

Abstract
Ubiquitination, the structured degradation and turnover of cellular proteins, is regulated by the ubiquitin–proteasome system (UPS). Most proteins that are critical for cellular regulations and functions are targets of the process. Ubiquitination is comprised of a sequence of three enzymatic steps, and aberrations in the pathway can lead to tumor development and progression as observed in many cancer types. Recent evidence indicates that targeting the UPS is effective for certain cancer treatment, but many more potential targets might have been previously overlooked. In this review, we will discuss the current state of small molecules that target various elements of ubiquitination. Special attention will be given to novel inhibitors of E3 ubiquitin ligases, especially those in the SCF family.
Keywords: : cancer therapeutics, deubiquitinase, E3 ligase inhibition, high-throughput virtual screening, hot spots, in silico modeling, SCF complex, Skp2 inhibitors, small molecule inhibitors, ubiquitination
Ubiquitination, a step in the nonlysosomal degradation of proteins, is a crucial post-translational modification in eukaryotic organisms. Rapid and timely degradation of transcriptional regulators and other proteins by the ubiquitin–proteasome system (UPS) regulates a wide variety of cellular processes [1]. Ubiquitination involves covalent attachment of ubiquitin, a small 8-kDa protein, to a substrate and results in recognition and shuttling of the substrate to the 26S proteasome complex for degradation [2]. It is important to note that the ubiquitination process combined with the proteasome complex step is also referred to as the ubiquitin–proteasome system (UPS) or ubiquitin proteasome pathway (UPP).
The ubiquitination process is tightly controlled by three families of enzymes: ubiquitin-activating enzymes (E1s), ubiquitin-conjugating enzymes (E2s), and finally ubiquitin-protein enzymes (E3s). There exists two E1 enzymes with ubiquitin-activating capability: UBA1 being the primary E1 and the recently discovered UBA6 with unclear functions and uncharacterized regulations [3,4]. In contrast to the small number of E1s, there are approximately 40 E2s [5,6] and 500–1000 human E3 ligases, providing both specificity and versatility [7]. The three steps of the ubiquitination process (Figure 1) have been reviewed previously [8,9]. Briefly, the activation step requires binding of both ATP and ubiquitin and links the α-carboxyl group of the C-terminal glycine residue of ubiquitin to a cysteine residue on E1, and a thioester linkage is formed between the ubiquitin and E1.
Figure 1. . Select targeting strategies for the ubiquitin proteasome pathway.

Broad targeting of E1, E2 and proteasome are possible, but targeting the E3 enzymes offers specificity. Here the E3 ligase is represented by the SCFSKp2, an E3 that has multiple regions on which small molecules have been designed. Also of interest are DUB inhibitors.
DUB: Deubiquitination enzyme.
Then the E2 binds to both activated ubiquitin and the E1 enzyme and thus transfers the ubiquitin from E1 to the active site cysteine of the E2 via a trans(thio)esterification reaction. Finally, the E3 catalyzes the linking of ubiquitin to a lysine residue on the substrate. Repetitions of these sequential steps results in a long chains of ubiquitin (polyubiquitin) on the protein to be degraded, and the specific lysine residue on ubiquitin used for linking (e.g., K48, K63, etc.) results in different topologies [10]. Ubiquitination was originally described as a mechanism by which cells dispose of short-lived, damaged, or abnormal proteins, but more recent studies have revealed that it also plays a significant role in post translational modification. Ubiquitination can result in the addition of a single ubiquitin moiety, called monoubiquitination, rather than polyubiquitination. Generally, polyubiquitination reactions are formed on the K48 residue, and this process tags substrates for proteasomal degradation and recycling [11]. On the other hand, the K63-linked nonproteolytic ubiquitination spares proteins from degradation and regulates localization and activity of multiple kinases and pathways, such as PKB/Akt, TAK1, IKKγ/NEMO, TNFR, IRAK1, MLK3, IGF-1R, T-cell receptor (TCR), NOD-like receptor (NLR) and RIG-I-like receptor pathways. This type of ubiquitination can cooperate with other linkage types to achieve the physiologically required output of a signaling pathway [10,12–13] and, therefore, has been crucially implicated in diverse biological processes including signal transduction, transcriptional regulation, growth response, innate immune response and DNA repair and replication [12–14].
Ubiquitination in cancer
Ubiquitination can affect cancer development and progression in many ways. Both tumor suppressing and promoting pathways have elements that are tightly regulated by the process. One fundamental aspect of cancer is the deregulation of the cell cycle and checkpoint control [15], which is highly regulated through constant synthesis coupled to a particular timeframe of specific proteolysis of cyclins, cyclin-dependent kinases (CDKs) as well as CDK inhibitors (CKIs) executed by the UPS [16]. Another well-known example is the E3 ligase MDM2 which bind to the tumor suppressor protein p53 that is inactivated in more than 50% of human cancers. Also, mutations and alterations in ubiquitin ligases are found in a wide variety of tumor types and tremendously impact clinical outcomes [17–20].
In addition to the above proteolytic polyubiquitination, which may contribute to cancer development, it is worth mentioning that monoubiquitination has unique effects on cancer as well [13]. Monoubiquitin can serve as a recruitment signal to proteins that contain ubiquitin binding domains, and the functions of such nonproteolytic ubiquitination include, but not limited to: altered protein activity, subcellular localization, enzyme activation, DNA repair, chromatin dynamics [12,21–23] and transcriptional regulation [24,25]. These facts underscore the importance of ubiquitination in tumorigenesis and the resulting interest as a clinical target.
