

Abstract
Glial cells efficiently recognize and clear cellular debris after nervous system injury to maintain brain homeostasis, but pathways governing glial responses to neural injury remain poorly defined. We identify the Drosophila guanine-nucleotide exchange factor (GEF) complex Crk/Mbc/dCed-12, and the small GTPase Rac1 as novel modulators of glial clearance of axonal debris. We show Crk/Mbc/dCed-12 and Rac1 function in a non-redundant fashion with the Draper pathway—loss of either pathway fully suppresses clearance of axonal debris. Draper signaling is required early during glial responses, promoting glial activation, which includes increased Draper and dCed-6 expression and extension of glial membranes to degenerating axons. In contrast, the Crk/Mbc/dCed-12 complex functions at later phases promoting glial phagocytosis of axonal debris. Our work identifies new components of the glial engulfment machinery and shows that glial activation, phagocytosis of axonal debris, and termination of responses to injury are genetically separable events mediated by distinct signaling pathways.
Introduction
Nervous system injury or disease can lead to the production of significant amounts of neural waste material including neuronal cell corpses, as well as axonal, dendritic, and synaptic debris. In the mammalian CNS microglia and astrocytes are extraordinarily sensitive to neuronal health and in response to neurodegeneration undergo reactive gliosis, a process whereby glia invade the injury site and clear degenerating neural debris1–5. Rapid clearance of neuronal debris is thought to be essential to maintain brain health, suppress inflammation or autoimmunity, and promote functional recovery6–8. Rapid disposal of neuronal debris is also critical for synaptic plasticity: suppressing clearance of axonal/synaptic debris at the Drosophila NMJ potently suppresses synaptic expansion9. Despite widespread phagocytic roles for glia in then brain, we know surprisingly little about how neuronal debris is recognized, internalized and degraded by engulfing glial cell types10,11.
Molecular insights into cell corpse clearance came from studies of cell death abnormal (ced) mutants in C. elegans12,13. Two partially redundant pathways together promote the clearance of cell corpses during development and in the germline. In one pathway, CED-2, CED-5, and CED-12 act upstream of the small GTPase CED-10 (Rac1 in mammals) to promote cytoskeletal reorganization and corpse engulfment14,15. Subsequent work on the mammalian orthologs of CED-2/CED-5/CED-12, CrkII/Dock180/Elmo, revealed this complex acts as a novel guanine nucleotide exchange factor (GEF) that activates Rac116,17. In a second parallel genetic pathway CED-1 and CED-6 also promote the removal of cell corpses. CED-1 is a transmembrane receptor thought to act in corpse recognition18, degradation19, and through association with the PTB-domain protein CED-620 activate engulfment. Loss of the CED-2/CED-5/CED-12 or CED-1/CED-6 pathway leads to a partial suppression of cell corpse engulfment, while simultaneous inactivation of both pathways suppresses clearance of cell corpses much more strongly13.
The Drosophila ortholog of ced-1, draper, is required in embryonic glia for glial clearance of the neuronal cell corpses generated during embryonic neurogenesis21. However roles for Draper in recognizing and engulfing neural debris in Drosophila have now been extended to developmentally pruned axons22,23 and dendrites24, shed presynaptic NMJ debris9, and axons undergoing Wallerian degeneration25. Drosophila CED-6 (dCed-6) and the Src family signaling cascade composed of the non-receptor tyrosine kinases Src42a and Shark act downstream of Draper to activate engulfment events26,27. The closest mammalian sequence orthologs of Draper, Jedi-1 and MEGF10, are expressed in the precursors of dorsal root ganglion satellite glial cells where they modulate glial clearance of cell corpses28. Thus, at the level of Draper signaling, the molecular mechanisms by which glia recognize and engulf neural debris appear well-conserved.
Loss of Draper signaling blocks all glial responses to axon injury in vivo—even initial activation, which entails up-regulation of engulfment genes and extension of glial membranes to injury sites25–27. These observations argue for an early role for Draper in activating glial responses to brain injury. How later steps in glial responses to axon injury are regulated (e.g. phagocytosis of axons and termination of glial responses) remains less clear. Here we explore the role of the Drosophila orthologs of the CED-2/CED-5/CED-12 complex, CED-10, Crk/Mbc/dCed-12 and Rac1 in Wallerian degeneration. We show that each of these molecules are required for clearance of degenerating axonal debris, and the Draper and Crk/Mbc/dCed-12 pathways act in a non-redundant fashion. We demonstrate each pathway acts at different temporal phases during glial response to brain injury: Draper is required for glial activation during very early glial responses to axotomy, while the Crk/Mbc/dCed-12 complex is specifically required for activation of the glial phagocytic phenotype, a later stage of the glial response. These data shed new light on the function of these well-conserved signaling pathways in modulating glial responses to axotomy, and demonstrate that glial recruitment to degenerating axons and phagocytosis of axonal debris are genetically separable events mediated by distinct molecular pathways.
Results
Crk/Mbc/dCed-12 and Draper drive engulfment of axon debris
To identify new genes governing glial responses to brain injury we explored the role of the GEF complex Crk/Mbc/dCed12 and the small GTPase Rac1 in glial clearance of axons undergoing Wallerian degeneration. We labeled olfactory receptor neurons (ORNs) with GFP (GFP+, using OR85e-mCD8::GFP), knocked down crk, mbc, dced12 and rac1 in glial cells using the glial-specific repo-Gal4 driver and gene-specific UAS-RNAi constructs, severed ORN axons, and assayed degeneration and clearance of GFP+ axons. Strikingly, glial-specific knockdown of either crk, dced12, or rac1 potently suppressed clearance of axonal debris, with the vast majority persisting at 5, 10, 15 and even 30 days after axotomy (Figure 1a,b). Similar results were found with additional UAS-crkRNAi and UAS-rac1RNAi lines (Supplementary Figure 1a,b). Glial knockdown of mbc resulted in lethality, necessitating our use of adult- and cell type-specific knockdown of mbc in glia to assay its function (see below). Severed axons fragmented within 1 day after axotomy in crkRNAi, dced12RNAi, and rac1RNAi backgrounds, indicating their depletion in glia does not affect fragmentation of axons. Clearance of degenerating axons occurred normally in RNAi-alone controls (Supplementary Figure 2a,b). Thus depletion of glial Crk, dCed-12, or Rac1 is sufficient to block clearance of degenerating axons for weeks after axotomy.
Figure 1. Drosophila Crk, dCed-12, and Rac1 are required for glial clearance of severed axons from the CNS.
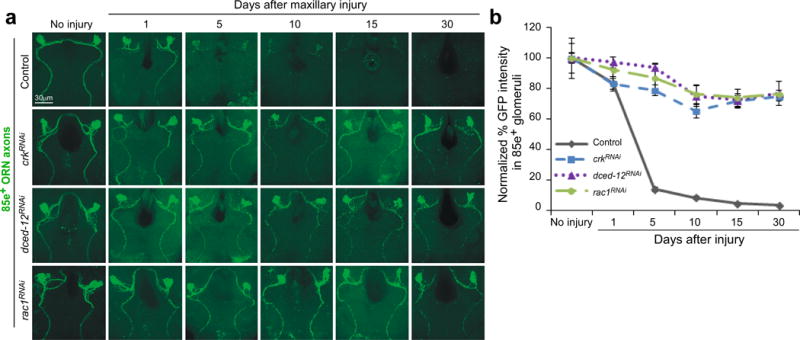
(a) OR85e-expressing ORNs were labeled with mCD8::GFP in control (w;OR85e-mCD8::GFP/+; repo-Gal4/+) and glial RNAi for crkRNAi (w;OR85e-mCD8::GFP/UAS-crk19061RNAi; repo-Gal4/+), dced-12RNAi (w;OR85e-mCD8::GFP/+; repo-Gal4/UAS-dced-1210455RNAi), and rac1RNAi (w, UAS-rac149247RNAi/+ ;OR85e-mCD8::GFP/+; repo-Gal4/+). Maxillary palps were bilaterally ablated and clearance of axonal debris from the CNS was assayed with anti-GFP antibody stains (green) 1, 5, 10, 15, and 30 days after injury.
(b) Normalized quantification of data to control from (a) Error bars represent s.e.m.; n>10 for all.
draper null mutations are known to suppress glial clearance of degenerating axons 5 days after injury, but whether clearance of severed axons is simply delayed in draper null mutants, as in developmental axon pruning22,23, or completely blocked is unknown. We therefore assayed axon clearance in draper animals at 5, 10, and 27 days after axotomy. Remarkably, the majority of axonal debris remained in draperΔ5 mutants even 27 days after axotomy (Supplementary Figure 3a,b). Thus Draper is absolutely required for clearance of severed axons in the adult brain. Since we observe a near complete suppression of axon clearance when either pathway is perturbed, our results indicate that Draper signaling and Crk/dCed-12 act in a non-redundant fashion during glial engulfment of severed axons.
