GRA6 protein and presented by H-2Ld MHC class I molecules. The generation of HF10–H-2Ld complexes and protective CD8+ T cell responses depended critically on ERAAP-mediated proteolysis of antigenic precursors in the endoplasmic reticulum. ERAAP is therefore a factor determining the susceptibility of mice to toxoplasmosis and is essential for protective immunity to this parasite.
T. gondii belongs to the phylum Apicomplexa, which includes other clinically important pathogens such as Plasmodium spp. (malaria) and Cryptosporidium spp. (cryptosporidiosis), as well as major animal pathogens such as Theileria spp. (East Coast fever in cattle). T. gondii parasites infect an extremely wide range of hosts from mammals to birds and have a complex life cycle in the host. In acute infection in mice or humans, T. gondii exists in a fast-replicating tachyzoite stage that during chronic infection converts into a slower-growing, cyst-forming bradyzoite stage. By restricting parasite replication, the CD8+ T cell response protects infected hosts from T. gondii9–11 as well as Plasmodium spp.35.
We have identified GRA6 as an immunodominant antigen that elicited a protective CD8+ T cell response in T. gondii–infected mice. Furthermore, we found that the HF10 decapeptide of GRA6 was naturally processed in infected cells and was presented by H-2Ld MHC class I molecules. Our results resolve the longstanding mystery of why the H-2Ld locus is associated with fewer brain cysts in T. gondii–infected mice13. We identified the GRA6 antigen by an unbiased expression cloning approach that did not rely on predictive algorithms based on the peptide-binding characteristics of MHC molecules28. Indeed, the unusual length of the HF10 decapeptide makes it unlikely to be identified in a search for the canonical nonamer peptides typically presented by H-2Ld MHC class I molecules. Accordingly, the HF10 decamer was over 100-fold more active than the PF9 nonamer (PGSVNEFDF).
The CD8+ T cell response of H-2d mice infected by the type II T. gondii strain seemed to be targeted entirely to the single HF10–H-2Ld complex. Notably, similar focusing of the CD8+ T cell response to a single antigen from the circumsporozoite protein has been reported in mice infected with Plasmodium yoelii36. Likewise, during infection with Trypanosoma cruzi, another intracellular protozoan parasite, CD8+ T cells ‘preferentially’ recognize a small subset of epitopes encoded by only one family of trans-sialidase genes37. Given the genetic complexity of these eukaryotic organisms, the dominance of a single antigen is very notable and contrasts with the diversity of antigens used in viral infections (for example, by mouse cytomegalovirus)38. One possibility is that these differences in antigenic complexity may arise because of the distinct mechanisms of pathogen replication. Viruses replicate by using the translational machinery of the host to synthesize their proteins, whereas parasites replicate independently in specialized intracellular compartments. The latter may limit access of microbial proteins to the antigen-processing pathway. Notably, the carboxy-terminal region of GRA6 including the HF10 peptide is highly polymorphic among the various T. gondii strains31. It is possible that GRA6 evolved under selective pressure, as suggested by the failure of HF10-specific T cells to recognize the corresponding GRA6 peptide from a type I strain (data not shown).
Our findings have also provided insight into the antigen-transport pathways involved in antigen processing. These pathways have so far only been inferred from parasites engineered to express model antigens. As judged by microscopy, GRA6 is located exclusively in the parasitophorous vacuole39, unlike some components of the secreted T. gondii rhoptries, such as ROP1–2 (ref. 40) and ROP18 (refs. 41,42), which are found in the cytoplasm, or ROP16 (ref. 42) and PP2C43, which reach the nucleus. Nevertheless, GRA6 or its antigenic fragments must reach the cytoplasm because presentation of HF10–H-2Ld complexes is inhibited by the blockade of proteasome activity or TAP function. The nature of these antigenic precursors and the point at which they intersect with the endogenous antigen-processing pathway remain to be determined29,44,45. The identification of GRA6 as a precursor of the naturally processed HF10 peptide is important for addressing these issues. We suggest that the ability of exogenous antigens to access the MHC class I processing pathway is a critical parameter determining whether proteins serve as sources for peptide-MHC ligands that elicit CD8+ T cell responses.
