

Abstract
Ventricular hypertrophy is an ominous escalation of hemodynamically stressful conditions such as hypertension and valve disease. The pathophysiology of hypertrophy is complex and multifactorial, as it touches on several cellular and molecular systems. Understanding the molecular background of cardiac hypertrophy is essential in order to protect the myocardium from pathological remodeling, or slow down the destined progression to heart failure. In this review we highlight the most important molecular aspects of cardiac hypertrophic growth in light of the currently available published research data.
MeSH Keywords: Adaptor Proteins, Signal Transducing; Cardiomyopathy, Hypertrophic; Cell Adhesion Molecules
Etiology and Risks
Hypertrophic growth of the heart is an adaptive response to hemodynamic stress, which is believed to have a compensatory role to enhance cardiac performance and diminish ventricular wall tension and oxygen consumption [1]. Physiological hypertrophy of the heart can ensue as a result of exercise or pregnancy, and is deemed mild and/or reversible [2,3]. However, in the presence of chronic stressful conditions such as hypertension and valvular disease, a form of pathological hypertrophy develops, which is characterized by excessive increase in ventricular dimensions, accompanied by myocardial dysfunction and fibrosis [4,5]. These are foreboding signs of the development heart failure and pathological remodeling [6]. Additionally, myocardial supply-demand mismatch secondary to increased myocardial oxygen consumption of the hypertrophic heart further predisposes to multiple cardiovascular ailments, including arrhythmias, myocardial infarction, cerebrovascular events, and sudden death [7]. Ventricular hypertrophy is hence considered as a predictor of cardiovascular morbidity and mortality [7].
Pathogenesis
Mechanical stress
At the cellular level, cardiomyocyte hypertrophy is characterized by an increase in cell size, enhanced protein synthesis, and heightened organization of the sarcomere [8,9]. Mechanical stress is thought to induce a hypertrophic response downstream of mechanosensitive molecules. The sarcomeric Z-disc and its associated proteins have been suggested to drive mechanical stress-induced signal transduction, a process referred to as “mechanotransduction” [4]. A good example of mechanosensitive molecules that have gained attention in recent years are a family of Z-disc-specific proteins called calsarcins, also known as myozenins [10]. Calsarcins were shown to couple the cardiac skeletal apparatus to signaling molecules that can directly influence gene expression. They do this by binding to the Z-disc myofilament anchor proteins, α-actinin and telethonin, and tethering them to calcineurin, a calcium-dependent phosphatase that was shown to directly induce cardiomyocyte hypertrophy by downstream transcriptional pathways [10,11].
Humoral stimuli
Humoral stimuli, on the other hand, act on cell surface receptors, triggering downstream second messenger cascades, finally culminating in cellular hypertrophic response and the associated gene expression program. Based on their target receptor, humoral stimuli can be nested under 2 major groups. The first are targeted by growth factors, such as insulin-like growth factor-1 (IGF-1) and transforming growth factor beta (TGF-β), which act on tyrosine kinase-coupled receptors (RTKs) and are responsible for the eutrophic, as well as adaptive (physiological), myocyte growth. On the other hand, G-protein-coupled receptors (GPCR)-activating molecules, such as catecholamines, angiotensin II, and endothellin-1, are linked to the ominous progression to heart failure, and hence have been the target of many pharmacological antagonists [12,13]. This strongly suggests that a cardiomyocyte undergoing physiological hypertrophy uses different signaling pathways than another one undergoing pathological hypertrophy [8]. A good example supporting this notion is the calcineurin-nuclear factor of activated T cells (NFAT) signaling axis, which was shown to activate pathological hypertrophy [14]. Sustained elevation of calcium ions (Ca+2) downstream of GPCR (αq/α11 subclass) is sensed by calmodulin (Cam) and conveyed to calcineurin, an obligate dimer of regulatory and catalytic subunits with phosphatase activity. Once activated, calcineurin tightly binds and dephosphorylates conserved serine residues at the N-terminus of cytoplasmic NFAT transcription factors, permitting their translocation to the nucleus and activation of pathological hypertrophic gene expression [15].