Strategies of targeting the UPS pathway
The three steps in the ubiquitination pathway are commonly considered to be enzymatic reactions, and many strategies could be used to target different stages of the UPS pathway (Figure 1). The most straightforward strategy is to target the substrate binding site. Along this line, multiple proteasome inhibitors have been successfully developed and approved [26–29]. Theoretically targeting the E1 enzyme is also possible, however, due to the lack of specificity (only two E1 enzymes), inhibition of this stage is not therapeutically sound and there are few E1 inhibitors reported [30–32]). Despite this, two compounds from Millennium/Takeda, MLN7243 and MLN4924, which are reported as UBA1 and NEDD8-activating enzyme (NAE1) inhibitors [33–35], respectively, are currently in Phase I/II and Phase I clinical trials (ClinicalTrials.gov identifiers: NCT02045095 and NCT02122770). It is worth noting that protein–protein interactions play an important role in E2 and E3 functions. Therefore, one popular strategy is to disrupt the protein–protein interactions (PPIs) that are the interfaces of these ubiquitination complexes. However, discovery of PPI small molecule inhibitors requires a rigorous understanding of the structures and energetics of the proteins involved. As the energy contribution of all residues in a PPI is not uniform, some residues contribute more than others to the energy of binding and they are referred to as hot spot residues. Technically speaking these residues contribute more than 2 kcal/mol to the total binding energy of the complex. While experimental techniques such as alanine scanning can determine hot spots, it is slow and costly. Many computational techniques are available to predict hot spot residues, and they can be used to aid the design of PPI inhibitors [36].
Targeting the proteasome
The first therapeutic proteasome inhibitor tested in humans was bortezomib (Velcade), which was first synthesized in 1995, entered clinical trials in 1998, and approved by the FDA for use in multiple myeloma (MM) in 2003 (Table 1) [26]. The clinical antitumor activity of bortezomib is well established as both a single-use agent and combination in the treatment of MM and other hematological malignancies. Bortezomib also exhibits efficacy in nonsmall cell lung cancer and pancreatic cancer [37,38], and more recently it was expanded for use in patients with mantle cell lymphoma (MCL). High affinity and specificity of binding of bortezomib is achieved partly through the boron atom to the 26S proteasome’s catalytic site [39–41]. Consequently, proteasome inhibition alters the balance of all intracellular peptides, increasing those that require cleavage at acidic and hydrophobic sites and causing side effects such as neuropathy and autophagy in certain conditions [42–45]. As is an issue with many cancer therapeutics, resistance can develop quickly and this occurs with bortezomib on average in about one year [24,46,47]. Another notable proteasome inhibitor approved recently by the US FDA is carfilzomib (marketed under the trade name Kyprolis), and is used for relapsed and refractory MM who have been previously treated with bortezomib [48,49]. Carfilzomib is also approved for MM and was derived from epoxomicin, a natural product that was shown to contain potent anti-inflammatory activity and proteasome inhibition [50].
Table 1. . Selected compounds targeting elements of the ubiquitin–proteasome system and their current status in the drug development pipeline.
| Class | Drug name | Target | Status | Structure | Ref. |
|---|---|---|---|---|---|
| Proteasome inhibitors | Bortezomib | 20S proteasome | US FDA approved for MM and mantle cell lymphoma | 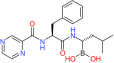 |
[26] |
| Carfilzomib | 20S proteasome | FDA approved for relapsed and refractory MM | [48,49] | ||
| Marizomib | 20S proteasome | Phase I | [28] | ||
| Ixazomib (MLN-9708) | 20S proteasome | Phase I |  |
[29] | |
| |
CEP-18770 |
20S proteasome |
Phase II |
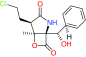 |
[51] |
| E2 | Leucettamol A | Ubc13–Uev1A interaction | Preclinical/research | [52,53] | |
| |
CC0651 |
Cdc34 |
Preclinical/research |
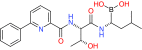 |
[54] |
| E3 immunomodulators | Thalidomide | CRBN | FDA approved for treatment of MM | 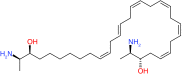 |
[55] |
| Lenalidomide | CRBN | FDA approved for treatment of MM | 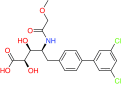 |
[56] | |
| |
Pomalidomide |
CRBN |
FDA approved for refractory MM |
 |
[57] |
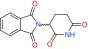
| E3-p53 potentiators | Nutlin-3a | MDM2/MDMX antagonist | Multiple Phase I trials | 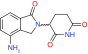 |
[58,59] |
| RG7112 | MDM2/MDMX | Phase I | 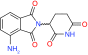 |
[60] | |
| ATSP-7041 | Dual inhibition of MDM2 and MDMX | Preclinical/research | 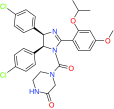 |
[61,62] | |
| |
NSC207895 |
Dual inhibition of MDM2 and MDMX |
Preclinical/research |
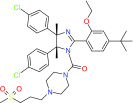 |
[63] |
| E3-F-box protein antagonists | NSC689857 | Skp2 | Preclinical/research | 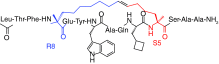 |
[64] |
| NSC681152 | Skp2 | Preclinical/research | [64] | ||
| SCF-I2 | Cdc4 | Preclinical/research | 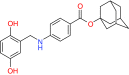 |
[65] | |
| C1, C2, C16, C20 | Skp2 | Preclinical/research |  |
[66] | |
| Compound A | Skp2 | Preclinical/research | 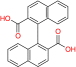 |
[67] | |
| Compound ZL25 | Skp2 | Preclinical/research | 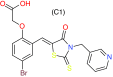 |
[68] | |
| GS143 | βTrCP | Preclinical/research | 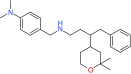 |
[69] | |
| Erioflorin | βTrCP | Preclinical/research | 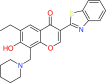 |
[70] | |
| BC-1215 | Fbxo3 | Preclinical/research | 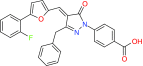 |
[71] | |
| |
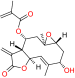
SMER3 |
Met30 |
Preclinical/research |
 |
[72] |
| DUB inhibitors | WP1130 | USPs: (5, 9, 14), UCH-L5 | Preclinical/research | 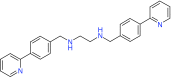 |
[73] |
| IU1 | USP14 | Preclinical/research | [74] | ||
| NSC632839 | USP2, USP7 | Preclinical/research | 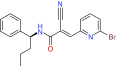 |
[75] | |
| PR-619 | Broad-spectrum DUB inhibitor | Preclinical/research |  |
[76] |
CRBN: Cereblon; DUB: Deubiquitination enzyme; MM: Multiple myeloma.