ERAAP was required for the efficient generation of HF10–H-2Ld complexes in infected ‘professional’ APCs as well as other cells (data not shown). This observation indicates that the precursor of the HF10 peptide contained additional amino-terminal flanking residues that were trimmed by ERAAP in the endoplasmic reticulum. Because ERAAP does not trim peptides containing the ‘X-P’ motif (where ‘X’ is any amino acid) at the amino terminus46, and TAP does not transport peptides with proline residues at the p1 or p2 position15, the 11–amino acid peptide LF11 (LHPGSVNEFDF) is probably the precursor of HF10 in the endoplasmic reticulum. A test of this hypothesis requires monitoring of the proteolytic intermediates of GRA6 in T. gondii–infected cells, as done earlier in studies of ovalbumin peptides29.
During in vivo infection, the magnitude of the CD8+ T cell response was compromised in ERAAP-deficient mice, probably because of impaired expression of HF10–H-2Ld complexes in the absence of proteolysis in the endoplasmic reticulum. Alternatively, the lower number of T. gondii–specific CD8+ T cells may have also resulted from an absence or deficit of HF10-specific precursors in the T cell repertoire of ERAAP-deficient mice. These possibilities are not mutually exclusive.
Although the function of ERAAP in shaping the peptide–MHC class I repertoire has been established16,20, this is the first report to our knowledge demonstrating that ERAAP can serve as a ‘susceptibility factor’ for an infectious organism. Most notably, vaccination with DCs pulsed with the HF10 peptide protected mice from lethal toxoplasmosis in an MHC-dependent way. In this context, it is notable that H-2Ld is the only MHC class I molecule in laboratory mice that presents peptides with the ‘X-P’ motif; this feature is shared by approximately 20% of all known human MHC molecules28. Thus, our analysis of H-2d mice could be directly applicable to the development of a T. gondii vaccine for humans. Finally, progress in understanding the development of protective CD8+ T cell immunity to T. gondii provides a framework with which to study protective immunity to apicomplexan parasites, including the malaria-causing parasite Plasmodium falciparum.
METHODS
Mice and parasites
C57BL/6J, B10.D2-Hc1 H-2d H-2T18c/nSnJ (B10.D2), BALB/cJ, CBA/J and B6.129S2-Tap1tm1/Arp/J (TAP-deficient) mice were from the Jackson Laboratory. H-2b ERAAP-deficient mice on the C57BL/6 background have been described16. H-2d ERAAP-deficient mice were obtained by the crossing of H-2b ERAAP-deficient with B10.D2 mice. Sex- and age-matched 8- to 20-week-old ERAAP-heterozygous littermates and wild-type B10.D2 mice were used as controls. As no difference was generally noted in wild-type versus ERAAP-heterozygous mice, both genotypes were pooled throughout the study for comparison with ERAAP-deficient mice. Mice were used with the approval of the Animal Care and Use Committee of the University of California.
In vivo infection and immunization
The parental Prugniaud strain of T. gondii (PruΔhpt; hypoxanthine-xanthine-guaninephosphoribosyltransferase deficient) and the green fluorescent protein (GFP)-expressing derivative (Pru-GFP) were gifts from J. Boothroyd. Tachyzoites were maintained by passage in confluent monolayers of human foreskin fibroblasts. Cysts were obtained from the brains of CBA mice infected 3–6 weeks earlier with 3 × 103 tachyzoites. Mice were infected intraperitoneally with tachyzoites in 100 μl PBS, passed through 3-μm filter and released from a syringe, or orally by gavage with 300 μl crude brain homogenate containing five cysts. For induction of protection, B10.D2 BMDCs were activated for 24 h with LPS (100 ng/ml; Sigma), were incubated for 90 min with 1 μM synthetic peptide, were washed twice with PBS and were used for immunization. Mouse footpads were injected with 5 × 106 peptide-loaded BMDCs and mice were infected 7 d later. Where indicated, mice were depleted of CD8+ cells 36 h before immunization by one intraperitoneal injection of 250 μg depleting CD8-specific antibody (YTS169.4; Hybridoma and Monoclonal Antibody Core, University of California, San Francisco).
