

Abstract
We sought to identify quantitative trait loci (QTLs) by genome-wide linkage analysis for BMI and waist circumference (WC) exploring various strategies to address heterogeneity including covariate adjustments and complex models based on epistatic components of variance. Because cholesterol-lowering drugs and diabetes medications may affect adiposity and risk of coronary heart disease, we excluded subjects medicated for hypercholesterolemia and hyperglycemia. The evidence of linkage increased on 2p25 (BMI: lod = 1.59 vs. 2.43, WC: lod = 1.32 vs. 2.26). Because environmental and/or genetic components could mask the effect of a specific locus, we investigated further whether a QTL could influence adiposity independently of lipid pathway and dietary habits. Strong evidence of linkage on 2p25 (BMI: lod = 4.31; WC: lod = 4.23) was found using Willet’s dietary factors and lipid profile together with age and sex in adjustment. It suggests that lipid profile and dietary habits are confounding factors for detecting a 2p25 QTL for adiposity. Because evidence of linkage has been previously detected for BMI on 7q34 and 13q14 in National Heart, Lung, and Blood Institute Family Heart Study (NHLBI FHS), and for diabetes on 15q13, we investigated epistasis between chromosome 2 and these loci. Significant epistatic interactions were found between QTLs 2p25 and 7q34, 2q37 and 7q34, 2q31 and 13q14, and 2q31–q36 and 15q13. These results suggest multiple pathways and factors involving genetic and environmental effects influencing adiposity. By taking some of these known factors into account, we clarified our linkage evidence of a QTL on 2p25 influencing BMI and WC. The 2p25, 2q24–q31, and 2q36–q37 showed evidence of epistatic interaction with 7q34, 13q14, and 15q13.
INTRODUCTION
The prevalence of obesity has grown sharply in the United States (1). Obesity is a risk factor for type 2 diabetes mellitus, hypertension, and cardiovascular diseases (CVDs), and associated with dyslipidemia, nonalcoholic fatty liver disease, gallbladder disease, osteoarthritis, and several cancers (2–5). Although BMI is a measure of overall adiposity, waist circumference (WC) reflects abdominal fat accumulation, which is more strongly associated with metabolic syndrome, insulin resistance, and CVD.
Complex genetic and environmental interactions regulate the accumulation and distribution of body fat. The majority of studies report heritabilities of 40–65% for BMI and WC (6–8). Several quantitative trait loci (QTLs) have been uncovered along all autosomal chromosomes for obesity-related phenotypes, in different population (9). In families participating in the National Heart, Lung, and Blood Institute Family Heart Study (NHLBI FHS), we reported strong evidence of QTLs on chromosomes 7q32 (lod = 4.7) and 13q14 (lod = 3.2) for BMI (6). Other studies have reported other locations for QTLs influencing BMI and/or obesity (9). The lack of replication among studies could be due to population stratification, a difference on allele frequency, gene penetrance, sample size, and density of genotyping. Moreover, interaction among genes and with environmental factors play an important role in determining BMI and WC levels and, consequently, may play a major confounding role in detecting regions by linkage analysis. Additional covariate adjustment, subset analysis, and/or complex genetic modeling may aid identifying linked regions and also assist gene discovery. Epistatic models may enhance linkage signals between interacting QTLs and have been reported for BMI, WC, and percent fat (10,11), and for height (12) and hip circumference (13). In the current study, we extended our genome-wide scan analysis for BMI and WC exploring adjustments for different covariates as well as a subsample selection multipoint variance components linkage analysis. We tested gene–gene interactions using two-locus epistatic models in linkage analysis applied to families of the NHLBI FHS.
METHODS AND PROCEDURES
Study design and measurements
The NHLBI FHS was proposed to identify and evaluate the genetic and nongenetic determinants of CVD and atherosclerosis. The design and objectives of this study were described previously (14). In summary, subjects were invited for a clinic visit that included measurements of body size, blood pressure, lipids, lipoproteins, hemostatic factors, insulin, glucose, and routine blood chemistries. Additional information on health history, medication use, lifestyle, and dietary habits was obtained by questionnaires. BMI was calculated as weight (in kilograms) divided by the square of height (in meters). WC was taken at the level of the smallest circumference on the torso below the sternum (in cm). The NHLBI FHS collected data on dietary habits by using a shortened version of the 61-item semiquantitative food frequency questionnaire developed by Willett et al. http://circ.ahajournals.org/cgi/content/full/111/22/2921-R21-165596#R21-165596 (ref. 15). The intake of specific nutrients was computed by multiplying the frequency of consumption of an item by the nutrient content of specified portions. Composition values for nutrients were obtained from the Harvard University Food Composition Database derived from US Department of Agriculture sources (1963–1988) and manufacturer information. Use of cholesterol-lowering and hyperglycemic medications was obtained by self-report.