To provide genetic evidence in support of a role for Crk/Mbc/dCed12 and Rac1 in glial engulfment activity, we next assayed the effects of loss of function mutants for these genes on glial engulfment function. Null alleles of mbc, dced-12, and rac1 are lethal during development, precluding an analysis of loss of function mutants at adult stages. We therefore assayed for dominant genetic interactions between these loci in axotomy assays. Axonal debris was cleared normally mbcC1/+, rac1J11/+, or dced-12KO/+ animals, indicating that none of these mutations dominantly affect clearance of degenerating ORN axons (Figure 2a,b). However, we observed a modest, but significant delay in axon clearance in transheterozygous mutant combinations of mbcC1/rac1J11, dced-12KO/+; mbcC1/+, and dced-12KO/+; rac1J11/+. Moreover, in the triple transheterozygote dced-12KO/+; mbcC1/rac1J11 animals, we found that ~60% of axonal debris remained in the brain 5 days after axotomy (Figure 2a,b). These data, coupled with our glial-specific RNAi data for crk, dced-12, mbc and rac1, indicate that Crk/Mbc/dCed12 and Rac1 function together in glial cells to promote clearance of degenerating axons from the brain.
Figure 2. mbc, dced-12, and rac1 mutants exhibit dominant genetic interactions in ORN axon clearance assays.
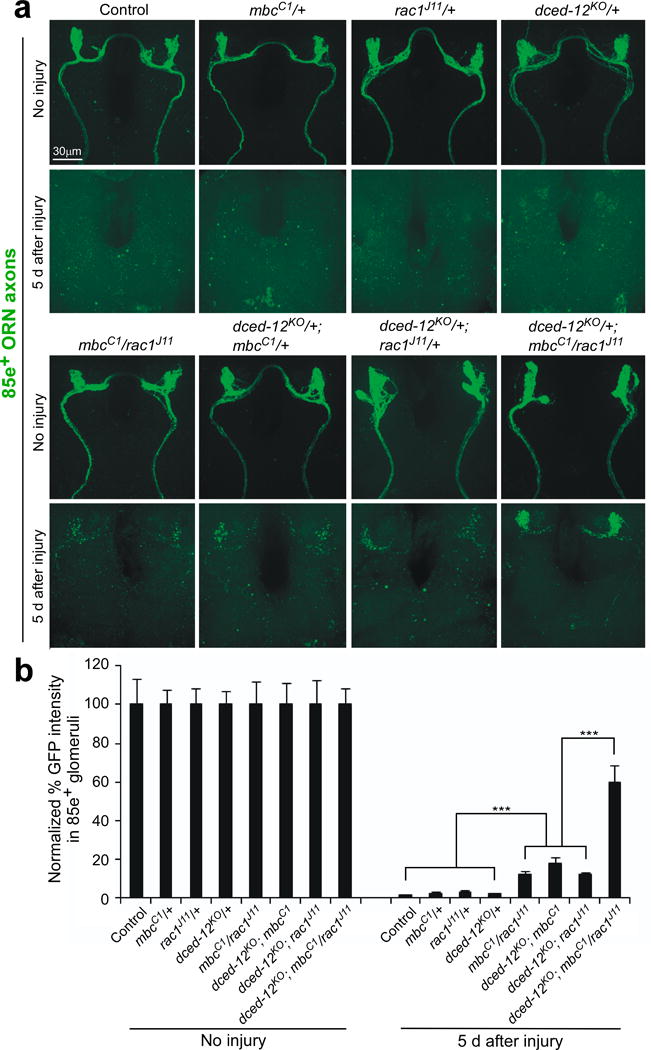
(a) OR85e-expressing ORN axons were labeled in control (w;OR85e-mCD8::GFP/+) animals and in mbcC1, dced-12KO, or rac1J11 heterozygous mutant backgrounds, maxillary palps were ablated, or left uninjured, and clearance of axons was assayed after 5 days (anti-GFP, green).
(b) Normalized quantification of data to uninjured cohorts from (a) Error bars represent s.e.m.; n>10; *** p<0.0001.
Crk/Mbc/dCed-12 promote axon debris clearance in the adult
Driving RNAis with repo-Gal4 results in a chronic knockdown throughout development, raising the possibility that glial engulfment defects could be developmental in origin. Indeed we found that driving UAS-mbcRNAi or a UAS-regulated dominant negative version of Rac1, UAS-rac1N17, with repo-Gal4 resulted in pupal lethality. We therefore performed adult-specific RNAi for crk, mbc, dced-12, and rac1, and also drove adult-specific expression of UAS-rac1N17. We generated lines bearing each of these UAS-regulated transgenes, repo-Gal4 and tub-Gal80ts. In tub-Gal80ts animals, Gal80ts suppresses the Gal4-dependent expression of UAS-regulated constructs at 18°C. However at 30°C, Gal80ts is inactivated, Gal4 activity is de-repressed, and UAS-regulated constructs are activated. We raised the above animals at 18°C, shifted them to 30°C for one week after eclosion, and performed axotomy assays. We found that adult-specific glial knockdown of mbc, crk, dced-12, or rac1 with RNAi constructs, or expressing racN17 in mature glia led to a robust suppression of glial clearance of axonal debris (Supplementary Figure 4a,b). These data indicate that Crk, Mbc, dCed-12, and Rac1 function during adult stages in glia to promote engulfment of degenerating axons.
There are two primary subtypes of neuropil-associated glial cells in the adult brain antennal lobe. Ensheathing glia express components of the Draper signaling pathway and are essential for clearance of degenerating ORN axons. Astrocyte-like glia are also present in the adult brain, but these do not express Draper, nor do they respond morphologically to ORN axon injury26. We used drivers specific to ensheathing (mz0709-Gal4) or astrocyte-like (alrm-Gal4) glia to drive crk, mbc, dced-12, and rac1 RNAi constructs and assayed ORN axon clearance after axotomy. Ensheathing glia-specific knockdown of each of these genes strongly suppressed clearance of axonal debris, while knockdown in astrocyte-like glia had no effect (Supplementary Figure 4c,d). We next drove expression of mbcRNAi, crkRNAi, dced-12RNAi, rac1RNAi, or sharkRNAi (i.e. to block the Draper pathway) in ensheathing glia using the additional ensheathing glia-specific TIFR-Gal4 driver and observed a strong suppression of glial clearance of degenerating axons (Supplementary Figure 5a,b). We conclude that all components of the Crk/Mbc/dCed-12 GEF complex and Rac1 are required in ensheathing glia for clearance of degenerating axons from the brain.
To determine whether the GEF complex composed of Crk/Mbc/dCed-12 might be dynamically recruited to severed axons after axotomy, we assayed localization of dCed-12 during glial responses to axon injury. dCed-12 immunoreactivity was not detectable on maxillary nerves control animals. However, at 1 or 3 days after axotomy we found dCed-12 enriched on the degenerating maxillary nerve (Supplementary Figure 6a,b). This staining was specific to dCed-12 since glial-specific knockdown of dCed-12 eliminated injury-induced localization of dCed-12 to severed axons. We conclude that glial-expressed dCed-12 rapidly localizes to degenerating axons in response to axotomy. Since we fail to detect significant levels of expression of dCed-12 prior to injury, glial expression of dCed-12, like that of Draper and dCed-625,26, appears to be up-regulated in glia following axotomy.
Rac1 is downstream of Crk/Mbc/dCed-12 and Draper
We suspected that Rac1 functioned primarily downstream of the Crk/Mbc/dCed-12 GEF. We therefore drove the expression of a constitutively activated version of Rac1 (RacV12) in glia where we also blocked glial Crk function. Expression of RacV12 in glia during development resulted in animal lethality, so we again used a conditional approach using Gal80ts to restrict the expression to adult brain glia (Figure 3b). We found adult-specific expression of crkRNAi in ensheathing glia potently suppress glial clearance of degenerating axons 11 days after axotomy (Figure 3a,c). However, co-expression of RacV12 resulted in normal clearance of degenerating axonal debris compared to controls (Supplementary Figure 7a,b,c). These data argue that activation of Rac1 is a primary function of Crk/Mbc/dCed-12 during glial engulfment of degenerating axons.
Figure 3. Constitutively active Rac rescues both Draper signaling and GEF pathway engulfment defects.
