

Abstract
Hyperkalemia is a potentially life-threatening condition, and patients who have chronic kidney disease, who are diabetic, or who are taking renin–angiotensin–aldosterone system inhibitors are at increased risk. Therapeutic options for hyperkalemia tend to have limited effectiveness and can be associated with serious side effects. Colonic potassium secretion can increase to compensate when urinary potassium excretion decreases in patients with renal impairment, but this adaptation is insufficient and hyperkalemia still results. Patiromer is a novel, spherical, nonabsorbed polymer designed to bind and remove potassium, primarily in the colon, thereby decreasing serum potassium in patients with hyperkalemia. Patiromer has been found to decrease serum potassium in patients with hyperkalemia having chronic kidney disease who were on renin–angiotensin–aldosterone system inhibitors. Results of nonclinical studies and an early phase clinical study are reported here. Two studies with radiolabeled drug, one in rats and the other in dogs, confirmed that patiromer was not absorbed into the systemic circulation. Results of an in vitro study showed that patiromer was able to bind 8.5 to 8.8 mEq of potassium per gram of polymer at a pH similar to that found in the colon and had a much higher potassium-binding capacity compared with other resins, including polystyrene sulfonate. In a study in hyperkalemic rats, a decrease in serum potassium was observed via an increase in fecal potassium excretion. In a clinical study in healthy adult volunteers, a significant increase in fecal potassium excretion and a significant decrease in urinary potassium excretion were observed. Overall, patiromer is a high-capacity potassium binder, and the chemical and physical characteristics of patiromer may lead to good clinical efficacy, tolerability, and patient acceptance.
Keywords: patiromer, hyperkalemia, chronic kidney disease, RAASi, potassium
Introduction
Potassium is an essential dietary mineral that is precisely balanced by internal and external homeostatic mechanisms. Internal balance, largely provided by muscle and liver tissue, is maintained through a defined distribution of potassium between intracellular (98%) and extracellular fluids (2%). In contrast, net gain or loss of potassium from the body comprises external balance and is primarily regulated by the kidney, along with extrarenal mechanisms present in the gastrointestinal (GI) tract.1,2
Potassium ingested in the diet totals, on average, ∼100 mEq/d. More than 90% of this dietary intake is absorbed, with the primary absorption site being the jejunum and upper ileum of the small intestine.3 Normally, potassium balance is regulated by the kidneys, which excrete 90% to 95% of dietary potassium, with the remaining 5% to 10% excreted in the colon.3 Several physiologic signals promote potassium secretion into the urine, including aldosterone, high distal sodium delivery, high urine flow rate, high potassium concentration in tubule cells, and metabolic alkalosis.4 The large intestine is the site of additional potassium secretion, which occurs by 2 mechanisms, passive secretion through a paracellular route1 and active secretion through BK channels.5 Accordingly, potassium has a relatively high concentration in the colon compared to other cations.6,7
When kidney function is compromised, as found in patients with chronic kidney disease (CKD) and end-stage renal disease, urinary excretion of potassium is decreased and colonic secretion of potassium is markedly increased.8–10 Both active and passive secretory mechanisms are used to increase colonic potassium secretion to levels as high as 20 to 80 mEq/d in these patients.3,11,12 This adaptive response to increased serum potassium involves an upregulation of the number of BK channels (KCNMA1, commonly known as “big potassium” channels) present on the apical surface of colonic epithelial cells, as well as an alteration in the regulatory signals that promote potassium secretion.13,14 This adaptive response, however, may not be sufficient to maintain normal serum potassium levels, due in part to variation in colonic secretion capacity and the central role provided by the kidney in potassium excretion.
As a result, incidental hyperkalemia rates as high as 26% in patients with CKD stages 3 to 5 have been reported in one study,15 and persistent hyperkalemia rates of 4% in patients with CKD stage 3 and of 13% in patients with CKD stage 4 have been reported in another study.16 There is a significant clinical risk associated with hyperkalemia, and this risk is of particular significance in patients with CKD, where its occurrence increases the odds of mortality within 1 day of an observed hyperkalemic event.15 Other clinical risk factors for hyperkalemia include diabetes mellitus and older age.17,18 The use of renin–angiotensin–aldosterone system inhibitors (RAASi), such as angiotensin-converting enzyme inhibitors (ACEIs), angiotensin II receptor blockers, aldosterone antagonists, and renin inhibitors, can also reduce potassium excretion.19–22
More than 50 years ago, the cation-exchange resin sodium polystyrene sulfonate (SPS; Kayexalate; Sanofi-Aventis, Bridgewater, New Jersey) became available for clinical use in the United States.23 However, randomized, controlled clinical data to demonstrate the effectiveness of SPS are limited, and it is generally not well tolerated in patients, thus limiting chronic use.24 Administration of the drug can be associated with life-threatening side effects including intestinal necrosis.24,25 Further, because sodium is used as the counterion in SPS, caution is advised in patients who cannot tolerate even a small increase in sodium loads, such as patients with heart failure, severe hypertension, or marked edema, all of which are common in patients with CKD.25–29
Patiromer
Patiromer, a novel next-generation spherical nonabsorbed polymer, is approved for the treatment of hyperkalemia. Patiromer was developed to provide higher potassium-binding capacity relative to the polystyrene sulfonate polymer. It possesses improved physical properties such as a low swelling ratio allowing only minimal water absorption and, importantly, patiromer uses calcium, not sodium, as the exchange cation.
Patiromer was designed to be fully ionized at the physiological pH of the colon for optimal ion exchange where the concentration of potassium in the GI tract is the highest. Clinical trials assessing the safety, tolerability, and efficacy of patiromer in treating patients with CKD having hyperkalemia have been completed with all end points (including safety, reduction and sustained control of serum potassium within the normal range, and reduction in recurrent hyperkalemia) having been met.30,31 Here, we describe the structure of the active moiety (ie, the polymer anion) and the synthesis, physical properties, in vitro potassium-binding capacity, and putative mechanism of action of patiromer. In addition, the physical stability and potassium fecal and urine excretion profile of patiromer in humans are reported.
Methods and Materials
All animal experiments were conducted in accordance with the Guide for the Care and Use of Laboratory Animals from the Institute for Laboratory Animal Research, National Research Council. All study protocols were approved by an Institutional Animal Care and Use Committee prior to the initiation of the studies. Human studies were reviewed by an institutional review board prior to study initiation. Written, informed consent was obtained from all participants before conducting any study activity.
As noted in the following methods of the nonclinical studies and the early clinical study of patiromer presented here, the drug substance used, RLY5016, was the polymer anion with calcium counterion, except for one nonclinical study that used protonated patiromer (RLY5016H). Subsequently, a more stable drug substance was developed. This drug substance, comprising the polymer anion and calcium-sorbitol counterion (RLY5016S), was used in later studies, including a phase 1 onset of action study,32 a phase 3 2-part trial,30 and a phase 2 1-year study.31 Importantly, sorbitol was not added to induce an osmotic diarrhea as is the case with sorbitol added to SPS. In addition, the amount of sorbitol in RLY5016S is approximately 5- to 10-fold lower than that commonly administered in a dose of SPS. Regardless of differences in the drug substances, the active moiety (ie, the polymer anion) was unchanged throughout the nonclinical and clinical development program. In the clinical study discussed here, patiromer doses are expressed as the amount of the polymer anion.
