

Abstract
Small cerebral infarcts, i.e. microinfarcts, are common in the aging brain and linked to vascular cognitive impairment. However, little is known about the acute growth of these minute lesions and their effect on blood flow in surrounding tissues. We modeled microinfarcts in the mouse cortex by inducing photothrombotic clots in single penetrating arterioles. The resultant hemodynamic changes in tissues surrounding the occluded vessel were then studied using in vivo two-photon microscopy. We were able to generate a spectrum of infarct volumes by occluding arterioles that carried a range of blood fluxes. Those resulting from occlusion of high-flux penetrating arterioles (flux of 2 nL/s or higher) exhibited a radial outgrowth that encompassed unusually large tissue volumes. The gradual expansion of these infarcts was propagated by an evolving insufficiency in capillary flow that encroached on territories of neighboring penetrating arterioles, leading to the stagnation and recruitment of their perfusion domains into the final infarct volume. Our results suggest that local collapse of microvascular function contributes to tissue damage incurred by single penetrating arteriole occlusions in mice, and that a similar mechanism may add to pathophysiology induced by microinfarcts of the human brain.
Keywords: Ischemia, microcirculation, cerebral blood flow, two-photon microscopy, venous thrombosis
Introduction
Cerebral microinfarcts1 have emerged as a potential determinant of cognitive decline, as they are one of the most wide-spread forms of tissue infarction in the aging brain.2,3 The prevalence of microinfarcts is estimated to be twice as high in those affected by vascular cognitive impairment,4 suggesting a path between small vessel disease and brain dysfunction.2 Although individually small, microinfarcts may exist in sufficient numbers to disrupt the function of neural networks. However, the development of these minute lesions and the local tissue pathologies they elicit has remained difficult to understand from cross-sectional neuropathological examination. Studies of microinfarct pathology during life have been hampered by the small size of these lesions, which typically range between only 0.1 and 3 millimeters in diameter. While recent studies have shown the feasibility of detecting large microinfarcts using 3T and 7T MRI,5 it remains likely that many more go undetected due to limitations in imaging resolution or lack of signal contrast. In this respect, animal models may be useful in understanding the impact of microinfarcts on local brain integrity because neural and vascular function can be visualized in greater detail using fluorescence imaging.
Microinfarcts are thought to arise from the thromboembolic occlusion of arteriosclerotic6 or amyloid-burdened7 cerebral arterioles, as they share many histopathological features with macroscopic infarcts caused by large vessel occlusion. Loss of flow to penetrating arterioles, which feed small and discrete domains of tissue, is a particularly likely mechanism for the pathogenesis of human microinfarcts.8 Optical methods to visualize and manipulate small cortical vessels in the laboratory setting have enabled the precise induction of such an event in the rodent brain.9 Building on the principles of photothrombotic vascular occlusion,10 Nishimura and colleagues11 demonstrated that penetrating arterioles in rodent cortex could be selectively occluded using a focused green laser to activate a circulating photosensitizer, Rose Bengal. These occlusions resulted in columnar regions of ischemia that gradually matured into small cortical infarcts,8 which were similar to human cortical microinfarcts in terms of their absolute size, shape and location. This is because the perfusion domains of cortical penetrating arterioles in mouse12 and man13 are comparable in scale and architecture. The bottleneck-like topology of penetrating arterioles and apparent lack of communicative flow between neighboring arterioles provided an explanation for why blood flow from surrounding tissues was unable to compensate for the flow loss.11
In past studies performed in rat cortex, we also observed an intriguing phenomenon that microvascular pathology, i.e. blood flow impairment, oxidative endothelial damage and blood–brain barrier (BBB) damage, appeared to extend beyond the perfusion domain of the targeted arteriole, encroaching upon territories of neighboring arterioles that were independently sourced from the pial surface.8 Since all blood flowing to the brain must pass through a dense capillary network before exiting the venous system,14 it was postulated that impaired subsurface flow contributed to the spread of microvascular damage and infarct growth. At the scale of capillaries, blood flow is particularly sensitive to change in lumen diameter given the close match in size between blood cells and the intraluminal space.15 Pathological processes that constrict and/or obstruct capillaries can profoundly impact brain perfusion, and this process has been well documented in classic models of large-scale stroke.16–18
The current study was designed to fulfill two goals. The first was to characterize the effect of occluding single penetrating arterioles in mouse cortex, which could be imaged with two-photon laser-scanning microscopy (TPLSM) through a thinned-skull window that prevented disruption of the intracranial environment.19 Specifically, we examined the range of infarcts that could be produced, and the extent of non-specific vascular damage caused by the photothrombotic procedure itself. The second goal was to use TPLSM to track hemodynamic changes in tissues surrounding an occluded penetrating arteriole such that the evolution of ischemic microvascular pathology could be better understood. Building on past observations,8 we tested the hypothesis that infarct growth involved a collapse of local microcirculatory function, with flow resistance originating at the capillary level.
Materials and methods
The Institutional Animal Care and Use Committee at the Medical University of South Carolina approved the procedures used in this study. The University has accreditation from the Association for Assessment and Accreditation of Laboratory Animal Care International, and all experiments were performed within its guidelines. All data were analyzed and reported according to ARRIVE guidelines.
Animals and surgery
All reagents were obtained from Sigma-Aldrich, unless otherwise noted. Male C57BL/6 mice, ranging from 3 to 6 months of age, were used in this study. All animals were obtained from the Jackson laboratories (Stock number 000664) and maintained in standard cages on a 12-h light–dark cycle. Anesthesia was induced with isoflurane (Patterson Veterinary) at 4% mean alveolar concentration in 100% oxygen and maintained at 1 to 2% during surgery. Body temperature was maintained at 37℃ with a feedback-regulated heat pad (FHC). PoRTs windows were generated over the left cerebral hemisphere to gain optical access to the sensorimotor cortex,9,19 under guidance of a stereoscope (SXZ10; Olympus). Two-photon imaging was performed within 24 to 48 h of window construction. All animals were administered buprenorphine prior to or immediately following the surgery at a concentration of 0.05 mg/kg for analgesia.
In vivo two-photon imaging
Imaging was performed with a Sutter moveable objective microscope (MOM) and a Coherent Ultra II Ti:Sapphire laser source. Mice were maintained under light isoflurane (0.5–0.75%) supplied in medical air (20–22% oxygen and 78% nitrogen, moisturized by bubbling through water; AirGas Inc.) over the duration of imaging. Body temperature was maintained at 37℃ with a feed-back regulated heat pad. The blood serum was labeled by retro-orbital vein injection of 0.03 mL of 2 MDa fluorescein-dextran (FD2000S; Sigma-Aldrich) prepared at a concentration of 5 % (w/v) in sterile saline. Intraperitoneal injections of 5% (w/v) glucose in 0.2 mL saline were given every 2 h for rehydration. Pulse oximetry (MouseOx; Starr Life Sciences) was used to monitor blood oxygen saturation and heart rate in a cohort of mice (n = 5) to ensure that imaging conditions did not deteriorate cardiovascular function (Table S1). These mice were not included in the imaging portion of the study but were matched for age and sex. Animals were maintained under 0.75% MAC isoflurane in medical air for the entire duration of physiological testing, as was done with imaging (Figure 4). The fur over the right thigh was removed with depilatory cream, and readings were collected using a thigh cuff before vessel occlusion and 4 and 8 h following occlusion. Blood gases and arterial pressure were not collected in these studies. However, we note that the small size of the infarcts, lack of injury in subcortical structures and absence of morbidity and/or mortality suggests that aberrant change in systemic physiological variables are less likely compared to large stroke.
Figure 4.