Phenotypic covariate adjustments
The adjustments were carried out by stepwise regression analysis separately by sex, using up to a cubic polynomial in age and clinical center. Terms significant at the 5% level were retained in the model. The residual variances were also examined by regressing the squared residuals from the mean age regression on another similar regression model in a stepwise manner and retaining significant terms. We also employed a second multivariate stepwise regression adjustment using the total dietary intake of fats, carbohydrates, proteins, vitamins, salts, and minerals; lipid levels (high-density lipoprotein-cholesterol (HDL-C), low-density lipoprotein-cholesterol, total cholesterol, and triglycerides (TGs)) in addition to the standard covariates (age, sex, and field centers), retaining the significant terms at the 5% level.
Genotype data
Detailed description on NHLBI FHS genotype data and quality control has been previously published (6). Two groups of families were geno-typed: (i) 988 white subjects in 267 sibships were typed for 243 autosomal short tandem repeat markers by the Utah Molecular Genetics Laboratory, and (ii) 2,666 white subjects distributed among 397 largest pedigrees were typed for 402 markers by the Mammalian Genotyping Service. Approximate chromosomal locations were ascertained from the Genetic Location Database and the locations in cM were used from the Marshfield map.
Genetic analysis
Linkage analyses using a variance component approach were implemented by the computer program SOLAR version 2.1.4 (Southwest Foundation for Biomedical Research, San Antonio, TX) (16). The multipoint identity-by-descent probabilities were estimated using GeneHunter software (version 2.0; Whitehead Institute for Biomedical Research, Cambridge, MA) and imported into SOLAR. The variance due to an additive genetic contribution of a QTL (), a polygenic component (), and an environmental component not shared among family members (e2) are estimated. The linkage hypothesis is tested by likelihood-ratio contrasting no-linkage model () and the alternative (). The difference in minus twice the log likelihoods of the two hypotheses is asymptotically distributed as a ½: ½ mixture of χ2 with one degree of freedom and a point mass at zero. The lod score is calculated by dividing the χ2 value by 2 × log10. The bivariate multipoint linkage approach is an extension of the univariate analysis and tests whether the correlation pattern between two quantitative traits in families is due to pleiotropic genetic effects (17). The pleiotropy is modeled as cross-trait correlations caused by a QTL (ρq), a residual additive genetic effect (ρg), and a random environmental effect (ρe). The hypothesis of pleiotropy was tested by the complete model (ρq >0) against the null that the two traits has no common QTL effect (ρq = 0), using likelihood-ratio tests (18). We also performed analyses for epistatic additive interactions using a two-locus multipoint linkage analysis. This approach is an extension of the univariate analysis that estimates two-locus ( and ) component variances with an additive epistasis variance component (). To test an epistatic interaction effect, comparison of (i) the likelihood of two-locus model with only additive effects for each pair of loci () and (ii) the likelihood of two-locus model with additive effects as well as an epistatic term for interaction between the two loci was performed. Significance test was determined by likelihood-ratio test using a χ2 statistic with one degree of freedom. The false discovery rate (FDR) (19) was calculated to measure the global error for multiple tests, i.e., the expected proportion of false rejections of the null hypothesis among the total number of rejections. FDR was estimated separately by each phenotype (WC or BMI) and model (univariate, bivariate, and epistatic) linkage analysis using the SAS package (SAS Institute, Cary, NC). The P value adjusted for FDR was denoted here by FDR P value.
RESULTS
Table 1 shows the descriptive statistics for unadjusted phenotypes and covariates. Plasma levels of HDL-C and TG were the major predictors of BMI and WC and accounted for 10–16% in their phenotypic variances, whereas age, field centers, and dietary intake factors (animal protein, fat calorie, fatty acid, total fat, polyunsaturated fat, carotene, iron, sodium, sucrose, zinc, and vitamin B6) accounted for 4–9% of the variances in BMI and WC. Significant phenotypic correlations were detected for age- and sex-adjusted BMI and WC with HDL-C (−0.26, for both traits), TG (0.27 and 0.29, respectively), and dietary intake factors (proteins: 0.08; fats: 0.04–0.10; carbohydrates, vitamins, salts, and minerals: 0.01–0.05, for both traits). The heritabilities were over 40% for BMI and WC (Table 2).
Table 1.