Other next generation proteasome inhibitors are in clinical trials and have the potential to achieve a better therapeutic ratio and reduced probability of inherent and acquired resistance. An example is the structurally and pharmacologically unique marizomib (also called salinosporamide A) currently in Phase I trials [28]. It is a natural product produced from marine bacteria, and its mechanism of action is unique in that it irreversibly and covalently modifies the active site threonine residues of the 20S proteasome [77,78]. Moreover, a Phase Ib clinical study of the combination of marizomib with vorinostat has just completed but the results have not yet been posted with nonsmall lung cancer patients (ClinicalTrials.gov identifier NCT00667082).
Finally, two other reversible peptide boronic acid-based proteasome inhibitors in different stages of development are CEP-18770 and MLN-9708 [27,79]. CEP-18770 has been tested in Phase I trials for solid tumors and non-Hodgkin’s lymphoma, and entered Phase II trials for relapsed and refractory MM by itself and in combination with lenalidomide (a thalidomide derivative) and dexamethasone (ClinicalTrials.gov identifiers NCT00572637, NCT01023880 and NCT01348919). It has shown in vitro antiangiogenic activity and potently represses RANKL-induced osteoclastogenesis, and is not cytotoxic to normal human epithelial cells and bone marrow-derived stromal cells [51]. MLN-9708 (also called Ixazomib) has shown great promise and advanced to Phase III trials; it is also being considered in combination with lenalidomide and dexamethasone (ClinicalTrials.gov identifier NCT01564537). In Phase II trials, while showing adverse side effects, it was generally well tolerated and the majority of patients (58%) had a very good partial response or better [29].
Targeting E2 enzymes
While few therapies targeting the E2 enzymes are in development (relative to proteasome and E3 inhibition), there are some recent examples worth mentioning that are in preclinical stages (Table 1). Leucettamol A, which was isolated from the sea sponge Leucetta aff. Microrhaphis, was shown to inhibit the Ubc13-Uev1A interaction, blocking the E1-E2 complex formation [52]. It was determined that, contrary to an earlier assessment that this was a racemic compound, it is in fact a chiral and optically active compound [53]. Another marine sponge, Lissodendryx fibrosa was the source for manadosterols A and B which inhibited the same Ubc13–Uev1A interaction with higher potency than leucettamol A [80]. CC0651 was found to allosterically inhibit the E2 enzyme Cdc34 and cause accumulation of p27 by inserting itself into a cryptic pocket distant from the catalytic site, causing displacement of secondary structural elements [54]. However, targeting the E2 is still lacking sufficient specificity compared with E3 inhibitors as described in the following sections.
Targeting E3 ligases
E3 ubiquitin ligases represent a diverse set of enzymes and provide the specificity of the ubiquitination reaction, and have significant roles in many different diseases, especially cancer. The E3 ligases are currently broadly classified into two major families from their structural motifs: Homologous to the E6-AP Carboxyl Terminus (HECT) type and Really Interesting New Gene (RING) type; RING domain ligases are the most common [81]. There are also two RING-finger derivative domains: UFD2-homology domain (U-box) and plant homeo domain (PHD) E3 types, but RING-finger types are the largest groups of E3 ligases.
E3 elements in the UPS are considered to be primarily responsible for the specific recognition of a large number of target proteins, acting as the substrate recognition component of the UPS pathway [2]. This specificity is crucial when considering the prospect of designing drugs for the entire ubiquitination pathway, as they can be designed to target specific substrates of the E3 ligase without affecting other substrates. As a result, the E3 ubiquitin ligases are drug targets with the most potential for cancer therapies as they have fewer targets and offer a higher specificity of the system [82,83]. There are several notable E3 ligase targeting agents that are worthy of mentioning, and there have been many recent developments that show promising results in preclinical testing (Table 1).
Thalidomide & its derivatives
Thalidomide was originally developed as a sedative and an agent to cure hyperemesis gravidarum in pregnant women, and became infamous for its limb formation birth defects. Although banned after discovery of these effects in 1962, it had off-label use for patients of erythema nodosum leprosum (ENL); it was this use that led to clinical trials of its assessment and characterization of its immunomodulatory effects. Its primary target has been identified to be cereblon (CRBN), which forms an E3 ligase complex with DDB1 and Cul4A that is important for limb development, and binding of thalidomide to this complex appears to be the mechanism for the teratogenic effects in embryonic development [55]. Two optimized second-generation derivatives of thalidomide, lenalidomide and pomalidomide [56,57], are also used for MM by targeting CRBN and modulate its specificity as a substrate receptor, not by inhibition, rather by selective enhancement of the ubiquitination and degradation of Ikaros 1 and 3 (IKZF1 and IKZF3) zinc finger transcription factors [84]. A crystal structure of lenalidomide in complex with CBRN shows the binding mode and that it binds to the substrate binding domain of CRBN and blocks ubiquitination [85–87].