Similarly, activation of the mitogen-activated protein kinases (MAPK) c-Jun N-terminal kinase (JNK) was also shown to contribute to pathological hypertrophic maladaptive gene expression. On the other hand, physiological hypertrophy, such as that induced by exercise, utilizes a different signaling pathway, mainly through phosphoinositide 3-kinase (PI3K), Akt protein kinase B (PKB), and the mammalian target of rapamycin (mTOR) [15]. Studies from genetically modified animal models corroborate this notion, as Akt1−/− mice were shown to be defective in exercise-induced cardiac hypertrophy [4]. However, one should be careful in interpreting this phenomenon, This is because cellular signaling pathways are highly intricate and interconnected, and pathways activated by either physiological and pathological pathways can converge downstream of different stimuli, as in the case of the MAPK signaling cascade, which can be initiated by GPCRs, RTKs, receptor serine/threonine kinases, and glycoprotein receptors (e.g., gp130), as well as stretching stress [4]. Further research is still needed to better delineate the signaling pathways that govern the development of either adaptive or non-adaptive cardiac hypertrophy, as well as those driving the transition to heart failure.
Figure 1 illustrates some major intracellular signaling pathways controlling cardiac hypertrophy.
Figure 1.
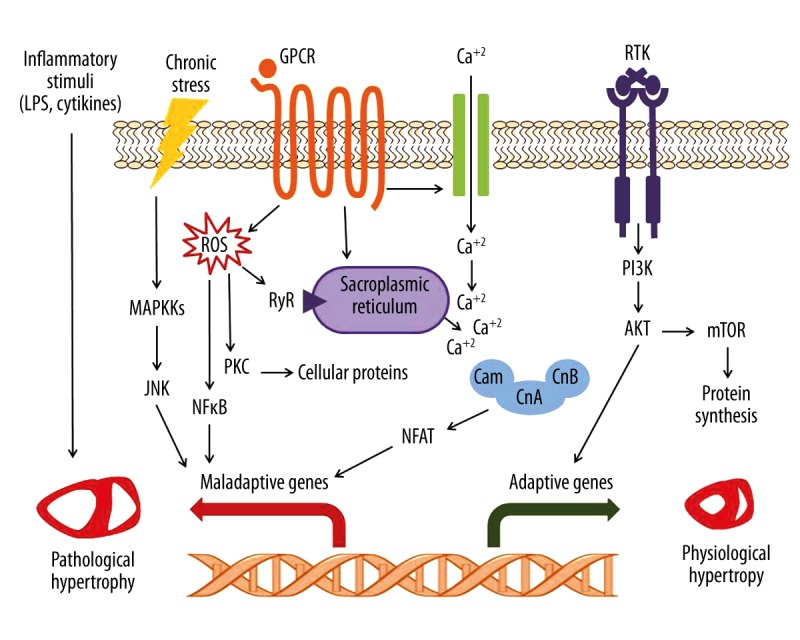
Diagrammatic representation of main intracellular signaling pathways regulating cardiac hypertrophy. Elevated calcium ion levels downstream of GPCRs, either through activation of voltage-gated calcium channels or from intracellular stores, is sensed by calmodulin, which activates calcineurin. Calcineurin in turn dephosphorylates NFAT transcription factor, leading to its nuclear translocation, where it activates gene transcription. ROS can contribute to hypertrophy by direct interaction with cellular proteins and subsequent changes in cellular contraction and/or induction of apoptosis, or by activation of NFκB-mediated gene transcription. Inflammatory stimuli can exacerbate the disease condition by inducing interstitial inflammatory cell infiltration and fibrosis. Stress signals can trigger the activation MAP kinase cascades, which activate a number of downstream targets such as JNK, finally leading to transcriptional activation. Growth hormones induced by physiological cues, such as exercise or pregnancy, bind to and activate downstream signaling of RTKs, which, on the other hand, leads to adaptive hypertrophic growth. GPCR – G-protein-coupled receptor; RTK – receptor tyrosine kinase; MAPKKs – mitogen-activated protein kinase kinases; ROS – reactive oxygen species; RyR – Ryanodine receptor; PI3K – phosphoinositide-3 kinase; JNK – c-Jun N-terminal kinase; NFκB – nuclear factor kappa B; PKC – protein kinase C; Cam – calmodulin; CnA – calcineurin A; CnB – calcineurin B; NFAT – nuclear factor of activated T-cell; Akt – protein kinase B (PKB); mTOR – mammalian target of rapamycin; LPS – lipopolysaccharide.