Descriptive analysis for unadjusted variables in the NHLBI FHS
| Complete data |
Nonmedicated dataa |
|||||
|---|---|---|---|---|---|---|
| Variables | N | Mean | s.d. | N | Mean | s.d. |
| Age (years) | 5,710 | 52.6 | 14.1 | 4,363 | 50.8 | 13.9 |
| BMI (kg/m2) | 5,072 | 27.5 | 5.4 | 4,363 | 27.0 | 5.3 |
| Waist circumference (cm) | 5,076 | 97.0 | 15.2 | 4,363 | 95.9 | 15.1 |
| High-density lipoprotein-cholesterol (mg/dl) | 5,620 | 50.3 | 15.2 | 4,363 | 50.8 | 15.2 |
| Total cholesterol (mg/dl) | 5,620 | 205.5 | 40.5 | 4,363 | 203.7 | 39.3 |
| Triglycerides (mg/dl) | 5,620 | 150.5 | 107.6 | 4,363 | 143.5 | 103.6 |
| Energy intake (kJ/day) | 4,883 | 7,316 | 3,137 | 4,194 | 7,410 | 3,123 |
| Energy from saturated fat (%) | 4,883 | 11.5 | 4,194 | 11.8 | ||
| Energy from monounsaturated fat (%) | 4,883 | 12.1 | 4,194 | 12.2 | ||
| Energy from protein (%) | 4,883 | 18.1 | 4,194 | 17.9 | ||
| Energy from carbohydrates (%) | 4,883 | 51.2 | 4,194 | 52.5 | ||
| Iron (g/day) | 4,883 | 0.012 | 0.006 | 4,194 | 0.012 | 0.007 |
| Sodium (g/day) | 4,883 | 1.64 | 0.74 | 4,194 | 1.65 | 0.75 |
| Sucrose (g/day) | 4,883 | 0.051 | 0.035 | 4,194 | 0.053 | 0.036 |
| Zinc (g/day) | 4,883 | 0.011 | 0.006 | 4,194 | 0.012 | 0.006 |
| Vitamin B6 (mg/day) | 4,883 | 2.0 | 0.9 | 4,194 | 2.0 | 0.9 |
| Carotene (IU) | 4,883 | 8,651.2 | 10,313.4 | 4,194 | 8,569.2 | 10,344.9 |
| Self-reported use of cholesterol-lowering drugs | 5,710 | 13% | ||||
| Self-reported use of diabetes medication | 5,710 | 6% | ||||
NHLBI FHS, National Heart, Lung, and Blood Institute Family Heart Study.
Nonmedicated data: subjects taking medications for high cholesterol and/or diabetes were excluded.
Table 2.
Linkage analysis results on chromosome 2p25 in the NHLBI FHS
| Sample and adjustment |
Complete data |
Nonmedicated dataa |
|
|---|---|---|---|
| Basicb | Basicb | Extendedc | |
| BMI | |||
| Overall additive genetic heritability |
0.43 ± 0.04 | 0.44 ± 0.04 | 0.48 ± 0.04 |
| Lod (location) | 1.59 (18 cM) | 2.43 (15 cM) | 4.31 (13 cM) |
| P value | 0.00343 | 0.00041 | 0.0000042 |
| FDR P value | 0.02366 | 0.00453 | 0.0000512 |
| WC | |||
| Overall additive genetic heritability |
0.41 ± 0.04 | 0.42 ± 0.04 | 0.45 ± 0.04 |
| Lod (location) | 1.32 (18 cM) | 2.26 (16 cM) | 4.23 (13 cM) |
| P value | 0.00677 | 0.00063 | 0.0000050 |
| FDR P value | 0.03187 | 0.00559 | 0.0000568 |
FDR, false discovery rate; NHLBI FHS, National Heart, Lung, and Blood Institute Family Heart Study; WC, waist circumference.
Nonmedicated data: subjects taking medications high cholesterol and/or diabetes were excluded.
Basic adjustment: age, field centers, and sex.
Extended adjustment: age, field centers, sex, lipid–lipoprotein levels, and dietary intake factors.
Because lipid-lowering drugs and diabetes medications may affect adipose tissue and/or be related to subjects with higher values for CVD risk factors than the average population, we did linkage analysis excluding subjects with self-reported use of cholesterol-lowering medications and/or hyperglycemic medications. On chromosome 2p25 (at marker GATA116B01), the lod scores were higher for BMI (2.43 vs. 1.59, Table 2, Figure 1) and WC (2.26 vs. 1.32), in spite of the reduction in sample size (the number of sib-pairs decreased 15%). Besides lipid-lowering drugs, dietary habits and deregulation in lipid pathway affect the lipolysis in adipocytes that may develop in fat store. However, there is a possibility that a QTL influences adiposity independently of lipid pathway and/ or dietary habits, and these factors may play a confounding role to detect the locus. To test this hypothesis, we added lipid profile and Willett’s dietary intake factors in the basic (age, sex, and field centers) covariate adjustment. We found strong evidence of linkage at the same 2p25 location using this extended adjustment in nonmedicated subjects. The lod scores increased further from 2.43 to 4.31 (FDR P value = 0.0000512) for BMI and from 2.26 to 4.23 (FDR P value = 0.0000568) for WC. These results suggest that drugs, dietary habits, and lipid profile (TG and HDL-C levels) are confounding factors to detect a 2p25 QTL for adiposity traits.
Figure 1.