Evolution of acute capillary pathology in tissues surrounding an occluded penetrating arteriole. (a) Wide-field two-photon image showing the location of high-resolution imaging (cyan square) relative to an occluded penetrating arteriole (green circle; target). The approximate border of the resulting infarct, as measured using histology 24 h post-occlusion, is demarcated with a dotted line. Note that three other penetrating arterioles exist within this boundary (yellow arrowheads). (b) Projections of high-resolution image stacks, spanning 150 µm in depth from the pia, collected at various times post-occlusion. Vessels with the absence of flowing RBCs are pseudocolored in yellow. (c to f) Each capillary segment, defined as a length of capillary extending between two branchpoints, was visually examined for features of pathology. The observed pathology was categorized into four groups: constriction, dilation, obstruction and BBB leak. Further, each capillary received a binary measure of flow or no-flow, based on the presence of streaks caused by moving blood cells within the lumen. Examples for each scenario are shown. Regions of constriction and dilation are marked with yellow arrowheads. Obstructing objects and puncta of plasma leakage are marked with yellow arrows. Note the presence of streaks in vessels of pre-occlusion images formed by the movement of RBCs while the image was scanned. (g) Matrices showing occurrence of each pathological feature for all capillaries examined as a function of post-occlusion time. The matrices were first sorted along the y-axis to highlight constrictive or dilatory events with respect to the time of first emergence and duration (left panel, purple and red). All other panels were sorted with the same capillary index as defined in the left panel, to show subsequent loss of flow (black), lumen obstruction (blue) and BBB leak (green). This data comprises 199 capillary segments repeatedly sampled from four separate infarcts from four mice. (h) Overall evolution of capillary constriction (red curve), dilation (purple curve), obstruction (blue curve) and BBB breakdown (green curve) in the vascular beds of the stroke periphery. Data are shown as a proportion of total capillaries examined with 0.95 confidence intervals at each time-point. (i) Constricted capillary segments detected in an early phase (2 to 4 h post-occlusion) of stroke were significantly more likely to be non-flowing (left) or obstructed (right) in the later phase of stroke (4 to 6 h post-occlusion). (j) Dilation of capillaries in early phase did not affect the likelihood of loss of flow or lumen obstruction in the late phase. Data for panels (i) and (j) are provided as proportion of total capillaries with 0.95 confidence intervals.
Procedures for blood flow imaging and analysis have been described previously.9 Wide-field images were collected using a 4-X, 0.13 NA objective lens (Olympus UPLFLN 4X) to generate vascular maps of the entire window for navigational purposes. High-resolution imaging of microvessels was performed using a 20-X, 1.0 NA objective lens (Olympus XLUMPLFLN 20XW). High-resolution image stacks of the vasculature were collected across a 326 by 326 µm field and a depth of 150 µm from the pial surface. Lateral sampling was 0.3 to 0.6 µm per pixel and axial sampling was 1 µm per pixel.
To establish image quality with the 20-X, 1.0 NA objective, we measured the point-spread function of sub-resolution yellow-green FluoSpheres (0.2 µm; F8811; Life Technologies) that were pressure-injected directly into cortex through a small hole flanking the PoRTs window. Image stacks were acquired across a 12 by 12 µm lateral field over a depth of 100 to 140 µm below the pial surface with lateral sampling of 0.06 µm per pixel and axial sampling of 0.2 µm per pixel to produce a 3-dimensional intensity distribution of the TPLSM signal from the bead. The lateral and axial resolutions measured in vivo were 0.7 ± 0.1 µm and 1.7 ± 0.2 µm, respectively. The lateral value is the full width at half-maximal (FWHM) intensity along either the x- or y-axis in the plane of highest peak intensity, while the axial value is the FWHM along the central axial line through the bead in the longitudinal slice of highest intensity of the rotated image stack. By comparison, the lateral and axial resolutions achieved by imaging beads embedded in 1.5% agarose overlaid with a cover glass was 0.6 ± 0.1 and 1.2 ± 0.1 µm, respectively, indicating a loss in axial resolution but not lateral resolution, when imaging upper cortex through an acutely prepared PoRTs window. Accordingly, all measurements of vessel diameter were made in the lateral plane.
Targeted photothrombotic occlusions of single penetrating vessels
To induce targeted photothrombotic clots in single penetrating vessels, the camera port of the MOM was adapted to introduce a 532 nm continuous wave laser (Beta Electronics) into the imaging beam path. This green laser was directed toward the objective by toggling the primary dichroic away from the imaging beam path. The beam greatly underfilled the back aperture of the objective, leading to a fixed, circular point of illumination spanning 20 µm in diameter at the plane of focus during imaging, which roughly corresponded to the diameter of a mouse cortical penetrating arteriole (Figure 1c). A clot was initiated by irradiating a targeted penetrating vessel for 30-s immediately following infraorbital administration of ∼0.025 mL of 1.25% (w/v) Rose Bengal (330000; Sigma-Aldrich) dissolved in sterile saline. The power of the green laser at the sample was 1 mW; this power was titrated to be just over the threshold necessary to stably occlude the target vessel, and well below the power that would lead to direct clotting of neighboring, non-targeted arterioles/venules. While penetrating venule clots generated in this fashion were stable, clots placed in penetrating arterioles occasionally required 2 to 3 rounds of irradiation to be stably occluded (with no additional injection of Rose Bengal). For control off-target irradiation experiments, we used an identical Rose Bengal dose, irradiation time (1 to 3 rounds of 30-s exposure) and laser intensity as for on-target penetrating vessel occlusions, but placed the focus of irradiation away from the descending neck of the penetrating vessel (Figure S5). For repeated imaging experiments lasting 8 h, the stability of the clot was checked regularly. For experiments that did not involve extended imaging of capillaries, the stability of the clots was verified for at least 2 h post-occlusion. In these experiments, 20% of the penetrating arteriole occlusions had reperfused when re-examined at 24 h post-occlusion, but an infarct was still present indicating sufficient time for stroke development. None of the venules occluded had reperfused at 24 h post-occlusion. To examine the potential for heat damage induced by the 532 nm laser, the temperature at the focus of the laser was examined with a thermocouple (IT-24P; Physitemp) embedded in a thin layer of 1% agarose in artificial cerebral spinal fluid.9 Temperature shifts of 1.37 ± 0.19, 0.68 ± 0.05 and -0.13 ± 0.04℃ were measured for 3 mW, 1 mW and no irradiation, respectively. Assuming similar brain and body temperatures, the brief and localized temperature shift induced by 1 mW power puts brain temperature just above the normothermic range, 36.5 to 37.5℃, but below temperatures used to induce pathological hyperthermia, 40℃.20
Figure 1.

In vivo vasodynamics of individual penetrating vessels in mouse cortex. (a) In vivo two-photon image of cortical pial vasculature. Arterioles are pseudocolored in red and venules in blue. The image is composed of a montage of maximally projected stacks spanning a depth of 75 µm from the pial surface. Subsurface capillaries within this imaging depth are shown in white, as are dural vessels. The point of entry for a single penetrating arteriole (PA) into the cortical parenchyma is marked with a red circle and the exit of a single penetrating venule (PV) from the parenchyma is marked with a blue circle. (b) 3-D rendering of an in vivo image stack providing a side view of a PA and PV. This stack was taken in vivo following removal of the overlying skull to show penetrating vessels at depth. (c) Scatter plot and histograms of centerline RBC velocity, v(0), plotted as a function of lumen diameter, d, for individual penetrating vessels. The crossbars show mean ± SEM for all data points in each vessel group. (d) Histogram showing the distribution of RBC volume flux, F, for PAs and PVs. A vertical line marks the average flux for each vessel type and the range for SEM is shaded.
Quantification of microvascular pathology
For data presented in Figure 4, the morphology and patency of individual capillary segments, i.e. a length of microvessel between two branchpoints, were quantified from high-resolution in vivo image stacks. These image stacks were acquired prior to penetrating vessel occlusion and at 0.25, 2, 4, 6 and 8 h post-occlusion. Since each frame within the stack shifted only 1 µm in depth from the previous frame, the movement of red blood cells (RBCs) could be tracked in individual capillaries, similar to a movie collected from a fixed plane. The time span that a capillary could be observed within this stack, before the imaging plane shifted beyond the vessel, was approximately 5 s. In this way, “flow” or “no flow” was quantified as the presence or absence of moving RBC shadows (visible as streaks in the lumen) within the segment under consideration. The diameter of a capillary segment was measured manually across the lumen at the peak intensity of the profile using Fiji software. These measurements were made at the narrowest point along the segment, which was not necessarily the same location along the capillary at each imaging time-point. Constrictions and dilations were defined as a 20% decrease or increase from baseline diameter, respectively. A capillary was categorized as obstructed when any dark object was lodged within the lumen. In these analyses, we did not distinguish between different types of obstructions, i.e. clot, RBC or leukocyte, as all could have contributed to impairment of flow. Finally, BBB breakdown was identified when intravascular FITC-dextran dye had extravasated into the surrounding tissue. The quantification of these pathological features was performed by an individual blinded to the post-occlusion time-points.
For data presented in Figures 5 and 6, the patency of penetrating arterioles and venules at the brain surface was determined through direct observation in movies collected at 24 h post-occlusion. As with capillaries, pial vessels that had stagnated were devoid of dark streaks caused by movement of RBCs through the lumen and were often brighter due to buildup of fluorescent plasma.
Figure 5.

