

Abstract
Fibrocytes were initially described in 1999 and since that time there has been a growing body of literature to suggest their importance in a number of chronic lung diseases. It is now well established that fibrocytes derive from the bone marrow and circulate within the peripheral blood. However, when injury occurs, fibrocytes can travel to the site of damage via chemokine-mediated recruitment. Recent studies suggest that fibrocyte numbers increase within the lung or circulation during numerous disease processes. Although fibrocytes readily differentiate into fibroblasts in vitro, whether they do so in vivo is still unknown. The variety of pro-fibrotic mediators that are secreted by fibrocytes makes it likely that they act via paracrine functions to influence the behavior of resident lung cells. This review summarizes recent insights regarding fibrocytes in asthma, scleroderma and IPF.
Keywords: fibrosis, bone marrow, fibroblasts, fibrocytes, chemokines, lung
PULMONARY FIBROSIS: DESCRIPTION AND CLASSIFICATION
Pulmonary fibrosis is characterized by epithelial cell injury and hyperplasia, variable degrees of inflammatory cell infiltrate, fibroblast proliferation and accumulation, and the relentless deposition of extracellular matrix (ECM) (1). The end results of this process are a loss of lung elasticity and loss of alveolar surface area, leading to severe compromises in pulmonary function and respiratory failure (1–5). In humans, there are many types of fibrotic lung diseases (6). Among the diffuse parenchymal lung diseases (DPLDs) are the diseases of known cause (e.g. drug-related or those associated with collagen vascular disease), the idiopathic interstitial pneumonias (IIPs), granulomatous DPLDs (e.g. sarcoidosis) and non-categorized diseases, such as lymphangioleiomyomatosis. Idiopathic pulmonary fibrosis (IPF) is the most common disease within the category of IIPs, and is histopathologically identified as usual interstitial pneumonia (UIP). Additional diseases within the IIP category include desquamative interstitial pneumonia, respiratory bronchiolitis interstitial lung disease, acute interstitial pneumonia, cryptogenic organizing pneumonia, lymphocytic interstitial pneumonia and nonspecific interstitial pneumonia (NSIP). IPF carries a poor prognosis, with a mean survival time of less than 5 years following diagnosis (4, 7–9). The fact that biopsies from a single patient can show heterogenous patterns consistent with both UIP and NSIP (8, 10, 11), have led to a further reevaluation of the pathobiologic mechanisms of pulmonary fibrosis, and may suggest that NSIP precedes development of UIP.
In addition to the DPLDs, there are other diseases of the lung in which fibroproliferative changes have been described. These include late stages of acute respiratory distress syndrome, airway remodeling associated with asthma and vasculopathies, such as primary pulmonary hypertension. In this document, we will consider the pathobiology of IPF, scleroderma and asthma.
PATHOGENESIS OF FIBROSIS
The pathogenesis of all fibrotic lung diseases involves the accumulation and activation of fibroblasts. Fibroblasts are mesenchymal cells capable of synthesizing collagen types I and III and fibronectin, the predominant ECM proteins deposited in IPF lungs. Fibroblasts that actively secrete ECM proteins can be termed “effector” fibroblasts. Effector fibroblasts can acquire the expression of α-smooth muscle actin (α-SMA) and non-muscle myosins. The expression and organization of these proteins into stress fibers confers contractile properties to these cells, referred to as myofibroblasts (12–16). Myofibroblasts are potent producers of ECM proteins and these cells are considered to be the principal collagen-producing cells within fibrotic lesions (12, 13). The fibrotic foci that are a distinguishing feature of UIP histopathology are actually clusters of myofibroblasts that represent sites of active collagen synthesis (8, 11, 17). However, the origins of effector fibroblasts and myofibroblasts in pulmonary fibrosis are still uncertain.
ORIGIN OF INTERSTITIAL FIBROBLASTS
Several mechanisms have been proposed explaining the origin of the effector fibroblasts and myofibroblasts that are seen in fibrotic lung disease. For years, the prevailing theory was that they could arise from tissue-specific mesenchymal precursors and certainly local smooth muscle, pericytes and endothelial cells may also contribute to ECM deposition (18–20). More recently, studies have suggested that organ specific epithelial cells may generate local fibroblasts via epithelial-mesenchymal transition (EMT) (21–23), although this is controversial (24). Finally, circulating bone marrow (BM)-derived progenitor cells (fibrocytes) may migrate via the peripheral circulation to sites of tissue injury and participate in organ-specific fibrogenesis (25, 26). Animal models of bleomycin induced pulmonary fibrosis suggest that effector fibroblasts arise via all three of these proposed mechanisms (27). In one study, EMT-derived fibroblasts accounted for approximately 1/3 of lung S100A4-expressing effector fibroblasts following bleomycin administration whereas fibrocytes accounted for 1/5th of this total (27). Interestingly, in this study myofibroblasts did not readily arise from EMT or bone marrow-derived progenitors. In contrast, in a transforming growth factor (TGF)β-driven model of fibrogenesis, EMT contributed substantially to the vimentin positive cells (presumably effector fibroblasts) within the lung (28). Fibrocytes can differentiate to fibroblasts readily in vitro; however, in vivo, it is likely that they may contribute to fibrogenesis via secretion of paracrine mediators (20), something that will be discussed below.
FIBROCYTE CHARACTERIZATION
Fibrocytes are a population of cells which circulate in peripheral blood and express both fibroblast and leukocyte markers (26, 29, 30). They are adherent, can be cultured in vitro, and synthesize fibroblast derived proteins collagen I, collagen III, and fibronectin (26). In addition, fibrocytes express a variety of leukocyte markers such as the common leukocyte antigen, CD45; the pan-myeloid antigen, CD13; the hematopoietic stem cell antigen, CD34; and the class II major histocompatibility complex (MHC) antigens. Fibrocytes do not express epithelial or endothelial markers, and are negative for non-specific esterases and the monocyte/macrophage markers CD14 and CD16 (26). In addition, they are negative for the Langerhan’s cell marker CD1a, dendritic-cell-associated CD25, CD10 and CD38 and the pan B cell antigen CD19 (26). It has been estimated that this population comprises about 0.5% of human peripheral blood leukocytes in healthy volunteers (26). Since we last reviewed this topic in 2005(31), in vitro, animal and human studies have provided new insights into the role circulating fibrocytes play in a wide variety of fibrotic lung diseases.