Structure and Synthesis of the Patiromer Polymer
The active moiety, patiromer, is a carboxylic acid compound substituted at the alpha position with fluorine, comprising a copolymer of 2-fluoroacrylic acid, divinylbenzene, and 1,7-octadiene (Figure 1). The patiromer polymer has a lower molecular weight than polystyrene sulfonate per potassium-binding site, providing a higher absolute binding capacity. Because of the strong electron-withdrawing characteristic of the fluorine that is positioned close to the carboxylate group, the pKa is reduced relative to acrylic acid,33 resulting in an acid that is strong enough to maintain its binding capacity at the physiological pH of the small and large intestines.
Figure 1.
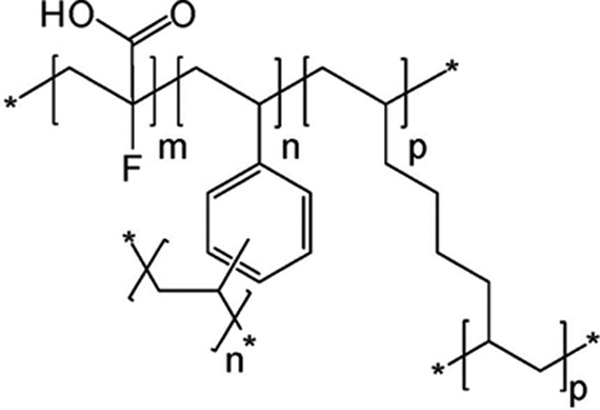
Chemical structure of the active moiety of patiromer CAS#: 1260643-52-4. CAS name: 2-propenoic acid, 2-fluoro-, polymer with diethenylbenzene and 1,7-octadiene. m indicates number of 2-fluoro-2-propenoic acid groups, m = 0.91; n, p, number of cross-linking groups, n + p = 0.09. *An extended polymeric network.
Patiromer is made in a high-yield 2-step process, with the first being polymerization and the second being hydrolysis. Standard suspension polymerization techniques form the patiromer beads by mixing a homogeneous organic phase containing the starting materials methyl 2-fluoroacrylate, divinylbenzene, and 1,7-octadiene in an immiscible aqueous medium that contains suspending agent, salt, an inhibitor, and a buffer system. This process creates an emulsion, with the organic phase dispersed into spheres containing the monomer and reactive cross-linkers that form the polymer. The size of the spheres can be carefully controlled by altering the reactor conditions, thus ensuring the formation of uniform beads with a defined size. The spherical beads resulting from the polymerization reaction (step 1) undergo hydrolysis with sodium hydroxide in a water:methanol mixture at elevated temperatures to cleave the methyl ester group and form the corresponding carboxylic acid sodium form. This carboxylic acid sodium form is then converted in situ to the calcium form by ion exchange with calcium chloride solution.
Determination of Patiromer Particle Size Distribution
The particle size and size distribution of patiromer were determined via laser light diffraction with a Malvern Mastersizer 2000 (Malvern Instruments, Malvern, UK). RLY5016 sample (150-170 mg) was placed into a vial, and 2 mL of water was added. The suspension was sonicated for 5 minutes. An aliquot of this suspension was then used to measure the particle size. Results for distribution D (0.50), D (0.05), and D (0.95) are presented (in μm) as the mean value of the 3 replicate measurements.
Patiromer Absorption, Distribution, Metabolism, and Excretion Studies in Rats and Dogs
RLY5016 was radiolabeled with 14C during synthesis using a [1-14C]-1,7-octadiene cross-linking agent that is a component of the finished polymer beads. The specific activity of the radiolabeled beads, determined by combustion analysis, was 343 μCi/g of RLY5016. Final water washes of the radiolabeled beads yielded a specific activity of 0.702 μCi/g polymer (ie, approximately 0.2% of the total specific activity of 343 μCi/g polymer). An analysis of impurities confirmed the radioactivity in water washes was from unincorporated radiolabeled cross-linker ([1-14C]-1,7-octadiene). Plasma, fecal, and urinary monitoring was conducted to assess possible absorption and distribution of the labeled drug in rats and dogs.
Seventeen male Sprague Dawley rats were assigned to 3 groups and administered a single dose of 14C-RLY5016 at 313 mg/kg (107 μCi/kg) via oral gavage. Urine and feces were collected at designated predose and postdose time intervals from group 1 (n = 3). Cage wash/wipe samples and tissue from the residual carcasses of these rats were collected upon killing. Blood was collected from group 2 (n = 9) with 3 rats per collection time point, at 0.25, 0.5, 1, 2, 4, 8, 24, 48, and 72 hours postdose. In group 3 (n = 5), rats were immediately prepared for quantitative whole-body autoradiography (QWBA) following blood collections (1 rat per collection time point) at 0.25, 1, 4, 24, and 72 hours postdose. Briefly, for QWBA, rats were euthanized and immersed in a hexane/dry ice bath following blood collections, embedded in chilled carboxymethylcellulose, and frozen into blocks. Sections were prepared from 5 levels of interest in the sagittal plane that encompassed all major tissues, organs, and biological fluids.
Six purebred beagle dogs (3 males and 3 females) were administered a single oral dose (capsule) of 14C-RLY5016 at 350 mg/kg (105 μCi/kg). Blood samples were collected at 0.25, 0.5, 1, 2, 4, 8, 24, 48, 72, 96, 120, and 168 hours postdose. Urine and feces were collected at various intervals through 168 hours postdose. Following the final urine and fecal collections, animals were killed and cage wipe/wash samples were also collected for radioanalysis.
Whole blood, plasma, feces, urine, vomitus, homogenized tissue, and cage wash samples were analyzed for radioactivity by liquid scintillation counting. Tissue distribution of radioactivity associated with single-dose administration of 14C-RLY5016 was measured in rats using QWBA and analysis of tissue samples.
In Vitro Potassium-Binding Capacity of Patiromer
Comparative in vitro binding capacities of different polymers were measured using the proton (acid) form of each polymer (4 mg/mL) incubated for 24 hours in triplicate at room temperature in a simple potassium buffer (200 mmol/L 2-[morpholino]ethanesulfonic acid, 150 mmol/L potassium) at pH 6.5. Depletion of potassium from the buffer supernatant was measured by ion chromatography and used to calculate potassium-binding capacity. Protonated patiromer (RLY5016H) was prepared by exposure to 4 N hydrochloric acid (HCl), washing in water to remove excess HCl, and drying. Proton-form polystyrene sulfonate (polystyrene sulfonate H) was similarly prepared from commercially obtained SPS. Amberlite IRP-64 H and Dowex 50WX4-200 were obtained already in the proton form from a commercial vendor (Sigma-Aldrich, St Louis, Missouri).