Capillary flow impedance associated with a growing infarct stagnates flow of neighboring penetrating arterioles and venules. (a) Wide-field image collected by two-photon microscopy showing the location of an occluded penetrating arteriole (green circle). High-resolution movies were collected within this larger field to assess RBC flow in pial vessels (inset, see panel d). (b, c) Vessel tracing of the imaged region in panel a with non-flowing vascular segments highlighted in yellow. Note the marked increase in non-flowing neighboring penetrating venules and arterioles at 24 h post-occlusion. (d, e) Montages from high-resolution movies showing the extent of flow loss in the pial vascular network. In panel (e), a surface arteriole coursing through the imaging field remains patent, while two neighboring penetrating arterioles (arrows) that are sourced by the arteriole no longer support flow. A neighboring penetrating venule completely collapses and is no longer visible at 24 h (arrowhead). This is consistent with an increase in capillary resistance that prevents the influx and efflux of blood from the cortical parenchyma. (f) Scatter plot of the number of stagnant neighboring penetrating venules and arterioles as a function of the pre-occlusion RBC flux of a targeted penetrating arteriole (n = 12 separate occlusions). (g) Distance of stagnant neighboring penetrating vessels relative to the border of the infarct, as demarcated by loss of NeuN staining in post hoc histology. An inset shows the cumulative distribution plot of infarct radius size.
Figure 6.

Occlusion of a single penetrating venule is sufficient to stagnate neighboring penetrating arterioles. (a) Schematic depicting the placement of the green laser focus for occlusion of single cortical penetrating venules. (b) Wide-field image collected by two-photon microscopy showing the location of an occluded penetrating venule (green circle). (c, d) Vessel tracings from the region imaged in panel (b) with non-flowing vascular segments highlighted in yellow. Note the loss of flow in two neighboring penetrating arterioles at 24 h post-occlusion (white arrows). (e) Scatter plot of the number of stagnant neighboring penetrating arterioles and venules as a function of the pre-occlusion RBC flux of a targeted penetrating venule (n = 11 separate occlusions). (f) Distance of stagnant neighboring penetrating vessels relative to the border of the infarct, as demarcated by loss of NeuN staining in post hoc histology. Inset shows cumulative distribution plot of infarct radius size. (G) Left panel, scatter plot of the infarct volume versus the pre-occlusion RBC volume flux of individual penetrating venules. Venule occlusions that resulted in secondary stagnation of neighboring penetrating arterioles are shown as solid circles, while those that did not are shown as open circles. Infarct volume was not correlated with pre-occlusion flux (n = 10). The crossbars show mean ± SEM for all data points. Right panel, infarcts that involved stagnation of a neighboring arteriole resulted in significantly larger infarcts (*p = 0.02, Mann-Whitney test).
For experiments presented in Figure S5, we measured capillary diameters with a different approach than with data in Figure 4. We collected in vivo high-resolution image stacks flanking the site of irradiation with a lateral sampling resolution of 0.3 µm per pixel and axial sampling of 1 µm per pixel. Each stack, extending from the pial surface to 150 µm in depth, was obtained five times in succession before and 4 h following photothrombotic occlusion to obtain a variance for the diameters of individual capillary segments. Capillary diameters were calculated as the FWHM of the intensity profile across the capillary width. The intensity profiles were measured from single-image planes positioned at the lumen center of the segment under examination. Diameters measured at 4 h were then compared to baseline values for each capillary using a two-tailed Student’s t-test to identify statistically significant constrictions or dilations.
Magnetic resonance imaging
MRI was performed with a small animal 7 Tesla Bruker Biospec scanner (Bruker Biospin, Germany). Images were acquired either in the coronal plane or tangential to the surface of the cortex to provide a plane similar to that obtained with TPLSM. For T2-weighted (T2W) images, turbo-rapid acquisition relaxation enhancement (Turbo-RARE) was acquired at TR = 2500 ms and TE = 33 ms, voxel dimension: 0.078 × 0.078 × 0.5 mm3 (Figure 3a,b) or 0.1 × 0.1 × 0.3 mm3 (Figure 3d), RARE Factor: 8, and number of averages: 2. For diffusion imaging (Figure 3c), echo planar imaging was acquired at TR = 3000 ms and TE = 30 ms, voxel dimension: 0.2 × 0.2 × 0.3 mm3, b-values 0, 1000, 2000 s/mm2 with 64 diffusion encoding directions. Mean diffusivity maps were calculated with Diffusional Kurtosis Estimator.21 For data of Figure 3(a), we imaged the same mouse at 2, 4, 6 and 24 h post-occlusion. For data of Figures 3(c–e), we imaged the same mice at 3, 24 and 72 h post-occlusion. No pre-occlusion data was collected because the cranial window preparation did not lead to noticeable differences in signal contrast with either structural or diffusion-weighted sequences. For consistency between all mice (six infarct regions: five mice with one mouse having two well-separated infarcts) and time points (3, 24 and 72 h), all image contrasts were set between 0 and 3 mm2/s for the mean diffusivity (MD) maps and between 0 and 25,000 units for T2W maps when manually drawing the regions of interests using MRIcron software. The ROIs were drawn on the hypointensities of MD maps and hyperintensities of T2W image and the areas were statistically compared using SPSS software (IBM SPSS Statistics version 22).
Figure 3.

Radial expansion of infarcts generated by occlusion of high-flux penetrating arterioles. (a) Repeated T2-weighted (T2W) MRI to track infarct growth. Two penetrating arterioles, one with flux of 2.3 nL/s (high-flux) and the other with 0.3 nL/s (low flux) were occluded within a single imaging window. MRI was then performed in a plane tangential to the cortical surface at various times post-occlusion. (b) Coronal view of infarcts (green arrowhead) resulting from occlusion of the low flux (top) and high-flux (bottom) arterioles at 24 h post-occlusion. (c, d) Time course showing change in mean diffusivity of water (MD, upper row) and T2W signal (lower row) following occlusion of a single high-flux penetrating arteriole (1.8 nL/s). Note that a homogeneous region of reduced MD was present before detectable T2W hyperintensity at 3 h post-occlusion. (e) Area and diameter of affected tissues, as quantified from MD and T2W images, plotted as a function of post-occlusion time (n = 6 high-flux penetrating arteriole occlusions). Data provided as mean ± SEM. MD and T2W curves are statistically different (p < 0.001, Friedman’s two-way ANOVA), and pairwise comparisons at each time-point revealed a significant difference at the 3 h post-occlusion time-point (*p < 0.05, Mann-Whitney test adjusted for multiple comparison across time with the Dunn-Sidak post hoc test). Note that the term “infarct” was used for consistency with other figures, but MRI signal change does not necessarily report the region of true tissue infarction. (f) Representative images of immunohistochemical staining for tissue infarction and vascular leakage in mice sacrificed at various times post-occlusion. The boundary of each infarct was delineated by the interface between the presence and absence of NeuN-stained neuronal nuclei (red). Note that retention of circulating FITC-dextran dye in the tissue (green), which corresponds to regions of capillary BBB leakage,31 occurs in visually normal tissues outside the delineated infarct boundary.
Immunohistology
For data of Figure 3(f), animals were killed for histological examination at 2, 4, 6 and 24 h post-occlusion. For data of Figure 2 animals were killed at 24 h post-occlusion. All animals were perfusion-fixed through a trans-cardiac route. This involved an initial washout of blood by perfusing with PBS. Once the perfusion fluid exiting the right atrium became clear, the animals were subsequently perfused with ∼20 mL of 4 % (w/v) paraformaldehyde in PBS. The perfusion fluid was osmotically balanced with respect to electrolytes, but not to equivalent protein levels found in plasma. Brains were extracted and post-fixed overnight at 4℃. The whole brain was then mounted for vibratome sectioning such that it could be sliced tangential to the cortical surface at the location of the infarct. Brain sections were collected at a thickness of 50 µm and stored in PBS with 0.02% sodium azide. For demarcation of infarct borders, anti-NeuN primary antibody from guinea pig host (ABN90P; 1:1000 dilution; Millipore) was diluted in buffer that consisted of 10 % (v/v) goat serum (Vector Labs), 2 % (v/v) Triton X-100 and 0.2 % (w/v) sodium azide. Free-floating sections were then incubated overnight under slow nutation at room temperature. The following day, sections were washed in PBS for 30 min on an orbital shaker, incubated with anti-guinea pig Alexa 594 secondary antibody (A11076l; 1:1000 dilution; Life Technologies) for 2 h in the same buffer as the primary antibody. Slices were then washed again in PBS, mounted and dried on slides for 30 min. All slides were then sealed with Fluoromount-G (Southern Biotechnology Associates Inc.) and a No. 1 glass coverslip (Corning).
Figure 2.