FIBROCYTES ARE BONE MARROW DERIVED
The hematopoietic origin of fibrocytes was originally controversial because female mice that received male bone marrow (BM) transplants following 800 rads of total body irradiation (TBI) did not produce fibrocytes bearing significant levels of the male SRY gene. (26) However, these studies were likely misinterpreted because the dose of irradiation used was insufficient to effectively deplete host hematopoietic precursors. More recently studies using green fluorescent protein (GFP) BM-chimeric mice have provided evidence that pulmonary fibroblasts can arise from radiosensitive bone marrow precursors (32–35). In GFP-BM chimeric mice (wild-type mice that have received a BM transplant from a GFP transgenic mouse), GFP-expressing fibroblasts have been identified in fibrotic regions of lungs exposed to irradiation (32), and in fibrotic regions of bleomycin-exposed lungs (33). Similarly, fibrocytes are increased in number in BM of bleomycin-treated mice (36) and mice that received 10 Gy of TBI also showed evidence of BM-derived fibroblasts following lung irradiation (32) or bleomycin administration (33, 34). These studies suggest that the progenitor that gives rise to fibrocytes/fibroblasts is indeed a relatively radio-resistant cell, which is eliminated by higher levels of irradiation (~10 Gy of TBI). Despite the evidence that these cells may become fibroblasts, there is little evidence that they can become myofibroblasts (33, 35).
THE ROLE OF FIBROCYTES IN FIBROTIC DISEASE
Fibrocytes were described in 1990 but their role in human fibrotic disease of the lung was not identified until 2003 when they were found in biopsies from remodeled airways of human asthmatic patients (37). Within the past year fibrocytes have also been implicated in several new murine models of human disease including acute lung injury (ALI) (38),bronchiolitis obliterans (39) and sickle cell lung disease (40). Increased fibrocyte numbers predicted lung transplant patients that would go on to develop bronchiolitis obliterans in a recent clinical study as well (41). For the remainder of this review, we will focus on the recent developments regarding fibrocytes in asthma, scleroderma and IPF.
FIBROCYTES IN HUMAN ASTHMA
Airway remodeling is a long-term complication of chronic asthma and histologically is characterized by subepithelial fibrosis. In 2003, Schmidt et al successfully used immunohistochemistry to identify fibrocytes in biopsy samples from the remodeled airways of asthmatics. Cells expressing CD34 co-localized with cells expressing collagen 1 mRNA and localized in areas of collagen deposition and basement membrane thickening, suggesting a pathogenic role for these cells in the development of subepithelial fibrosis (37). Further, using an animal model of allergen challenge, labeled fibrocytes in the peripheral circulation were shown to increase in number at 2h and 24h after allergen exposure, home to bronchial mucosa, and show the ability to differentiate locally and express α-SMA (37). This was one of the first studies to demonstrate that fibrocyte recruitment to the lung correlates with the development of fibrosis.
Nihlberg et al. were able to make further correlations between fibroblasts in BAL fluid of asthmatic patients, histopathological changes, and clinical alteration of lung function. In this 2006 study, asthmatic patients with fibroblasts identified in BAL fluid were also shown to have an increase in fibrocytes expressing co-localizing markers (CD34, CD45RO, pro-collagen I and α-SMA) in close proximity to the basement membrane (42). The increased number of subepithelial fibrocytes was also associated with significant thickening of the basement membrane (42). Finally, this group also showed that levels of circulating fibrocytes in the nonadherent non-T cell fraction of leukocytes were highest in patients with airflow obstruction compared to patients without airflow obstruction. The percentage of fibrocytes within the non-adherent non-T cell fraction also correlated with the severity of decline in pulmonary function as measured by FEV1(42). This study suggests that fibrocytes play a role in the airway remodeling that can be seen in uncontrolled asthma.
Further studies by several groups have confirmed the correlation between elevated levels of circulating fibrocytes in the peripheral blood and lung tissue in severe asthmatics compared to healthy controls (43, 44). Saunders et al. specifically demonstrated localization of fibrocytes in proximity to airway smooth muscle (ASM) bundles on histological examination. ASM has an effect on fibrocyte chemotaxis and chemokinesis in a platelet-derived growth factor dependent mechanism (44). Wang et al. were also able to show that TGF-β1 is able to transform fibrocytes from humans with chronic airway obstruction into myofibroblasts in vitro (43). Together, these studies provided further evidence supporting mechanistic ways in which fibrocytes may leave the peripheral circulation and have an effect in the fibrotic response to tissue injury in the lung.
More recently, investigators have explored specifically what cells and factors are responsible for fibrocyte migration and local differentiation. In patients with allergic asthma, in addition to fibrosis, there is a significant subepithelial inflammatory infiltrate characterized primarily by Th2 and Th17 cells. There are increased levels of Th2 and Th17 derived factors IL-4, IL-13, and IL-17A in the sputum of human asthmatics with elevated levels of circulating fibrocytes (45). Interestingly, in vitro studies suggested that there were differing effects of these factors on fibrocyte behavior. In response to stimulation with Th2 derived factors IL-4 and IL-13 fibrocytes produce increased levels of collagenous and non-collagenous ECM proteins. In contrasts, fibrocytes were seen to proliferate, produce pro-inflammatory cytokines which promote neutrophil recruitment and increase α-SMA production in response to stimulation with IL-17A (45). These results provide further evidence of the pluripotent potential of circulating fibrocytes depending on the local environment to which they migrate.
FIBROCYTES IN SYSTEMIC SCLEROSIS
Systemic sclerosis, or scleroderma, is a multisystem autoimmune disease which results in both cutaneous and visceral fibrosis. Within the lung, scleroderma manifests as an interstitial lung disease with areas of fibrosis and inflammation replacing the normal parenchymal architecture which impairs alveolar gas exchange and ultimately results in respiratory failure. Given the systemic nature of the fibrosis that occurs in scleroderma, it is reasonable to think that circulating fibrocytes are likely playing a role in the local tissue fibrosis that occurs in scleroderma associated ILD (SSc-ILD). A study by Mathai et al. in 2010, was the first to look at fibrocytes in human subjects with systemic sclerosis. In this study, peripheral blood mononuclear cells were isolated from Ficoll-Paque gradients, stained for fibrocyte markers and analyzed by flow cytometry (46). There was no difference in the percentage of circulating CD45+/procollagen I+ cells in patients with SSc-ILD compared to healthy controls, however there was an overall increased number of total circulating cells in the scleroderma patients with a resultant significant increase in the absolute number of fibrocytes in SSc-ILD patients relative to controls (46).