MDM2/p53
The tumor suppressor protein p53 has been called the ‘guardian of the genome’ due to its critical role in inducing cell cycle arrest and apoptosis in response to DNA damage. There exist over ten E3s associated with the regulation of p53, but the one with the most unquestionable importance has been MDM2 (murine double minute 2) [88]. The p53-binding domain of MDM2 is at the N-terminus, and the RING domain in the C-terminus acts as the ubiquitin ligase to promote rapid degradation of p53 after its export from the nucleus [89–91]. Inhibition of the MDM2/p53 interaction has been achieved by the small molecule nutlins, which have completed Phase I clinical trials and [58,59]. Nutlins are the general name given to compounds based on a cis-imidazoline scaffold, and their derivatives offer better activities but still retain elements of the core structure. Serdemetan was tested in a Phase I trial and showed good p53 induction, but cardiac construction effects were observed [71,72,92]. Nutlin-3a (a more promising agent) was optimized for better pharmacological properties to become RG7112 (also known as RO5045337); it stabilized p53 and activated the p53 pathway in cancer cells, and is being tested both as a monotherapy and in combination and some Phase I trials are still ongoing (NCT01677780) [60,93]. Nutlins and their derivatives are competitive inhibitors and structural mimics of p53 (via Phe19, Trp23 and Leu26) [58]. MDMX (murine double minute X – also known as HDMX in humans, or also MDM4 or HDM4) shares significant homology with MDM2 and is also a negative regulator of p53, but nutlin-3 has shown decreased effectiveness in inhibiting MDMX-p53 interactions due to its differential binding [94]. This highlights one limitation of MDM2-targeting agents such as nutlins: tumors with high MDMX and low MDM2 expression respond poorly to MDM2 inhibition [61]. To combat this problem, a dual inhibitor of MDM2 and MDMX (ATSP-7041) was discovered and activates the p53 pathway in vitro and in vivo [61,62]. Another compound overcoming the limitation, NSC207895, was found to target MDMX specifically and acted additively with nutlin-3a to activate p53 and decrease cancer cell viability [63].
SCF E3 ligases
The RING-finger type SCF (Skp1/cullin/F-box) E3 ligases are the largest family of E3 ubiquitin ligases, and they consist primarily of three core subunits: an F-box protein that contributes to the specificity of the SCF, Skp1 that acts as a bridging protein and binds to the F-box, and Cul1 (one of the seven family members in the Cullin family) that forms the major structural scaffold of the SCF. The majority of the substrates of SCF E3 ligases are involved in regulating cell cycle progression, gene transcription, DNA replication and signal transduction [7,95,96]. A growing body of evidence suggests that phenotypes such as genomic instability, uncontrolled proliferation and cancer result from the dysregulation of these E3s [7]. Their potential as drug targets has been only been uncovered very recently, and as the F-box proteins provide the specificity of the SCF complex, many small molecules have been designed to target specific members (Table 1).
SMER3 was discovered from a yeast-based screen of rapamycin enhancers, and blocks the SCFMet30 in vivo and in vitro but not the closely related SCFCdc4. SMER3 was also demonstrated to directly bind to Met30 and prevents degradation of Met4, an antiproliferative transcriptional activator [97]. Small molecule screens identified SCF-I2, which inhibits the F-box protein Cdc4 in yeast but not its human ortholog Fbxw7 [65]. Its specificity can be explained by an allosteric mechanism: it binds between two beta strands in the WD40 domain of Cdc4 that is 25Å away from the substrate binding site. While SCF-I2 failed to inhibit Cdc4 activity in vivo, it did show the potential of using allosteric inhibition of the WD40 domain.
Another example is BC-1215 which was recently synthesized to inhibit Fbxo3 and blocks degradation of another F-box protein (Fbx12) which, in turn, degrades the TNF receptor-associated factor (TRAF) adaptor proteins that are responsible for cytokine secretion [71]. This resulting TRAF inhibition by BC-1215 dampens NF-κB activation through the TNF signaling cascade [98].
βTrCP is an E3 ligase that binds to Skp1 and promotes degradation of a breadth of key regulatory protein elements in cancer biogenesis, including: pro-caspase-3, WEE1, MCL1, p100, p105 and CD4. In most cases, it functions as an oncoprotein and has been found with upregulated mRNA levels in many cancer types [99,100]. GS143 and Erioflorin both block interaction between βTrCP and their targets: phospho-IκBα and Pdcd4, respectively [69,70]. Inhibition of βTrCP can result in tumor cell arrest at multiple points in the cell cycle at nanomolar concentrations and the inhibitors show promising results in vitro [101,102].
Skp2, a notable F-box protein
Skp2 (S-phase-kinase-associated protein-2, also called FBXL1) is another highly significant F-box protein and our lab has recently reported that it is an ideal target for cancer therapeutics development. Skp2 binds to Skp1 in the SCFSkp2 complex, and it regulates cell cycle progression, proliferation and apoptosis by promoting the ubiquitination and degradation of several negative cell cycle regulator such as p130, p57, and most notable are the p27 and p21 (also known as CDKN1A) CKIs [7,95,96,103]. Recently some novel biological functions of Skp2 were identified and it can activate Akt through nonproteolytic K63-linked ubiquitination for glycolysis (via activating Glut1 expression) [104]. It has been reported that many human cancers have overexpression of Skp2, and this overexpression promotes cancer invasion and metastasis [105–107]. Also, a recent report shows that Skp2 activates LKB1 (via K-63 ubiquitination) for cancer cell survival under energy stress via oncogenic Ras upstream of Skp2 [108]. This indicates that the Ras/Skp2/LKB1 axis can be another potential means for cancer therapy.