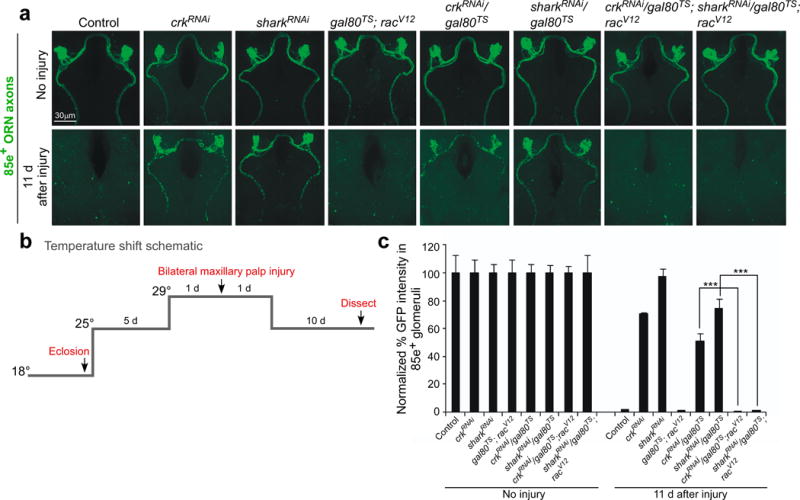
(a) OR85e-expressing ORNs were labeled with mCD8::GFP in control (w;OR85e-mCD8::GFP/+;TIFR-Gal4/+) and glial RNAi for crk (w;OR85e-mCD8::GFP, UAS-crk106498RNAi /+;TIFR-Gal4/+) and shark (w;OR85e-mCD8::GFP, UAS-shark6bRNAi /+;TIFR-Gal4/+) either containing gal80TS (w;OR85e-mCD8::GFP; UAS-crk106498RNAi /gal80TS;TIFR-Gal4/+ and w;OR85e-mCD8::GFP, UAS-shark6bRNAi /gal80TS;TIFR-Gal4/+) or with constitutive active racV12 (w;OR85e-mCD8::GFP; UAS-crk106498RNAi /gal80TS;TIFR-Gal4/racV12 and w;OR85e-mCD8::GFP, UAs-shark6bRNAi /gal80TS;TIFR-Gal4/racV12 and w;OR85e-mCD8::GFP/gal80TS;racV12/+).
Maxillary palps were bilaterally ablated and clearance of axonal debris was assayed with anti-GFP antibody stains (green) 11 days after injury, according to the temperature shift protocol outlined in (b).
(c) Normalized quantification of data to uninjured cohorts from (a) Error bars represent s.e.m.; n>10; *** p<0.0001.
We next drove expression of a sharkRNAi construct in adult brain ensheathing glia in the presence or absence of RacV12. As expected, we found that adult-specific knockdown of Shark was sufficient to block axon clearance after axotomy (Figure 3a,c). However, we made the surprising observation that co-expression of Racv12 in the sharkRNAi background restored normal glial engulfment activity (Figure 3a,c). This observation suggests Rac1 activation is sufficient to bypass requirements for the Draper signaling pathway. We note that we found no evidence for dominant genetic interactions between Draper and Crk/Mbc/dCed-12 (Supplementary Figure 8a,b). In summary, these data indicate that activation of Rac1 is sufficient to overcome loss of Crk/Mbc/dCed-12 or loss of Draper signaling pathway components. Thus Rac1 appears to function downstream of Crk/Mbc/dCed-12, Draper, or both during glial engulfment of axonal debris in vivo.
Glial respond to injury in the absence of Crk/Mbc/dCed-12
Drosophila adult brain glia become activated within 4–6 hours after axon injury, phagocytose axonal debris within 5–7 days, and then retreat from the injury site. Loss of components of the Draper signaling pathway completely blocks these events25,26,30. To further define how the Crk/Mbc/dCed-12 complex modulates these events we assayed the extension of glial membranes to sites of injury using Draper and dCed6 as a fiduciary markers for glial membranes. One day after axotomy in controls Draper accumulated on severed axons and in antennal lobe glomeruli innervated by maxillary palp ORNs (Figure 4a,d). Draper remained on these axons until 3–5 days after injury (at which time clearance of axons is complete), and by 10 days Draper was no longer enriched at these sites and returned to baseline levels (Figure 4a,d). Glial-specific knockdown of crk or dced-12 slightly delayed, but did not block the accumulation of Draper on severed axons. In glial crkRNAi or dced-12RNAi animals Draper was detectable on maxillary palp-innervated glomeruli within 3 days after axotomy, remained on these glomeruli 5 days after injury, and returned to baseline levels by 10 days after injury (Figure 4b,c,d). We observed a similar dynamic movement and retreat of glial membranes in these genetic backgrounds when we labeled them with membrane-tethered tdTomato (see below) or dCed-6. Thus depletion of Crk or dCed-12 from glia does not block activation or termination of glial responses. Rather, glia with depleted Crk or dCed-12 are recruited to severed axons, although they are slightly delayed, remain on axons for 5–7 days, fail to engulf axons, and then retreat from the injury site. These data indicate Crk/Mbc/dCed-12 are specifically required for glial internalization of axonal debris. Moreover, these observations reveal, surprisingly, that glia are capable of terminating reactive responses after a certain window of time regardless of whether axonal debris has been phagocytosed.
Figure 4. Crk and dCed-12 are not required for activation of glia after axotomy.
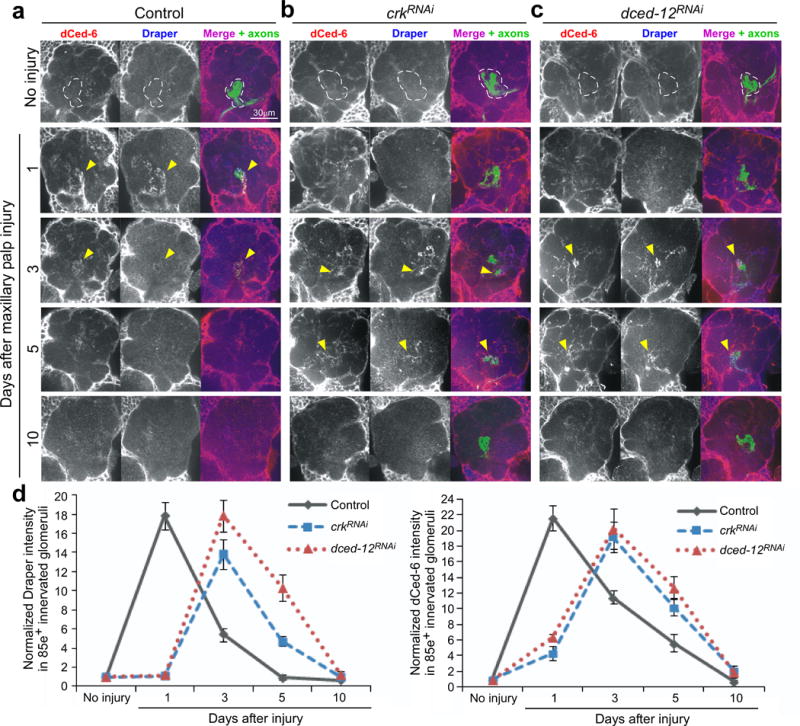
(a, b, and c) OR85e-expressing ORNs were labeled with mCD8::GFP in control (w;OR85e-mCD8::GFP/+; repo-Gal4/+) and those with glia-specific knockdown of crk and dced-12 (w;UAS-crk19061RNAi/ OR85e-mCD8::GFP;repo-Gal4/+ and w; OR85e-mCD8::GFP/+; repo-Gal4/UAS-dced-1210455RNAi). Animals were assayed for injury-induced recruitment of dCed-6 (red, pseudo-colored grey, left panel) and Draper (blue, pseudo-colored grey middle panel) to severed axons in maxillary-palp innervated glomeruli (arrows) 1, 3, 5, and 10 days after injury.
(d) Normalized quantification to uninjured cohorts of Draper and dCed-6 intensity in 85e-innervated glomeruli (right panel, dotted outline) from (a, b, and c). Error bars represent s.e.m.; n>10 for all.
Rac1 phenocopies draper mutants
Since Crk/Mbc/dCed-12 can act as a GEF for Rac1 we expected that depleting Rac1 function would result in a phenotype similar to that seen in crkRNAi and dced-12RNAi animals. However, we found that knockdown of rac1 in glia completely suppressed activation of glia and the recruitment of glial membranes to severed axons (Supplementary Figure 9a,b,c,d,e). This is precisely what is observed when Draper signaling is blocked25–27. Thus Rac1, in contrast to Crk/Mbc/dCed-12, appears to act very early in glial responses to axon injury and phenocopies draper mutants. This observation is consistent with our observed rescue of Draper signaling pathway inhibition by activated Rac1, and the previous observation that CED-10 also acts downstream of CED-1 in C. elegans cell corpse engulfment31. Since the Rac1 loss of function phenotype differs so dramatically with respect to glial recruitment to severed axons from that of Crk/Mbc/dCed-12, we would predict that an alternative unidentified GEF must activate Rac1 downstream of Draper during activation of glial responses to axotomy.