Serum Potassium and Potassium Fecal Excretion With Patiromer in an In Vivo Hyperkalemic Rat Model
We developed a rat hyperkalemia model, designated NADR-TQ, in which chronic renal failure was induced in male Sprague Dawley rats via subtotal (5/6) nephrectomy (Nx) followed by a single intravenous injection of doxorubicin hydrochloride (Adriamycin, 3.5 mg/kg), and a hyperkalemic state was induced by treatment with trimethoprim (T; 0.3% wt/wt in the chow, which reduces renal potassium excretion, similar to the effect of a potassium-sparing diuretic) and quinapril (Q; an ACEI; 30 mg/L in the drinking water) at 2 weeks post-Nx.34 Sprague Dawley rats with normal renal function have a serum potassium level of 6.0 ± 0.65 mEq/L (data on file). NADR rats that were not exposed to Q and T showed a slight but significant elevation in serum potassium at 2 weeks postdoxorubicin injection, 6.58 ± 0.65 mEq/L (P < .01 relative to normal controls). The combination of T and Q as supplements to the diet of the NADR rats further exacerbated the hyperkalemia, resulting in a persistent and progressive hyperkalemia. The serum potassium level in the NADR-TQ rats at days 7 and 14 was 7.6 ± 0.7 mEq/L and 7.3 ± 0.7 mEq/L, respectively (P < .001 relative to normal controls).
To examine the effect of patiromer on hyperkalemia, animals were randomly assigned to receive RLY5016 (4% wt/wt [2.6 g/kg/d] in chow) or chow alone (10 per group). Serum samples were collected from all rats 5 days prior to doxorubicin injection and on days 7 and 14 postdoxorubicin injection. Twenty-four-hour urine and fecal samples were collected, and body weight and food and water consumption were assessed at day −1 and days 7 and 14 postdoxorubicin injection.
Potassium Fecal and Urine Excretion and Patiromer Structural Stability in Healthy Adults Administered Patiromer
The extent of potassium fecal and urine excretion following RLY5016 treatment was determined in a phase 1 single- and multiple-dose escalation study. In this study, 33 healthy adult volunteers were randomized to 1 of the 4 treatment groups in which 6 of the 8 participants received RLY5016: at 0.8, 4.2, 8.4, or 16.8 g patiromer, administered 3 times daily (TID), and 2 of the 8 participants received placebo, for 8 days (days 12-19 of the study). Participants were required to consume a controlled diet throughout the course of the study that provided a consistent amount of daily elemental potassium. Urine and feces were collected over 24-hour periods over days 5 to 11 following screening (baseline) and during days 13 to 19 of each treatment period (end point). Urine aliquots were assayed by Bronson Methodist Hospital Laboratory, Kalamazoo, Michigan. Each pooled 24-hour fecal collection was weighed and frozen at −20°C until analysis; homogenized fecal aliquots were analyzed by Battelle Toxicology Northwest, Richland, Washington. Mean values at baseline were compared among treatment groups using a 1-way analysis of variance fixed-effects model. Analysis of covariance with the baseline value as the covariate was used for the end point and for the change from baseline to end point analyses of urinary and fecal potassium.
The structural stability of patiromer was assessed by recovering polymer beads from fecal samples obtained from healthy subjects in the phase 1 study described above. Patiromer polymer beads were separated from fecal samples, and images of beads were taken using an Olympus BX40F-3 microscope under bright field conditions.
Results
Uniform and Controlled Particle Size of Patiromer
Patiromer manufactured with the suspension polymerization process resulted in insoluble spherical beads of uniform and controlled size. Particle size and volume-based particle size distribution of patiromer were determined by laser diffraction. A Malvern chromatograph of a representative patiromer batch (Figure 2) showed that 90% of the bead particles were within the size range of 74 to 179 μm, with a median particle size of 118 μm (D [0.05] = 74 μm; D [0.50] = 118 μm; and D [0.95] = 179 μm; ie, 95% of the particles were larger than 74 μm and 95% were smaller than 179 μm).
Figure 2.

Malvern chromatograph of a representative RLY5016 batch. Particle size and size distribution of RLY5016 were determined via laser light diffraction.
A light microscopy image of patiromer beads and SPS crystals is shown in Figure 3. In contrast to patiromer, SPS particles have characteristic irregular, jagged-shaped fragments and a broad particle size distribution with many small fines.
Figure 3.

Light microscopy images (×100 magnification) of RLY5016S (left) and sodium polystyrene sulfonate (right).
Patiromer Is not Systemically Absorbed
The nonabsorbed nature of the patiromer polymer was shown in rat and dog studies using radiolabeled drug. Following the administration of radioactively labeled RLY5016 to rats, radioactivity was assessed in urine, feces, cage wash, cage wipe, and carcasses. Elimination kinetics through urine and feces are shown in Figure 4. Following oral administration of 313 mg/kg 14C- RLY5016 (107 μCi/kg) to rats, the mean total recovery of radioactivity was 84.3%, with 84.1% excreted in feces and 0.15% in urine. The proportion of radioactivity detected in the urine was consistent with unincorporated radiolabeled cross-linker ([1-14C]-1,7-octadiene) present in a water wash of the polymer (0.2%). Most of the radioactivity was excreted within 48 hours of dosing. There was no detectable radioactivity by 168 hours postdose. The lower-than-expected fecal recovery of radioactivity in this study was attributed to difficulty collecting and homogenizing the rat fecal samples; this is corroborated by the negative (outside the GI tract) results of QWBA analysis, which demonstrated that radioactivity was limited to the GI tract (stomach, small intestine, large intestine, and cecum), and no detectable levels of radioactivity were seen in any other tissues or organs at any time point after dosing (Table 1).
Figure 4.
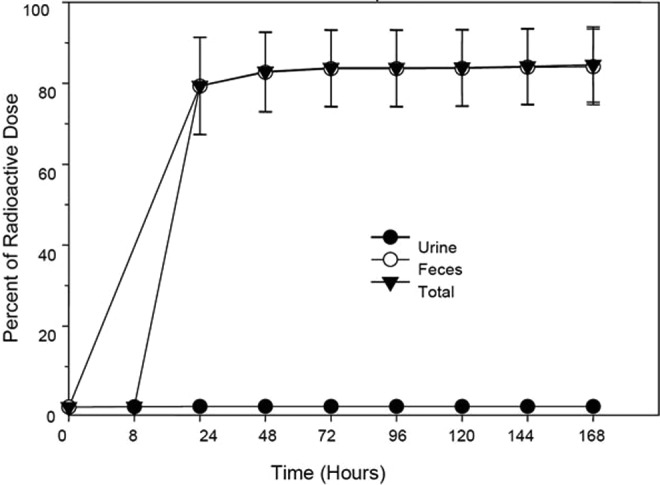
Radioactivity in urine and feces after 14C-RLY5016 administration to rats. Rats (n = 3) were administered with a single oral dose of 14C-RLY5016 (313 mg/kg; 107 μCi/kg). Data are shown as mean ± standard deviation; N = 3.