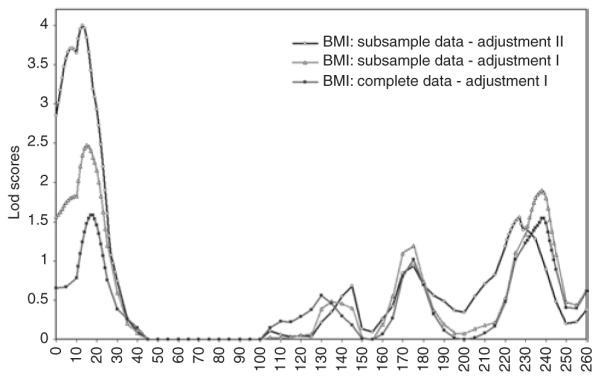
Lod plots for BMI on chromosome 2. Nonmedicated data: subjects taking medications for high cholesterol and/or diabetes were excluded. Basic adjustment: age, field centers, and sex; extended adjustment: age, field centers, sex, lipid levels, and dietary intake factors.
As the maximum lod scores for BMI and WC are located on the same chromosome 2p25 region, we also performed bivariate analysis to explore to what extent their correlation could be explained by that QTL. The bivariate lod slightly increased to 4.53 (2p25) for age-, sex-, lipid-, and dietary factors–adjusted BMI_WC (vs. 4.31 and 4.23 for BMI and WC, respectively). The trait correlation attributable to the QTL (ρq) bounded to 1, suggesting that both traits are influenced by the same underlying gene(s) at this location. The trait correlation due to residual additive genetic effects (ρg) was 0.74. We also investigated whether a pleiotropic genetic effect on chromosome 2 influenced adiposity and lipid profile. On 2p25, the bivariate lod scores for age- and sex-adjusted cross-trait correlations did not increase compared with the univariate lod scores (lodBMI_HDL-C = 0.87, lodBMI_TG = 0.94, lodWC_HDL-C = 0.68, and lodWC_TG = 0.71 vs. lodBMI = 1.59, lodWC = 1.32, lodHDL-C = 0.58, and lodTG = 0.02). These results suggest that there was not a common QTL on 2p25 affecting the mean levels of adiposity, HDL-C, and TG.
Because other QTLs have been detected on chromosomes 7q34 and 13q14 for BMI in the NHLBI FHS (6), we tested for epistasis between loci on chromosome 2 conditioning on the linkage evidence on 7q34 and 13q14. The epistasis variance component linkage analysis was carried out for age- and sex-adjusted BMI and WC in the complete NHLBI FHS data. There was significant evidence of epistasis between QTLs on 2p25 and 7q34 (BMI: , P = 0.0128 and WC: , P = 0.0153, Table 3, Figure 2), 2q37 and 7q34 (BMI: , P = 0.0120), and 2q31 and 13q14 (BMI: , P = 0.0217 and WC: , P = 0.0343).
Table 3.
Summary of linkage analysis for oligogenic and epistatic models in the complete NHLBI FHS data
| Interaction |
||||||
|---|---|---|---|---|---|---|
| Oligogenic (I) |
Epistatic (II) |
Test: I vs. II |
||||
| Chromosome (cM) | Lod | Loda | P valueb | FDR P value | P valuec | |
| BMI | ||||||
| 7q34 (140) and 2p25 (19) | 1.35 | 4.04 | 0.33 | 0.0000079 | 0.00017 | 0.0128 |
| 7q34 (140) and 2q37 (238) | 1.65 | 4.39 | 0.35 | 0.0000034 | 0.00015 | 0.0120 |
| 13q14 (42) and 2p25 (19) | 1.20 | 2.74 | 0.27 | 0.0001901 | 0.00335 | 0.0596 |
| 13q14 (42) and 2q31 (178) | 1.07 | 3.36 | 0.34 | 0.0000417 | 0.00218 | 0.0217 |
| 15q13 (22) and 2q31 (174) | 1.22 | 3.15 | 0.27 | 0.0000698 | 0.00229 | 0.0349 |
| WC | ||||||
| 7q34 (140) and 2p25 (16) | 1.09 | 3.64 | 0.31 | 0.0000211 | 0.00080 | 0.0153 |
| 13q14 (42) and 2p25 (18) | 0.94 | 2.46 | 0.26 | 0.0003862 | 0.00610 | 0.0617 |
| 13q14 (42) and 2q31 (176) | 1.07 | 2.57 | 0.31 | 0.0002912 | 0.00610 | 0.0343 |
| 15q13 (22) and 2q31 (173) | 0.81 | 3.13 | 0.30 | 0.0000735 | 0.00171 | 0.0208 |
| 15q13 (22) and 2q36 (221) | 0.92 | 2.90 | 0.28 | 0.0001282 | 0.00182 | 0.0325 |
FDR, false discovery rate; , epistatic variance component; NHLBI FHS, National Heart, Lung, and Blood Institute Family Heart Study; WC, waist circumference.