Ex vivo cell preparations
Mice were killed 10–13 d after infection. Peritoneal cells were recovered by lavage with 8 ml cold PBS. Spleens, livers and brains were collected and were stored at −80 °C for further DNA extraction or were immediately processed as follows. Spleens were dissociated into single-cell suspensions in complete RPMI medium (Invitrogen) supplemented with 10% (vol/vol) FCS (Hyclone). Samples were depleted of erythrocytes with AKC lysis buffer (100 μM EDTA, 160 mM NH4Cl and 10 mM NaHCO3). Mononuclear cells from the brain were prepared according to a published protocol47. Brains were minced and were digested for 1 h at 37 1C with collagenase type IA (1 mg/ml; Sigma) and DNaseI (100 μg/ml; Roche) in complete RPMI medium. Brains were then dissociated, were filtered through 70-μm cell strainers and were centrifuged for 10 min at 200g. Cells were resuspended in 60% (vol/vol) Percoll (GE Healthcare), were overlayed on 30% (vol/vol) Percoll and were centrifuged 20 min at 1,000g. 0 cells were lysed with 0.83% (wt/vol) NH4Cl. Cells were washed twice in complete RPMI medium before analysis.
Parasite load analysis
The number of infected splenocytes and peritoneal macrophages was determined by measurement of the percentage of GFP+ cells by flow cytometry. Results from two samples with over 2 × 105 events collected per tube were averaged for each mouse. Parasite burden in spleen, liver and brain was assessed by semiquantitative PCR as described48. Organs were crushed in liquid nitrogen with a mortar and pestle. Genomic DNA was extracted from less than 10 mg ground tissue with the DNeasy Blood & Tissue kit (Qiagen). A fragment of the T. gondii B1 gene was amplified by PCR along with increasing amounts of a competitive internal standard DNA added in the same reaction. The parasite load was estimated by comparison with samples containing known numbers of tachyzoites. The B1 internal control DNA was a gift from R. McLeod.
Generation of T. gondii–specific T cell hybridomas
Female BALB/c mice were immunized by two intraperitoneal injections of 1 ×106 irradiated Pru tachyzoites (14,000 rads) 21 d apart. Mice were killed 1 week after the second challenge and spleens were collected. T. gondii–specific T cell populations were expanded in vitro by one restimulation with irradiated syngeneic splenocytes (2,000 rads) followed by weekly restimulations with irradiated BALB/c-derived J774 macrophages (3,500 rads). The proportion of T. gondii–specific cells was monitored weekly by intracellular staining of IFN-γ on CD4+ or CD8α+ cells. APCs used for restimulation and IFN-γ assays were infected the day before with γ-irradiated tachyzoites at a multiplicity of infection of 4 or were incubated with recombinant surface antigen 1 (10 μg/ml; ProSpec-Tany) previously dialyzed against PBS. Responding cells were fused to the TCRαβ-negative lacZ-inducible BWZ.36.CD8α fusion partner as described24. The specificity of the resulting hybridomas was assessed by overnight incubation with infected or uninfected J774 cells. TCR-mediated induction of β-galactosidase was quantified with the chromogenic substrate CPRG (chlorophenol red–β-D-galactopyranoside; Roche). Cleavage of CPRG by β-galactosidase releases a purple product; absorbance was monitored at 595 nm with a reference at 655 nm.