Table 1.
Concentrations of Radioactivity in Tissues Determined by QWBA at Specified Times After a Single Oral Administration of 350 mg/kg 14C-RLY5016 to Male Rats.
| Time Postdose, hours | μg equivalents 14C-RLY5016/g | |
|---|---|---|
| Contents in GI Tract (Stomach, Small Intestine, Large Intestine, and Cecum)a | All Other Tissues/Organs | |
| 0.25 | 76,800-84,900 | ND |
| 1 | 47,900-60,100 | ND |
| 4 | 10,500-19,400 | ND |
| 24 | ND | ND |
| 72 | ND | ND |
Abbreviations: GI, gastrointestinal; ND, not detectable; QWBA, quantitative whole-body autoradiography.
aValues represent the range of concentrations of radioactivity measured within the individual sections of the GI tract.
Following the administration of radioactively labeled RLY5016 to dogs, radioactivity in urine, feces, vomitus/emesis, cage debris, cage wash, and cage wipe was measured. Oral administration of 14C-RLY5016 (350 mg/kg; 105 μCi/kg) showed that radioactivity was readily eliminated, with most of the signal excreted from the animals by 48 hours postdose. Elimination kinetics through urine and feces are shown in Figure 5. After oral administration of radiolabeled RLY5016, the mean total recovery of radioactivity was 102% in male dogs and 107% in female dogs, with 99.9% of the radioactivity excreted in the feces and <0.1% excreted in the urine.
Figure 5.
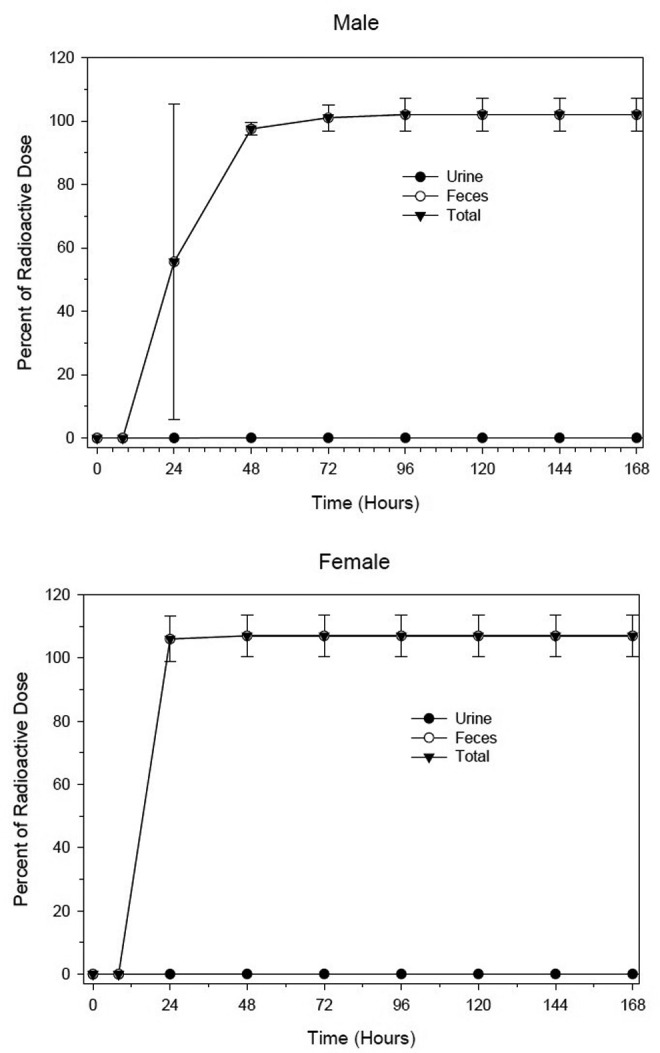
Radioactivity in urine and feces after 14C-RLY5016 administration to dogs. Three male and 3 female beagle dogs were administered a single oral dose of 14C-RLY5016 (350 mg/kg; 105 μCi/kg). Data are shown as mean ± standard deviation.
Analysis showed that 0.004% and 0.002% of the administered radiolabel was present in plasma in the rat and dog, respectively. Chromatographic data indicated that the small amount of radiolabel detected in plasma was from unincorporated labeled cross-linker ([1-14C]-1,7-octadiene) and not from the labeled polymer itself. Collectively, these findings demonstrated the nonabsorbed nature of the patiromer polymer and its lack of systemic bioavailability.
Patiromer Has High In Vitro Potassium-Binding Capacity
To compare the potassium-binding capacity of RLY5016H with traditional cation exchange polymers at a pH typical of the colon, protonated (H) versions of the products were incubated in a buffer at pH 6.5 in the presence of excess (150 mmol/L) potassium. Under these conditions, RLY5016H bound 8.5 to 8.8 mEq of potassium per gram of polymer (Table 2). The maximum binding capacity for patiromer (ie, under highly basic conditions [100 mmol/L KOH, pH 12]) was 8.4 to 10 mEq/g. Therefore, at physiological pH, ≥85% of the total binding capacity of patiromer was available for cation exchange.
Table 2.
Potassium Binding by Protonated Polymers Under Conditions Mimicking Large Intestine pH and Residence Time.
| Polymer (4 mg/mL) | Potassium Bounda (mEq/g) (Mean ± SD) | Final pHb (Mean ± SD) |
|---|---|---|
| Experiment 1 (N = 3) | ||
| RLY5016Hc | 8.53 ± 0.13 | 6.21 ± 0.02 |
| Amberlite IRP-64H | 3.62 ± 0.28 | 6.42 ± 0.02 |
| Polystyrene sulfonate H | 5.09 ± 0.05 | 6.35 ± 0.02 |
| Dowex 50WX4 | 5.25 ± 0.11 | 6.34 ± 0.02 |
| Experiment 2 (N = 3) | ||
| RLY5016Hc | 8.77 ± 0.35 | 6.23 ± 0.01 |
| Amberlite IRP-64H | 3.75 ± 0.05 | 6.43 ± 0.02 |
| Polystyrene sulfonate H | 4.80 ± 0.3 | 6.36 ± 0.01 |
| Dowex 50WX4 | 4.97 ± 0.03 | 6.35 ± 0.02 |
Abbreviation: SD, standard deviation.
aPotassium (K) bound to the test polymers (mEq/g) was calculated as (Kstart − Keq) × dilution factor/polymer concentration, where Kstart is the potassium ion concentration (mmol/L) measured in the diluted control sample, and Keq is the potassium ion concentration (mmol/L) in the diluted test samples after incubation with polymer.
bStarting pH was 6.5.
cRLY5016H is the protonated form of RLY5016.