Inhibitors disrupting Skp2–Skp1 interactions
Using the high-quality crystal structures of Skp2 in complex with Skp1 [109,110] as a basis for hot spot analysis, along with our unique in silico modeling/screening work flow (Figure 2), we discovered a series of active compounds, with compound ZL25 being selected as the lead compound for further studies. Compound ZL25 binds directly to the F-box domain of Skp2 and disrupts the binding of Skp2 to Skp1. This compound phenocopies the effects seen with Skp2 deficiency: augmenting p27-mediated apoptosis/senescence and impairing Akt-regulated glycolysis [68]. Compound ZL25 was able to stabilize expression of p27 and p21, but not other F-box proteins including Fbw7 and β-TrCP, indicating its selectivity to the SCFSkp2 complex. Skp2 mutagenesis experiments confirmed that the critical residues for binding were Trp97 and Asp98, as these mutants were unable to bind to compound ZL25, and structure-activity relationship (SAR) studies validated the chemical components of compound ZL25 that are needed for its PPI disruption qualities [68] (Figure 3). As senescence and glycolysis are significant for cancer stem cells (CSC), compound ZL25 was also shown to reduce CSC populations in a dose dependent manner. Finally, antitumor activity of compound ZL25 was validated in (but not necessarily limited to) breast, lung and prostate mouse xenograft cancer models [68]. We expect that compound ZL25 will be active in other solid tumors, and we are continuing to examine the mechanism of binding in the SCFSkp2 complex and design derivatives that will have higher potency. Further preclinical development of compound ZL25 is currently ongoing and we will modify the structure to increase both the binding affinity and E3 ligase inhibition. Our latest data and SAR analysis demonstrates that the benzothiazole position of the chromone core is critical for its binding to Skp2 and modifications to a comparable moiety such as benzoimidazole retained the activities. The ethyl group also contributes significantly to the enzymatic activities, and changes to a larger moiety such as butyl clearly enhance the activity of disrupting Skp2–Skp1 interactions. This is further corroborated by observations that the activity is also improved by substitution with piperidinyl and piperazinyl groups. In addition, the chromone core of compound ZL25 can be altered to chromen-2-one which exhibited comparable ability to the lead (ZL25) in preventing Skp2–Skp1 interactions. Moreover, we are investigating the potential of compound ZL25 that may have an impact on obesity and other metabolic disorders, as genetic ablation of Skp2 protects mice from obesity due to adipocyte reduction [111,112].
Figure 2. . Hit identification work flow used by our group.

Figure 3. . Compound ZL25 in complex with the Skp2 F-box domain.

Mutational analysis confirmed that the critical residues for binding are Trp97 and Asp98, as compound #25 failed to inhibit E3 ligase activity of these mutant proteins (W97A, D98A).
Targeting the Skp2/Cks1 interaction
While the SCFSkp2 complex requires that Skp1 bind to Skp2 in the F-box domain, the accessory protein Cdc kinase subunit 1 (Cks1) must also bind to Skp2 in the leucine-rich region (LLR) on Skp2 to recruit substrate including p27 [113]. Binding of p27 to Cks1/Skp2 occurs after p27 is phosphorylated on Thr187 by cyclin E-CDK2 [66]. Phosphorylation of Thr187 on p27 is required for its ubiquitination, and the critical Glu185 and phospho-Thr187 residues on p27 create a pocket on Cks1/Skp2 to which small molecules can bind [110,114,115]. Additionally, our group had recent success in screening small molecules that target the Skp2-Cks1/p27 interface (unpublished). These compounds have binding affinities in the high nanomolar concentration, and interact with both pockets occupied by Glu185 and the Thr187 of p27. They effectively inhibited p27 ubiquitination/degradation and significantly restored the p27 levels from cytoplasm to nucleus in a variety of cancer cell types. Similarly another group identified a series of inhibitors through in silico screens (called C1, C2, C16 and C20) that target the binding interface for p27 on Cks1/Skp2 [66]. Increased p27 protein levels and half-life were reported to increase in a Skp2-dependent manner in metastatic melanoma cell lines upon inhibitor treatment. Independently, two compounds (NSC689857 and NSC681152) were identified through an AlphaScreen assay and were validated by a structure–activity relationship analysis. They were shown to disrupt the Skp2/Cks1 site and inhibit p27 ubiquitination in vitro [64].
Other groups have also previously discovered small molecules targeting Skp2 mediated p27 ubiquitination through different mechanisms of targeting Skp2. Compound A (CpdA) was discovered via a high-throughput screen and prevents Skp2 incorporation into the SCF. CpdA induced cell-cycle arrest but the mechanism has not been fully elucidated [67].
DUB inhibitors
Ubiquitination is a dynamic and reversible process; the addition of ubiquitin is facilitated by the E1, E2 and E3 enzymes, while the reverse reaction and regulation of ubiquitination is controlled by the deubiquitination enzymes (DUBs). Thus, DUBs oppose E3 ligase activity and can rescue substrates from proteasomal degradation (Figure 1). The 79 identified DUB enzymes belong to the protease superfamily, and there exist five protease classes that can be classified into two distinct families: cysteine proteases and metalloproteases. The cysteine proteases can be further divided into ubiquitin-specific proteases (USPs), ubiquitin C-terminal hydrolases (UCHs), Machado-Josephin domain proteases (MJDs) and ovarian tumor proteases (OTU). The metalloprotease family contains only the Jab1/Mov34/Mpr1 Pad1 N-terminal+ (commonly referred to as JAMM/MPN+) proteases [116]. The 58 human USPs [82], which represent more than half of the known DUBs, have been the subject of significant consideration recently due to their implications in cancer and other pathologies [117–119].
For instance, USP14 has been recently indicated as required for WNT/β-catenin signaling and clinically relevant as well. The severity of gastrointestinal cancer and lung cancer mortality correlates with high levels of USP14 as well [120,121]. An inhibitor called IU1 targeting USP14 was discovered through high-throughput screening. Treating cultured cells with IU1 enhances proteasome efficiency, and the USP14 inhibition accelerated degradation of oxidized proteins and offered resistance to oxidative stress [74]. While some groups have developed specific DUB inhibitors, there have been numerous attempts at deriving small molecules as inhibitors of multiple DUB enzymes. Here we will discuss several examples, but this is by no means a complete list. WP1130 (degrasyn) was derived from a Janus-activated kinase (JAK2) inhibitor called AG490, and has been shown to be active towards at least five DUBs including USP9x, USP5, USP14 and UCH-L1 [73]. WP1130 was also found to activate Bcr/Abl destruction and induce apoptosis of CML cell lines as well as for its potential use in B-cell malignancies [122]. It was recently found to block autophagy and is currently in preclinical development and awaiting entry into clinical trials [122,123]. Proteasomal-dependent degradation of c-Myc is also activated by WP1130 in MM and other tumor cell lines using signaling processing domains not previously associated with c-Myc regulation [124]. Another screening study identified NSC632839, which targets USP2 and USP7 and triggers a unique apoptotic mechanism by upregulation of BH2-only protein Noxa, which induces mitochondrial fragmentation and caspase activation. It was shown that this compound can initiate a Bcl-2 dependent but apoptosome-independent pathway of caspase activation [75]. Finally, PR-619 is a reversible, cell permeable pan-DUB inhibitor but maintains selectivity towards DUBs over other proteases, such as calpain 1 or cathepsins [76]. It has some interesting properties when incubated with oligodendroglial cells: upregulation of heat shock proteins (HSPs), stabilization of the microtubule network (possibly through tau phosphorylation modulation), protein aggregate formation, and also the activation of the autophagic pathway [125,126].