Recently, Gan et al. explored role of semaphorin-7a in a model of TGF-β1 induced pulmonary fibrosis. Using a BM transplant model, they showed that fibrocyte recruitment was dependent on the expression of semaphorin-7a on the hematopoietic cells (47). Neutralization of β1 integrin prevented fibrocyte recruitment and fibrosis (47). They were able to verify some of these findings in studies examining human peripheral blood monocytes from patients with scleroderma. They demonstrated increased expression of mRNA for semaphorin-7a and its receptors in fibrocytes (47). Scleroderma patients showed enhance fibrocyte outgrowth from the peripheral blood, and treatment of normal peripheral blood monocytes with recombinant semaphorin-7a enhanced fibrocyte differentiation (47).
Finally, Tourkina et al. published a series of experiments both confirming the increased presence of fibrocytes in SSc-ILD lung tissue, but also proposing a mechanistic rationale for increased fibrocyte migration from the circulation into SSc-ILD lungs. First, they demonstrated increased numbers of fibrocytes in the lungs of SSc-ILD patients compared to normal lungs (48). In particular, the subset of fibrocytes expressing CXCR4+/ColI+ were more prevalent than subsets expressing CD34+/ColI+ and CD45+/ColI+ (48). Both CXCR4 and its ligand CXCL12 are also upregulated in lung tissue from SSc-ILD patients, a finding that has previously been attributed to a deficiency of caveolin-1 in the peripheral blood monocytes of scleroderma and IPF patients (49). Peripheral monocytes from scleroderma patients demonstrate enhanced chemotaxis toward CXCL12 compared to normal monocytes as a result of the increased CXCR4 expression (48). When SSc-ILD and normal monocytes are treated with caveolin scaffold domain (CSD) peptide, an intervention that compensates for the deficiency of caveolin-1, migration in response to CXCL12 is almost completely inhibited (48). Conversely, treatment with TGF-β has the opposite effect, causing a decrease in caveolin-1, increased expression of CXCR4, and enhanced migration of fibrocytes in response to CXCL12 (48). This series of experiments identifies a specific defect in systemic sclerosis (caveolin) which may explain the enhanced CXCR4-dependent fibrocyte migration from the peripheral circulation into tissue and explain the propensity for these patients to develop ILD.
FIBROCYTES IN IPF
As we have previously discussed, several studies have established a link between circulating fibrocytes and their ability to migrate to areas of tissue injury in animal models of pulmonary fibrosis. While the mechanisms are still not well understood, recent studies in human IPF patients have also made progress in establishing a correlation between circulating peripheral fibrocyte numbers in IPF patients, prognosis and clinical severity of disease.
The first study to analyze peripheral fibrocyte markers examined a series of 5 patients who underwent open lung biopsy for the diagnosis of interstitial lung disease (50). Histological findings were consistent with UIP in 4 of the patients and fibrotic NSIP in the fifth. Plasma levels of CXCL12 and percentages of circulating fibrocytes were compared to levels in 5 normal control patients. Plasma CXCL12 levels were 2.4-fold higher in fibrotic patients than in the normal volunteers (50). This correlated with prior work showing CXCL12 accumulation as measured by immunohistochemistry in archived samples of human fibrotic lung tissue (50). Further, circulating leukocytes were isolated from the buffy coat of peripheral blood samples and analyzed by flow cytometry for expression of fibrocyte markers CD45, collagen 1, CXCR4 and α-SMA. The percentage of circulating leukocytes with fibrocyte markers in patients with fibrotic lung disease was an order of magnitude higher than in normal patients (6–10% vs. 0.5%) (50). Interestingly, only a small percentage of circulating fibrocytes expressed the myofibroblast marker α-SMA, suggesting that fibrocytes remain relatively undifferentiated prior to tissue migration (50). Although this study examined a small number of patients, it was the first to clearly show a correlation between altered number of circulating fibrocytes and IPF, support the notion of the CXCR4/CXCL12 axis in fibrocyte migration and the relatively undifferentiated potential of the circulating fibrocyte. This study was somewhat remarkable for the high percentages of fibrocytes that were noted in the circulation. As discussed below, differences in methodologies and patient poplations have led to highly variable measures of fibrocyte percentages in more recent studies.
Andersson-Sjöland et al. used histochemical analysis of lung tissue to confirm the presence of fibrocytes in the lung tissue of patients with IPF (51). Analysis of different combinations of fibrocyte markers (CXCR4, CD34, CD45RO, prolyl-4-hydroxylase and α-SMA confirmed the presence of fibrocytes in 8 of 9 IPF patients (51). The density of fibrocytes per mm2 varied from 1.3 to 10.3 depending on the combination of markers analyzed, but interestingly, the density of CXCR4+/prolyl-4-hydroxylase+ cells correlated positively with the number of fibroblastic foci per cm2 of lung tissue (51). One caveat to these studies is that the CXCR4/prolyl-4-hydroxylase markers could simply represent resident mesenchymal cells rather than cells of fibrocyte origin. Definitive identification as fibrocytes would require co-staining with CD45 or CD34. Additionally, these studies failed to show the presence of fibrocytes in bronchoalveolar lavage fluid (BALF) of IPF patients or in normal lung tissue (51). These results are somewhat contradictory to murine studies which have shown the presence of fibrocytes in normal lungs and in the BALF post-fibrotic injury(34).