Amberlite IRP-64H, a weak, carboxylic acid (poly[methacrylic acid]) polymer, had a total capacity of 10.4 mEq/g under basic conditions (100 mmol/L KOH, pH 12) but bound only 3.6 to 3.8 mEq of potassium per gram of product at physiological pH, corresponding to only 36% of the total binding capacity for the product. Polystyrene sulfonate H and Dowex 50WX4 polymers contain strongly acidic sulfonate groups, which are fully ionized in the physiological pH range and use their full capacity to bind potassium under these conditions. However, the total capacity of these 2 polymers is only 4.8 to 5.3 mEq/g. Under in vitro conditions mimicking the pH and potassium content of the colon, patiromer demonstrated a 1.5- to 2.5-fold improvement over the other polymers tested.
Patiromer Decreases Serum Potassium via an Increase in Fecal Excretion in a Hyperkalemic Rat Model
In a rat model of hyperkalemia, NADR-TQ, 5/6 nephrectomized Sprague Dawley rats were administered doxorubicin, T, and Q to create a progressive hyperkalemic state. NADR-TQ rats were treated with RLY5016 (4% wt/wt [2.6 g/kg/d]) in chow or chow alone for 14 days. After 14 days of treatment, NADR-TQ rats receiving RLY5016 had significantly (P < .001) greater fecal potassium excretion than untreated NADR-TQ rats (0.33 ± 0.06 mEq potassium/d vs 0.15 ± 0.08 mEq potassium/d; Figure 6A). RLY5016 induced an additional increase in fecal potassium excretion through the feces, despite the fact that fecal potassium excretion in the NADR-TQ rats was already increased 3-fold due to renal insufficiency. In contrast, there was no effect of RLY5016 treatment on urinary potassium excretion in NADR-TQ rats (Figure 6B). Thus, the increased fecal potassium excretion observed in RLY5016-treated animals was not a compensation for decreased urinary secretion.
Figure 6.
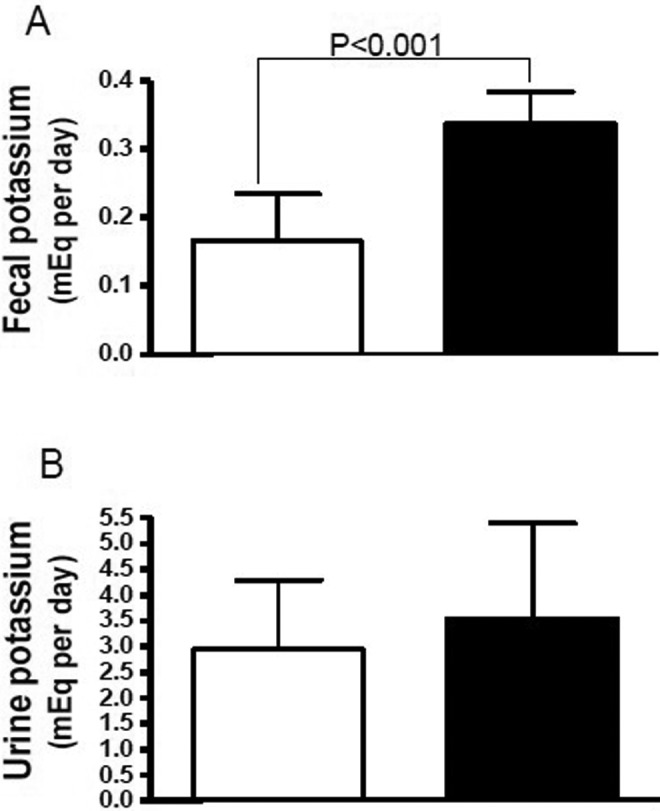
Patiromer effects on fecal (A) and urinary (B) potassium excretion in NADR-TQ rats at day 14 of treatment. Male Sprague Dawley rats with chronic renal failure and hyperkalemia (N = 10 per group) were randomly assigned to receive RLY5016 (4% wt/wt in chow; closed bars) or chow alone (open bars). Data are shown as mean ± standard deviation.
The net effect of increased fecal potassium excretion in RLY5016-treated animals was reduction in total body potassium and normalization of serum potassium levels. At day 7, RLY5016-treated NADR-TQ rats demonstrated significantly lower serum potassium levels than untreated NADR-TQ rats (6.4 ± 0.8 mEq/L vs 7.6 ± 0.7 mEq/L, P < .001). Thus, serum potassium levels were within the normal range (6.00 ± 0.65 mEq/L) in the treated animals, whereas untreated NADR-TQ rats were hyperkalemic (>6.7 mEq/L; Figure 7). At day 14, the serum potassium levels in the RLY5016-treated group were also lower than untreated NADR-TQ rats, but the difference was not statistically significant (data not shown).
Figure 7.
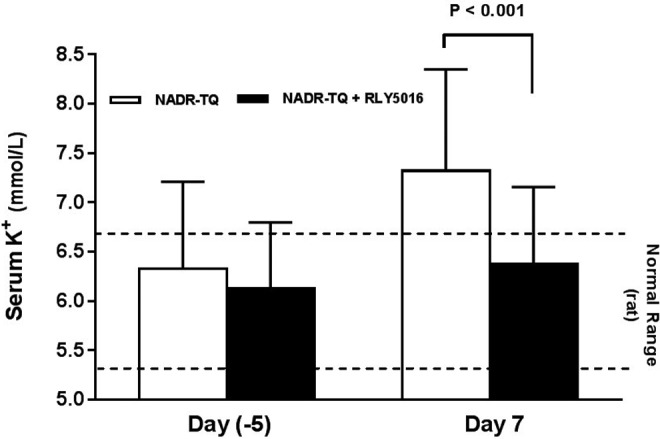
Patiromer attenuates hyperkalemia in NADR-TQ rats. Male Sprague Dawley rats with chronic renal failure and hyperkalemia (N = 10 per group) were randomly assigned to receive RLY5016 (4% wt/wt in chow) or chow alone. Serum potassium levels were measured in all rats 5 days prior to doxorubicin injection and on days 7 postdoxorubicin injection. Data are shown as mean ± standard deviation. Horizontal dotted lines mark the normal serum potassium range for male Sprague Dawley rats of the same age. K+ indicates potassium; NADR-TQ, 5/6 nephrectomy (Nx) followed by a single intravenous injection of doxorubicin hydrochloride (Adriamycin; 3.5 mg/kg), and a hyperkalemic state was induced by treatment with trimethoprim (T; 0.3% wt/wt in the chow, which reduces renal potassium excretion, similar to the effect of a potassium-sparing diuretic) and quinapril (Q; an angiotensin-converting enzyme inhibitor [ACEI]; 30 mg/L in the drinking water) at 2 weeks postnephrectomy.
Patiromer increases fecal and decreases urinary potassium excretion and remains physically stable during passage through the GI tract in healthy adults
Fecal and urinary potassium excretion was assessed in the phase 1 study of 33 healthy participants administered RLY5016 at 0.8, 4.2, 8.4, or 16.8 g patiromer TID for 8 days. Fecal potassium excretion increased and urinary potassium excretion decreased relative to placebo for the 3 highest RLY5016 dose groups, and the effect appears to be dose proportional (P < .001 vs placebo for 8.4 and 16.8 g patiromer TID; Table 3).