The epistatic lod value is for chromosome 2 conditioned on chromosome 7, 13, or 15.
P value: epistatic lod P value.
P value of χ21 = (model I (oligogenic log-like)–model II (epistatic log-like)).
Figure 2.
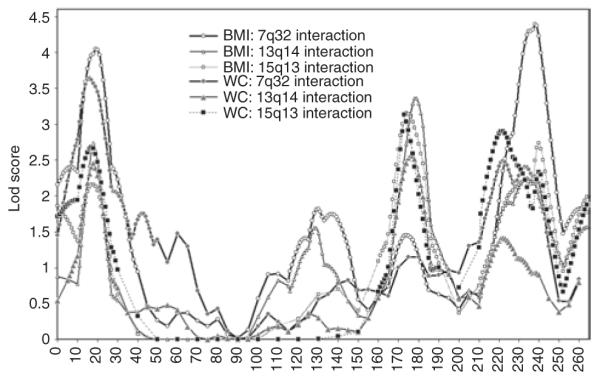
Lod plots on chromosome 2 with epistatic additive interactions with chromosomes 7q32, 13q14, and 15q13. The analyses were performed on age-, sex-, and field centers–adjusted BMI and WC in the complete National Heart, Lung, and Blood Institute Family Heart Study data. WC, waist circumference.
Because interactions between loci on chromosomes 2 and 6q (ref. 12), and 15q (ref. 20) have been reported in the literature for obesity-related traits, we investigated further the possibility of epistasis with QTLs influencing BMI and WC at these locations. There was no suggestion of genetic interaction between QTLs on chromosomes 2 and 6 in the current data. However, significant evidence of epistasis between QTLs on 2q31 and 15q13 (BMI: , P = 0.0349 and WC:, P = 0.0208), and 2q36 and 15q13 (WC: , P = 0.0325) were found. Moreover, the evidence of linkage increased significantly on 2p25, 2q31, and 2q36–q37 locations when the variance component due to epistasis was modeled into the two-locus linkage analysis (Table 3, Figure 2). Compared with oligogenic lod scores, the epistatic lod scores were significantly higher on 2p25 (and 7q34 QTL: lod = 4.04 vs. 1.35), 2q31 (and 13q14 QTL: lod = 3.36 vs. 1.07), and 2q37 (and 7q34 QTL: lod = 4.39 vs. 1.65), for BMI as exempla. We also tested for epistasis between loci on chromosomes 7, 13, and 15 conditioning on the linkage evidence on chromosome 2q. The epistatic lod scores increased on chromosomes 13q14 (from 2.72 to 3.03) and 15q21 (from 1.55 to 2.87). These results suggest that besides significant epistatic interactions between these QTLs, the epistatic model conditioning on 2q also improved the evidence of linkage on chromosomes 13 and 15.
DISCUSSION
There is strong evidence that chromosome 2 harbors at least three QTLs (2p25, 2q24–q31, and 2q36–q37) influencing the variation of BMI and WC levels in families participating in the NHLBI FHS. On chromosome 2p25, the linkage signal was enhanced when subjects taking medications for hypercholesterolemia and/or hyperglycemia were excluded. Medicated subjects for hypercholesterolemia and/or hyperglycemia are associated with higher CVD risk factors than the average population and may act as confounders in the linkage analysis. Some lipid-lowering drugs (21) and antidiabetic medications (22) have been reported to be associated with weight change. Niacin is a lipid-lowering drug and has been shown to affect the adipose tissue lipolysis raising adiponectin and leptin (21,23). Also, recent findings have reported that lipid-lowering drugs, diet, and impairment of lipase enzymes (e.g., hormone-sensitive lipase and adipose triglyceride lipase) influence triglyceride lipolysis in adipocytes that in turn may affect development of fat stores (21,24–26). Dietary factors, especially fats and carbohydrates (3), influence the development of obesity and may alter the expression of several genes in adipose tissue (24). However, the exact metabolic pathways involving drugs, diet, and lipids in the adipocytes are unclear.
We hypothesized that the interaction of dietary and genetic factors may act as major confounders in detecting QTLs and also may mask the effect of a QTL influencing adiposity independently of lipid pathway. When we tested this hypothesis, by adjusting for lipid profile and dietary factors as well as age and sex effects, the evidence for linkage to 2p25 location for BMI and WC in nonmedicated subset was strengthened. Thus, our data suggest that these lifestyle and genetic effects as well as their interactions are involved in a complex metabolic pathway of obesity and related CVD risk factors. By controlling for these confounding factors, evidence for a QTL on 2p25 affecting adiposity independent of dietary habits and lipid pathway was enhanced.