A spectrum of cortical infarct volumes induced by targeted photothrombotic occlusion of single penetrating arterioles. (a) Schematics depicting the placement of the green laser focus for occlusions of cortical penetrating arterioles (on-target; right) or control irradiations (off-target; left). (b) In vivo two-photon images of the pial surface before and after targeted irradiation. Non-flowing vascular segments are colored in yellow. Only the pre-occlusion image is shown for the off-target irradiation because no loss of pial vessel flow was observed. (c) NeuN immunohistology to quantify the volume of tissue infarct resulting from single penetrating arteriole occlusions or off-target irradiations. The white matter of the external capsule is labeled, wm, to show the base of the cortex. (d to f) Scatter plot of infarct diameter, depth and volume versus the pre-occlusion RBC volume flux of individual penetrating arterioles. Infarct diameter and volume, but not depth, were significantly correlated with flux (n = 18). Off-target irradiations generated infarcts volumes over an order of magnitude smaller than penetrating arteriole occlusions (p < 0.001, Mann-Whitney test). Since no penetrating vessel was occluded with off-target irradiation, there is no associated flux to report. The crossbars show mean ± SEM for all data points in each irradiation group. Regression analyses of penetrating arteriole occlusion data were performed on log10-transformed values. (g) Schematic showing relative infarct size between off-target irradiations and on-target occlusions, based on average values from panels (d) to (f).
To quantify infarct volumes we used anti-NeuN stained tissue sections. Images of each infarct were taken from slices separated by 100 µm intervals using an epi-fluorescence microscope (BX53; Olympus). The infarct boundary was delineated as the border between normal tissue exhibiting dense neuronal nuclei and infarcted regions devoid of nuclei. The area of tissue damage was measured in each tissue section and the total infarct volume, Vt, was calculated using the following formula: Vt = (A1 + A2 + … + An)h, where An was the area of damage in the nth slice, and h was the distance between adjacent sections.
For detection of platelet aggregation, the same immunohistochemistry protocol was used, but the primary antibody solution contained anti-CD41 primary antibody from rat (553847; 1:250 dilution; BD Biosciences) and anti-collagen IV antibody from rabbit (ab19808; 1:250 dilution; Abcam). The secondary antibodies used were anti-rat Alexa 594 (A11007; 1:1000 dilution; Life Technologies) and anti-rabbit Alexa 647 (A21245; 1:1000 dilution; Life Technologies), respectively. Images were collected on an EVOS Cell Imaging System (Life Technologies).
Statistics
All statistical analyses were performed using custom code written with MATLAB or SPSS software. Vasodynamic parameters between penetrating arterioles and venules in Figure 1(c) were analyzed with a Student’s t-test. Figures 1(c), 2(d) to 2(f), 5(f), 6(e) and 6(g) were analyzed with Pearson’s correlation. Figures 1(d) and S5C were analyzed with the Kolmogorov-Smirnov test. Figures 2(f) and 6(g) (right panel) were analyzed with the Mann-Whitney test. Figure 3(e) was analyzed using a Friedman’s two-way ANOVA to compare the areas measured on the MD map and T2W image at the various time points. Pairwise comparison at the different time points was performed by using Mann-Whitney tests and adjusted for multiple comparisons across time with the Dunn-Sidak post hoc test. Figure 4(h) was analyzed using a maximum likelihood estimate of probability and 0.95 confidence intervals were calculated with normal approximation. The data used for the analysis consisted of binomial measurements of capillary dysfunction, i.e. flow or no flow, constriction or no constriction etc., for each of the 199 capillaries observed at each post-occlusion time-point. The 0.95 confidence intervals for proportions in Figures 4(i), (j) and S5(d) were calculated using normal approximation.
Results
We imaged vasculature in the sensory cortex of isoflurane anesthetized adult male C57Bl/6 mice. The blood plasma was intravenously labeled with a high molecular weight fluorescein-dextran (2 MDa) to allow visualization of the intraluminal space during TPLSM. To gain optical access to the brain surface, we made use of wide-field, glass-reinforced thinned-skull windows, which reduced mechanical disruption and preserved native microcirculatory function.19 Despite the presence of a thin layer of bone, blood vessels of the cortex could be clearly imaged from the pial surface to capillaries 250 µm deep.
Occlusion of penetrating arterioles generate a broad range of infarct volumes in mouse cortex. We first documented the range and reproducibility of cortical infarcts generated by occlusion of individual penetrating arterioles (Figure 1a). Penetrating arterioles descend from the pial arterial network to perfuse capillaries in columns of cortical tissue,11 while penetrating venules route blood from the capillary beds back to the brain surface22 (Figure 1b). Consistent with data from the mouse cortical angiome,12 penetrating venules were found to outnumber penetrating arterioles by a factor of 2, with 18 ± 1 penetrating venules and 9 ± 1 penetrating arterioles per square millimeter of cortical area (mean ± SEM; n = 3 mice). To characterize flow in individual vessels, we acquired measurements of lumen diameter, d, and RBC velocity, v(0) (Figure 1c).9 While the average diameter between penetrating arterioles and venules was not different (18.6 ± 1.0 µm for arterioles vs. 19.8 ± 0.8 µm for venules; p = 0.37, mean ± SEM, Student’s t-test), RBC velocity was significantly lower in venules (10.5 ± 0.5 mm/s for arterioles vs. 3.4 ± 0.2 mm/s for venules; p < 0.001, Student’s t-test). As a result, penetrating venules possess a nearly two-fold lower RBC volume flux, F (1.78 ± 0.2 nL/s for arterioles vs. 0.73 ± 0.1 nL/s for venules; p < 0.001, Kolmogorov-Smirnov test; Figure 1d). These vasodynamics are consistent with recent measurements from mouse cortex using high-speed reflectance imaging,23 yet are in contrast to our past work using rats where penetrating venules and penetrating arterioles share a similar magnitude of flux due to larger venular diameters (Figure S1).8
The introduction of a fixed green laser beam into the imaging path of our TPLSM system allowed for focal photothrombotic occlusion of individual penetrating vessels immediately before their descent into the cortex, i.e. on-target occlusion (Figure 2a and b, right panels).9 We attenuated the power of the green laser such that it generated negligible change in heat (see methods) and dissipated rapidly after contact with the brain surface in order to minimize direct clotting of the underlying capillaries. This method refined the pioneering approach of Watson et al.,10 in order to generate ischemia only in the perfusion domain of the targeted penetrating vessel by blocking influx11 or efflux22 of blood at the brain surface.
We first probed the relationship between penetrating arteriole flux and the resulting infarct volumes. Tissue infarction was demarcated as a region of contiguous neuronal loss, i.e. absence of NeuN immunostaining, in histological slices at 24 h post-occlusion.8,24 Occlusion of a single penetrating arteriole was sufficient to generate a cylindrical (or inverted cone-like) region of tissue infarction spanning many cortical layers (Figure 2c, right panel). Pre-occlusion arteriolar RBC volume flux was positively correlated with infarct volume, consistent with past studies in rat (Figure 2f).8 This correlation was largely dictated by the diameter of the infarct (Figure 2d), rather than the depth it reached below the pial surface (Figure 2e). While occlusion of many penetrating arterioles led to small infarcts, similar to past work in rats,8 we noted that the occlusion of high-flux arterioles, i.e. 2 nL/s or higher, could generate larger infarcts, spanning ≥1 mm in diameter. The volume of these large infarcts matched well with the perfusion volumes of the largest penetrating arterioles calculated from the mouse cortical angiome,12 suggesting that a subset of high-flux arterioles perfused large volumes of cortical tissue that overlapped with domains of neighboring penetrating arterioles (Figure S2). These high-flux arterioles were targeted in subsequent experiments to study the spread of ischemic injury.
Consistent with the infarcts being a result of ischemia generated in the perfusion territory of the target penetrating arteriole, off-target control irradiations over pial areas devoid of penetrating vessels led to infarct volumes an order of magnitude smaller, due to a small amount of direct thrombosis of superficial capillaries (Figure 2a–f, left panel, and 2 g; c.f. black and red volumes).