Table 3.
Fecal and Urinary Excretion of Potassium in a Phase 1 Study of RLY5016 in Healthy Adults.
| RLY5016 Dose (TID) | Placebo (N = 8) | ||||
|---|---|---|---|---|---|
| 0.8 g (N = 6) | 4.2 g (N = 6) | 8.4 g (N = 6) | 16.8 g (N = 6) | ||
| Fecal potassium excretion (mg/d), mean ± SD | |||||
| Baselinea | 758.1 ± 91.5 | 794.0 ± 198.9 | 676.5 ± 114.5 | 862.0 ± 172.6 | 768.1 ± 297.9 |
| End pointb | 901.7 ± 127.6 | 1411.1 ± 279.9 | 1746.1 ± 246.6 | 2791.4 ± 309.2 | 937.4 ± 708.2 |
| Change from baseline to endpoint | 143.6 ± 96.6 | 617.2 ± 132.8c | 1069.7 ± 218.1d | 1929.4 ± 416.5d | 169.3 ± 477.2 |
| Urinary potassium excretion (mg/d), mean ± SD | |||||
| Baselinea | 3493.7 ± 570.4 | 3447.2 ± 446.6 | 3765.5 ± 325.8 | 3748.8 ± 248.5 | 3644.9 ± 468.3 |
| End pointb | 3491.8 ± 539.3 | 2914.7 ± 452.8 | 2622.4 ± 221.1 | 1792.9 ± 384.3 | 3389.3 ± 669.2 |
| Change from baseline to end point | −1.9 ± 136.6 | −532.5 ± 234.3 | −1143.2 ± 316.9d | −1955.8 ± 277.3d | −255.6 ± 518.6 |
Abbreviations: SD, standard deviation; TID, 3 times daily.
aBaseline data were collected on days 5 to 11.
bEnd point data were collected on days 13 to 19. Patiromer doses are expressed as the amount of the polymer anion.
cP < .02 versus placebo.
dP < .001 versus placebo.
Patiromer beads were recovered from the phase 1 study human fecal samples. The physical stability of the patiromer beads during transit through the GI tract was evaluated through microscopic analysis, which confirmed that the beads recovered from feces remained as intact spheres of approximately 100 μm (Figure 8).
Figure 8.

Microscopic analysis of patiromer beads isolated from the feces of healthy subjects. Healthy subjects (n = 6 per group) received RLY5016 (0.8, 4.2, 8.4, or 16.8 g patiromer 3 times daily [TID]) orally for 8 days. Complete 24-hour fecal collections were performed on days 13 to 19. Patiromer beads isolated from the fecal samples remained as intact spheres of approximately 100 μm.
Discussion
The principle behind controlling serum cation levels (eg, sodium and potassium) using the oral administration of cation-exchange resins was first introduced in 1946 in the context of increasing sodium excretion and fluid management in the prediuretic era.35 More than a decade of intensive study followed, where different acid-containing resins (ie, polymers) were administered with different counterions (ie, salt forms), to examine their efficacy in treating fluid overload, hypernatremia, or hyperkalemia. Sodium polystyrene sulfonate was approved more than 50 years ago as a potassium binder, however, it has not been widely used to treat chronic hyperkalemia, possibly because of the risk for life-threatening adverse effects and fluid overload as a consequence of the sodium counterion.24–29 Because of these troubling features of SPS, there has been a long-felt medical need for a new polymer to treat hyperkalemia.
Patiromer has been developed as a next-generation, nonabsorbed, high-capacity potassium-binding polymer with optimal physical characteristics of the polymer beads such as uniform spherical shape and defined particle size. Patiromer has been shown in randomized clinical studies to significantly reduce serum potassium in patients with CKD (with or without diabetes) on RAASi.30–32,36 Furthermore, the use of calcium rather than sodium (which is the exchange cation in SPS)25 as the exchange counterion may make patiromer a good choice for patients who cannot tolerate even small increases in sodium load.
The fact that the patiromer polymer is not absorbed is a major contributing factor for the safety profile of patiromer. With an average bead size of ∼100 µm, patiromer particles are too large to be absorbed during transit through the GI tract.37 This has been confirmed by animal studies using 14C-RLY5016, in which recovery of the radiolabel in feces and urine of dogs was 99.9% and <0.1%, respectively. Radiolabel studies in rats showed a slightly lower recovery in feces and similar recovery in urine (84.1% and 0.15%, respectively); however, the lower-than-expected fecal recovery was attributed to losses during the collection and homogenization of the rat fecal samples. Quantitative whole-body autoradiography of rats treated with 14C-RLY5016 did not detect any labeled polymer outside the GI tract.
A number of chemical features of patiromer were designed to enhance efficacy. Patiromer takes advantage of the high-binding capacity of carboxylic acid resins, but the unique design feature that incorporates an electron-withdrawing fluorine enables the carboxylates in patiromer to retain their binding capacity in the small and large intestine. The carboxylic acid binding sites in the patiromer polymer (Figure 1) have higher binding capacity than the polystyrene sulfonate binding sites on SPS. In the studies measuring potassium-binding capacity under physiologically relevant pH conditions, patiromer was able to bind 8.5 to 8.8 mEq of potassium per gram of polymer at a pH (6.5) similar to that found in the colon. More importantly, patiromer has a much higher potassium-binding capacity (1.5- to 2.5-fold improvement) than other polymers tested under these conditions.
The electron-withdrawing ability of the fluorine molecule (Figure 1) lowers the pKa of carboxylate compounds, which increases the acidity of the carboxylate. When fluorine replaces the methyl group in methacrylic acid, the pKa drops from 4.7 to approximately 2.6.33 Consequently, the presence of fluorine at the α-carbon adjacent to the acrylate carbonyl group in patiromer lowers the pKa of the carboxyl group in the polymer to a pH of 6.0, which is lower than the average colon pH of 6.6.38 Thus, patiromer should exist mostly in the ionized form in the colon. Moreover, as potassium is the most abundant cation in the colon6,7 and the residence time of the polymer is the longest in this segment of the GI tract,39,40 patiromer is believed to act primarily at this site, although potassium binding may occur in the upper GI tract as well. The presence of colonic BK channels and their upregulation in the setting of reduced renal function further highlight the colon as a physiologically important locus for potassium binding by patiromer.14 In patients with advanced kidney failure and hyperkalemia, secretion of potassium into the colon is upregulated 2-fold to 8-fold via these channels. By binding potassium in the colonic lumen, a concentration gradient is established that increases secretion of potassium by colonic BK channels. In addition, reabsorption of potassium in the rectosigmoid colon that might attenuate this secretion process is prevented, promoting additional potassium elimination.