Another member of the DUB family that is very promising is USP15. USP15 regulates both tumor growth and antitumor immunity with the capacity to stabilize TGF-β and MDM2 [127,128]. High expression of USP15 correlates with high TGF-β activity, and it is found amplified in glioblastoma, breast and ovarian cancer [127]. Knockdown of USP15 in cancer cells results in rapid degradation of MDM2 and simultaneous upregulation of p53, causing increased apoptosis and attenuated xenograft tumor growth. Finally, USP15 deficiency promoted T-cell activation in vitro and a knockout model further showed enhancement of T-cell activation and immune responses to both bacterial infections and tumor growth. These USP15 knockout mice displayed resistance to transplanted B16 melanoma tumors, producing profoundly higher levels of effector T cells infiltrating tumors [129]. The above information indicates the significance of USP15 as an attractive target for both cancer immunotherapy and small molecule treatment. Although the complete structure of USP15 has yet to be elucidated, its homologues, including USP4 and USP8, are available and can form the basis for structure-based screening models for hit identification. Despite a low sequence similarity overall, USPs share a structurally conserved catalytic domain with a well-defined catalytic cleft, thus it may be possible to develop a general structure-based strategy for inhibitor development [130]. We are currently conducting a similar study to discover USP15 inhibitors in which our model of USP15 based on the structures of USP4 and USP8 (Figure 4) is being used for in silico screening of USP15 inhibitors. The hits from this screen are being validated in a novel deubiquitination assay and the hits will be further examined for their effects on the degradation of endogenous MDM2, stimulation of p53-target genes, and enhancement of apoptosis.
Figure 4. . Comparison of USP4 and USP8 structures.

Several USP family proteins have high similarity of sequence and structures. Critical residues binding to ubiquitin have been elucidated. Yellow: USP8; cyan: USP4; magenta: ubiquitin. The labeled residues (sticks) are conserved in several USP families and involved in ubiquitin binding.
Future perspective
With increased understanding of the ubiquitination, especially the E3 ligase structures/functions, we will improve the rational design of selective therapeutics and tool compounds by targeting this critical pathway. It has been reported that there exists more than 1000 proteins in the entire ubiquitin–proteasome system, and the ubiquitin ligase system has a larger number of these proteins relative to other elements in the UPS, with a high potential of druggable target identification and validation [101]. Additionally, to date there has been no effort to perform a comprehensive analysis of the mutations in the UPS in human cancer, and this will certainly open another avenue to study ubiquitination in cancer development [131]. Studies continue to be conducted to showcase new connections of E3 ligases in diseases (especially cancer). Of the ˜70 F-box proteins in the human genome, only a few are well characterized [99,132]. A global protein stability profiling study of SCF E3 ligases found over 350 potential substrates involved in cell cycle, apoptosis and signaling pathways [133,134]. It should be noted that recent work has uncovered transitory intermediate states of E3 proteins that appear to be catalytically ‘primed’ before they can function normally, and these might also be considered when targeting ubiquitination [135]. One such example is the neural precursor cell expressed, developmentally downregulated (NEDD) family of proteins. In a recent study, the HECT family E3 ligase NEDD4 has been reported to bind the C-terminal tail of HER3 and negatively regulates HER3/ErbB3 [136]. NEDD4 has been previously shown to ligate fibroblast growth factor-1 and epithelial sodium channels, regulating their signaling, ubiquitination and degradation [137–140]. Previously, Nrdp1 (a RING finger E3 ligase) was shown to also ubiquitinate HER3 and regulate levels of HER3, but bound to the juxtamembrane domain or kinase domain of HER3, not the C-terminal tail [141]. This study suggests that NEDD4 might act as a marker for the development of antibody therapies targeting HER3 and that independent E3 ligases appear to regulate HER3 ubiquitination in unique cellular contexts [136].
As described with USP15, an emerging field is cancer immunotherapy. It has been reported that several E3 ubiquitin ligases play a crucial role in establishing the threshold for T-cell activation and controlling peripheral T-cell tolerance via multiple mechanisms. In addition to USP15, for instance, accumulating evidence suggests that Cbl-b also regulates innate immunity responses and is important in host defense to pathogens [142]. Understanding the signaling pathways in innate and adaptive immune cells is, therefore, essential for efficient manipulation of Cbl-b in emerging immunotherapies for human disorders such as autoimmune diseases, infections and cancer [143,144]. At the same time, such E3 ligases can be targeted with small molecules as showcased in our USP15 project. Therefore, combining immunotherapy and small molecule inhibitors to target the ubiquitination pathway represents another promising avenue for cancer treatment.
Finally, similar to the large chemical library screening that we performed to target Skp2, the ‘big data’ analysis will become an emerging technique for ubiquitination studies. For instance, Udeshi et al. identified and quantified over 10,000 ubiquitination sites in a single proteomics experiment [145]. As such studies continue, it is possible and imperative to develop prediction models that can be scaled to big data analysis [146].