Multimodal MRI reveals a gradual infarct expansion. To understand the time-course of infarct expansion, we initially used repeated 7 Tesla T2W MRI. After occlusion of a high-flux penetrating arteriole (2.3 nL/s), a gradual and radial expansion of the hyperintense infarct core was observed over 24 h, indicating a significant peripheral zone that was recruited into the infarction. For instance, a doubling of the infarct diameter from 0.5 to 1 mm between 4 and 24 h corresponded to a quadrupling of infarct volume (Figure 3a and b; lower infarct). In comparison, an infarct resulting from the occlusion of a low-flux arteriole (0.3 nL/s) exhibited limited expansion over the same time period (Figure 3a and b; upper infarct). We then probed early and late tissue changes following occlusion of high-flux arterioles using paired diffusion-weighted and T2W imaging (n = 6 occlusions, 5 mice). At 3 h post-occlusion, maps of mean diffusivity (MD) revealed uniform hypointense regions following all occlusions. In contrast, T2W hyperintensities were rarely observed at this time-point. That is, in 4 of 6 cases, little or no T2W contrast could be discerned for quantification at 3 h (Figure 3d). Very restricted hyperintense regions were observed in the remaining two cases (Figure 3a). At 24 h post-occlusion, regions of MD hypointensity and T2W hyperintensity were substantially larger and well-matched in size between imaging modalities, as expected for complete tissue infarction. Signal contrast at 72 h was similar to 24 h with a slight reduction in infarct area. A delayed emergence of T2W contrast relative to MD is also observed with large-scale focal stroke models, i.e. MCAo, which suggests the tissues affected by penetrating arteriole occlusion experience a gradual response to ischemia caused by a distal occlusion.25 This is contrasted to simultaneous changes in MD and T2W that occur when photothrombosis directly occludes underlying capillary beds, initiating more rapid vascular leakage.26,27
Finally, to ensure that T2W signal increase involved tissue infarction, we performed NeuN immunohistology on mice receiving occlusions to penetrating arterioles of similar flux, but sacrificed at varying post-occlusion times (Figure 3f). In agreement with MRI data, a gradual increase of infarct size was observed between 2 to 24 h (Figure 3d and e). Critically, the retention of circulating FITC-dextran in the capillary endothelium, an effect of severe BBB leakage,31 was always observed beyond the border of the infarct. This suggested that capillary dysfunction preceded overt neuronal loss during infarct growth (Figure 3f, see 4 and 6 h post-occlusion).
Evolution of microvascular pathology at the infarct border. We next used TPLSM to repeatedly image the patency and structure of subsurface capillaries within the receding peri-infarct tissues following occlusion of high-flux arterioles. The vessels examined were primarily capillaries, ranging from 3 to 7 µm in diameter, but may have included small subsurface arterioles and venules (Figure S4). High-resolution image stacks were collected in a fixed region spanning the first 350 µm from the occluded target vessel (Figure 4a; cyan square). The imaged region was positioned such that interface between ischemic and non-ischemic tissues could be visualized as the infarct grew radially outward. These infarcts often grew into the perfusion domains of neighboring arterioles, which were independently perfused from the pial vasculature (Figure 4a, yellow arrowheads). In total, 199 individual capillary segments, i.e. capillary regions between branchpoints, were tracked for 8 h at 2-h intervals in the periphery of four separate infarcts in four mice (Figure 4b). Each capillary segment was then categorized based on the forms of pathology exhibited, including (1) lack of RBC flow, (2) lumen constriction, (3) lumen dilation, (4) lumen obstruction and (5) BBB leakage (Figure 4c–f). Since imaging was performed in regions flanking the target penetrating arteriole (Figure 4a) and restricted to the upper 150 µm of cortex, the capillaries examined typically could not be traced back to the targeted arteriole for analyses of topology.14 However, since capillary inputs to the ascending venules are denser in upper cortical layers,12 most vessels examined likely resided near the venular pole of the capillary bed. Importantly, capillary resistance anywhere along the vascular supply chain is capable of impacting the influx and efflux of blood from cortex.
When capillary responses were examined on a vessel-by-vessel basis over the imaged period, we observed substantial heterogeneity in both the timing and duration of constrictive and dilatory events (Figure 4g). To understand the overall dynamics of capillary response over time, we first considered the capillaries as a population (Figure 4h). We found that an average of 50% of the total capillaries measured lost flow over 8 h, as assessed by the cessation of movement of RBCs within the lumen (Figure 4h; black line). This gradual loss of flow was specific to the stroke periphery and did not result from a run-down of the animal preparation, as only subtle and transient loss of flow was observed in more distant image stacks, collected 350 to 500 µm from the occluded arteriole (Figure S3). To determine the mechanism of this flow loss, we first considered changes in the early phase of stroke. We found that constriction of the capillary lumen was a surprisingly early event, becoming evident as quickly as 15 min post-occlusion (Figure 4c, h, red line, and S4). The proportion of constricted vessels plateaued by 4 h, affecting 30% of the total capillaries examined. In contrast, dilation was detected in 9% of total vessels at 15 min post-occlusion (Figure 4d), and this proportion did not change appreciably with post-occlusion time (Figure 4h, purple line, and S4). Approximately one-third of the dilated vessels were determined to be small primary or secondary subsurface branches of the targeted arteriole.28 These branches were previously shown to be ensheathed by smooth muscle-like cells, consistent with a capacity for dilation,29 but other dilating capillaries may have represented thoroughfare channels.30 With regard to the spatial sensitivity of our imaging approach, we defined constrictions or dilations as decreases or increases greater than or equal to 20% from baseline diameter, respectively. This equated to a change of 0.8 µm from the mean baseline diameter of 4.1 ± 0.1 µm (Figure S4). Given the lateral point-spread function measured through a PoRTs window (0.7 ± 0.1 µm, see methods) this was a discernable change for our in vivo measurements.
Constriction was followed by a gradual increase in the proportion of capillaries that were obstructed by circulating blood elements (Figure 4e and h, blue line). These appeared as dark particles, of cellular or embolic origin, trapped within the lumen. The proportion of obstructed vessels increased linearly over the entire duration of imaging, eventually affecting 40% of all the vessels at 8 h post-occlusion. All obstructed vessels were, by definition, non-flowing. Finally, the degradation of the BBB and leakage of circulating FITC-dextran31 into the parenchyma was a comparatively late event, becoming evident only at 6 h post-occlusion and affecting 6% of the total microvessels (Figure 4f and h, green line). BBB leakage was only detected in non-flowing vessels. Since vascular leakage was a late event in the evolution of capillary dysfunction, and leakage was observed outside the infarct border in histology (Figure 3c), the capillary dysfunction observed in vivo preceded and likely contributed to the expansion of the infarct.
Given the spatiotemporal heterogeneity of capillary diameter during infarct growth (Figure 4g), comparisons of lumen diameter versus flow at single time-points would not adequately explain the impact of diameter change over time. This motivated an analysis to determine how early diameter changes impacted flow in the same capillaries later in stroke. We divided the data into two time-periods: An early post-occlusion phase (2 to 4 h) when constrictions and dilations had occurred but flow was largely present, and a late phase (6 to 8 h) when obstructions and flow loss was more substantial. Indeed, capillaries that were constricted in the early post-occlusion phase were more likely to be obstructed and non-flowing in the late phase (Figure 4i). In contrast, early dilation did not increase the likelihood of flow or decrease obstruction in the same capillaries later in stroke (Figure 4j). This finding suggests that early capillary constriction, rather than dilation, contributes to flow resistance that progressively worsens microcirculation within the peri-infarct tissues.
Capillary constriction is a response to ischemia within the arteriole perfusion territory rather than non-specific actions of Rose Bengal activation. To test whether the observed capillary constriction could have resulted from non-specific actions of photothrombosis, we compared capillary responses between off-target and on-target photothrombotic occlusions. In this case, however, off-target occlusions were formed in the surface arteriole immediately upstream of a target penetrating arteriole, using the same Rose Bengal dose, laser power and irradiation time as for on-target occlusions (Figure S5a and S5b). Since blood flow to the penetrating arteriole was preserved by flow reversal from downstream arterioles,32 the ischemic component of photothrombosis could be dissociated from potential downstream effects of intraluminal photosensitization, such as aberrant endothelial signaling, oxidative injury or release of micro-emboli. We restricted imaging time-points to pre-occlusion and 4 h post-occlusion, when constrictive events normally peaked. We found that off-target occlusions led to a shift toward dilation of capillaries, rather than constriction (p < 0.001, Kolmogorov-Smirnov test, n = 118 capillaries from two mice for off-target occlusions and n = 94 capillaries from two mice for on-target occlusions; Figure S5c and d). This confirmed that early capillary constriction seen with on-target occlusion was a specific effect of ischemia in the penetrating arteriole perfusion territory.
Infarct growth involves stagnation of neighboring penetrating arterioles. What is the consequence of the observed capillary pathology on flow of neighboring penetrating arterioles? Since each imaged region was supplied by numerous penetrating arterioles, each independently sourced by the redundant pial network,14,32 a logical hypothesis would be the continued flow of neighboring arterioles and a compensation for perfusion deficits incurred by the occluded arteriole. Neighboring penetrating venules should therefore also remain patent to serve the efflux from penetrating arterioles. Our findings were contrary to this hypothesis, as over 24 h, the flow of both penetrating venules and arterioles flanking a targeted penetrating arteriole completely stagnated due to increased capillary resistance (Figure 5; compare panel b and c). Upstream leptomeningeal collaterals, however, remained perfused (Figure 5e; white arrowheads), effectively bypassing penetrating arterioles located within the infarcted territory (Figure 5; compare white arrows in panels d and e). This was consistent with an inability of blood to pass through the underlying capillary beds and confirmed that upstream flow input was unaffected. Histological examination of capillaries at 4 and 24 h post-occlusion revealed intravascular platelet aggregation within and immediately beyond the infarct border, as detected by CD41 staining, suggesting that platelet aggregation was either an inducer or consequence of flow reduction (Figure S6). Further, occlusion of penetrating arterioles with higher pre-occlusion flux led to stagnation of a greater number of neighboring penetrating vessels, and generated larger infarcts (Figure 5f).