Recent genome-wide association studies have identified novel loci for obesity (27–29). A review of these findings reveals significant variants of transmembrane protein 18 gene residing on 2p25, that could, in part, account for the linkage signal. To test this hypothesis, we investigated the effect of transmembrane protein 18 variants in a subset of 856 unrelated subjects from the NHLBI FHS data that has been typed using the Illumina 550K chip (Illumina, San Diego, CA). Selecting 89 tag-single-nucleotide polymorphisms across 324.4 kilobases of transmembrane protein 18 (from 471,504 to 795,871 base pairs), we found three single-nucleotide polymorphisms significant at P < 0.0007, confirming the association of the locus with BMI in our data. These three single-nucleotide polymorphisms collectively accounted for 3% of variability in BMI, suggesting that although these loci may indeed account for some of the linkage signal, there are likely other genes in the 2p25 region.
The three loci on chromosomes 2p25, 2q24–q31, and 2q36–q37 appear to interact with chromosomes 7q34, 13q14, and 15q13. The evidence of epistatic interactions was consistent across BMI and WC. Also, the epistatic variance component two-locus linkage model enhanced significantly the evidence of linkage on 2p25, 2q31, and 2q36–q37 locations for both BMI and WC phenotypes. The epistatic lod scores remained significant even after correction for multiple tests (FDR P value, Table 3). Several independent linkage studies have suggested susceptibility loci to obesity-related phenotypes on chromosome regions 2p25, 2q24–q31, 2q36–q37, 7q34, 13q14, and 15q13 (ref. 9). Moreover, epistasis has been reported between 2p25–p24 and 13q13–q21 for BMI, percent fat, and WC in European-American families (11), and between 2q37 and 15q21 for diabetes in Mexican Americans (20).
There are several candidate genes on chromosome 2 (proopiomelanocortin (POMC), insulin receptor substrate 1, calpain 10, among others), on 7q31 (leptin), on 13q14 (5-hydroxytryptamine, serotonin; forkhead box-O1A), and on 15q (LIPC, hepatic lipase), which are linked and/or associated with obesity-related phenotypes. POMC (2p23) encodes a polypeptide hormone precursor that undergoes extensive, tissue-specific, posttranslational processing via cleavage by subtilisin-like enzymes known as prohormone convertases. The encoded protein is synthesized mainly in corticotroph cells of the anterior pituitary. Adrenocorticotrophin, essential for normal steroidogenesis and the maintenance of normal adrenal weight, and β-lipotropin are the major end products. Mutations in POMC have been associated with obesity (30), whereas calpain 10 (2q37) and insulin receptor substrate 1 (2q36) genes have shown to be associated with diabetes (31,32). The calpain 10 encodes a ubiquitously expressed member of the calpain-like cysteine protease family (31). IRS-1 mediates the control of various cellular processes by insulin and has shown an important role in insulin-induced metabolism in the muscle and adipose tissue in animal model (32).
It is worth mentioning that gene–gene interactions have been reported in animal models (33,34). Recently, Kitamura et al. (34) found that Foxo1 promotes opposite patterns of co-activator–corepressor exchange at the Pomc and agouti-related protein promoters, resulting in activation of agouti-related protein and inhibition of Pomc. Foxo1 represents a shared component of pathways integrating food intake and peripheral metabolism (34). Also, epistatic interaction between two nonstructural loci influencing hepatic lipase activity was reported in an obese mouse model (35). Thus, these current results showing evidence of epistatic interaction on chromosomes 2 with 7q34, 13q14, and 15q may bring new insights for discovery of gene networks and obesity-metabolism pathways.
In conclusion, adiposity phenotypes interacting with lipids, diabetes, and environmental exposures may play a major confounding role in identifying regions that likely harbor QTL influencing BMI and WC. We observed that excluding subjects taking medications for hypercholesterolemia and/or hyperglycemia, and also including lipid levels and dietary intake factors in the basic covariate adjustment improved effectively the linkage signals. We also found evidence of three QTLs on chromosome 2 (2p25, 2q24–q31, and 2q36–q37) with epistatic interaction with chromosomes 7q34, 13q14, and 15q13 influencing the levels of BMI and WC. Thus, the current study shows the evidence of linkage on chromosome 2 enhanced when biological and behavioral effects involved in the obesity-related metabolism were accounted in the linkage analysis models. We believe these findings will guide further investigations in gene discovery and may shed light on genetic architecture of obesity.
ACKNOWLEDGMENTS
The current study is supported by NIH grant RO-1 HL068891-06 (Epidemiological and Genetic Studies of Body Mass Index in the Family Heart Study).
Footnotes
DISCLOSURE
The authors declared no conflict of interest.