As a follow-up to Mehrad et al’s, study from 2007, Moeller et al. examined the correlation between peripheral circulating fibrocytes and pulmonary fibrosis in a much larger group of IPF patients. In total peripheral blood was collected from 51 patients with stable IPF and 7 patients with acute exacerbation of IPF (AE-IPF) (52). The increased percentage of circulating fibrocytes was confirmed in IPF patients (2.72 ± 0.34%) compared to controls (1 ± 0.12%), although with a less drastic increase compared to the Mehrad study discussed above (50, 52). One of the most intriguing observations from this series of experiments were the findings that patients with AE-IPF actually had a significantly increased percentage of circulating fibrocytes (14.51 ± 2.53%) compared to stable IPF patients or normal controls (52). In addition, serum samples from 3 patients who recovered from and survived the acute exacerbation showed that circulating fibrocytes levels decreased dramatically in association with clinical recovery (52). On further analysis, patients with circulating fibrocyte percentage of >5% had worse prognosis with life expectancy of 7.5 versus 27 months (52). The patients experiencing AE-IPF are largely responsible for the increased percentages noted in this analysis. Taken together, while fibrocytes are present and slightly increased in the circulation of patients with stable IPF, significant increases in total percentage may be highly associated with AE-IPF, a condition known to be highly correlated with poor prognosis (53). It is important to note that while these studies suggested that fibrocyte levels in the circulation may correlate with more active fibroproliferation and a worse prognosis, they were unable to correlate these findings with objective measurements of pulmonary function including; forced vital capacity (FVC), total lung capacity (TLC), diffusion capacity for carbon monoxide (DLCO) or 6-minute hallwalk (52).
Finally, in a recently published study, Fujiwara et al. attempted to further establish a correlation between increased numbers of circulating fibrocytes and lung diseases with a course characterized by progressive pulmonary fibrosis. This study examined a total of 41 patients with interstitial lung disease, including 20 patients with IPF, 10 with collagen vascular disease associated interstitial pneumonitis and 11 with sarcoidosis compared to 7 healthy volunteers (54). Flow cytometry of circulating leukocytes confirmed that fibrocytes are increased in all patients with ILD compared to controls, and in particular are elevated in patients with IIPs and collagen vascular disease associated ILD (54). Further, supporting the idea that fibrocytes are increased specifically in processes which lead to interstitial fibrosis and not just inflammation in general, circulating fibrocytes were increased in the subsets of patients with histological patterns described as UIP and NSIP but not in patients with histology consistent with cryptogenic organizing pneumonia (54). When measured by ELISA, plasma levels of the chemokines CCL2 and CXCL12 were also noted to be elevated in ILD patients compared to controls (54). Similar to the work of Moeller et al. discussed above, serial follow-up of 2 individual patients, one with dermatomyositis associated-ILD and another with AE-IPF, again demonstrated a fall in circulating fibrocytes levels with clinical recovery after exacerbation (54). Additionally, this was the first study to clearly show correlation with measurable clinical parameters, as circulating fibrocyte levels were inversely associated with percent-predicted values of FVC and DLCO (54).
Together, all of these studies in human IPF patients have established a strong correlation between circulating fibrocytes and fibroproliferative activity. Moeller and Fujiwara were even able to show a correlation between circulating fibrocytes and AE-IPF, suggesting that circulating fibrocyte analysis may serve as a prognostic bio-marker for pulmonary function and even mortality. Unfortunately, there are still many holes in our understanding of fibrocyte recruitment, migration and differentiation. Evaluation of individual subsets of patients show that elevated levels of circulating fibrocytes is not a universal finding and there is actually a wide overlap in circulating percentages even with normal patients. Thus, while these studies have given us significant insight, they also serve as reminders of the heterogeneity of IPF.
FIBROCYTE FUNCTIONS
All of the studies mentioned thus far suggest that fibrocytes correlate with worsened fibrotic outcomes. However, there are a paucity of studies which have actually examined the fibrotic potential of fibrocytes directly. This is largely due to the fact that fibrocytes cannot be sorted from human tissues or blood. The definitive identification of fibrocytes requires that cells be fixed and permeabilized to allow for the intracellular staining of collagen 1 or pro-collagen 1. This kills the cells and precludes further functional analysis. What is possible, is to culture fibrocytes either from lung tissue or peripheral blood for a period of at least 2 weeks. By this time, leukocyte populations have largely died off and the adherent fraction that remains consists almost entirely of CD45+, collagen 1+ cells from the blood, or a mixture of fibrocytes and fibroblasts when cultured from murine lung tissue (34). Fibrocytes make up 15–25% of the cells which are cultured from murine lungs. It is possible to purify a CD45+ fraction from these cultures of murine lungs using magnetic beads. This approach allows for subsequent functional analysis of fibrocyte actions. Our laboratory was the first to demonstrate that adoptive transfer of fibrocytes isolated from cultured lungs could enhance fluorescein isothiocyanate (FITC)-mediated fibrosis whereas transfer of fibroblasts or spleen cells did not(55). Fibrocytes are recruited to sites of tissue injury via chemokine gradients. Fibrocytes are known to express multiple chemokine receptors including CXCR4, CCR7 and CCR2 and these receptors can mediate migration of the fibrocytes (34, 36, 48, 55). The FITC model of lung fibrosis is highly dependent on CCR2-mediated recruitment of inflammatory cells and CCR2−/− mice are protected from experimental fibrosis (34). When testing adoptive transfer of fibrocytes, wild type fibrocytes could augment fibrosis whereas transfer of fibrocytes which lacked CCR2 could not augment FITC-induced fibrosis(55). Chemokines can also transmit activation signals to fibrocytes. For instance, CCL2, a ligand for CCR2, is known to upregulate collagen synthesis in fibrocytes(34).
Studies on cultured fibrocytes readily demonstrate that fibrocytes can differentiate into fibroblasts and myofibroblasts in vitro. Once fibrocytes transition into fibroblasts in vitro, this is associated with loss of chemokine receptors (34). It was speculated that this might facilitate retention of the fibroblasts within the local tissue environment to promote fibrogenesis if the same thing occurs in vivo. However, in our murine models, we were unable to show that adoptively transferred fibrocytes lost expression of CD45 in vivo (unpublished observation). These observations suggested that the profibrotic nature of fibrocytes may relate more to paracrine functions.
Fibrocytes may be uniquely poised to support fibrogenesis via paracrine functions. The fact that these cells can be localized to areas of tissue injury suggests that release of profibrotic factors may influence resident epithelial or mesenchymal cells to differentiate to myofibroblasts to promote fibrogenesis. Such a mechanism would be consistent with the observations that bone-marrow derived cells rarely acquire myofibroblast markers in animal models of fibrosis (33, 35), yet adoptive transfer of fibrocytes worsens disease (55). Fibrocytes are known to secrete a variety of growth and differentiation factors that may promote fibrogenesis. Table 1 provides a list of known fibrocyte mediators. Secretion of factors like TGFβ and periostin are likely important in driving proliferation, migration and extracellular matrix production by resident fibroblasts and epithelial cells. (56)
Table 1.