The loss of flow to neighboring arterioles further suggests that their perfusion domains were recruited into the ischemic core. To examine this, we plotted the distance of the non-flowing neighboring vessels relative to the border of the infarct for each case at 24 h post-occlusion (Figure 5g). This analysis indicated that nearly all affected vessels resided within the infarcted zone. It was consistent with past work showing that the distance between nearest neighbor penetrating arterioles is shorter than the radius of the average infarct produced by penetrating arteriole occlusion.12
Selective occlusion of penetrating venules is sufficient to stagnate neighboring penetrating arterioles. The stagnation of surrounding penetrating venules was always observed following occlusion of a penetrating arteriole (Figure 5f and g). Since cortical blood flow relies upon adequate blood efflux, we reasoned that blockage of venular flow could directly impair flow through adjacent penetrating arterioles as an intermediary step in the propagation microvascular collapse. Indeed, direct photothrombotic occlusion of a single penetrating venule was sufficient to stagnate neighboring penetrating arterioles (Figure 6). The neighboring vessels affected by the targeted venular occlusion resided within the final infarct radius, consistent with their contribution to tissue injury (Figure 6f). Critically, venule occlusions with secondary stagnation of arterioles generated infarcts that were ∼5 times larger than those that did not affect neighboring arterioles (612 ± 210 nL versus 112 ± 45 nL, p = 0.02, Mann-Whitney test; Figure 6g, right panel). Finally, we note that the secondary effect on penetrating arterioles also explains why no correlation was found between infarct volumes and the pre-occlusion flux of the target venule itself (Figure 6g, left panel).
Discussion
We have characterized the spectrum of small cortical infarcts that can be generated by direct photothrombotic occlusion of single penetrating arterioles in cerebral cortex of male C57Bl/6 mice. As previously observed with rats, these infarcts resembled aspects of human microinfarcts with regard to their absolute size, their shape and orientation within the cortex.8 On one end of this spectrum, we found that occlusion of high-flux penetrating arterioles (≥2 nL/s) generated relatively large infarcts that expanded radially and encroached on perfusion territories of neighboring penetrating arterioles. Two-photon imaging of hemodynamic changes in tissues surrounding the occluded vessel revealed a gradual collapse of microcirculatory function that contributed to infarct expansion (Figures 5 to 7). These data suggest that large microinfarcts in the human brain may elicit considerable vascular damage in surrounding tissues and occupy the perfusion domains of several penetrating arterioles and venules.
Figure 7.
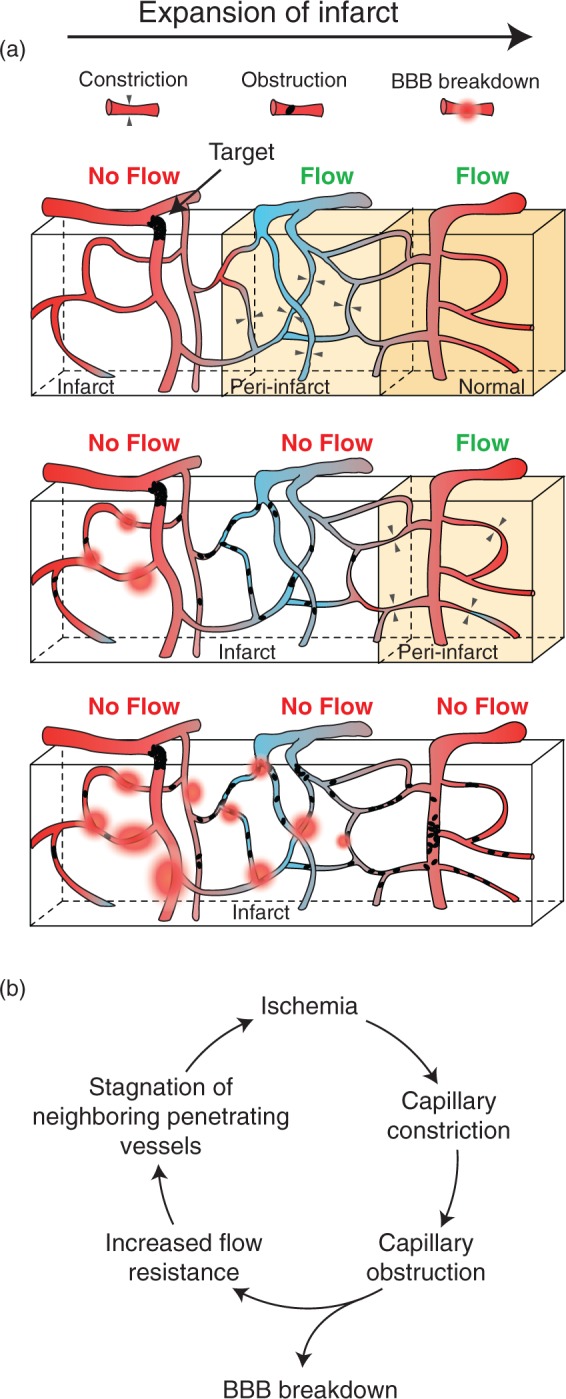
Schematic depicting the evolution of microvascular changes in the periphery of a growing microinfarct. (a) Three snapshots in time depicting the progression of microvascular pathology relative to the infarct core and its consequence on flow in neighboring vessels. Capillary constriction is an early event in the periphery of the growing infarct (upper panel). This constriction increases the likelihood of obstruction by blood elements leading to loss of flow in cortical domains perfused by neighboring penetrating venules (middle panel). The walls of capillaries degrade and leak closer to the infarct core, while those more distant to the core begin to constrict. The expansion of the infarct involves stagnation of neighboring, independently sourced penetrating arterioles, which rely on penetrating venules for blood efflux (bottom panel). (b) Cycle of vascular events leading to growth of the infarct core.
Recent studies have also modeled cerebral microinfarcts in mice using a high-throughput approach of injecting cholesterol crystals33 or beads34 through the internal carotid artery. These intravascular agents obstruct small arterioles on their way to the capillary bed, leading to discrete ischemic infarcts. In general, we find that the infarcts generated using these approaches are similar in size to those created by photothrombotic occlusion of low-flux arterioles, i.e. <2 nL/s. These studies may not have observed larger infarcts because obstructions were limited to arterioles below a threshold size. For instance, occluded vessels were ∼12 µm in lumen diameter in a study by Silasi et al.;34 for comparison low-flux and high-flux arterioles in the current study were 13 ± 1 µm (n = 29) and 26 ± 1 µm (n = 21) in diameter, respectively (Figure 1c). Another important distinction is that infarcts caused by circulating embolic agents tend to be contained completely within the cortical parenchyma, presumably because the obstruction is lodged deeper in the penetrating arteriole tree. Interestingly, Wang et al. also reported a delayed neuronal death that initiated 3 days following cholesterol crystal injection.33 This may be due to a partial or transient occlusion that results in milder ischemia compared to abrupt flow loss from photothrombotic occlusion. Of note, mouse models of small vessel disease using chronic bilateral carotid artery stenosis35 or partial deletion of eNOS36 have reported larger microinfarcts that likely span the perfusion domains of multiple penetrating arterioles. Nevertheless, the range of models available that produce microinfarcts, either induced or spontaneous, are able recapitulating the diversity of microinfarcts observed in human neuropathological studies. Further investigations are necessary to examine the potential disparities in infarct evolution and impact between these models. The advantages of the model described here are that infarcts can be induced with high spatiotemporal precision, and that the ischemic volume can be titrated based on the pre-occlusion flux of the targeted arteriole. The disadvantages are that infarcts are restricted to cortical gray matter and that occlusion of multiple arterioles in sequence takes time.