Aged axonal debris remains a viable engulfment target
If engulfment cues presented by degenerating axons are labile, it is possible that they could degrade within the first 1–2 days, so that by the time glial membranes from crkRNAi or dced-12RNAi animals arrive at the injury site, “eat me” cues are no longer present. To address this possibility we allowed severed axons to age in vivo without being engulfed and asked—how long does axonal debris retain its ability to activate glia and promote its own engulfment? We labeled ORN axons with GFP and drove glial-specific expression of the temperature-sensitive dynamin, shibirets, which at restrictive temperature can block glial recruitment to sites of axon injury26. Axotomies were performed at restrictive temperature (30°C), cohorts of flies were maintained for 1 or 7 days at 30°C after axotomy before being then shifted back to permissive temperature (18°C), and clearance was allowed to occur for 7, 11, or 30 days. Impressively, even when severed axons were “aged” for 7 days, glial membranes were still recruited to severed axons and axonal debris was cleared from the CNS after animals were returned to permissive temperatures (Figure 5a,b; Supplementary Figure 10a,b,). These data argue strongly that the clearance defect we see in crkRNAi and dced-12RNAi animals is not likely due to the delay in glial arrival at the injury site. Moreover, these data indicate that axonal debris retains its ability to re-activate local glia and promote its own clearance from the CNS. Thus we predict that at least some of the “come and get me” and “eat me” signals exposed by severed axons are quite stable.
Figure 5. Axons remain viable engulfment targets capable of activating glia for over a week after axotomy.
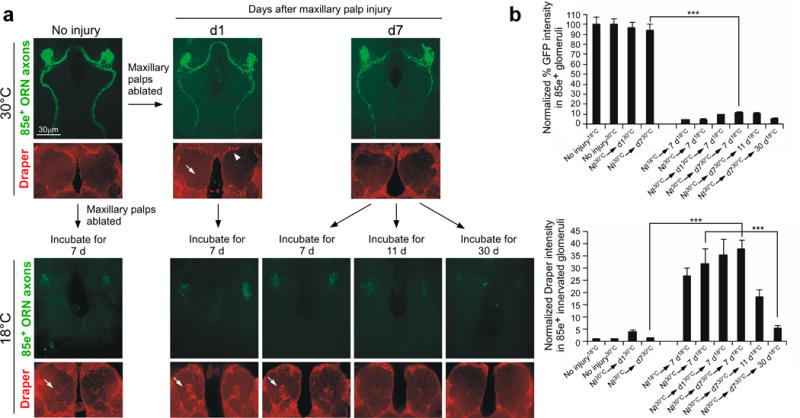
(a) Experimental animals (w;OR85e-mCD8::GFP/UAS-shibirets; mz0709-Gal4/+) were raised at the permissive temperature, 18°C, and kept at 18°C for 7 days after eclosion. Adult animals were then shifted to the restrictive temperature, 30°C, and either left uninjured or injured via bilateral maxillary palp ablation. Left, expression of Shibirets in ensheathing glia where animals were returned to 18°C, injured, and allowed 7 days to clear axonal debris. Cohorts kept at 30°C (where glial engulfment activity was “frozen”) were assayed for changes in glial Draper expression (red) around maxillary-innervated glomeruli (arrows) and clearance of axonal debris (anti-GFP, green) at 1 (D1) or 7 (D7) days after injury at 30°C. Arrowhead; appearance of Draper+ puncta only at periphery of antennal lobe. These same cohorts were returned to 18°C and allowed 7, 11, or 30 additional days to clear degenerating axons.
(b) Normalized quantification of data to cohort controls from (a) Error bars represent s.e.m.; n>10; ***, P<0.0001.
Despite the fact that glia are blocked from invading sites of injury (while at 30°C in shits backgrounds), we noted that glia surrounding the antennal lobe exhibited a marked increase in Draper 1 day after axotomy (Figure 5; Supplementary Figure 10a,b). Several of these glia (especially those in the dorsal-lateral regions of the antennal lobe) are not in direct physical contact with severed maxillary palp axons26. This observation implies that severed axons can send “eat- or find-me” signals to glia over considerable distances in the brain, which lead to increased Draper expression, and ultimately activation of glial responses.
Crk/Mbc/dCed-12 promotes glial phagocytosis of axon debris
Reactive glia are highly phagocytic, but how phagocytic pathways are genetically activated, and how closely their activation is coupled to initial glial responses (e.g. recruitment to injury sites) remains poorly defined. We therefore developed several new tools to examine activation of the glial phagocytic phenotype, and glial internalization of degenerating axonal debris. First, we assayed lysosomal activity within glial cells in uninjured brains using Lysotracker. Lysosomal activity in the uninjured brains was nearly undetectable; however within 1 day after axotomy of antennal ORNs we found a dramatic increase in Lysotracker+ puncta in antennal lobe, which localized to ensheathing glia (Figure 6a,b). We suspect these represent glial phagolysosomes, the product of phagosome fusion with acidifying lysosomes. Interestingly, we also note the presence of a large population of vesicles within glia that do not stain for Lysotracker and are only present after injury (Figure 6b, arrows). We suspect these represent phagosomes that have not yet fused with lysosomes and are therefore not acidified. This is supported by our observation that many of the vacuoles marked by TIFR-Gal4 house axonal debris (see below).
Figure 6. Axotomy-induced activation of phagolysosomes in engulfing glia.
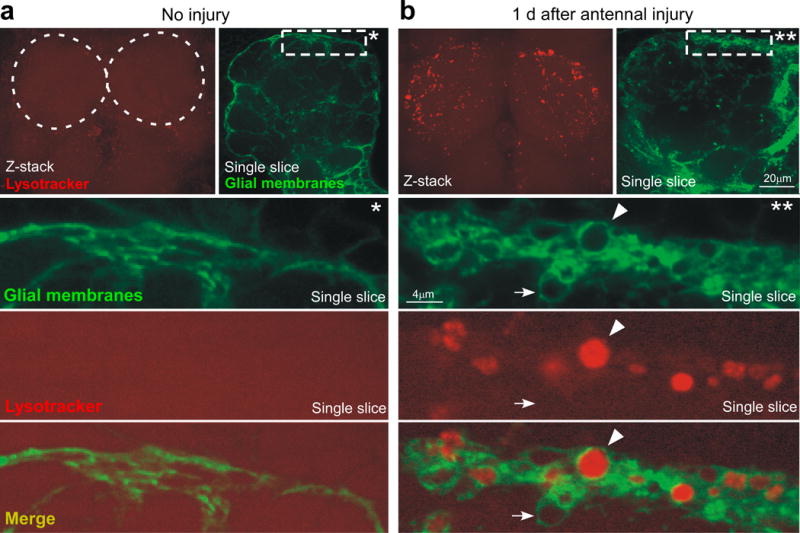
(a) Uninjured animals (w;UAS-mcd8::GFP/+;TIFR-Gal4/+) were stained for Lysotracker Red (confocal Z-stack, top) and visualized for GFP+ glial membranes (single confocal slice, top). Dotted circles; location of antennal lobes. *; high magnification view of the marked rectangle in a single confocal section.
(b) Animals (w;UAS-mcd8::GFP/+;TIFR-Gal4/+) shown 1 day after bilateral antennal injury were stained for Lysotracker Red (Z-stack top) and visualized for GFP+ glial membranes (single slice, top). **; high magnification view of the marked rectangle in a single confocal section. Arrow; an injury-induced glial vesicle. Arrowhead; glial vesicle positive for lysosomal activity.
Lysotracker signal in the brain was specific to regions where severed axons were degenerating. For example, ablation of maxillary palps led to the preferential accumulation of Lysotracker+ puncta along the maxillary nerve (Figure 7b) and glomeruli housing degenerating maxillary palp ORN axons within the antennal lobe (Figure 7a), but not elsewhere at appreciable levels. Only a subset of vesicles were Lysotracker positive, but all appeared to be surrounded by Draper (Figure 7a,b), and all were in regions containing of GFP+ axon debris. We found that Lysotracker+ puncta were observed maximally in the antennal lobe 1 day after axotomy, decreased at day 3 and approached baseline levels by 5 days after axotomy (Figure 8a,e). We conclude that lysosomal activity is strongly up-regulated in ensheathing glia in response to axonal injury, and propose Lysotracker+ signal represents the maturation of phagosomes into phagolysosomes.
Figure 7. Glial phagolysosomes are decorated with Draper and preferentially accumulate around degenerating axons.