Transcriptional switch to the fetal gene program
A characteristic feature of pathological cardiac hypertrophy is the switch to the fetal gene expression profile, including upregulation of cardiac myosin heavy chain-beta (MHC-β) in lieu of the adult predominant alpha isoform (MHC-α), skeletal alpha actin (SKA), and atrial natriuretic factor (ANF) genes [16]. Additionally, cardiomyocytes switch to carbohydrate-dependent energetic machinery instead of fatty acid oxidation, which in turn necessitates alterations in expression levels of metabolic genes [17]. Interestingly, exercise-induced physiological hypertrophy was associated with downregulation of the pathological fetal gene program and suppression of NFAT activity [18].
The role of inflammation
Importantly, inflammation was shown to be a prominent hallmark of ventricular hypertrophy [19]. Interstitial inflammatory cell infiltration involving macrophages, T-lymphocytes, fibrosis, high expression levels of cytokines such as interleukins (IL)-6, IL-1β, IL-1RA, and tumor necrosis factor-alpha (TNF-α), and activation of inflammatory signaling pathways such as nuclear factor kappa B (NF-κB) are all characteristic hallmarks of a pathologically hypertrophied heart [20,21]. The pathogenic role inflammation plays is not clearly understood; however, it most probably exacerbates the disease condition. For example, IL-6 was shown to directly induce hypertrophy both in vitro and in vivo [22,23]. Furthermore, macrophage microRNA-155, induced by pro-inflammatory stimuli, including lipopolysaccharide (LPS), TNF-α, and interferon-gamma (INF-γ), promotes cardiac hypertrophy and failure [24]. Additionally, targeting inflammatory cell receptors and mediators was shown to modify the disease process and might preserve cardiac function [25,26].
The role of inflammatory cells in cardiac hypertrophy is not to be overlooked. A good example which merits further elaboration is macrophages Mϕ. Mϕ are mononuclear phagocytes widely distributed throughout the body performing important active and regulatory functions in innate and adaptive immunity, as well as a crucial role in tissue remodeling and repair [27,28]. Two distinct phenotypes of Mϕ can be found in the heart: classically activated pro-inflammatory M1, and alternatively activated anti-inflammatory M2 [28,29]. The former (M1) agitates inflammation in the heart by liberating cytokines and accelerating apoptosis, and contributes to cardiac remodeling [28,30,31]. The latter (M2), on the other hand, thwarts inflammation and stimulates cardiac reparative pathways and angiogenesis [31]. A strong link between Mϕ and hypertrophy was established; however, studies have shown that Mϕ depletion aggravates cardiac dysfunction upon hypertrophy, suggesting a crucial, yet-to-be-understood role in both disease process and outcome [28]. Taken together, inflammation is an attractive target for studying disease progression and developing new therapeutic interventions [26,32].
The role of redox signaling
The role of oxidative stress was shown to be strongly involved in the pathogenesis of ventricular hypertrophy. Reactive oxygen species (ROS) were shown to activate a plethora of signaling pathways implicated in hypertrophic growth and remodeling, including tyrosine kinases, protein kinase C (PKC), and MAPK, among others [33,34]. Furthermore, ROS were shown to mediate angiotensin II, as well as norepinephrine-induced hypertrophy downstream of GPCR [35,36]. Anti-oxidant treatment was shown to abolish TNF-α-induced hypertrophy via NF-κB, suggesting an important role of redox signaling in inflammation-induced hypertrophy [37]. Moreover, ROS contribute to contractile dysfunction by direct modification of proteins central to the excitation-contraction coupling (e.g., the Ryanodine receptor) [38]. Importantly, ROS are involved in the fibrotic remodeling of the heart due to their interaction with extracellular matrix and their activation of matrix metalloproteinase by posttranslational modifications [39]. Finally, ROS can contribute to the loss of myocardial mass upon cardiac remodeling by inducing cardiomyocyte apoptosis [33].