Conclusion
Data from a myriad of disciplines of biochemistry, structural biology, cell and regulatory biology, animal studies and clinical patient data, all provide evidence of the critical importance that the ubiquitination pathway plays in the tightly regulated cellular processes, and that their dysregulation contributes to tumorigenesis. Therefore, the UPS provides a valuable repository of druggable targets to treat cancer. However, the complexity of this system leads to significant challenges, and more research should be carried out to explore the entirety of ubiquitination and deubiquitination pathways. Further understanding of ubiquitination in cancer development, progression and metastasis opens a new avenue to expand the available drug-target space, and targeting this pathway represents a unique, promising strategy which will propel the field from hypothesis-driven science forward to clinical applications and thus decrease the mortality associated with cancer.
Key terms.
Ubiquitination: The molecular ‘kiss of death’ process for a protein. It is a post-translational modification that involves the attachment of ubiquitin to a substrate protein. The substrate usually becomes inactivated and is tagged for proteasome degradation. Ubiquitin is a 76-amino acid protein that is found in almost all cellular tissues in humans and other eukaryotic organisms. During ubiquitination, it is covalently attached to proteins via lysine residues in either a monoubiquitin or polyubiquitin manner. Ubiquitination plays a major role in the regulation of proteins, and thus involved in a wide variety of cellular processes and disease development, including cancer.
Ubiquitin–proteasome system (USP): The collective name for all of the steps in the ubiquitin-dependent degradation of proteins. This includes the activation of ubiquitin by E1 enzymes, the ubiquitin-conjugation via the E2s, the transfer of ubiquitin to the target protein substrate by the E3 ubiquitin ligases, and finally the proteolytic degradation of the tagged proteins via the proteasome complex.
SCF E3 ligase complex: A particular multi-protein complex that transfers ubiquitin to a substrate protein for proteasomal degradation. The SCF complex is made up of three core constituents, and multiple less critical components. The core constituents also make the name SCF: Skp1 – an adapter protein, Cullin – a major structural scaffold that binds to Skp1, and an F-box protein (e.g., Skp2) – a protein with at least one F-box domain that provides the specificity of the SCF complex and binds to the tagged protein target. Other elements found in some SCF complexes include Rbx1, to which the E2-ubiquitin conjugate binds, and Cks1, which is required to bind to Skp2 in order to tag p27 for destruction.
F-Box proteins: A class of proteins that contain at least one F-box domain. The F-box domain is a specific structural motif that is about 49 amino acids long and responsible for interacting with the SCF protein Skp1. F-box proteins are commonly found with other interaction motifs, such as leucine-rich repeats and WD (tryptophan-aspartic acid) repeats that are responsible for binding other substrates to the SCF complex. There are approximately 69 F-box proteins in the human genome.
Deubiquitinating enzymes (DUBs): Proteases that catalyze the removal of ubiquitin from native conjugates, thereby reversing the ubiquitination process and rescuing proteins from destruction via the proteosome. DUBs assemble into distinct complexes to process the numerous and diverse monoubiquitin and polyubiquitin tags and they also process ubiquitin precursors and scavenge ubiquitin from tagged substrates. DUBs have been implicated in several important pathways including cell growth and differentiation, transcriptional regulation, and oncogenesis.
Executive summary.
Ubiquitination plays an essential role in both normal cellular processes and pathological development.
Given that the ubiquitin–proteasome system manages tumor suppressors and oncogenic proteins, dysregulation of the ubiquitin–proteasome system is commonly seen in many different types of cancers.
There have been many attempts to develop small molecules targeting ubiquitination.
Proteasome inhibitors have been approved by the US FDA for certain cancer treatment, but limitations are significant.
The E3 ubiquitin ligase inhibitors provide the specificity of targeting ubiquitination and have attracted significant attentions for drug development.
Skp2 E3 ligase has multiple regions that can be targeted by small molecule inhibitors.
Deubiquitination enzymes are also promising targets for drug design. Some of them can be combined with immunotherapeutic strategies for cancer treatment.
Acknowledgements
The authors thank the OpenEye Scientific Software and ChemAxon for their free academic licenses. The authors also thank the high-performance computing resources from Texas Advanced Computing Center.
Footnotes
Financial & competing interests disclosure
Shuxing Zhang is partially supported by CPRIT DP150086 and RP140244 as well as NSF CHE-1411859, NIH/NIGMS GM070737, and NIH/NCI P30CA016672. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
- 1.Hershko A, Ciechanover A. The ubiquitin system. Annu. Rev. Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 2.Hershko A. Ubiquitin: roles in protein modification and breakdown. Cell. 1983;34(1):11–12. doi: 10.1016/0092-8674(83)90131-9. [DOI] [PubMed] [Google Scholar]
- 3.Handley PM, Mueckler M, Siegel NR, Ciechanover A, Schwartz AL. Molecular cloning, sequence, and tissue distribution of the human ubiquitin-activating enzyme E1. Proc. Natl Acad. Sci. USA. 1991;88(1):258–262. doi: 10.1073/pnas.88.1.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schulman BA, Harper JW. Ubiquitin-like protein activation by E1 enzymes: the apex for downstream signalling pathways. Nat. Rev. Mol. Cell Biol. 2009;10(5):319–331. doi: 10.1038/nrm2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jentsch S. The ubiquitin-conjugation system. Annu. Rev. Genet. 1992;26:179–207. doi: 10.1146/annurev.ge.26.120192.001143. [DOI] [PubMed] [Google Scholar]
- 6.Berndsen CE, Wolberger C. New insights into ubiquitin E3 ligase mechanism. Nat. Struct. Mol. Biol. 2014;21(4):301–307. doi: 10.1038/nsmb.2780. [DOI] [PubMed] [Google Scholar]
- 7.Nakayama KI, Nakayama K. Ubiquitin ligases: cell-cycle control and cancer. Nat. Rev. Cancer. 2006;6(5):369–381. doi: 10.1038/nrc1881. [DOI] [PubMed] [Google Scholar]
- 8.Hershko A, Ciechanover A. The ubiquitin system for protein degradation. Annu. Rev. Biochem. 1992;61:761–807. doi: 10.1146/annurev.bi.61.070192.003553. [DOI] [PubMed] [Google Scholar]
- 9.Ciechanover A. The ubiquitin-proteasome proteolytic pathway. Cell. 1994;79(1):13–21. doi: 10.1016/0092-8674(94)90396-4. [DOI] [PubMed] [Google Scholar]; • One of the earliest review articles of the ubiquitin–proteasome system as a major, selective proteolytic and regulatory pathway of biological processes.