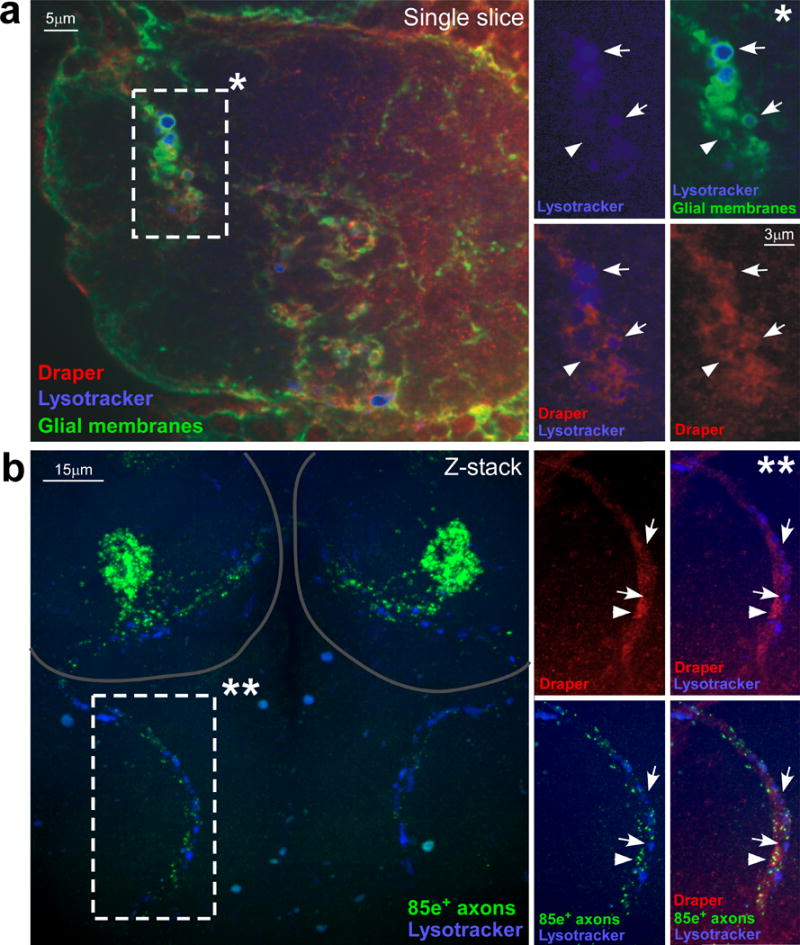
(a) Animals (w;UAS-mcd8::GFP/+;TIFR-Gal4/+) shown 1 day after bilateral maxillary palp injury were stained for Draper (pseudo-colored red), Lysotracker Red (pseudo-colored blue), and visualized for GFP+ glial membranes. *; high magnification view of the marked rectangle in a single confocal section. Arrows; injury-induced vesicles positive for Draper recruitment and lysosomal activity. Arrowhead; vesicles positive for Draper recruitment but lacking lysosomal activity.
(b) Animals (w;OR85e-mCD8::GFP/+) shown 1 day after bilateral maxillary palp injury were stained for GFP (green), Draper (pseudo-colored red) and Lysotracker Red (pseudo-colored blue). Circles; location of antennal lobes. **; high magnification view of the marked rectangle in a confocal Z-stack. Arrows; areas of lysosomal activity with little to no remaining GFP+ axon debris. Arrowhead; areas where groups of axon debris co-localize with Draper along the degenerating maxillary palp nerve.
Figure 8. Crk and dCed-12 are required for internalization of axonal debris and phagolysosome formation.
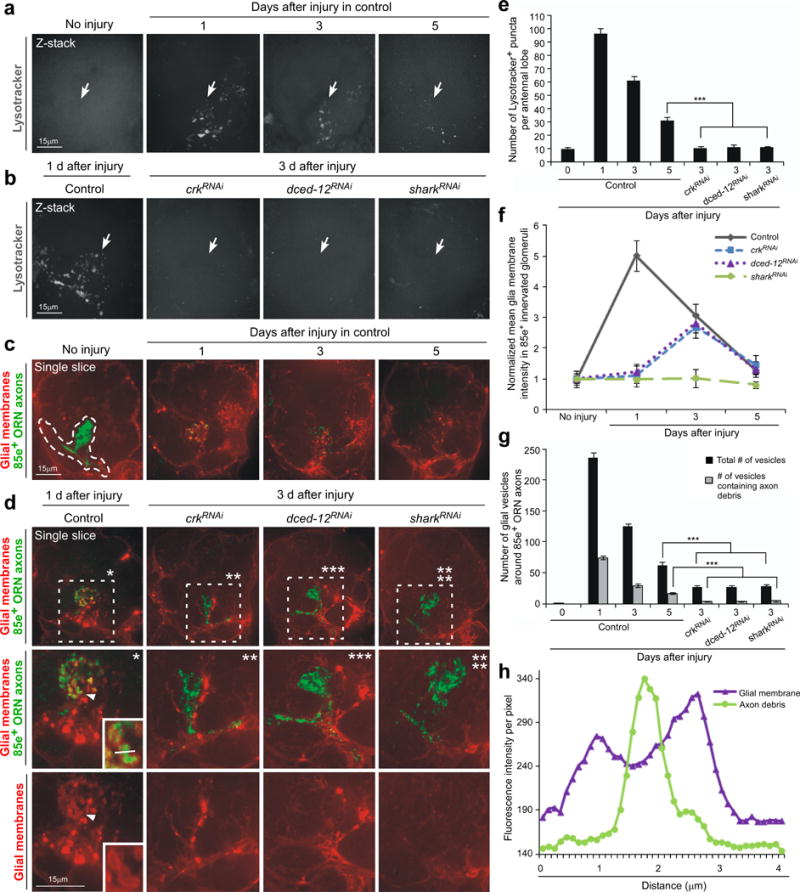
(a) Lysosomal activity was monitored with Lysotracker Red (pseudo-colored grey) in control (w; TIFR-Gal4/+) animals in individual antennal lobes (pictured) 1, 3, and 5 days after maxillary palp injury. Arrows; location of 85e-innervated maxillary-palp glomeruli. Representative single confocal Z-sections are shown for all.
(b) Lysosomal activity was monitored in control (w; TIFR-Gal4/+) animals 1 day after maxillary palp injury, and 3 days after injury in glial RNAi animals for crk106498RNAi, dced-1210455RNAi, and shark6bRNAi. Arrows; location of 85e-innervated maxillary-palp glomeruli.
(c) Glial membranes were labeled with tdTomato, OR85e-expressing axons (anti-GFP, green) were labeled with mCD8::GFP (w;OR85e-mCD8::GFP/UAS-cd4-tdTomato;TIFR-Gal4/+) and we monitored glial membrane recruitment to axonal debris, and the formation of vesicles within ensheathing glia 0, 1, 3, and 5 days after maxillary palp injury. Outlined; 85e-innervated maxillary-palp glomerulus used to quantify glial invasion and vesicle formation into injured area.
(d) Glial membranes and axons were labeled as in (d). Recruitment of glial membranes to degenerating axons, and internalization of axonal debris by ensheathing glia was scored in control and glial crk106498RNAi, dced-1210455RNAi,and shark6bRNAi animals. (a’-d’); high magnification views of marked rectangle areas. Arrowhead; red glial vesicle containing axon debris. Lower right corner box in a’: high magnification view of debris-containing glial vesicles.
(e) Quantification of antennal lobe lysosomal puncta data from (a, b).; Error bars represent s.e.m.; n>10; ***, P<0.0001.
(f) Normalized quantification to control from (d, e) of glial membrane infiltration around degenerating 85e+ axons.
(g) Quantification from (d, e) of the total number of glial vesicles and number of vesicles containing axonal debris around 85e-innervated maxillary-palp glomeruli.; Error bars represent s.e.m.; n>10; ***, P<0.0001.
(h) Example from corner box in (a’) of fluorescence intensity of the glial membrane and axon debris along line drawn through an individual glial vesicle.
We next assayed for roles for the Crk/Mbc/dCed-12 complex in maturation of Lysotracker+ vesicles in engulfing glia. Strikingly, while glial membranes were recruited closely to severed axons in crkRNAi and dced-12RNAi backgrounds (see also above), there was a complete failure to induce lysosomal activity in these backgrounds (Figure 8b,e). Thus Crk and dCed-12 are essential for activation of lysosomal activity during glial internalization of degenerating axons. Activation of lysosomal activity is also ultimately downstream of Draper signaling since sharkRNAi animals also failed to induce lysosomal activity after axotomy (Figure 8b,e), and similar results were found in glial rac1RNAi animals (Supplementary Figure 9f,g).