Fibrotic Mediators Secreted by Fibrocytes
| Interleukin-1 (IL-1β) |
| Tumor Necrosis Factor (TNFα) |
| CCL2, 3, 4, CXCL2 |
| Platelet derived growth factor (PDGFα) |
| Transforming growth factor (TGFβ) |
| Matrix metalloproteinases (MMP1 and MMP9) |
| Periostin |
| Vascular endothelial growth factor (VEGF) |
FIBROCYTES AS BIOMARKERS
Although recent studies in scleroderma and IPF suggest that elevated numbers of fibrocytes can be found in circulation and that absolute numbers may predict poor prognosis (48, 50, 52), overall the studies have demonstrated significant overlap between percentages of fibrocytes found in patients with lung disease versus normal controls. Furthermore, the fact that fibrocytes make up such a small percentage of total peripheral blood cells in either population make these cells difficult to identify. Finally, the current methodology for identifying fibrocytes requires intracellular staining to look for collagen production, and yet, production of collagen may be limited in circulation and only upregulated once cells migrate into tissues. These issues make standardization of fibrocyte enumeration in the blood problematic. In our own studies, we have not been able to detect increased percentages of fibrocytes in peripheral blood of IPF patients enrolled in the PANTHER clinical trial or in an observational cohort of IPF patients (unpublished observations). The reasons for the discrepancy between our studies and those of Moeller et al. may reflect methodological differences or differences in patient populations. This is highlighted by the fact that the highest levels of circulating fibrocytes were noted in patients undergoing acute exacerbations in the Moeller study (52), whereas our population of IPF patients experienced few acute exacerbations during our study period. While these issues may mean that fibrocyte numbers in circulation may be a challenging biomarker, they in no way are meant to discount the idea that fibrocytes may regulate fibrogenesis. Even if the percentages are similar in control and IPF patients, function of the cells could be very different. This is exemplified by our finding that periostin production is readily noted in fibrocytes cultured from IPF, but not control subjects (56).
SUMMARY
Fibrocytes have received significant attention in recent studies of lung fibrosis in both humans and animal models. The fact that fibrocyte numbers are increased in fibrotic lungs and have been elevated in circulation of patients with fibrotic lung diseases suggests these cells are pathogenic. Adoptive transfer studies in mice verify that these cells can promote fibrogenesis in the lung. Recent revelations that fibrocytes cultured from lungs of IPF patients produce increased levels of pro-fibrotic periostin also suggests that the paracrine function of these cells may be more critical than their ability to directly secrete ECM. The fact that fibrocyte functions can be tightly regulated in both positive and negative fashions (for example by serum amyloid P)(57) highlights the central importance these cells may have in driving fibrogenesis. Figure 1 summarizes known regulators of fibrocyte functions and highlights the paracrine actions of these cells. However, more research is needed to understand the longitudinal changes in fibrocyte phenotypes that may ultimately regulate lung homeostasis vs. fibrosis.
Figure 1. Schematic representation of fibrocyte actions.
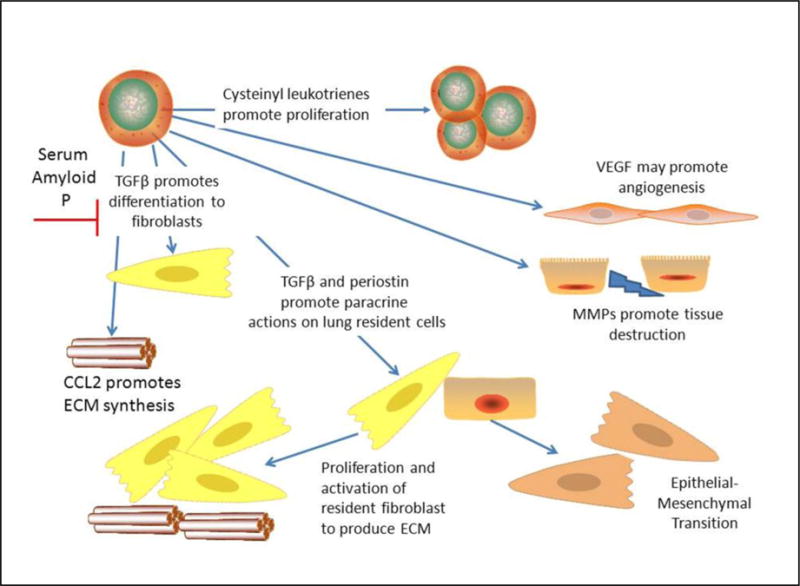
Fibrocytes can be positively regulated via cysteinyl leukotrienes to undergo proliferation. Factors such as CCL2 and TGFβ can promote extracellular matrix (ECM) secretion and myofibroblast differentiation (at least in vitro). Differentiation is negatively impacted by serum amyloid P. Fibrocytes may influence angiogenesis via secretion of VEGF and may promote tissue destruction and migration of resident cells via MMPs. Finally, fibrocytes likely secrete factors such as TGFβ and periostin to promote activation of resident lung fibroblasts and to promote EMT in lung epithelial cells.