Insights from therapy-oriented studies
At first it might seem obvious that in order to prevent, or at least, halt the progression of cardiac hypertrophy to its more pernicious stages, a correction of the predisposing hemodynamic stress and unloading the encumbered heart, by correction of blood pressure or valve disease, is crucial. However, and based on the above-illustrated molecular nature, cardiac hypertrophy and heart failure are seen as endocrine diseases. Due to the strong role of humoral stimuli in the disease pathology, targeting GPCRs by adrenergic antagonists, renin-angiotensin system modulators such as angiotensin-converting enzyme (ACE) inhibitors, or angiotensin receptor blockers, has been the criterion standard therapeutic approaches for decades [40]. However, a growing body of evidence has shown that such treatment might have a ceiling effect, characterized by lack of efficacy, and even regression, in some patients [13]. A recently published study has intriguingly shown that interference with the non-canonical pathways of the transforming growth factor beta (TGFβ) by Puerarin led to attenuation of hypertrophy in an AngII-induced heart hypertrophy mouse model [41]. The molecular knowledge gained from basic science has shed the lights on calcineurin as a central key player in the development of cardiac hypertrophy [14]. However, in vivo studies using calcineurin inhibitors such as Cyclosporin A have shown great discrepancies [9]. On the other hand, targeting inflammation has also been sought as a potential treatment for cardiac hypertrophy [26]. Cytokine inhibitors such as TNF-alpha antagonists have been clinically investigated for safety and efficacy, but with no apparent success so far in humans [13]. Due to the probably labyrinthine nature of inflammatory processes, a novel approach is currently under investigation that relies on the use of mesenchymal stem cells as modulators of inflammation, which are also capable of controlling inflammatory cells such as macrophages [31]. Such cell therapy-based approaches are now receiving increased attention in cardiovascular disease research.
Conclusions
Ventricular hypertrophy is a compensatory attempt of the heart to enhance its performance; however, it risks the development of heart failure or even sudden death. At the molecular level, hypertrophic growth of the myocardium is a multifaceted entity that demonstrates a high degree of cellular and molecular intricacy across multiple signaling pathways. Furthermore, the development of either physiological or pathological hypertrophy utilizes distinct molecular machinery, if not influencing each other, a phenomenon that needs extensive research. Indeed, this knowledge was made possible by virtue of genetically modified animal models. We encourage further implementation of these models, which serve as powerful tools in intricate signaling studies, and are able to guide the development of highly targeted therapies. On the other hand, the traditional mainstream rodent models, in which hypertrophy is surgically induced by aortic banding or pharmacological induction, do not fully represent the features seen in humans. Hence, in order to better study the effect of hemodynamic load and/or neurohormonal stress, there is a need for larger and longer-lived models.
With regard to therapeutic interventions, the use of ACE-inhibitors and sympatholytics has indeed afforded significant clinical benefits [9]. However, the limited clinical success seen in many patients (ceiling effect) is probably due to the nature of cardiac hypertrophy as an adaptive response, which does not need to be fully inhibited per se [9,13]. Hence, a delicate dissection of the molecular pathways involved, well-tailored drug dosing, and targeted therapy are needed to thwart the deleterious ramifications while maintaining heart homeostasis. Better understanding of the disease molecular foundations shall help us design more targeted and clinically efficacious therapeutic approaches.
Footnotes
Competing interests
The authors report no competing interests.
Source of support: Self financing
References
- 1.Berenji K, Drazner MH, Rothermel BA, Hill JA. Does load-induced ventricular hypertrophy progress to systolic heart failure? Am J Physiol Heart Circ Physiol. 2005;289(1):H8–H16. doi: 10.1152/ajpheart.01303.2004. [DOI] [PubMed] [Google Scholar]