It is possible that glia depleted for Crk/Mbc/dCed-12 indeed engulf degenerating axons, but fail to digest them through fusion of lysosomes with phagosomes. To explore this possibility we next labeled glial membranes with membrane-tethered tdTomato (UAS-cd4::tdTomato) using the TIFR-Gal4 driver, axons with mCD8::GFP (OR85e-mCD8::GFP), and assayed glial internalization of degenerating axons after axotomy in control, crkRNAi, dced-12RNAi, and sharkRNAi animals. In control animals we found 1 day after axotomy the vast majority of mCD8::GFP-labeled axonal debris was found within cd4::tdTomato-labeled vacuoles in glia (Figure 8c,d,g,h). The number of vesicles housing axonal material peaked in control animals 1 day after axotomy, decreased at 3 days, and approached baseline by 5 days after axotomy (Figure 8c,g)—this mirrored our findings with Lysotracker+ vesicles (above) and provides direct evidence that ensheathing glial cells internalize axonal debris. While tdTomato-labeled glial membranes were found in close proximity with axonal debris 3 days after axotomy in crkRNAi and dced-12RNAi animals, we found no evidence for internalization of GFP-labeled axonal debris (Figure 8d,f). Notably, in sharkRNAi animals we found, consistent with our previous work26,30, no evidence for recruitment of tdTomato-labeled glial membranes to severed axons. In addition, we found that sharkRNAi animals failed to internalize degenerating axonal debris.
In summary, our data show that loss of Draper signaling pathway components lead to very early defects in activation of glial responses to axonal injury—in the absence of Draper signaling, glial cells are not recruited to severed axons and fail to respond morphologically to axonal injury. In contrast, animals depleted for glial Crk/Mbc/dCed-12 exhibit normal glial activation and recruitment to severed axons, but they fail to activate phagocytic activity, and do not internalize axonal debris.
Discussion
Unique pathways regulate glial activation and phagocytosis
We identify Crk/Mbc/dCed-12 and Rac1 as new components of the glial engulfment machinery. There are several notable differences between the genetic requirements for these pathways in the engulfment of degenerating severed axons in Drosophila versus cell corpses in C. elegans. First, rather than acting in a partially redundant fashion as they do in cell corpse engulfment in C. elegans, both pathways (Draper/dCed-6/Src42a/Shark, and Crk/Mbc/dCed-12 and Rac1) are absolutely required for glia to clear severed axons from the adult Drosophila brain. It is interesting to note that in draper null mutants clearance of developmentally pruned mushroom body γ axons is delayed by only a few days, but all axons are ultimately cleared by adulthood22,23. This observation argues for the presence of pathways that act in a genetically redundant fashion with Draper during neurite pruning. The Crk/Mbc/dCed-12 complex and Rac1 are excellent candidates, but to date these have not been studied in the context of developmental axon pruning.
A second key point revealed by our work is that Crk/Mbc/dCed-12 and Draper each regulate distinct steps in the glial response after nerve injury. All known components of the Draper signaling pathway, as well as Rac1, act very early in the glial response and are required during glial activation for the up-regulation of engulfment genes and recruitment of glial membranes to severed axons. In contrast, glial-specific depletion of Crk, Mbc, or dCed-12 does not block glial recruitment to severed axons or termination of glial responses; rather it specifically blocks the ability of glia to activate a phagocytic phenotype and internalize axonal debris. These observations indicate that glial activation and phagocytosis of degenerating axons are genetically separable events mediated by distinct molecular pathways. Notably, our observations are contrary to what one would predict based on our understanding of CED-2/CED-5/CED-12 and CED-10 function in C. elegans. Specifically, since loss of CED-2/CED-5/CED-12 or CED-10, but not CED-1/CED-6, profoundly inhibits cell migration in C. elegans32,33 one might have predicted a role for Crk/Mbc/dCed-12 in the extension of glial membranes to severed axons. However, our observation that extension of glial membranes to severed axons is largely normal in animals depleted of Crk/Mbc/dCed-12 points to an unexpected but quite specific role for this complex in cellular dynamics underlying phagocytosis of degenerating axons. Moreover, since Draper robustly decorates severed axons in crk and dced-12 knockdown animals, our data also raise important new questions regarding Draper function during debris internalization: since these axons are decorated with Draper but they are not cleared, is Draper acting during phagocytosis of axonal debris? Were this the case it seems likely that the Crk/Mbc/dCed-12 GEF acts downstream of Draper. If not, perhaps the primary role for Draper is to specifically activate glia after sensing an injury (i.e. up-regulating engulfment genes and guiding glial membranes to injury sites) and then terminate glial responses in a timely fashion (i.e. returning glia to a resting state)34.
Finally, since Rac1 knockdown and dominant negative constructs phenocopy draper mutants, our data argue strongly in favor of Rac1 acting genetically downstream of the Draper receptor. Indeed, previous observations in C. elegans suggest that CED-10 (Rac1) acts downstream of CED-1 (Draper)31. Consistent with a role for Rac1 downstream of Draper, we showed that simply resupplying activated Rac1 in glia was sufficient to rescue loss of the Draper signaling pathway component Shark. However, based on the fact that Rac1 also appears to function at later phases with Crk/Mbc/dCed-12, we propose that Rac1 activation immediately downstream of Draper likely occurs via a GEF complex that is either distinct from, or redundant with, Crk/Mbc/dCed-12 (Supplementary Figure 11). Alternatively, during phagocytosis of degenerating axons the Crk/Mbc/dCed-12 complex may function downstream of an unidentified receptor that functions in parallel to Draper. Indeed, Crk-II/Dock180/Elmo and CED-2/CED-5/CED-12 have been shown to bind directly to the phosphatidylserine-binding receptor Bai1 and indirectly to the frizzled-like receptor MOM-5 to promote phagocytosis of cell corpses in mammals and C.elegans35,36. However no clear sequence orthologs of Bai1 or MOM-5 exist in the Drosophila genome.
Signaling between engulfing glial cells and axonal debris
After almost any brain injury glia rapidly invade the injury site and remain there to manage brain responses to trauma37–41. The neuron-glia signaling events that mediate the behavior of glial cells at injury sites remain poorly defined, but examples of glial cells rapidly abandoning an injury site prior to axonal debris having been cleared have not been reported to our knowledge. It is therefore striking that glial cells lacking Crk/Mbc/dCed-12 function retreated from glomeruli housing severed axons within days after axotomy, leaving behind the vast majority of unengulfed axonal debris. These data indicate that the presence of axonal debris is not sufficient to retain glial cells at an injury site. Is this because the cues that promote glial recruitment to and engulfment of degenerating axons are labile and degrade over time? The presence of an early and transient axon→glia signal that stimulates Draper expression is suggested by the fact that we see a transient induction of Draper 1 day after axotomy even in glial cells not in contact with severed axons, and frozen in their morphological invasion of the injury site (with shits). The observation that Draper is eventually down-regulated to baseline levels suggests that either this signal is lost or that glia adapt to its presence. It would appear that the chemical properties of axonal debris, specifically the cues required to recruit glia to injury sites, do not change for at least one week after axotomy: we were able to reversibly block glial recruitment to severed axons using shits for up to 7 days, yet still observe largely normal engulfment of axons when glia were allowed to re-activate engulfment behavior. Together these data argue for the possible presence of multiple injury signals from severed axons during activation of glia. First, we predict the presence of a signal that promotes increased Draper expression in glia, and this would be expected to act over some distance in the brain. Second, a signal that autonomously tags axonal debris in such a way that glia are able recognize it as an engulfment target, extend membranes to, and internalize it.
Why do glia leave the injury site in animals lacking glial Crk/Mbc/dCed-12? A key difference between our RNAi experiments with Crk/Mbc/dCed-12 and the shits blockade of engulfment activity is that in the former situation glial cells were able to interact directly with severed axons for several days. This contact with axons could have allowed glia to alter the surface properties of axonal debris. For example, glia could bind so-called “eat me” or “come get me” cues generated by axons, or otherwise tag axonal debris in such a way that made it no longer capable of attracting engulfing glial cell types. Alternatively, essential recognition receptors in glia which lack Crk/Mbc/dCed-12 could become adapted to the presence of axonal debris over time, thereby decreasing or eliminating their sensitivity to this engulfment target.