The capillary bed is a highly interconnected network that allows blood flow to efficiently re-route around local vascular defects, as shown by Schuier et al.37 using microbeads and Nishimura et al.38 using laser-induced capillary clots. However, at the scale of stroke produced by single penetrating arteriole occlusions, homeostatic mechanisms to redistribute blood appear limited in several ways. Predictions from large-scale vascular reconstructions suggest that collateral flow between penetrating arterioles is minimized by low-resistance venular shunts that divert flow away from the ischemic zone.12 In vivo imaging studies have also shown that neighboring penetrating arterioles do not dilate at the pial surface to compensate for the flow deficit.28 Although dilation was observed at the capillary level in the current study, as well as a past study in rats,28 this may have been an unsuccessful attempt to recruit flow through thoroughfare channels.30 Our results suggest that the long-term result of brain and blood-borne responses to the ischemia is the collapse of microvascular patency in tissues surrounding the occluded penetrating arteriole. We posit that over the course of 24 h, capillary resistance first stagnates flow of immediately neighboring penetrating venules, which would normally efflux blood supplied by the target arteriole (Figure 7a). This then leads to a secondary loss of flow in neighboring penetrating arterioles, which also rely upon the same venules for flow output. Consistent with this idea, seminal studies by Woolsey and colleagues39 demonstrated in rat cortex that individual penetrating venules serve as shared outputs by more than one penetrating arteriole, thus providing a “link” for propagation of microvascular resistance between neighboring penetrating arterioles. Overall, the cycle of events depicted in Figure 7(b) is believed to be the mechanism by which ischemic injury propagates at the border of small cortical infarcts, particularly those resulting from high-flux arterioles (Figure 7b). This process likely underlies our recent observation that multiple small infarcts placed within close proximity tend to coalesce, leading to a non-linear growth of tissue damage.8 Microinfarcts located within white matter may elicit different effects on surrounding microvessels due to a sparser vascular topology.
An issue with photothrombotically-induced occlusions is the potential for diffuse singlet oxygen production that can cause non-specific endothelial damage. A specific goal of this study was therefore to determine the extent to which this influenced microvascular responses when the technique was applied to mouse cortex. Our findings provide evidence that ischemia resulting from loss of arteriole perfusion can be dissociated from non-specific oxidation at some level. First, a control experiment with off-target occlusion of an upstream surface arteriole, but maintained flow in the target penetrating arteriole, resulted in a shift toward capillary dilation rather than constriction (Figure S5). Also, the occlusion of penetrating venules22 could generate infarcts that were comparable in size to penetrating arteriole occlusion, despite an opposite direction of flow (Figure 6). These results indicate that dispersal of singlet oxygen through the penetrating vessel tree could not alone explain the capillary changes observed. Second, off-target irradiations placed away from penetrating vessels resulted in infarcts that were an order of magnitude smaller in volume (Figure 2), while on-target occlusions resulted in infarct sizes that were correlated with pre-occlusion flux (Figure 2d, right panel), and consistent with perfusion volumes calculated from mouse vascular reconstructions (Figure S2). Finally, on-target occlusions generated a rapid drop in MD, but delayed T2W contrast, suggesting that changes in intracellular water diffusion preceded vasogenic edema (extracellular water from vascular leakage that affects T2). This is contrasted to the simultaneous increase in T2 and MD contrast observed when direct capillary thrombosis is induced, i.e. earlier vascular leakage, and indicates a more gradual response to ischemia is occurring after penetrating arteriole occlusion.26,27 Collectively, this suggests that ischemia caused by loss of flow to the targeted arteriole is the principal factor eliciting the observed microvascular responses. However, we cannot rule out the possibility that there was sub-threshold endothelial damage caused by photosensitization that augmented capillary responses to ischemia.
This study did not delineate the source of capillary constriction or obstruction. Though, concepts from animal models of large-scale stroke may apply16,18 For example, studies using the transient MCAo model suggest that pericyte dysfunction leads to aberrant and sustained capillary contraction. Indeed, pericytes appear highly sensitive to oxidative/nitrosative stress40 and increased extracellular glutamate.41 We believe that most constrictions here were occurring in true capillaries, as opposed to smooth muscle actin-expressing pre-capillary arterioles, because the latter vessel type is relatively sparsely distributed compared to capillaries.29 Other extraluminal forces implicated in capillary compression during ischemia include edematous swelling of astroglial endfeet and endothelial cells.42 Astrocyte swelling and reduced capillary RBC flow correlates with cortical spreading depression (CSD),43 and CSD events have been observed following single penetrating arteriole occlusions.8 This should be examined in future studies, as no electrophysiology was performed in the current study. Finally, it is also possible that reduced lumen diameter is a passive result of intraluminal pressure loss due to upstream vessel blockade. Further experiments are required to test the contribution of these putative mechanisms. Additionally, while we did not distinguish between factors that could obstruct capillaries, we believe that leukocytes are involved given their larger size and ability to adhere to the endothelium during injury.17 The gradual ramping of capillary obstruction we observed is also in agreement with the delayed time-course for inflammatory cell infiltration (Figure 4h). However, obstruction by RBCs is also possible given that RBCs are far more common in blood, and may pack into slow-flowing rouleuxs.
Large-scale stroke models, such as distal MCAo, generate much larger infarcts. For comparison, the average infarct generated by occluding a penetrating arteriole is 1/100th the volume of that created by distal MCAo in the same mouse strain;44 infarcts resulting from occlusion of high-flux arterioles occupy 1/25th the volume. The progression of microvascular responses elicited by such large strokes may differ due to the broader gradient of ischemia applied over cortex. A greater intracranial pressure induced by a large stroke may also produce a different balance of constrictive and dilative capillary responses. Yet, tissues bordering larger strokes exhibit heterogeneous “mini-cores and mini-penumbras” that could reflect intermediary steps in the recruitment of new penetrating arteriole or venule domains into the infarct, similar to that observed here.45 Nevertheless, the capillary changes we observed are consistent with previous histological46,47 and in vivo40 findings that capillaries can constrict and become occluded during ischemia. A technical caveat to consider is that our quantifications of microvascular flow are based only on the movement of blood cells and not plasma, which can persist despite blockage of cellular flow to carry nutriment and oxygen to the tissue.48 Our measurements were also not sensitive to partial flow reduction in capillaries, because a binary flow-no flow metric was used to assess patency.
Conclusions
Acute human microinfarcts may involve a period of substantial growth, involving microvascular deficits in surrounding tissues that precede loss of neuronal viability. The gradual nature of this growth suggests that microinfarct-induced damage may be mitigated by interventions to protect neurons or capillaries.49 Therapeutics designed to treat acute large-scale stroke, but were limited by the short therapeutic window, could be effective for treating progressive and cumulative small-scale ischemic events in the aging brain.
Supplementary Material
Acknowledgements
We thank David Kleinfeld, Philbert Tsai and Pablo Blinder for providing data for Figure S2. We also thank David Hartmann and Manuel Levy for critical reading of the manuscript.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Our work is generously supported by grants to AYS from the NINDS (NS085402), the Dana Foundation, the American Heart Association (14GRNT20480366), and South Carolina Clinical and Translational Institute (UL1TR000062). AYS is also supported by an Institutional Development Award (IDeA) from the NIGMS under grant number P20GM12345.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
ZJT, ANW and AYS designed, executed and analyzed the two-photon imaging studies. AYS, ESH, XN RLD, JHJ and JAH designed, executed and analyzed the MRI studies. AYS wrote the manuscript with feedback from ZJT, ESH, JHJ and JAH.