Construction of cDNA library and plasmid constructs
A tachyzoite cDNA library was generated as described25. Poly(A)+ messenger RNA was isolated from about 6 × 108 Pru tachyzoites (that had been passed through a 3-μm filter) with the Oligotex Direct mRNA Midi/Maxi kit (Qiagen). The cDNA was generated with a Superscript cDNA Synthesis kit (Invitrogen) and oligo(dT) primers. The largest fragments were cloned into the mammalian expression vector pcDNA1 between the BstXI and NotI sites. In 96-well plates, 5 × 104 H-2Ld L cells per well were transfected with pools of cDNA (16 colony-forming units per well) by the diethylaminoethyl dextran method25. CTgEZ.4 T cell hybridomas were added (1 × 105 cells per well) followed by incubation overnight. The five most positive pools were subdivided and rescreened and the sequences of their cDNA inserts were ‘blasted’ against the T. gondii Me49 genome with the ToxoDB database (Toxoplasma gondii Genome Resource)31. All five had 100% identity to the gene 63.m00002, which encodes dense granule protein 6 (GRA6). GRA6 deletion mutants were obtained by subcloning of PCR products from the original GRA6 cDNA into pcDNAI with the BstXI and NotI sites. The appropriate NotI site (underlined in the following sequences) was introduced with the following primers: for full-length GRA6, T7 (forward) and 5′-TATGCGGCCGCTTAAAAATCAAACTCATTCACACTTCC-3′ (reverse); for the construct lacking the carboxy-terminal 55 amino acids, T7 (forward) and 5′-TATGCGGCCGCCTAGAAAGCGGTAAGCATTGCCACAGATACTGC-3′ (reverse). Putative candidate peptides as well as DF15 were introduced in the cytosol of transfected cells with a ubiquitin-based antigen-release system as described49. For each peptide sequence, the coding and cDNA strands containing the appropriate SacII and BamHI protruding ends were annealed together and were cloned in the pEGFP-Ub vector (a gift from F. Levy) with the SacII and BamHI sites.
BMDC and BMDM in vitro differentiation and infection
Bone marrow cells were obtained from mouse femurs and tibias. DCs were differentiated from bone marrow cells for 6 d with granulocyte-macrophage colony-stimulating factor (10 ng/ml; Biosource) in complete RPMI medium (purity, about 70% CD11c+). Primary macrophages were differentiated for 7 d in Petri dishes with RPMI medium supplemented with 20% (vol/vol) FCS and 10% (vol/vol) colony-stimulating factor–containing culture supernatant (purity, about 95% CD11b+). Colony-stimulating factor–producing 3T3 cells were a gift from R. Vance. H-2Ld MHC class I was introduced into C57BL/6J and TAP-deficient BMDMs by retroviral transduction. H-2Ld DNA was cloned into the pMSCV2.2 retroviral vector (a gift from W. Sha) and was used to transfect BOSC packaging cells. On days 3 and 4 during differentiation, BMDMs were transduced with the retroviral supernatant by two ‘spin infections’ (1 h at 1800g and 32 °C) with polybrene (5 μg/ml; Sigma) and were used at day 6. For proteasome inhibition, BMDMs were preincubated for 2 h with epoxomicin or lactacystin (Calbiochem). The drugs were left throughout the infection but were washed off during incubation with the CTgEZ.4 hybridoma. Cells were infected by mixture for 8 h or 16 h of APCs with irradiated tachyzoites (14,000 rads) at various multiplicities of infection. Cells were then washed twice for elimination of most of the extracellular parasites and were used in subsequent assays. The proportion of infected (GFP+) cells was controlled with Pru-GFP tachyzoites.
Peptide extracts and HPLC fractionation
Peptides were extracted from about 5 × 106 infected or control cells by boiling in 10% acetic acid solution. After centrifugation for 15 min at 16,000g, supernatants were filtered through Microcon filters with a cutoff of 10 kilodaltons. Filtrates were fractionated by HPLC on a C18 reverse-phase column (Vydac). Samples were separated for 29 min by a linear gradient of acetonitrile into water (15–35%) with 0.1% (vol/vol) trifluoroacetic acid as the ion-pairing agent. Synthetic peptides prepared (by D. King) by solid-phase synthesis were ‘spiked’ on J774 cell extracts, which were treated as other cell extracts were before HPLC fractionation.