- 2.Chung E, Leinwand LA. Pregnancy as a cardiac stress model. Cardiovasc Res. 2014;101(4):561–70. doi: 10.1093/cvr/cvu013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ellison GM, Waring CD, Vicinanza C, Torella D. Physiological cardiac remodelling in response to endurance exercise training: Cellular and molecular mechanisms. Heart. 2012;98(1):5–10. doi: 10.1136/heartjnl-2011-300639. [DOI] [PubMed] [Google Scholar]
- 4.Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7(8):589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- 5.Kannel WB, Dannenberg AL, Levy D. Population implications of electrocardiographic left ventricular hypertrophy. Am J Cardiol. 1987;60(17):85I–93I. doi: 10.1016/0002-9149(87)90466-8. [DOI] [PubMed] [Google Scholar]
- 6.Artham SM, Lavie CJ, Milani RV, et al. Clinical impact of left ventricular hypertrophy and implications for regression. Prog Cardiovasc Dis. 2009;52(2):153–67. doi: 10.1016/j.pcad.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 7.Kavey RE. Left ventricular hypertrophy in hypertensive children and adolescents: Predictors and prevalence. Curr Hypertens Rep. 2013;15(5):453–57. doi: 10.1007/s11906-013-0370-3. [DOI] [PubMed] [Google Scholar]
- 8.Carreño JE, Apablaza F, Ocaranza MP, Jalil JE. [Cardiac hypertrophy: molecular and cellular events]. Rev Esp Cardiol. 2006;59(5):473–86. [PubMed] [Google Scholar]
- 9.Frey N, Katus HA, Olson EN, Hill JA. Hypertrophy of the heart: A new therapeutic target? Circulation. 2004;109(13):1580–89. doi: 10.1161/01.CIR.0000120390.68287.BB. [DOI] [PubMed] [Google Scholar]
- 10.Frey N, Richardson JA, Olson EN. Calsarcins, a novel family of sarcomeric calcineurin-binding proteins. Proc Natl Acad Sci USA. 2000;97(26):14632–37. doi: 10.1073/pnas.260501097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Molkentin JD, Lu JR, Antos CL, et al. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell. 1998;93(2):215–28. doi: 10.1016/s0092-8674(00)81573-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hunter JJ, Chien KR. Signaling pathways for cardiac hypertrophy and failure. N Engl J Med. 1999;341(17):1276–83. doi: 10.1056/NEJM199910213411706. [DOI] [PubMed] [Google Scholar]
- 13.Mehra MR, Uber PA, Francis GS. Heart failure therapy at a crossroad: Are there limits to the neurohormonal model? J Am Coll Cardiol. 2003;41(9):1606–10. doi: 10.1016/s0735-1097(03)00245-6. [DOI] [PubMed] [Google Scholar]
- 14.Molkentin JD. Calcineurin-NFAT signaling regulates the cardiac hypertrophic response in coordination with the MAPKs. Cardiovasc Res. 2004;63(3):467–75. doi: 10.1016/j.cardiores.2004.01.021. [DOI] [PubMed] [Google Scholar]
- 15.Molkentin JD. Parsing good versus bad signaling pathways in the heart: Role of calcineurin-nuclear factor of activated T-cells. Circ Res. 2013;113(1):16–19. doi: 10.1161/CIRCRESAHA.113.301667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chien KR, Zhu H, Knowlton KU, et al. Transcriptional regulation during cardiac growth and development. Annu Rev Physiol. 1993;55:77–95. doi: 10.1146/annurev.ph.55.030193.000453. [DOI] [PubMed] [Google Scholar]
- 17.Taegtmeyer H, Sen S, Vela D. Return to the fetal gene program: A suggested metabolic link to gene expression in the heart. Ann NY Acad Sci. 2010;1188:191–98. doi: 10.1111/j.1749-6632.2009.05100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Konhilas JP, Watson PA, Maass A, et al. Exercise can prevent and reverse the severity of hypertrophic cardiomyopathy. Circ Res. 2006;98(4):540–48. doi: 10.1161/01.RES.0000205766.97556.00. [DOI] [PubMed] [Google Scholar]
- 19.Yang F, Dong A, Mueller P, et al. Coronary artery remodeling in a model of left ventricular pressure overload is influenced by platelets and inflammatory cells. PLoS One. 2012;7(8):e40196. doi: 10.1371/journal.pone.0040196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuusisto J, Kärjä V, Sipola P, et al. Low-grade inflammation and the phenotypic expression of myocardial fibrosis in hypertrophic cardiomyopathy. Heart. 2012;98(13):1007–13. doi: 10.1136/heartjnl-2011-300960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Erten Y, Tulmac M, Derici U, et al. An association between inflammatory state and left ventricular hypertrophy in hemodialysis patients. Ren Fail. 2005;27(5):581–89. doi: 10.1080/08860220500200072. [DOI] [PubMed] [Google Scholar]
- 22.Hilfiker-Kleiner D, Shukla P, Klein G, et al. Continuous glycoprotein-130-mediated signal transducer and activator of transcription-3 activation promotes inflammation, left ventricular rupture, and adverse outcome in subacute myocardial infarction. Circulation. 2010;122(2):145–55. doi: 10.1161/CIRCULATIONAHA.109.933127. [DOI] [PubMed] [Google Scholar]