Abbreviations
- AE-IPF
acute exacerbation of idiopathic pulmonary fibrosis
- ALI
acute lung injury
- ASM
airway smooth muscle
- α-SMA
alpha smooth muscle actin
- BM
bone marrow
- DLCO
diffusion capacity for carbon monoxide
- DPLDs
diffuse parenchymal lung diseases
- ECM
extracellular matrix
- ELISA
enzyme linked immunosorbent assay
- EMT
epithelial-mesenchymal transition
- FEV1
forced expiratory volume in 1 second
- FITC
fluorescein isothiocyanate
- FVC
forced vital capacity
- GFP
green fluorescent protein
- Gy
grey
- IIP
idiopathic interstitial pnuemonias
- ILD
interstitial lung disease
- IPF
idiopathic pulmonary fibrosis
- MHC
major histocompatibility complex
- NSIP
non-specific interstitial pneumonia
- SSc-ILD
scleroderma associated interstitial lung disease
- TBI
total body irradiation
- TGFβ
transforming growth factor beta
- TLC
total lung capacity
- UIP
usual interstitial pneumonia
- VEGF
vascular endothelial growth factor
References
- 1.Kuhn C. Pathology. In: Phan S, Thrall R, editors. Pulmonary Fibrosis. Marcek Dekker, Inc.; New York: 1995. pp. 59–83. [Google Scholar]
- 2.Sime PJ, O’Reilly KM. Fibrosis of the lung and other tissues: new concepts in pathogenesis and treatment. Clin Immunol. 2001;99:308–319. doi: 10.1006/clim.2001.5008. [DOI] [PubMed] [Google Scholar]
- 3.Kuhn C, Boldt J, King TE, Jr, Crouch E, Vartio T, McDonald J. An immunohistochemical study of architectural remodeling and connective tissue synthesis in pulmonary fibrosis. Am Rev Respir Dis. 1989;140:1693–1703. doi: 10.1164/ajrccm/140.6.1693. [DOI] [PubMed] [Google Scholar]
- 4.Coultas D, Zumwalt R, Black W, Sobonya R. The epidemiology of interstitial lung disease. Am J Respir Crit Care Med. 1994;150:967–972. doi: 10.1164/ajrccm.150.4.7921471. [DOI] [PubMed] [Google Scholar]
- 5.Adamson IY, Young L, Bowden DH. Relationship of alveolar epithelial injury and repair to the induction of pulmonary fibrosis. Am J Pathol. 1988;130:377–383. [PMC free article] [PubMed] [Google Scholar]
- 6.American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. Am J Resp Crit Care Med. 2002;165:277–304. doi: 10.1164/ajrccm.165.2.ats01. [DOI] [PubMed] [Google Scholar]
- 7.Flaherty KR, Thwaite EL, Kazerooni EA, Gross BH, Toews GB, Colby TV, Travis WD, Mumford JA, Murray S, Flint A, Lynch JP, 3rd, Martinez FJ. Radiological versus histological diagnosis in UIP and NSIP: survival implications. Thorax. 2003;58:143–148. doi: 10.1136/thorax.58.2.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Monaghan H, Wells AU, Colby TV, du Bois RM, Hansell DM, Nicholson AG. Prognostic implications of histologic patterns in multiple surgical lung biopsies from patients with idiopathic interstitial pneumonias. Chest. 2004;125:522–526. doi: 10.1378/chest.125.2.522. [DOI] [PubMed] [Google Scholar]
- 9.Perez A, Rogers R, Dauber J. The prognosis of idopathic pulmonary fibrosis. Am J Respir Cell Mol Biol. 2003;29:S19–26. [PubMed] [Google Scholar]
- 10.Flaherty KR, Travis WD, Colby TV, Toews GB, Kazerooni EA, Gross BH, Jain A, Strawderman RL, Flint A, Lynch JP, Martinez FJ. Histopathologic variability in usual and nonspecific interstitial pneumonias. Am J Respir Crit Care Med. 2001;164:1722–1727. doi: 10.1164/ajrccm.164.9.2103074. [DOI] [PubMed] [Google Scholar]
- 11.Katzenstein AL, Zisman DA, Litzky LA, Nguyen BT, Kotloff RM. Usual interstitial pneumonia: histologic study of biopsy and explant specimens. Am J Surg Pathol. 2002;26:1567–1577. doi: 10.1097/00000478-200212000-00004. [DOI] [PubMed] [Google Scholar]
- 12.Kuhn C, McDonald J. The roles of the myofibroblast in idiopathic pulmonary fibrosis: ultrastructural and immunohistochemical features of sites of active extracellular matrix synthesis. Am J Pathol. 1991;138:1257–1265. [PMC free article] [PubMed] [Google Scholar]
- 13.Phan S. The myofibroblast in pulmonary fibrosis. Chest. 2002;122:286S–289S. doi: 10.1378/chest.122.6_suppl.286s. [DOI] [PubMed] [Google Scholar]
- 14.Serini G, Gabbiani G. Mechanisms of myofibroblast activity and phenotypic modulation. Exp Cell Res. 1999;250:273–283. doi: 10.1006/excr.1999.4543. [DOI] [PubMed] [Google Scholar]
- 15.Vaughan MB, Howard EW, Tomasek JJ. Transforming growth factor-beta1 promotes the morphological and functional differentiation of the myofibroblast. Exp Cell Res. 2000;257:180–189. doi: 10.1006/excr.2000.4869. [DOI] [PubMed] [Google Scholar]
- 16.Thannickal VJ, Lee DY, White ES, Cui Z, Larios JM, Chacon R, Horowitz JC, Day RM, Thomas PE. Myofibroblast differentiation by transforming growth factor-beta1 is dependent on cell adhesion and integrin signaling via focal adhesion kinase. J Biol Chem. 2003;278:12384–12389. doi: 10.1074/jbc.M208544200. Epub 12003 Jan 12316. [DOI] [PubMed] [Google Scholar]
- 17.Flaherty KR, Colby TV, Travis WD, Toews GB, Mumford J, Murray S, Thannickal VJ, Kazerooni EA, Gross BH, Lynch JP, 3rd, Martinez FJ. Fibroblastic foci in usual interstitial pneumonia: idiopathic versus collagen vascular disease. Am J Respir Crit Care Med. 2003;167:1410–1415. doi: 10.1164/rccm.200204-373OC. Epub 2003 Feb 1420. [DOI] [PubMed] [Google Scholar]
- 18.Goumans MJ, van Zonneveld AJ, ten Dijke P. Transforming growth factor beta-induced endothelial-to-mesenchymal transition: a switch to cardiac fibrosis? Trends Cardiovasc Med. 2008;18:293–298. doi: 10.1016/j.tcm.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 19.Black JL, Burgess JK, Johnson PR. Airway smooth muscle–its relationship to the extracellular matrix. Respir Physiol Neurobiol. 2003;137:339–346. doi: 10.1016/s1569-9048(03)00157-5. [DOI] [PubMed] [Google Scholar]