Flow cytometry
Antibody to mouse CD4 (anti–mouse CD4; GK1.5), anti–mouse CD8 (53–6.7) and anti–mouse IFN-γ (XMG1.2) were from BD Biosciences. Surfaces were labeled according to standard procedures with flow cytometry buffer (3% (vol/vol) FCS and 1 mM EDTA in PBS). Intracellular IFN-γ in CD4+ or CD8α+ cells was detected with the Cytofix/Cytoperm kit (BD Pharmingen). DimerX H-2Ld:Ig (fusion protein of H-2Ld and immunoglobulin; BD Biosciences) was used according to the manufacturer’s instructions. DimerX was loaded overnight at 37 °C with a 160-molar excess of HF10 peptide or a control H-2Ld-binding peptide (QL9 or YL9). Cells (1 × 106) were stained for 1 h with 2 μg DimerX, followed by staining for 30 min with 0.05 μg phycoerythrin-coupled anti–mouse immunoglobulin G1 (A85-1; BD Pharmingen). These conditions gave the highest signal/noise ratio. All flow cytometry data were acquired on an XL Analyzer (Coulter) and were analyzed with FlowJo software (Tree Star).
Statistical analysis
Prism software (GraphPad) was used for statistical analyses. P values were calculated with the two-tailed Mann-Whitney (nonparametric) test unless otherwise specified.
Supplementary Material
Acknowledgments
We thank J. Boothroyd (Stanford University) for the parental Pru strain of T. gondii and the GFP-expressing derivative; R. McLeod (University of Chicago) for B1 internal control DNA; F. Levy (University of Epalinges) for the pEGFP-Ub vector; R. Vance (University of California, Berkeley) for the colony-stimulating factor–producing 3T3 cells; W. Sha (University of California, Berkeley) for the pMSCV2.2 retroviral vector; D. King (University of California, Berkeley) for peptide synthesis; J. Boothroyd and R. McLeod for discussions; the Center for Host-Pathogen Studies core facilities and S.-W. Chan for technical assistance; and B. Striepen for comments on the manuscript. Supported by the US National Institutes of Health (N.S. and E.A.R.) and the international Human Frontier Science Program (LT00841/2005-l to N.B.).
Footnotes
Note: Supplementary information is available on the Nature Immunology website.
AUTHOR CONTRIBUTIONS
N.B. designed and did all experiments in collaboration with F.G. (cDNA library), M.S. (brain cell isolation), N.T.J. (footpad immunization and measurement of the natural killer cell response), T.C. (processed peptide analysis) and A.J.S. (parasite burden measurements); E.A.R. and N.S. provided guidance and contributed to the experimental design; and N.B. and N.S. wrote the manuscript together with input from other authors.
References
- 1.Montoya JG, Liesenfeld O. Toxoplasmosis. Lancet. 2004;363:1965–1976. doi: 10.1016/S0140-6736(04)16412-X. [DOI] [PubMed] [Google Scholar]
- 2.Yap GS, Sher A. Cell-mediated immunity to Toxoplasma gondii: initiation, regulation and effector function. Immunobiology. 1999;201:240–247. doi: 10.1016/S0171-2985(99)80064-3. [DOI] [PubMed] [Google Scholar]
- 3.Gazzinelli RT, Eltoum I, Wynn TA, Sher A. Acute cerebral toxoplasmosis is induced by in vivo neutralization of TNF-α and correlates with the down-regulated expression of inducible nitric oxide synthase and other markers of macrophage activation. J Immunol. 1993;151:3672–3681. [PubMed] [Google Scholar]
- 4.Schluter D, et al. Both lymphotoxin-α and TNF are crucial for control of Toxoplasma gondii in the central nervous system. J Immunol. 2003;170:6172–6182. doi: 10.4049/jimmunol.170.12.6172. [DOI] [PubMed] [Google Scholar]