Supplementary material
Supplementary material for this paper can be found at http://jcbfm.sagepub.com/content/by/supplemental-data
References
- 1.Kövari E, Gold G, Herrmann FR, et al. Cortical microinfarcts and demyelination significantly affect cognition in brain aging. Stroke 2004; 35: 410–414. [DOI] [PubMed] [Google Scholar]
- 2.Smith EE, Schneider JA, Wardlaw JM, et al. Cerebral microinfarcts: the invisible lesions. Lancet Neurol 2012; 11: 272–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Westover MB, Bianchi MT, Yang C, et al. Estimating cerebral microinfarct burden from autopsy samples. Neurology 2013; 80: 1365–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gorelick PB, Scuteri A, Black SE, et al. Vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2011; 42: 2672–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Veluw, Zwanenburg SJ, Engelene-LJ JJ, et al. In vivo detection of cerebral cortical microinfarcts with high-resolution 7T MRI. J Cereb Blood Flow Metab. 2012; 33: 322–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zheng L, Vinters HV, Mack WJ, et al. Cerebral atherosclerosis is associated with cystic infarcts and microinfarcts but not Alzheimer pathologic changes. Stroke 2013; 44: 2835–2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Suter OC, Sunthorn T, Kraftsik R, et al. Cerebral hypoperfusion generates cortical watershed microinfarcts in Alzheimer disease. Stroke 2002; 33: 1986–1992. [DOI] [PubMed] [Google Scholar]
- 8.Shih AY, Blinder P, Stanley G, et al. The smallest stroke: occlusion of one penetrating vessel leads to infarction and a cognitive deficit. Nat Neurosci 2013; 6: 55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shih AY, Driscoll JD, Drew PJ, et al. Two-photon microscopy as a tool to study blood flow and neurovascular coupling in the rodent brain. J Cereb Blood Flow Metab 2012; 32: 1277–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Watson BD, Dietrich WD, Busto R, et al. Induction of reproducible brain infarction by photochemically initiated thrombosis. Ann Neurol 1985; 17: 497–504. [DOI] [PubMed] [Google Scholar]
- 11.Nishimura N, Schaffer CB, Friedman B, et al. Penetrating arterioles are a bottleneck in the perfusion of neocortex. Proc Natl Acad Sci USA 2007; 104: 365–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blinder P, Tsai PS, Kaufhold JP, et al. The cortical angiome: an interconnected vascular network with noncolumnar patterns of blood flow. Nat Neurosci 2013; 16: 889–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lauwers F, Cassot F, Lauwers-Cances V, et al. Morphometry of the human cerebral cortex microcirculation: general characteristics and space-related profiles. NeuroImage 2008; 39: 936–948. [DOI] [PubMed] [Google Scholar]
- 14.Bar T. The vascular system of the cerebral cortex. Adv Anat Embryol Cellular Biol 1980; 59: 1–62. [DOI] [PubMed] [Google Scholar]
- 15.Pries AR, Secomb TW, Gaehtgens P, et al. Blood flow in microvascular networks. Experiments and simulation. Circulation Res 1990; 67: 826–834. [DOI] [PubMed] [Google Scholar]
- 16.del Zoppo GJ, Mabuchi T. Cerebral microvessel responses to focal ischemia. J Cereb Blood Flow Metab 2003; 23: 879–894. [DOI] [PubMed] [Google Scholar]
- 17.del Zoppo GJ, Schmid-Schonbein GW, Mori E, et al. Polymorphonuclear leukocytes occlude capillaries following middle cerebral artery occlusion and reperfusion in baboons. Stroke 1991; 22: 1276–1283. [DOI] [PubMed] [Google Scholar]
- 18.Dalkara T, Arsava EM. Can restoring incomplete microcirculatory reperfusion improve stroke outcome after thrombolysis? J Cereb Blood Flow Metab 2012; 32: 2091–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Drew PJ, Shih AY, Driscoll JD, et al. Chronic optical access througha polished and reinforced thinned skull. Nat Meth 2010; 7: 981–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen H, Chopp M, Welch KM. Effect of mild hyperthermia on the ischemic infarct volume after middle cerebral artery occlusion in the rat. Neurology 1991; 41: 1133–1135. [DOI] [PubMed] [Google Scholar]
- 21.Tabesh A, Jensen JH, Ardekani BA, et al. Estimation of tensors and tensor-derived measures in diffusional kurtosis imaging. Magn Resonance Med 2011; 65: 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nguyen J, Nishimura N, Fetcho RN, et al. Occlusion of cortical ascending venules causes blood flow decreases, reversals in flow direction, and vessel dilation in upstream capillaries. J Cereb Blood Flow Metab 2011; 31: 2243–2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kazmi SM, Parthasarthy AB, Song NE, et al. Chronic imaging of cortical blood flow using multi-exposure speckle imaging. J Cereb Blood Flow Metab 2013; 33: 798–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu F, Schafer DP, McCullough LD. TTC, fluoro-Jade B and NeuN staining confirm evolving phases of infarction induced by middle cerebral artery occlusion. J Neurosci Meth 2009; 170: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hui ES, Du F, Huang S, et al. Spatiotemporal dynamics of diffusion kurtosis, mean diffusivity and perfusion changes in experimental stroke. Brain Res 2012; 1451: 100–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee VM, Burdett NG, Carpenter A, et al. Evolution of photochemically induced focal cerebral ischemia in the rat. Magnetic resonance imaging and histology. Stroke 1996; 27: 2110–2118. [DOI] [PubMed] [Google Scholar]
- 27.Carmichael ST. Rodent models of focal stroke: size, mechanism, and purpose. NeuroRX 2005; 2: 396–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nishimura N, Rosidi NL, Iadecola C, et al. Limitations of collateral flow after occlusion of a single cortical penetrating arteriole. J Cereb Blood Flow Metab 2010; 30: 1914–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hartmann DA, Underly RG, Grant RI, et al. Pericyte structure and distribution in the cerebral cortex revealed by high-resolution imaging of transgenic mice. Neurophotonics. 2015; 2: 041402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hudetz AG. Blood Flow in the cerebral capillary network: a review emphasizing observations with intravital microscopy. Microcirculation 1997; 4: 233–252. [DOI] [PubMed] [Google Scholar]
- 31.Chen B, Friedman B, Cheng Q, et al. Severe blood brain barrier disruption and surrounding tissue injury. Stroke 2009; 40: 666–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blinder P, Shih AY, Rafie CA, et al. Topological basis for the robust distribution of blood to rodent neocortex. Proc Natl Acad Sci USA 2010; 107: 12670–12675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang M, Iliff JJ, Liao Y, et al. Cognitive deficits and delayed neuronal loss in a mouse model of multiple microinfarcts. J Neurosci 2012; 32: 17948–17960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Silasi G, She J, Boyd JD, et al. A mouse model of small-vessel disease that produces brain-wide-identified microocclusions and regionally selective neuronal injury. J Cereb Blood Flow Metab 2015; 35: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Holland PR, Searcy JL, Salvadores N, et al. Gliovascular disruption and cognitive deficits in a mouse model with features of small vessel disease. J Cereb Blood Flow Metab 2015; 35: 1005–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tan XL, Xue YQ, Ma T, et al. Partial eNOS deficiency causes spontaneous thrombotic cerebral infarction, amyloid angiopathy and cognitive impairment. Mol Neurodegen 2015; 10: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schuier FJ, Vise M, Hossmann KA, et al. Cerebral microembolization. II. Morphological studies. Arch Neurol 1978; 35: 264–270. [DOI] [PubMed] [Google Scholar]
- 38.Nishimura N, Schaffer CB, Friedman B, et al. Targeted insult to individual subsurface cortical blood vessels using ultrashort laser pulses: three models of stroke. Nat Meth 2006; 3: 99–108. [DOI] [PubMed] [Google Scholar]
- 39.Woolsey TA, Rovainen CM, Cox SB, et al. Neuronal units linked to microvascular modules in cerebral cortex: Response elements for imaging the brain. Cereb Cortex 1996; 6: 647–660. [DOI] [PubMed] [Google Scholar]
- 40.Yemisci M, Gursoy-Ozdemir Y, Vural A, et al. Pericyte contraction induced by oxidative-nitrative stress impairs capillary reflow despite successful opening of an occluded cerebral artery. Nat Med 2009; 15: 1031–1037. [DOI] [PubMed] [Google Scholar]
- 41.Hall CN, Reynell C, Gesslein B, et al. Capillary pericytes regulate cerebral blood flow in health and disease. Nature 2014; 508: 55–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fischer EG, Ames A. Studies on mechanisms of impairment of cerebral circulation following ischemia: effect of hemodilution and perfusion pressure. Stroke 1972; 3: 538–542. [DOI] [PubMed] [Google Scholar]
- 43.Chuquet J, Hollender L, Nimchinsky EA. High-resolution in vivo imaging of the neurovascular unit during spreading depression. J Neurosci 2007; 27: 4036–4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sugimori H, Yao H, Ooboshi H, et al. Krypton laser-induced photothrombotic distal middle cerebral artery occlusion without craniectomy in mice. Brain Res Brain Res Protoc 2004; 13: 189–196. [DOI] [PubMed] [Google Scholar]
- 45.del Zoppo GJ, Sharp FR, Heiss WD, et al. Heterogeneity in the penumbra. J Cereb Blood Flow Metab 2011; 31: 1836–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Morris DC, Davies K, Zhang Z, et al. Measurement of cerebral microvessel diameters after embolic stroke in rat using quantitative laser scanning confocal microscopy. Brain Res 2000; 876: 31–36. [DOI] [PubMed] [Google Scholar]
- 47.Garcia JH, Liu K-F, Yoshida Y, et al. Brain microvessels: factors altering their patency after the occlusion of a middle cerebral artery (Wistar rat). Am J Pathol 1994; 145: 728–740. [PMC free article] [PubMed] [Google Scholar]
- 48.Rovainen CM, Woolsey TA, Blocher NC, et al. Blood flow in single surface arterioles and venules on the mouse somatosensory cortex measured with videomicroscopy, fluorescent dextrans, nonoccluding fluorescent beads, and computer-assisted image analysis. J Cereb Blood Flow Metab 1993; 13: 359–371. [DOI] [PubMed] [Google Scholar]
- 49.Gaudin A, Yemisci M, Eroglu H, et al. Squalenoyl adenosine nanoparticles provide neuroprotection after stroke and spinal cord injury. Nat Nanotechnol 2014; 9: 1054–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.