- 20.Duffield JS, Lupher M, Thannickal VJ, Wynn TA. Host Responses in Tissue Repair and Fibrosis. Annu Rev Pathol. 2012;22 doi: 10.1146/annurev-pathol-020712-163930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Willis BC, Liebler JM, Luby-Phelps K, Nicholson AG, Crandall ED, du Bois RM, Borok Z. Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-beta1: potential role in idiopathic pulmonary fibrosis. Am J Pathol. 2005;166:1321–1332. doi: 10.1016/s0002-9440(10)62351-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim KK, Kugler MC, Wolters PJ, Robillard L, Galvez MG, Brumwell AN, Sheppard D, Chapman HA. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc Natl Acad Sci U S A. 2006;103:13180–13185. doi: 10.1073/pnas.0605669103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest. 2002;110:341–350. doi: 10.1172/JCI15518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rock JR, Barkauskas CE, Cronce MJ, Xue Y, Harris JR, Liang J, Noble PW, Hogan BL. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc Natl Acad Sci U S A. 1475;108:28. doi: 10.1073/pnas.1117988108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abe R, Donnelly SC, Peng T, Bucala R, Metz CN. Peripheral blood fibrocytes: differentiation pathway and migration to wound sites. J Immunol. 2001;166:7556–7562. doi: 10.4049/jimmunol.166.12.7556. [DOI] [PubMed] [Google Scholar]
- 26.Bucala R, Spiegel LA, Chesney J, Hogan M, Cerami A. Circulating fibrocytes define a new leukocyte subpopulation that mediates tissue repair. Mol Med. 1994;1:71–81. [PMC free article] [PubMed] [Google Scholar]
- 27.Tanjore H, Xu XC, Polosukhin VV, Degryse AL, Li B, Han W, Sherrill TP, Plieth D, Neilson EG, Blackwell TS, Lawson WE. Contribution of epithelial-derived fibroblasts to bleomycin-induced lung fibrosis. Am J Respir Crit Care Med. 2009;180:657–665. doi: 10.1164/rccm.200903-0322OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim KK, Kugler MC, Wolters PJ, Robillard L, Galvez MG, Brumwell AN, Sheppard D, Chapman HA. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc Natl Acad Sci U S A. 2006;103:13180–13185. doi: 10.1073/pnas.0605669103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chesney J, Bacher M, Bender A, Bucala R. The peripheral blood fibrocyte is a potent antigen-presenting cell capable of priming naive T cells in situ. Proc Natl Acad Sci U S A. 1997;94:6307–6312. doi: 10.1073/pnas.94.12.6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chesney J, Metz C, Stavitsky AB, Bacher M, Bucala R. Regulated production of type I collagen and inflammatory cytokines by peripheral blood fibrocytes. J Immunol. 1998;160:419–425. [PubMed] [Google Scholar]
- 31.Moore BB, Thannickal VJ, Toews GB. Bone Marrow-Derived Cells in the Pathogenesis of Lung Fibrosis. Curr Respir Med Rev. 2005;1:69–76. doi: 10.2174/1573398052953613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Epperly M, Guo H, Gretton J, Greenberger J. Bone Marrow Origin of Myofibroblasts in Irradiation Pulmonary Fibrosis. Am J Resp Crit Care Med. 2003;29:213–224. doi: 10.1165/rcmb.2002-0069OC. [DOI] [PubMed] [Google Scholar]
- 33.Hashimoto N, Jin H, Liu T, Chensue S, Phan S. Bone marrow-derived progenitor cells in pulmonary fibrosis. J Clin Invest. 2004;113:243–252. doi: 10.1172/JCI18847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moore BB, Kolodsick JE, Thannickal VJ, Cooke K, Moore TA, Hogaboam C, Wilke CA, Toews GB. CCR2-mediated recruitment of fibrocytes to the alveolar space after fibrotic injury. Am J Pathol. 2005;166:675–684. doi: 10.1016/S0002-9440(10)62289-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tanjore H, Xu XC, Polosukhin VV, Degryse AL, Li B, Han W, Sherrill TP, Plieth D, Neilson EG, Blackwell TS, Lawson WE. Contribution of epithelial-derived fibroblasts to bleomycin-induced lung fibrosis. Am J Respir Crit Care Med. 2009;180:657–665. doi: 10.1164/rccm.200903-0322OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Phillips RJ, Burdick MD, Hong K, Lutz MA, Murray LA, Xue YY, Belperio JA, Keane MP, Strieter RM. Circulating fibrocytes traffic to the lungs in response to CXCL12 and mediate fibrosis. J Clin Invest. 2004;114:438–446. doi: 10.1172/JCI20997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schmidt M, Sun G, Stacey M, Mori L, Mattoli S. Identification of Circulating Fibrocytes as Precursors of Bronchial Myofibroblasts in Asthma. J Immunol. 2003;170:380–389. doi: 10.4049/jimmunol.171.1.380. [DOI] [PubMed] [Google Scholar]
- 38.Garibaldi BT, D’Alessio FR, Mock JR, Files DC, Chau E, Eto Y, Drummond MB, Aggarwal NR, Sidhaye V, King LS. Regulatory T cells reduce acute lung injury fibroproliferation by decreasing fibrocyte recruitment. Am J Respir Cell Mol Biol. 2013;48:35–43. doi: 10.1165/rcmb.2012-0198OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harris DA, Zhao Y, Lapar DJ, Emaminia A, Steidle JF, Stoler M, Linden J, Kron IL, Lau CL. Inhibiting CXCL12 blocks fibrocyte migration and differentiation and attenuates bronchiolitis obliterans in a murine heterotopic tracheal transplant model. J Thorac Cardiovasc Surg. 2012 doi: 10.1016/j.jtcvs.2012.03.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Field JJ, Burdick MD, DeBaun MR, Strieter BA, Liu L, Mehrad B, Rose CE, Jr, Linden J, Strieter RM. The role of fibrocytes in sickle cell lung disease. PLoS One. 2012;7:e33702. doi: 10.1371/journal.pone.0033702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.LaPar DJ, Burdick MD, Emaminia A, Harris DA, Strieter BA, Liu L, Robbins M, Kron IL, Strieter RM, Lau CL. Circulating fibrocytes correlate with bronchiolitis obliterans syndrome development after lung transplantation: a novel clinical biomarker. Ann Thorac Surg. 1016;92:470–477. doi: 10.1016/j.athoracsur.2011.04.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nihlberg K, Larsen K, Hultgardh-Nilsson A, Malmstrom A, Bjermer L, Westergren-Thorsson G. Tissue fibrocytes in patients with mild asthma: a possible link to thickness of reticular basement membrane? Respir Res. 2006;7:50. doi: 10.1186/1465-9921-7-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang CH, Huang CD, Lin HC, Lee KY, Lin SM, Liu CY, Huang KH, Ko YS, Chung KF, Kuo HP. Increased circulating fibrocytes in asthma with chronic airflow obstruction. Am J Respir Crit Care Med. 2008;178:583–591. doi: 10.1164/rccm.200710-1557OC. [DOI] [PubMed] [Google Scholar]