- 5.Suzuki Y, Orellana MA, Schreiber RD, Remington JS. Interferon-γ: the major mediator of resistance against Toxoplasma gondii. Science. 1988;240:516–518. doi: 10.1126/science.3128869. [DOI] [PubMed] [Google Scholar]
- 6.Goldszmid RS, et al. TAP-1 indirectly regulates CD4+ T cell priming in Toxoplasma gondii infection by controlling NK cell IFN-γ production. J Exp Med. 2007;204:2591–2602. doi: 10.1084/jem.20070634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khan IA, et al. CCR5 is essential for NK cell trafficking and host survival following Toxoplasma gondii infection. PLoS Pathog. 2006;2:e49. doi: 10.1371/journal.ppat.0020049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lutjen S, Soltek S, Virna S, Deckert M, Schluter D. Organ- and disease-stage-specific regulation of Toxoplasma gondii-specific CD8-T-cell responses by CD4 T cells. Infect Immun. 2006;74:5790–5801. doi: 10.1128/IAI.00098-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buzoni-Gatel D, Lepage AC, Dimier-Poisson IH, Bout DT, Kasper LH. Adoptive transfer of gut intraepithelial lymphocytes protects against murine infection with Toxoplasma gondii. J Immunol. 1997;158:5883–5889. [PubMed] [Google Scholar]
- 10.Gazzinelli RT, Hakim FT, Hieny S, Shearer GM, Sher A. Synergistic role of CD4+ and CD8+ T lymphocytes in IFN-gamma production and protective immunity induced by an attenuated Toxoplasma gondii vaccine. J Immunol. 1991;146:286–292. [PubMed] [Google Scholar]
- 11.Suzuki Y, Remington JS. Dual regulation of resistance against Toxoplasma gondii infection by Lyt-2+ and Lyt-1+, L3T4+ T cells in mice. J Immunol. 1988;140:3943–3946. [PubMed] [Google Scholar]
- 12.Parker SJ, Roberts CW, Alexander J. CD8+ T cells are the major lymphocyte subpopulation involved in the protective immune response to Toxoplasma gondii in mice. Clin Exp Immunol. 1991;84:207–212. doi: 10.1111/j.1365-2249.1991.tb08150.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown CR, et al. Definitive identification of a gene that confers resistance against Toxoplasma cyst burden and encephalitis. Immunology. 1995;85:419–428. [PMC free article] [PubMed] [Google Scholar]
- 14.Hammer GE, Kanaseki T, Shastri N. The final touches make perfect the peptide-MHC class I repertoire. Immunity. 2007;26:397–406. doi: 10.1016/j.immuni.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 15.Blanchard N, Shastri N. Coping with loss of perfection in the MHC class I peptide repertoire. Curr Opin Immunol. 2008;20:82–88. doi: 10.1016/j.coi.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hammer GE, Gonzalez F, Champsaur M, Cado D, Shastri N. The aminopeptidase ERAAP shapes the peptide repertoire displayed by major histocompatibility complex class I molecules. Nat Immunol. 2006;7:103–112. doi: 10.1038/ni1286. [DOI] [PubMed] [Google Scholar]
- 17.Yan J, et al. In vivo role of ER-associated peptidase activity in tailoring peptides for presentation by MHC class Ia and class Ib molecules. J Exp Med. 2006;203:647–659. doi: 10.1084/jem.20052271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Firat E, et al. The role of endoplasmic reticulum-associated aminopeptidase 1 in immunity to infection and in cross-presentation. J Immunol. 2007;178:2241–2248. doi: 10.4049/jimmunol.178.4.2241. [DOI] [PubMed] [Google Scholar]
- 19.York IA, Brehm MA, Zendzian S, Towne CF, Rock KL. Endoplasmic reticulum aminopeptidase 1 (ERAP1) trims MHC class I-presented peptides in vivo and plays an important role in immunodominance. Proc Natl Acad Sci USA. 2006;103:9202–9207. doi: 10.1073/pnas.0603095103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hammer GE, Gonzalez F, James E, Nolla H, Shastri N. In the absence of ERAAP, the MHC class I molecules present many unstable and highly immunogenic peptides. Nat Immunol. 2007;8:101–108. doi: 10.1038/ni1409. [DOI] [PubMed] [Google Scholar]