The data in animals reported here support the effectiveness of patiromer in lowering serum potassium via an increase in fecal potassium excretion, an important mechanistic finding. In hyperkalemic rats (NADR-TQ), where secretion of potassium into the colon is elevated to compensate for renal insufficiency, fecal potassium excretion increased 3-fold over that of normal rats. Despite the already elevated fecal potassium excretion in these animals, patiromer treatment resulted in an additional 2-fold increase in fecal potassium excretion compared to untreated NADR-TQ rats, thereby reducing serum potassium levels to within the normal range.
Fecal removal of potassium by patiromer was also confirmed in a phase 1 clinical study, in which patiromer significantly increased fecal potassium excretion and significantly decreased urinary potassium excretion in healthy adult volunteers. Analysis of the patiromer beads recovered from the feces of subjects in this study showed that the beads remained as intact spheres of approximately 100 µm, indicating that patiromer maintains its structure during passage through the GI system.
The optimized physical characteristics of patiromer (ie, uniform spherical shape, defined polymer bead size, and a low swelling ratio) may improve patient tolerability of administration and minimize undesirable GI effects. It was reported previously that bead polymers with similar physical characteristics had improved GI tolerability in patients compared with irregular-shaped polymers.41 In a 52-week randomized trial of patiromer, 304 patients with CKD maintained on RAASi were evaluated, and the most common adverse event noted over 52 weeks of treatment was mild or moderate constipation (6.3%); only 2 patients discontinued patiromer therapy due to constipation.32
Conclusion
Patiromer is a nonabsorbed, high-capacity polymer specifically designed to bind potassium in the colon, where the highest concentration of potassium is available for excretion via the feces. In vitro and animal studies confirmed that patiromer is nonabsorbed, has higher binding capacity than SPS in vitro, and decreases serum potassium via an increase in fecal excretion in a hyperkalemic rat model. In healthy humans, patiromer significantly increases fecal and decreases urine potassium excretion compared with placebo and, during passage through the GI tract, retains its structural integrity. The chemical and physical characteristics of patiromer contribute to its demonstrated efficacy and may lead to good tolerability and patient acceptance.
Footnotes
Authors’ Note: Editorial assistance was provided by Julie Obeid of Relypsa, Inc, and Brian Catton, PharmD, of AlphaBioCom, LLC, funded by Relypsa, Inc.
Author Contributions: L. Li contributed to conception or design, contributed to acquisition, analysis, or interpretation of data, drafted the manuscript, and critically revised the manuscript. S. Harrison contributed to conception and critically revised the manuscript. J. Cope contributed to conception and design, contributed to analysis and interpretation, and critically revised the manuscript. C. Park contributed to design, contributed to acquisition, analysis, and interpretation, and critically revised the manuscript. L. Lee contributed to conception, contributed to acquisition, analysis, and interpretation, and critically revised the manuscript. F. Salaymeh contributed to conception, contributed to acquisition and interpretation, and critically revised the manuscript. D. Madsen contributed to conception, contributed to analysis and interpretation, drafted the manuscript, and critically revised the manuscript. W. Benton contributed to conception, contributed to interpretation, drafted the manuscript, and critically revised the manuscript. L. Berman contributed to conception, contributed to analysis, and critically revised the manuscript. J. Buysse contributed to conception and design, contributed to analysis and interpretation, drafted the manuscript, and critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of work ensuring itegrity and accuracy.
Declaration of Conflicting Interests: The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article. Lingyun Li, Stephen D. Harrison, Craig Park, Lawrence Lee, Faleh Salaymeh, Deidre Madsen, Wade W. Benton, and Lance Berman are employees of Relypsa, Inc. Dr M. Jamie Cope has a consultant agreement with Relypsa, Inc. Drs M. Jamie Cope and Jerry Buysse were employed by Relypsa, Inc., when the studies described here were conducted.
Funding: The author(s) disclosed receipt of the following financial support for the research presented in this article: The studies were funded by Relypsa, Inc. Lingyun Li, Stephen D. Harrison, Craig Park, Lawrence Lee, Faleh Salaymeh, Deidre Madsen, Wade W. Benton, and Lance Berman disclosed receipt of the following financial support for authorship and/or publication of this article: Publication support was funded by Relypsa, Inc.
References
- 1. Binder HJ. Intestinal fluid and electrolyte movement In: Boron WF, Boulpaep EL. Medical Physiology: A Cellular and Molecular Approach. Philadelphia, PA: Elsevier Saunders; 2005:932–946. [Google Scholar]
- 2. Brown RS. Extrarenal potassium homeostasis. Kidney Int. 1986;30(1):116–127. [DOI] [PubMed] [Google Scholar]
- 3. Agarwal R, Afzalpurkar R, Fordtran JS. Pathophysiology of potassium absorption and secretion by the human intestine. Gastroenterology. 1994;107(2):548–571. [DOI] [PubMed] [Google Scholar]
- 4. Malnic GB, Baily MA, Giebisch GH. Control of renal potassium excretion In: Brenner B, Rector FC, editors. The Kidney. 7th ed London, UK: Saunders; 2004:453–496. [Google Scholar]
- 5. Sorensen MV, Matos JE, Praetorius HA, Leipziger J. Colonic potassium handling. Pflugers Arch. 2010;459(5):645–656. [DOI] [PubMed] [Google Scholar]
- 6. Fordtran JS, Locklear TW. Ionic constituents and osmolality of gastric and small-intestinal fluids after eating. Digest Dis Sci. 1966;11(7):503–521. [DOI] [PubMed] [Google Scholar]
- 7. Wrong O, Metcalfegibson A. The electrolyte content faeces. Proc R Soc Med. 1965;58(12):1007–1009. [PMC free article] [PubMed] [Google Scholar]
- 8. Bastl C, Hayslett JP, Binder HJ. Increased large intestinal secretion of potassium in renal insufficiency. Kidney Int. 1977;12(1):9–16. [DOI] [PubMed] [Google Scholar]
- 9. Hayes CP, Jr, McLeod ME, Robinson RR. An extravenal mechanism for the maintenance of potassium balance in severe chronic renal failure. Trans Assoc Am Physicians. 1967;80:207–216. [PubMed] [Google Scholar]
- 10. Martin RS, Panese S, Virginillo M, et al. Increased secretion of potassium in the rectum of humans with chronic renal failure. Am J Kidney Dis. 1986;8(2):105–110. [DOI] [PubMed] [Google Scholar]