It is also possible that glia might also be intrinsically programmed to remain at sites of engulfment for a particular time window before terminating their responses to injured axons regardless of the status of axonal debris clearance. We have recently identified a surprising new role for Draper-II, and alternative splice variant of Draper which specifically promotes termination of glial engulfment activity after axotomy in vivo34. Briefly, Draper-II is activated after the pro-engulfment receptor Draper-I and promotes a return to the glial resting state by 5–7 days after axotomy. Our observation that termination of glial responses occurs on schedule in animals lacking Crk/Mbc/dCed-12 suggests that Crk/Mbc/dCed-12 signaling is not critical for Draper-II function. Therefore Draper-dependent regulation of glial reactive status is likely genetically uncoupled from internalization of debris. Somehow the communication between axonal debris and glia must change in Crk/Mbc/dCed-12 backgrounds since glia retreated from the injury site after 5–7 days, and they did not appear to be re-recruited to debris based on the lack of clearance even 30 days after axotomy. Defining the molecular cues that mediate these cell-cell interactions will be interesting and relevant to neurological disease, since our data indicate that a failure by glia to efficiently clear engulfment targets during a critical window leads to the irreversible accumulation of potentially harmful neural debris in the mature brain.
Methods
Fly strains
The following Drosophila strains were used: repo-Gal442, mz0709-Gal443, alrm-Gal426, OR85e-mCD8::GFP (gift from B. Dickson, Research Institute of Molecular Pathology, Vienna, Austria), TIFR-Gal4 (a gift from H. Hing, U. Illinois, Urbana), draperΔ525, mbcC1 (Bloomington Drosophila Stock Center), rac1J1144, dced-12KO (a gift from Pernille Rørth, Institute of Molecular and Cell Biology, Singapore), UAS-shark6bRNAi30, UAS-rac1N17 (Bloomington Drosophila Stock Center), UAS-crkRNAi lines 19061 and 106498 (Vienna Drosophila RNAi Center), UAS-CD4::tdTomato45, UAS-mbcRNAi line 16044 (VDRC), UAS-dced12RNAi line 10455 (VDRC), UAS-Rac1RNAi lines 49246 and 49247 (VDRC), UAS-shibirets and UAS-tubulin gal80ts (gifts from S. Waddell, University of Massachusetts Medical School, Worcester MA).
Neuron injury protocol and temperature shift experiments
Olfactory receptor neurons were severed as previously described25 in animals aged at least one week after eclosion. After injury, animals were transferred to fresh food vials and aged at 18°C or 29°C (for Gal80ts and shits experiments), and 25°C for all other experiments. tub-Gal80ts flies were crossed with repo-Gal4 and RNAi lines including UAS-crkRNAi, UAS-mbcRNAi, UAS-dced-12RNAi, UAS-racN17 or UAS-rac1RNAi. Flies were raised at 18°C, shifted to 29°C upon eclosion and incubated at 29°C for at least 7 days before maxillary palp ablation, and maintained at 29°C after injury until ready for immunohistochemistry. UAS-shibirets flies were crossed with the mz0709-Gal4 driver line. Flies were raised and maintained for at least 7 days after eclosion at 18°C prior to ablation and/or temperature shifts described in Figure 5. For Rac rescue experiments, flies were crossed with the TIFR-Gal4 driver line and raised at 18°C. Once eclosed, flies were shifted to 25°C for 5 days, then shifted to 29°C for 1 day. Maxillary palps were bilaterally ablated and animals were left at 29°C for 1 day before being shifted down to 25°C for 10 days, when clearance of axonal debris was assayed with anti-GFP antibody stains, according to the temperature shift protocol outlined in Figure 4b.
Confocal microscopy
Adult brains were processed as previously described25. Primary antibodies were used at the following dilutions: 1:500 rabbit anti-Draper21, 1:200 mouse anti-GFP (Invitrogen), 1:200 rabbit anti-dCed-1246 and 1:500 rabbit anti-dCed-622 (both kindly provided by T. Awazaki, Janelia Farm, Chevy Chase, MD). All anti-IgG secondary antibodies were FITC, Cy3, or Cy5 conjugated (Jackson ImmunoResearch) and used at 1:200.
For Lysotracker stains, heads from live animals were removed and immediately immersed in chilled PBS, brains were dissected and bathed in Lysotracker Red (DND99, Invitrogen Inc) at a dilution of 1:5000. Brains were rocked for 15 minutes, washed 5 times in PBS for a total of 15 minutes, then fixed in PBS/0.1% Triton X-100/4% formaldehyde for 30 minutes with rocking in the dark. Brains were washed 5 times in PBS/0.1% Triton X-100 for a total 30 minutes, mounted in Vectashield antifade reagent (Vector Labs), and immediately imaged.
Quantification of Draper, dCed-6, glial membranes, and Lysotracker stains during glial engulfment of axons
For all experiments the entire antennal lobe was imaged in 1μm steps and scored for relevant phenotypes. In experiments in which cd8::GFP, cd4::tdTomato, Draper, or dCed-6 intensity in different genetic backgrounds was quantified, samples were fixed and stained side by side, and were imaged at the same time with standardized confocal settings for each series of experiments. Quantification of fluorescence intensity was performed on single Z sections in either the center of the relevant glomerulus (for Draper,dCed-6, and cd4::tdTomato the 85e-GFP marker was used to reproducibly identify the same glomerulus for quantification), at the same position of the maxillary nerve, or at the same dorsal position at the edge of the antennal lobe, with ImageJ software (http://rsb.info.nih.gov/ij/) as described in Doherty et al., 2009 and MacDonald et al., 2006. Lysotracker stains were quantified by counting the total number of positive puncta in an individual antennal lobe. To quantify the rates of internalization of 85e-GFP-labeled axonal debris by glia, tdTomato-labeled glial vesicles adjacent to the 85e-GFP-labeled debris were counted, and then were individually scored for presence or absence of GFP-labeled material within the vesicle. P values were generated using a two-tailed student’s t-test.
Supplementary Material
Acknowledgments
We thank P. Rørth, N. Franc, S. Waddell, T. Awasaki, H. Hing, Y-N Jan, and Y. Nakanishi for fly strains and antibodies. We thank L. Neukomm and A. Nicole Fox for critical reading of the manuscript and the entire Freeman laboratory for insightful discussions. This work was supported by NIH RO1 NS053538 to MRF, and MRF is an Early Career Scientist with the Howard Hughes Medical Institute.
Footnotes
Author contributions
JSZ and MRF designed the experiments; JSZ conducted the majority of the experiments; JD performed a subset of the Rac1 studies; MRF and JSZ wrote the manuscript.