REFERENCES
- 1.Centers for Disease Control and Prevention (CDC) Behavioral Risk Factor Surveillance System Survey Data. U.S. Department of Health and Human Services, Centers for Disease Control and Prevention; Atlanta, GA: 1984-2005. http://www.cdc.gov/brfss/technical_infodata/surveydata.htm. [Google Scholar]
- 2.Renehan AG, Tyson M, Egger M, Heller RF, Zwahlen M. Body-mass index and incidence of cancer: a systematic review and meta-analysis of prospective observational studies. Lancet. 2008;371:569–578. doi: 10.1016/S0140-6736(08)60269-X. [DOI] [PubMed] [Google Scholar]
- 3.Walker CG, Zariwala MG, Holness MJ, Sugden MC. Diet, obesity and diabetes: a current update. Clin Sci. 2007;112:93–111. doi: 10.1042/CS20060150. [DOI] [PubMed] [Google Scholar]
- 4.Bray GA, Bellanger T. Epidemiology, trends, and morbidities of obesity and the metabolic syndrome. Endocrine. 2006;29:109–117. doi: 10.1385/ENDO:29:1:109. [DOI] [PubMed] [Google Scholar]
- 5.Kavey RE, Allada V, Daniels SR, et al. Cardiovascular risk reduction in high-risk pediatric patients: a scientific statement from the American Heart Association Expert Panel on Population and Prevention Science; the Councils on Cardiovascular Disease in the Young, Epidemiology and Prevention, Nutrition, Physical Activity and Metabolism, High Blood Pressure Research, Cardiovascular Nursing, and the Kidney in Heart Disease; and the Interdisciplinary Working Group on Quality of Care and Outcomes Research: endorsed by the American Academy of Pediatrics. Circulation. 2006;114:2710–2738. doi: 10.1161/CIRCULATIONAHA.106.179568. [DOI] [PubMed] [Google Scholar]
- 6.Feitosa MF, Borecki IB, Rich SS, et al. Quantitative-trait loci influencing body-mass index reside on chromosomes 7 and 13: the National Heart, Lung, and Blood Institute Family Heart Study. Am J Hum Genet. 2002;70:72–82. doi: 10.1086/338144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bosy-Westphal A, Onur S, Geisler C, et al. Common familial influences on clustering of metabolic syndrome traits with central obesity and insulin resistance: the Kiel obesity prevention study. Int J Obes (Lond) 2007;31:784–790. doi: 10.1038/sj.ijo.0803481. [DOI] [PubMed] [Google Scholar]
- 8.Fox CS, Heard-Costa NL, Wilson PW, et al. Genome-wide linkage to chromosome 6 for waist circumference in the Framingham Heart Study. Diabetes. 2004;53:1399–1402. doi: 10.2337/diabetes.53.5.1399. [DOI] [PubMed] [Google Scholar]
- 9.Rankinen T, Zuberi A, Chagnon YC, et al. The human obesity gene map: the 2005 update. Obesity (Silver Spring) 2006;14:529–644. doi: 10.1038/oby.2006.71. [DOI] [PubMed] [Google Scholar]
- 10.Dong C, Wang S, Li WD, et al. Interacting genetic loci on chromosomes 20 and 10 influence extreme human obesity. Am J Hum Genet. 2003;72:115–124. doi: 10.1086/345648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dong C, Li WD, Li D, Price RA. Interaction between obesity-susceptibility loci in chromosome regions 2p25-p24 and 13q13-q21. Eur J Hum Genet. 2005;13:102–108. doi: 10.1038/sj.ejhg.5201292. [DOI] [PubMed] [Google Scholar]
- 12.Liu YZ, Guo YF, Xiao P, et al. Epistasis between loci on chromosomes 2 and 6 influences human height. J Clin Endocrinol Metab. 2006;91:3821–3825. doi: 10.1210/jc.2006-0348. [DOI] [PubMed] [Google Scholar]
- 13.Kissebah AH, Sonnenberg GE, Myklebust J, et al. Quantitative trait loci on chromosomes 3 and 17 influence phenotypes of the metabolic syndrome. Proc Natl Acad Sci USA. 2000;97:14478–14483. doi: 10.1073/pnas.97.26.14478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Higgins M, Province M, Heiss G, et al. NHLBI Family Heart Study: objectives and design. Am J Epidemiol. 1996;143:1219–1228. doi: 10.1093/oxfordjournals.aje.a008709. [DOI] [PubMed] [Google Scholar]
- 15.Djoussé L, Arnett DK, Carr JJ, et al. Dietary linolenic acid is inversely associated with calcified atherosclerotic plaque in the coronary arteries: the National Heart, Lung, and Blood Institute Family Heart Study. Circulation. 2005;111:2921–2926. doi: 10.1161/CIRCULATIONAHA.104.489534. [DOI] [PubMed] [Google Scholar]