- 44.Saunders R, Siddiqui S, Kaur D, Doe C, Sutcliffe A, Hollins F, Bradding P, Wardlaw A, Brightling CE. Fibrocyte localization to the airway smooth muscle is a feature of asthma. J Allergy Clin Immunol. 2009;123:376–384. doi: 10.1016/j.jaci.2008.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bellini A, Marini MA, Bianchetti L, Barczyk M, Schmidt M, Mattoli S. Interleukin (IL)-4, IL-13, and IL-17A differentially affect the profibrotic and proinflammatory functions of fibrocytes from asthmatic patients. Mucosal Immunol. 2012;5:140–149. doi: 10.1038/mi.2011.60. [DOI] [PubMed] [Google Scholar]
- 46.Mathai SK, Gulati M, Peng X, Russell TR, Shaw AC, Rubinowitz AN, Murray LA, Siner JM, Antin-Ozerkis DE, Montgomery RR, Reilkoff RA, Bucala RJ, Herzog EL. Circulating monocytes from systemic sclerosis patients with interstitial lung disease show an enhanced profibrotic phenotype. Lab Invest. 2010;90:812–823. doi: 10.1038/labinvest.2010.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gan Y, Reilkoff R, Peng X, Russell T, Chen Q, Mathai SK, Homer R, Gulati M, Siner J, Elias J, Bucala R, Herzog E. Role of semaphorin 7a signaling in transforming growth factor beta1-induced lung fibrosis and scleroderma-related interstitial lung disease. Arthritis Rheum. 2011;63:2484–2494. doi: 10.1002/art.30386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tourkina E, Bonner M, Oates J, Hofbauer A, Richard M, Znoyko S, Visconti RP, Zhang J, Hatfield CM, Silver RM, Hoffman S. Altered monocyte and fibrocyte phenotype and function in scleroderma interstitial lung disease: reversal by caveolin-1 scaffolding domain peptide. Fibrogenesis Tissue Repair. 2011;4:15. doi: 10.1186/1755-1536-4-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tourkina E, Richard M, Oates J, Hofbauer A, Bonner M, Gooz P, Visconti R, Zhang J, Znoyko S, Hatfield CM, Silver RM, Hoffman S. Caveolin-1 regulates leucocyte behaviour in fibrotic lung disease. Ann Rheum Dis. 2010;69:1220–1226. doi: 10.1136/ard.2009.117580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mehrad B, Burdick MD, Zisman DA, Keane MP, Belperio JA, Strieter RM. Circulating peripheral blood fibrocytes in human fibrotic interstitial lung disease. Biochem Biophys Res Commun. 2007;353:104–108. doi: 10.1016/j.bbrc.2006.11.149. [DOI] [PubMed] [Google Scholar]
- 51.Andersson-Sjoland A, de Alba CG, Nihlberg K, Becerril C, Ramirez R, Pardo A, Westergren-Thorsson G, Selman M. Fibrocytes are a potential source of lung fibroblasts in idiopathic pulmonary fibrosis. Int J Biochem Cell Biol. 2008;40:2129–2140. doi: 10.1016/j.biocel.2008.02.012. [DOI] [PubMed] [Google Scholar]
- 52.Moeller A, Gilpin SE, Ask K, Cox G, Cook D, Gauldie J, Margetts PJ, Farkas L, Dobranowski J, Boylan C, O’Byrne PM, Strieter RM, Kolb M. Circulating fibrocytes are an indicator of poor prognosis in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2009;179:588–594. doi: 10.1164/rccm.200810-1534OC. [DOI] [PubMed] [Google Scholar]
- 53.Collard HR, Moore BB, Flaherty KR, Brown KK, Kaner RJ, King TE, Jr, Lasky JA, Loyd JE, Noth I, Olman MA, Raghu G, Roman J, Ryu JH, Zisman DA, Hunninghake GW, Colby TV, Egan JJ, Hansell DM, Johkoh T, Kaminski N, Kim DS, Kondoh Y, Lynch DA, Muller-Quernheim J, Myers JL, Nicholson AG, Selman M, Toews GB, Wells AU, Martinez FJ. Acute exacerbations of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2007;176:636–643. doi: 10.1164/rccm.200703-463PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fujiwara A, Kobayashi H, Masuya M, Maruyama M, Nakamura S, Ibata H, Fujimoto H, Ohnishi M, Urawa M, Naito M, Takagi T, Kobayashi T, Gabazza EC, Takei Y, Taguchi O. Correlation between circulating fibrocytes, and activity and progression of interstitial lung diseases. Respirology. 2012;17:693–698. doi: 10.1111/j.1440-1843.2012.02167.x. [DOI] [PubMed] [Google Scholar]
- 55.Moore BB, Murray L, Das A, Wilke CA, Herrygers AB, Toews GB. The role of CCL12 in the recruitment of fibrocytes and lung fibrosis. Am J Respir Cell Mol Biol. 2006;35:175–181. doi: 10.1165/rcmb.2005-0239OC. Epub 2006 Mar 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Naik PK, Bozyk PD, Bentley JK, Popova AP, Birch CM, Wilke CA, Fry CD, White ES, Sisson TH, Tayob N, Carnemolla B, Orecchia P, Flaherty KR, Hershenson MB, Murray S, Martinez FJ, Moore BB. Periostin promotes fibrosis and predicts progression in patients with idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2012;303:L1046–L1056. doi: 10.1152/ajplung.00139.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pilling D, Buckley CD, Salmon M, Gomer RH. Inhibition of fibrocyte differentiation by serum amyloid P. J Immunol. 2003;171:5537–5546. doi: 10.4049/jimmunol.171.10.5537. [DOI] [PMC free article] [PubMed] [Google Scholar]