- 21.Karttunen J, Sanderson S, Shastri N. Detection of rare antigen presenting cells by the lacZ T-cell activation assay suggests an expression cloning strategy for T-cell antigens. Proc Natl Acad Sci USA. 1992;89:6020–6024. doi: 10.1073/pnas.89.13.6020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khan IA, Ely KH, Kasper LH. Antigen-specific CD8+ T cell clone protects against acute Toxoplasma gondii infection in mice. J Immunol. 1994;152:1856–1860. [PubMed] [Google Scholar]
- 23.Kim SK, Boothroyd JC. Stage-specific expression of surface antigens by Toxoplasma gondii as a mechanism to facilitate parasite persistence. J Immunol. 2005;174:8038–8048. doi: 10.4049/jimmunol.174.12.8038. [DOI] [PubMed] [Google Scholar]
- 24.Sanderson S, Shastri N. LacZ inducible peptide/MHC specific T-hybrids. Int Immunol. 1994;6:369–376. doi: 10.1093/intimm/6.3.369. [DOI] [PubMed] [Google Scholar]
- 25.Mendoza L, Malarkannan S, Shastri N. Methods in Molecular Biology. In: Solheim JC, editor. Antigen Processing and Presentation Protocols. Vol. 156. Humana Press; Totowa, NJ: 2000. pp. 255–264. [Google Scholar]
- 26.Lecordier L, et al. Characterization of a dense granule antigen of Toxoplasma gondii (GRA6) associated to the network of the parasitophorous vacuole. Mol Biochem Parasitol. 1995;70:85–94. doi: 10.1016/0166-6851(95)00010-x. [DOI] [PubMed] [Google Scholar]
- 27.Mercier C, et al. Biogenesis of nanotubular network in Toxoplasma parasitophorous vacuole induced by parasite proteins. Mol Biol Cell. 2002;13:2397–2409. doi: 10.1091/mbc.E02-01-0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rammensee H, Bachmann J, Emmerich NP, Bachor OA, Stevanovic S. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics. 1999;50:213–219. doi: 10.1007/s002510050595. [DOI] [PubMed] [Google Scholar]
- 29.Kunisawa J, Shastri N. Hsp90α chaperones large proteolytic intermediates in the MHC class I antigen processing pathway. Immunity. 2006;24:523–534. doi: 10.1016/j.immuni.2006.03.015. [DOI] [PubMed] [Google Scholar]
- 30.Kwok LY, et al. The induction and kinetics of antigen-specific CD8 T cells are defined by the stage specificity and compartmentalization of the antigen in murine toxoplasmosis. J Immunol. 2003;170:1949–1957. doi: 10.4049/jimmunol.170.4.1949. [DOI] [PubMed] [Google Scholar]
- 31.Gajria B, et al. ToxoDB: an integrated Toxoplasma gondii database resource. Nucleic Acids Res. 2008;36:D553–D556. doi: 10.1093/nar/gkm981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kong JT, Grigg ME, Uyetake L, Parmley S, Boothroyd JC. Serotyping of Toxoplasma gondii infections in humans using synthetic peptides. J Infect Dis. 2003;187:1484–1495. doi: 10.1086/374647. [DOI] [PubMed] [Google Scholar]
- 33.Gubbels MJ, Striepen B, Shastri N, Turkoz M, Robey EA. Class I major histocompatibility complex presentation of antigens that escape from the parasitophorous vacuole of Toxoplasma gondii. Infect Immun. 2005;73:703–711. doi: 10.1128/IAI.73.2.703-711.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dzierszinski F, et al. Presentation of Toxoplasma gondii antigens via the endogenous major histocompatibility complex class I pathway in nonprofessional and professional antigen-presenting cells. Infect Immun. 2007;75:5200–5209. doi: 10.1128/IAI.00954-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morrot A, Zavala F. Regulation of the CD8+ T cell responses against Plasmodium liver stages in mice. Int J Parasitol. 2004;34:1529–1534. doi: 10.1016/j.ijpara.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 36.Kumar KA, et al. The circumsporozoite protein is an immunodominant protective antigen in irradiated sporozoites. Nature. 2006;444:937–940. doi: 10.1038/nature05361. [DOI] [PubMed] [Google Scholar]