- 23.Hirota H, Yoshida K, Kishimoto T, Taga T, et al. Continuous activation of gp130, a signal-transducing receptor component for interleukin 6-related cytokines, causes myocardial hypertrophy in mice. Proc Natl Acad Sci USA. 1995;92(11):4862–66. doi: 10.1073/pnas.92.11.4862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heymans S, Corsten MF, Verhesen W, et al. Macrophage microRNA-155 promotes cardiac hypertrophy and failure. Circulation. 2013;128(13):1420–32. doi: 10.1161/CIRCULATIONAHA.112.001357. [DOI] [PubMed] [Google Scholar]
- 25.Velten M, Duerr GD, Pessies T, et al. Priming with synthetic oligonucleotides attenuates pressure overload-induced inflammation and cardiac hypertrophy in mice. Cardiovasc Res. 2012;96(3):422–32. doi: 10.1093/cvr/cvs280. [DOI] [PubMed] [Google Scholar]
- 26.Heymans S, Hirsch E, Anker SD, et al. Inflammation as a therapeutic target in heart failure? A scientific statement from the Translational Research Committee of the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail. 2009;11(2):119–29. doi: 10.1093/eurjhf/hfn043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11(11):723–37. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takeda N, Manabe I. Cellular interplay between cardiomyocytes and nonmyocytes in cardiac remodeling. Int J Inflam. 2011;2011:535241. doi: 10.4061/2011/535241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8(12):958–69. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fernandez-Velasco M, Gonzalez-Ramos S, Bosca L. Involvement of monocytes/macrophages as key factors in the development and progression of cardiovascular diseases. Biochem J. 2014;458(2):187–93. doi: 10.1042/BJ20131501. [DOI] [PubMed] [Google Scholar]
- 31.van den Akker F, de Jager SC, Sluijter JP. Mesenchymal stem cell therapy for cardiac inflammation: Immunomodulatory properties and the influence of toll-like receptors. Mediators Inflamm. 2013;2013:181020. doi: 10.1155/2013/181020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hofmann U, Frantz S. How can we cure a heart “in flame”? A translational view on inflammation in heart failure. Basic Res Cardiol. 2013;108(4):356. doi: 10.1007/s00395-013-0356-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kwon SH, Pimentel DR, Remondino A, et al. H(2)O(2) regulates cardiac myocyte phenotype via concentration-dependent activation of distinct kinase pathways. J Mol Cell Cardiol. 2003;35(6):615–21. doi: 10.1016/s0022-2828(03)00084-1. [DOI] [PubMed] [Google Scholar]
- 34.Tanaka K, Honda M, Takabatake T. Redox regulation of MAPK pathways and cardiac hypertrophy in adult rat cardiac myocyte. J Am Coll Cardiol. 2001;37(2):676–85. doi: 10.1016/s0735-1097(00)01123-2. [DOI] [PubMed] [Google Scholar]
- 35.Amin JK, Xiao L, Pimental DR, et al. Reactive oxygen species mediate alpha-adrenergic receptor-stimulated hypertrophy in adult rat ventricular myocytes. J Mol Cell Cardiol. 2001;33(1):131–39. doi: 10.1006/jmcc.2000.1285. [DOI] [PubMed] [Google Scholar]
- 36.Kuster GM, Pimentel DR, Adachi T, et al. Alpha-adrenergic receptor-stimulated hypertrophy in adult rat ventricular myocytes is mediated via thioredoxin-1-sensitive oxidative modification of thiols on Ras. Circulation. 2005;111(9):1192–98. doi: 10.1161/01.CIR.0000157148.59308.F5. [DOI] [PubMed] [Google Scholar]
- 37.Higuchi Y, Otsu K, Nishida K, et al. Involvement of reactive oxygen species-mediated NF-kappa B activation in TNF-alpha-induced cardiomyocyte hypertrophy. J Mol Cell Cardiol. 2002;34(2):233–40. doi: 10.1006/jmcc.2001.1505. [DOI] [PubMed] [Google Scholar]
- 38.Zima AV, Blatter LA. Redox regulation of cardiac calcium channels and transporters. Cardiovasc Res. 2006;71(2):310–21. doi: 10.1016/j.cardiores.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 39.Siwik DA, Colucci WS. Regulation of matrix metalloproteinases by cytokines and reactive oxygen/nitrogen species in the myocardium. Heart Fail Rev. 2004;9(1):43–51. doi: 10.1023/B:HREV.0000011393.40674.13. [DOI] [PubMed] [Google Scholar]
- 40.Willenbrock R, Philipp S, Mitrovic V, Dietz R. Neurohumoral blockade in CHF management. J Renin Angiotensin Aldosterone Syst. 2000;1(Suppl 1):24–30. doi: 10.3317/JRAAS.2000.030. [DOI] [PubMed] [Google Scholar]
- 41.Zhang X, Liu Y, Han Q. Puerarin attenuates cardiac hypertrophy partly through increasing mir-15b/195 expression and suppressing non-canonical transforming growth factor beta (Tgfβ) signal pathway. Med Sci Monit. 2016;22:1516–23. doi: 10.12659/MSM.895877. [DOI] [PMC free article] [PubMed] [Google Scholar]