References
- 1.Cuadros MA, Navascues J. The origin and differentiation of microglial cells during development. Prog Neurobiol. 1998;56:173–189. doi: 10.1016/s0301-0082(98)00035-5. [DOI] [PubMed] [Google Scholar]
- 2.Marin-Teva JL, Cuadros MA, Calvente R, Almendros A, Navascues J. Naturally occurring cell death and migration of microglial precursors in the quail retina during normal development. J Comp Neurol. 1999;412:255–275. [PubMed] [Google Scholar]
- 3.Parnaik R, Raff MC, Scholes J. Differences between the clearance of apoptotic cells by professional and non-professional phagocytes. Curr Biol. 2000;10:857–860. doi: 10.1016/s0960-9822(00)00598-4. [DOI] [PubMed] [Google Scholar]
- 4.Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- 5.Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009;32:638–647. doi: 10.1016/j.tins.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Savill J, Dransfield I, Gregory C, Haslett C. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat Rev Immunol. 2002;2:965–975. doi: 10.1038/nri957. [DOI] [PubMed] [Google Scholar]
- 7.Neumann H, Kotter MR, Franklin RJ. Debris clearance by microglia: an essential link between degeneration and regeneration. Brain. 2009;132:288–295. doi: 10.1093/brain/awn109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lalancette-Hebert M, Gowing G, Simard A, Weng YC, Kriz J. Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J Neurosci. 2007;27:2596–2605. doi: 10.1523/JNEUROSCI.5360-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fuentes-Medel Y, et al. Glia and muscle sculpt neuromuscular arbors by engulfing destabilized synaptic boutons and shed presynaptic debris. PLoS Biol. 2009;7:e1000184. doi: 10.1371/journal.pbio.1000184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Logan MA, Freeman MR. The scoop on the fly brain: glial engulfment functions in Drosophila. Neuron Glia Biol. 2007;3:63–74. doi: 10.1017/S1740925X07000646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luo L, O’Leary DD. Axon retraction and degeneration in development and disease. Annu Rev Neurosci. 2005;28:127–156. doi: 10.1146/annurev.neuro.28.061604.135632. [DOI] [PubMed] [Google Scholar]
- 12.Kinchen JM, Ravichandran KS. Journey to the grave: signaling events regulating removal of apoptotic cells. J Cell Sci. 2007;120:2143–2149. doi: 10.1242/jcs.03463. [DOI] [PubMed] [Google Scholar]
- 13.Reddien PW, Horvitz HR. The engulfment process of programmed cell death in caenorhabditis elegans. Annu Rev Cell Dev Biol. 2004;20:193–221. doi: 10.1146/annurev.cellbio.20.022003.114619. [DOI] [PubMed] [Google Scholar]
- 14.Hasegawa H, et al. DOCK180, a major CRK-binding protein, alters cell morphology upon translocation to the cell membrane. Mol Cell Biol. 1996;16:1770–1776. doi: 10.1128/mcb.16.4.1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou Z, Caron E, Hartwieg E, Hall A, Horvitz HR. The C. elegans PH domain protein CED-12 regulates cytoskeletal reorganization via a Rho/Rac GTPase signaling pathway. Dev Cell. 2001;1:477–489. doi: 10.1016/s1534-5807(01)00058-2. [DOI] [PubMed] [Google Scholar]
- 16.Brugnera E, et al. Unconventional Rac-GEF activity is mediated through the Dock180-ELMO complex. Nat Cell Biol. 2002;4:574–582. doi: 10.1038/ncb824. [DOI] [PubMed] [Google Scholar]
- 17.Gumienny TL, et al. CED-12/ELMO, a novel member of the CrkII/Dock180/Rac pathway, is required for phagocytosis and cell migration. Cell. 2001;107:27–41. doi: 10.1016/s0092-8674(01)00520-7. [DOI] [PubMed] [Google Scholar]
- 18.Zhou Z, Hartwieg E, Horvitz HR. CED-1 is a transmembrane receptor that mediates cell corpse engulfment in C. elegans. Cell. 2001;104:43–56. doi: 10.1016/s0092-8674(01)00190-8. [DOI] [PubMed] [Google Scholar]
- 19.Yu X, Lu N, Zhou Z. Phagocytic receptor CED-1 initiates a signaling pathway for degrading engulfed apoptotic cells. PLoS Biol. 2008;6:e61. doi: 10.1371/journal.pbio.0060061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu QA, Hengartner MO. Candidate adaptor protein CED-6 promotes the engulfment of apoptotic cells in C. elegans. Cell. 1998;93:961–972. doi: 10.1016/s0092-8674(00)81202-7. [DOI] [PubMed] [Google Scholar]
- 21.Freeman MR, Delrow J, Kim J, Johnson E, Doe CQ. Unwrapping glial biology. Gcm target genes regulating glial development, diversification, and function. Neuron. 2003;38:567–580. doi: 10.1016/s0896-6273(03)00289-7. [DOI] [PubMed] [Google Scholar]
- 22.Awasaki T, et al. Essential role of the apoptotic cell engulfment genes draper and ced-6 in programmed axon pruning during Drosophila metamorphosis. Neuron. 2006;50:855–867. doi: 10.1016/j.neuron.2006.04.027. [DOI] [PubMed] [Google Scholar]
- 23.Hoopfer ED, et al. Wlds protection distinguishes axon degeneration following injury from naturally occurring developmental pruning. Neuron. 2006;50:883–895. doi: 10.1016/j.neuron.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 24.Williams DW, Kondo S, Krzyzanowska A, Hiromi Y, Truman JW. Local caspase activity directs engulfment of dendrites during pruning. Nat Neurosci. 2006;9:1234–1236. doi: 10.1038/nn1774. [DOI] [PubMed] [Google Scholar]
- 25.MacDonald JM, et al. The Drosophila cell corpse engulfment receptor Draper mediates glial clearance of severed axons. Neuron. 2006;50:869–881. doi: 10.1016/j.neuron.2006.04.028. [DOI] [PubMed] [Google Scholar]
- 26.Doherty J, Logan MA, Tasdemir OE, Freeman MR. Ensheathing glia function as phagocytes in the adult Drosophila brain. J Neurosci. 2009;29:4768–4781. doi: 10.1523/JNEUROSCI.5951-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ziegenfuss JS, et al. Draper-dependent glial phagocytic activity is mediated by Src and Syk family kinase signalling. Nature. 2008 doi: 10.1038/nature06901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu HH, et al. Glial precursors clear sensory neuron corpses during development via Jedi-1, an engulfment receptor. Nat Neurosci. 2009;12:1534–1541. doi: 10.1038/nn.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ellis RE, Jacobson DM, Horvitz HR. Genes required for the engulfment of cell corpses during programmed cell death in Caenorhabditis elegans. Genetics. 1991;129:79–94. doi: 10.1093/genetics/129.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ziegenfuss JS, et al. Draper-dependent glial phagocytic activity is mediated by Src and Syk family kinase signalling. Nature. 2008;453:935–939. doi: 10.1038/nature06901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kinchen JM, et al. Two pathways converge at CED-10 to mediate actin rearrangement and corpse removal in C. elegans. Nature. 2005;434:93–99. doi: 10.1038/nature03263. [DOI] [PubMed] [Google Scholar]
- 32.Reddien PW, Horvitz HR. CED-2/CrkII and CED-10/Rac control phagocytosis and cell migration in Caenorhabditis elegans. Nat Cell Biol. 2000;2:131–136. doi: 10.1038/35004000. [DOI] [PubMed] [Google Scholar]
- 33.Wu YC, Tsai MC, Cheng LC, Chou CJ, Weng NYC. elegans CED-12 acts in the conserved crkII/DOCK180/Rac pathway to control cell migration and cell corpse engulfment. Dev Cell. 2001;1:491–502. doi: 10.1016/s1534-5807(01)00056-9. [DOI] [PubMed] [Google Scholar]
- 34.Logan MA, et al. Negative regulation of glial engulfment activity by Draper terminates glial responses to axon injury. Nat Neurosci. 2012 doi: 10.1038/nn.3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park D, et al. BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature. 2007;450:430–434. doi: 10.1038/nature06329. [DOI] [PubMed] [Google Scholar]
- 36.Cabello J, et al. The Wnt pathway controls cell death engulfment, spindle orientation, and migration through CED-10/Rac. PLoS Biol. 2010;8:e1000297. doi: 10.1371/journal.pbio.1000297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bechmann I, Nitsch R. Astrocytes and microglial cells incorporate degenerating fibers following entorhinal lesion: a light, confocal, and electron microscopical study using a phagocytosis-dependent labeling technique. Glia. 1997;20:145–154. doi: 10.1002/(sici)1098-1136(199706)20:2<145::aid-glia6>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 38.Kapadia SE, LaMotte CC. Deafferentation-induced alterations in the rat dorsal horn: I. Comparison of peripheral nerve injury vs rhizotomy effects on presynaptic, postsynaptic, and glial processes. J Comp Neurol. 1987;266:183–197. doi: 10.1002/cne.902660205. [DOI] [PubMed] [Google Scholar]
- 39.LaMotte CC, Kapadia SE. Deafferentation-induced alterations in the rat dorsal horn: II. Effects of selective poisoning by pronase of the central processes of a peripheral nerve. J Comp Neurol. 1987;266:198–208. doi: 10.1002/cne.902660206. [DOI] [PubMed] [Google Scholar]
- 40.Petersen MA, Dailey ME. Diverse microglial motility behaviors during clearance of dead cells in hippocampal slices. Glia. 2004;46:195–206. doi: 10.1002/glia.10362. [DOI] [PubMed] [Google Scholar]
- 41.Aldskogius H, Kozlova EN. Central neuron-glial and glial-glial interactions following axon injury. Prog Neurobiol. 1998;55:1–26. doi: 10.1016/s0301-0082(97)00093-2. [DOI] [PubMed] [Google Scholar]
- 42.Leiserson WM, Harkins EW, Keshishian H. Fray, a Drosophila serine/threonine kinase homologous to mammalian PASK, is required for axonal ensheathment. Neuron. 2000;28:793–806. doi: 10.1016/s0896-6273(00)00154-9. [DOI] [PubMed] [Google Scholar]
- 43.Ito K, Urban J, Technau GM. Distribution, classification and development of Drosophila glial cells in the late embryonic and early larval ventral nerve cord. Roux’s Arch Dev Biol. 1995;209:289–307. doi: 10.1007/BF02179499. [DOI] [PubMed] [Google Scholar]
- 44.Ng J, et al. Rac GTPases control axon growth, guidance and branching. Nature. 2002;416:442–447. doi: 10.1038/416442a. [DOI] [PubMed] [Google Scholar]
- 45.Han C, Jan LY, Jan YN. Enhancer-driven membrane markers for analysis of nonautonomous mechanisms reveal neuron-glia interactions in Drosophila. Proc Natl Acad Sci U S A. 2011;108:9673–9678. doi: 10.1073/pnas.1106386108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ishimaru S, Ueda R, Hinohara Y, Ohtani M, Hanafusa H. PVR plays a critical role via JNK activation in thorax closure during Drosophila metamorphosis. Embo J. 2004;23:3984–3994. doi: 10.1038/sj.emboj.7600417. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.