- 37.Martin DL, et al. CD8+ T-Cell responses to Trypanosoma cruzi are highly focused on strain-variant trans-sialidase epitopes. PLoS Pathog. 2006;2:e77. doi: 10.1371/journal.ppat.0020077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Munks MW, et al. Genome-wide analysis reveals a highly diverse CD8 T cell response to murine cytomegalovirus. J Immunol. 2006;176:3760–3766. doi: 10.4049/jimmunol.176.6.3760. [DOI] [PubMed] [Google Scholar]
- 39.Cesbron-Delauw MF, Gendrin C, Travier L, Ruffiot P, Mercier C. Apicomplexa in mammalian cells: trafficking to the parasitophorous vacuole. Traffic. 2008;9:657–664. doi: 10.1111/j.1600-0854.2008.00728.x. [DOI] [PubMed] [Google Scholar]
- 40.Hakansson S, Charron AJ, Sibley LD. Toxoplasma evacuoles: a two-step process of secretion and fusion forms the parasitophorous vacuole. EMBO J. 2001;20:3132–3144. doi: 10.1093/emboj/20.12.3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Taylor S, et al. A secreted serine-threonine kinase determines virulence in the eukaryotic pathogen Toxoplasma gondii. Science. 2006;314:1776–1780. doi: 10.1126/science.1133643. [DOI] [PubMed] [Google Scholar]
- 42.Saeij JP, et al. Polymorphic secreted kinases are key virulence factors in toxoplasmosis. Science. 2006;314:1780–1783. doi: 10.1126/science.1133690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gilbert LA, Ravindran S, Turetzky JM, Boothroyd JC, Bradley PJ. Toxoplasma gondii targets a protein phosphatase 2C to the nuclei of infected host cells. Eukaryot Cell. 2007;6:73–83. doi: 10.1128/EC.00309-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ravindran S, Boothroyd JC. Secretion of proteins into host cells by Apicomplexan parasites. Traffic. 2008;9:647–656. doi: 10.1111/j.1600-0854.2008.00723.x. [DOI] [PubMed] [Google Scholar]
- 45.Shastri N, Cardinaud S, Schwab SR, Serwold T, Kunisawa J. All the peptides that fit: the beginning, the middle, and the end of the MHC class I antigen-processing pathway. Immunol Rev. 2005;207:31–41. doi: 10.1111/j.0105-2896.2005.00321.x. [DOI] [PubMed] [Google Scholar]
- 46.Serwold T, Gonzalez F, Kim J, Jacob R, Shastri N. ERAAP customizes peptides for MHC class I molecules in the endoplasmic reticulum. Nature. 2002;419:480–483. doi: 10.1038/nature01074. [DOI] [PubMed] [Google Scholar]
- 47.Reichmann G, Villegas EN, Craig L, Peach R, Hunter CA. The CD28/B7 interaction is not required for resistance to Toxoplasma gondii in the brain but contributes to the development of immunopathology. J Immunol. 1999;163:3354–3362. [PubMed] [Google Scholar]
- 48.Kirisits MJ, Mui E, McLeod R. Measurement of the efficacy of vaccines and antimicrobial therapy against infection with Toxoplasma gondii. Int J Parasitol. 2000;30:149–155. doi: 10.1016/s0020-7519(00)00009-6. [DOI] [PubMed] [Google Scholar]
- 49.Levy F, Johnsson N, Rumenapf T, Varshavsky A. Using ubiquitin to follow the metabolic fate of a protein. Proc Natl Acad Sci USA. 1996;93:4907–4912. doi: 10.1073/pnas.93.10.4907. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.