- 11. Sandle G, Gaiger E, Tapster S, Goodship T. Enhanced rectal potassium secretion in chronic renal insufficiency: evidence for large intestinal potassium adaptation in man. Clin Sci. 1986;71(4):393–401. [DOI] [PubMed] [Google Scholar]
- 12. Sandle GI, Gaiger E, Tapster S, Goodship TH. Evidence for large intestinal control of potassium homoeostasis in uraemic patients undergoing long-term dialysis. Clin Sci (Lond). 1987;73(3):247–252. [DOI] [PubMed] [Google Scholar]
- 13. Sandle GI, Hunter M. Apical potassium (BK) channels and enhanced potassium secretion in human colon. QJM. 2009;103(2):85–89. [DOI] [PubMed] [Google Scholar]
- 14. Mathialahan T, Maclennan KA, Sandle LN, Verbeke C, Sandle GI. Enhanced large intestinal potassium permeability in end-stage renal disease. J Pathol. 2005;206(1):46–51. [DOI] [PubMed] [Google Scholar]
- 15. Einhorn LM, Zhan M, Hsu VD, et al. The frequency of hyperkalemia and its significance in chronic kidney disease. Arch Intern Med. 2009;169(12):1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang H-H, Hung C-C, Hwang D-Y, et al. Hypokalemia, its contributing factors and renal outcomes in patients with chronic kidney disease. PLoS One. 2013;8(7):e67140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Raml A, Schmekal B, Grafinger P, Biesenbach G. Risk for hyperkalemia during long-term treatment with angiotensin-converting enzyme inhibitors in insulin-dependent type 2 diabetics in relation to the glomerular filtration rate. Dtsch Med Wochenschr. 2001;126(47):1327–1330. [DOI] [PubMed] [Google Scholar]
- 18. Takaichi K, Takemoto F, Ubara Y, Mori Y. The clinically significant estimated glomerular filtration rate for hyperkalemia. Intern Med. 2008;47(14):1315–1323. [DOI] [PubMed] [Google Scholar]
- 19. Desai AS, Swedberg K, McMurray JJV, et al. Incidence and predictors of hyperkalemia in patients with heart failure. J Am Coll Cardiol. 2007;50(20):1959–1966. [DOI] [PubMed] [Google Scholar]
- 20. Indermitte J, Burkolter S, Drewe J, Krähenbühl S, Hersberger KE. Risk factors associated with a high velocity of the development of hyperkalaemia in hospitalised patients. Drug Saf. 2007;30(1):71–80. [DOI] [PubMed] [Google Scholar]
- 21. Ramadan FH, Masoodi N, El-Solh AA. Clinical factors associated with hyperkalemia in patients with congestive heart failure. J Clin Pharm Ther. 2005;30(3):233–239. [DOI] [PubMed] [Google Scholar]
- 22. Vereijken TLJ, Bellersen L, Groenewoud JMM, Kramers C. Risk calculation for hyperkalemia in heart failure patients. Br J Clin Pharmacol. 2007;63(4):507. [PubMed] [Google Scholar]
- 23. Flinn RB, Merrill JP, Welzant WR. Treatment of the oliguric patient with a new sodium-exchange resin and sorbitol; a preliminary report. N Engl J Med. 1961;264:111–115. [DOI] [PubMed] [Google Scholar]
- 24. Sterns RH, Rojas M, Bernstein P, Chennupati S. Ion-exchange resins for the treatment of hyperkalemia: are they safe and effective? J Am Soc Nephrol. 2010;21(5):733–735. [DOI] [PubMed] [Google Scholar]
- 25. Kayexalate (sodium polystyrene sulfonate) [Package Insert]. Sanofi-Aventis, Inc, Quebec, Canada, 2014. [Google Scholar]
- 26. Acelajado MC, Pisoni R, Dudenbostel T, et al. Refractory hypertension: definition, prevalence, and patient characteristics. J Clin Hypertens (Greenwich). 2012;14(1):7–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. McAlister FA, Ezekowitz J, Tonelli M, Armstrong PW. Renal insufficiency and heart failure: prognostic and therapeutic implications from a prospective cohort study. Circulation. 2004;109(8):1004–1009. [DOI] [PubMed] [Google Scholar]
- 28. Abboud H, Henrich WL. Stage IV chronic kidney disease. N Engl J Med. 2010;362(1):56–65. [DOI] [PubMed] [Google Scholar]
- 29. Murtagh FE, Addington-Hall JM, Edmonds PM, et al. Symptoms in advanced renal disease: a cross-sectional survey of symptom prevalence in stage 5 chronic kidney disease managed without dialysis. J Palliat Med. 2007;10(6):1266–1276. [DOI] [PubMed] [Google Scholar]
- 30. Weir MR, Bakris GL, Bushinsky DA, et al. Patiromer in patients with kidney disease and hyperkalemia receiving RAAS inhibitors. N Engl J Med. 2015;372(3):211–221. [DOI] [PubMed] [Google Scholar]
- 31. Bakris GL, Pitt B, Weir MR, et al. Effect of patiromer on serum potassium level in patients with hyperkalemia and diabetic kidney disease: The AMETHYST-DN randomized clinical trial. JAMA. 2015;314(2):151–161. [DOI] [PubMed] [Google Scholar]
- 32. Bushinsky DA, Williams GH, Pitt B, et al. Patiromer induces rapid and sustained potassium lowering in patients with chronic kidney disease and hyperkalemia. Kidney Int. 2015. doi:10.1038/ki.2015.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gokel G, Dean J. Dean’s Handbook of Organic Chemistry. 2nd ed New York, NY: McGraw-Hill; 2004. [Google Scholar]
- 34. Ishikawa I, Araya M, Hayama T, Sugano M, Yamato H, Ise M. Effect of oral adsorbent (AST-120) on renal function, acquired renal cysts and aortic calcification in rats with adriamycin nephropathy. Nephron. 2002;92(2):399–406. [DOI] [PubMed] [Google Scholar]
- 35. Dock W. Sodium depletion as a therapeutic procedure: the value of ion-exchange resins in withdrawing sodium from the body. Tr A Am Physicians. 1946;59(May):282. [Google Scholar]
- 36. Pitt B, Bakris GL, Bushinsky DA, et al. Effect of patiromer on reducing serum potassium and preventing recurrent hyperkalemia in patients with heart failure and chronic kidney disease on RAAS inhibitors. Eur J Heart Fail. 2015;17(10):1057–1065. doi:10.1002/ejhf.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jung T. Biodegradable nanoparticles for oral delivery of peptides: is there a role for polymers to affect mucosal uptake? Eur J Pharm Biopharm. 2000;50(1):147–160. [DOI] [PubMed] [Google Scholar]
- 38. Fallingborg J. Intraluminal pH of the human gastrointestinal tract. Dan Med Bull. 1999;46(3):183–196. [PubMed] [Google Scholar]
- 39. Read NW, Miles CA, Fisher D, et al. Transit of a meal through the stomach, small intestine, and colon in normal subjects and its role in the pathogenesis of diarrhea. Gastroenterology. 1980;79(6):1276–1282. [PubMed] [Google Scholar]
- 40. Rao SSC, Kuo B, McCallum RW, et al. Investigation of colonic and whole-gut transit with wireless motility capsule and radiopaque markers in constipation. Clin Gastroenterol Hepatol. 2009;7(5):537–544. [DOI] [PubMed] [Google Scholar]
- 41. Hatakeyama S, Murasawa H, Narita T, et al. Switching hemodialysis patients from sevelamer hydrochloride to bixalomer: a single-center, non-randomized analysis of efficacy and effects on gastrointestinal symptoms and metabolic acidosis. BMC Nephrol. 2013;14(1):222. [DOI] [PMC free article] [PubMed] [Google Scholar]