- 16.Almasy L, Blangero J. Multipoint quantitative-trait linkage analysis in general pedigrees. Am J Hum Genet. 1998;62:1198–1211. doi: 10.1086/301844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Williams JT, Van Eerdewegh P, Almasy L, Blangero J. Joint multipoint linkage analysis of multivariate qualitative and quantitative traits. I. Likelihood formulation and simulation results. Am J Hum Genet. 1999;65:1134–1147. doi: 10.1086/302570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Almasy L, Dyer TD, Blangero J. Bivariate quantitative trait linkage analysis: pleiotropy versus co-incident linkages. Genet Epidemiol. 1997;14:953–958. doi: 10.1002/(SICI)1098-2272(1997)14:6<953::AID-GEPI65>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 19.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc. 1995;57:289–300. [Google Scholar]
- 20.Cox NJ, Frigge M, Nicolae DL, et al. Loci on chromosomes 2 (NIDDM1) and 15 interact to increase susceptibility to diabetes in Mexican Americans. Nat Genet. 1999;21:213–215. doi: 10.1038/6002. [DOI] [PubMed] [Google Scholar]
- 21.Langin D. Adipose tissue lipolysis as a metabolic pathway to define pharmacological strategies against obesity and the metabolic syndrome. Pharmacol Res. 2006;53:482–491. doi: 10.1016/j.phrs.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 22.Malone M. Medications associated with weight gain. Ann Pharmacother. 2005;39:2046–2055. doi: 10.1345/aph.1G333. [DOI] [PubMed] [Google Scholar]
- 23.Westphal S, Borucki K, Taneva E, Makarova R, Luley C. Extended-release niacin raises adiponectin and leptin. Atherosclerosis. 2007;193:361–365. doi: 10.1016/j.atherosclerosis.2006.06.028. [DOI] [PubMed] [Google Scholar]
- 24.Viguerie N, Vidal H, Arner P, et al. Adipose tissue gene expression in obese subjects during low-fat and high-fat hypocaloric diets. Diabetologia. 2005;48:123–131. doi: 10.1007/s00125-004-1618-x. [DOI] [PubMed] [Google Scholar]
- 25.Berndt J, Kralisch S, Klöting N, et al. Adipose triglyceride lipase gene expression in human visceral obesity. Exp Clin Endocrinol Diabetes. 2008;116:203–210. doi: 10.1055/s-2007-993148. [DOI] [PubMed] [Google Scholar]
- 26.Jocken JW, Langin D, Smit E, et al. Adipose triglyceride lipase and hormone-sensitive lipase protein expression is decreased in the obese insulin-resistant state. J Clin Endocrinol Metab. 2007;92:2292–2299. doi: 10.1210/jc.2006-1318. [DOI] [PubMed] [Google Scholar]
- 27.Thorleifsson G, Walters GB, Gudbjartsson DF, et al. Genome-wide association yields new sequence variants at seven loci that associate with measures of obesity. Nat Genet. 2009;41:18–24. doi: 10.1038/ng.274. [DOI] [PubMed] [Google Scholar]
- 28.Willer CJ, Speliotes EK, Loos RJ, et al. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat Genet. 2009;41:25–34. doi: 10.1038/ng.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Renström F, Payne F, Nordström A, et al. Replication and extension of genome-wide association study results for obesity in 4923 adults from Northern Sweden. Hum Mol Genet. 2009;18:1489–1496. doi: 10.1093/hmg/ddp041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nillni EA. Regulation of prohormone convertases in hypothalamic neurons: implications for prothyrotropin-releasing hormone and proopiomelanocortin. Endocrinology. 2007;148:4191–4200. doi: 10.1210/en.2007-0173. [DOI] [PubMed] [Google Scholar]
- 31.Horikawa Y, Oda N, Cox NJ, et al. Genetic variation in the gene encoding calpain-10 is associated with type 2 diabetes mellitus. Nat Genet. 2000;26:163–175. doi: 10.1038/79876. [DOI] [PubMed] [Google Scholar]
- 32.Asano T, Fujishiro M, Kushiyama A, et al. Role of phosphatidylinositol 3-kinase activation on insulin action and its alteration in diabetic conditions. Biol Pharm Bull. 2007;30:1610–1616. doi: 10.1248/bpb.30.1610. [DOI] [PubMed] [Google Scholar]
- 33.Nonogaki K, Nozue K, Oka Y. Increased hypothalamic 5-HT2A receptor gene expression and effects of pharmacologic 5-HT2A receptor inactivation in obese Ay mice. Biochem Biophys Res Commun. 2006;351:1078–1082. doi: 10.1016/j.bbrc.2006.10.173. [DOI] [PubMed] [Google Scholar]
- 34.Kitamura T, Feng Y, Kitamura YI, et al. Forkhead protein FoxO1 mediates Agrp-dependent effects of leptin on food intake. Nat Med. 2006;12:534–540. doi: 10.1038/nm1392. [DOI] [PubMed] [Google Scholar]
- 35.Yi N, Chiu S, Allison DB, Fisler JS, Warden CH. Epistatic interaction between two nonstructural loci on chromosomes 7 and 3 influences hepatic lipase activity in BSB mice. J Lipid Res. 2004;45:2063–2070. doi: 10.1194/jlr.M400136-JLR200. [DOI] [PubMed] [Google Scholar]