- 10.Rieser E, Cordier SM, Walczak H. Linear ubiquitination: a newly discovered regulator of cell signalling. Trends Biochem. Sci. 2013;38(2):94–102. doi: 10.1016/j.tibs.2012.11.007. [DOI] [PubMed] [Google Scholar]
- 11.Komander D, Rape M. The ubiquitin code. Annu. Rev. Biochem. 2012;81:203–229. doi: 10.1146/annurev-biochem-060310-170328. [DOI] [PubMed] [Google Scholar]
- 12.Chen ZJ, Sun LJ. Nonproteolytic functions of ubiquitin in cell signaling. Mol. Cell. 2009;33(3):275–286. doi: 10.1016/j.molcel.2009.01.014. [DOI] [PubMed] [Google Scholar]
- 13.Yang WL, Zhang X, Lin HK. Emerging role of Lys-63 ubiquitination in protein kinase and phosphatase activation and cancer development. Oncogene. 2010;29(32):4493–4503. doi: 10.1038/onc.2010.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bach I, Ostendorff HP. Orchestrating nuclear functions: ubiquitin sets the rhythm. Trends Biochem. Sci. 2003;28(4):189–195. doi: 10.1016/S0968-0004(03)00055-0. [DOI] [PubMed] [Google Scholar]
- 15.Murray AW. Recycling the cell cycle: cyclins revisited. Cell. 2004;116(2):221–234. doi: 10.1016/s0092-8674(03)01080-8. [DOI] [PubMed] [Google Scholar]
- 16.Glotzer M, Murray AW, Kirschner MW. Cyclin is degraded by the ubiquitin pathway. Nature. 1991;349(6305):132–138. doi: 10.1038/349132a0. [DOI] [PubMed] [Google Scholar]
- 17.Hoeller D, Dikic I. Targeting the ubiquitin system in cancer therapy. Nature. 2009;458(7237):438–444. doi: 10.1038/nature07960. [DOI] [PubMed] [Google Scholar]; •• An excellent review of how the deregulation of components of the elaborate ubiquitination network leads to tumorigenesis and how this knowledge is to be exploited for both molecular diagnostics and the development of novel strategies to combat cancer.
- 18.Lipkowitz S, Weissman AM. RINGs of good and evil: RING finger ubiquitin ligases at the crossroads of tumour suppression and oncogenesis. Nat. Rev. Cancer. 2011;11(9):629–643. doi: 10.1038/nrc3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kirkin V, Dikic I. Ubiquitin networks in cancer. Curr. Opin. Genet. Dev. 2011;21(1):21–28. doi: 10.1016/j.gde.2010.10.004. [DOI] [PubMed] [Google Scholar]
- 20.Pal A, Young MA, Donato NJ. Emerging potential of therapeutic targeting of ubiquitin-specific proteases in the treatment of cancer. Cancer Res. 2014;74(18):4955–4966. doi: 10.1158/0008-5472.CAN-14-1211. [DOI] [PubMed] [Google Scholar]
- 21.Miranda M, Sorkin A. Regulation of receptors and transporters by ubiquitination: new insights into surprisingly similar mechanisms. Mol. Interv. 2007;7(3):157–167. doi: 10.1124/mi.7.3.7. [DOI] [PubMed] [Google Scholar]
- 22.Hicke L, Schubert HL, Hill CP. Ubiquitin-binding domains. Nat. Rev. Mol. Cell Biol. 2005;6(8):610–621. doi: 10.1038/nrm1701. [DOI] [PubMed] [Google Scholar]
- 23.Pickart CM. Ubiquitin enters the new millennium. Mol. Cell. 2001;8(3):499–504. doi: 10.1016/s1097-2765(01)00347-1. [DOI] [PubMed] [Google Scholar]
- 24.Sigismund S, Polo S, Di Fiore PP. Signaling through monoubiquitination. Curr. Top. Microbiol. Immunol. 2004;286:149–185. doi: 10.1007/978-3-540-69494-6_6. [DOI] [PubMed] [Google Scholar]
- 25.Hicke L. Protein regulation by monoubiquitin. Nat. Rev. Mol. Cell Biol. 2001;2(3):195–201. doi: 10.1038/35056583. [DOI] [PubMed] [Google Scholar]
- 26.Adams J, Kauffman M. Development of the proteasome inhibitor Velcade (bortezomib) Cancer Invest. 2004;22(2):304–311. doi: 10.1081/cnv-120030218. [DOI] [PubMed] [Google Scholar]
- 27.Dick LR, Fleming PE. Building on bortezomib: second-generation proteasome inhibitors as anti-cancer therapy. Drug Discov. Today. 2010;15(5–6):243–249. doi: 10.1016/j.drudis.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 28.Potts BC, Albitar MX, Anderson KC, et al. Marizomib, a proteasome inhibitor for all seasons: preclinical profile and a framework for clinical trials. Curr. Cancer Drug Targets. 2011;11(3):254–284. doi: 10.2174/156800911794519716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kumar SK, Berdeja JG, Niesvizky R, et al. Safety and tolerability of ixazomib, an oral proteasome inhibitor, in combination with lenalidomide and dexamethasone in patients with previously untreated multiple myeloma: an open-label Phase 1/2 study. Lancet Oncol. 2014;15(13):1503–1512. doi: 10.1016/S1470-2045(14)71125-8. [DOI] [PubMed] [Google Scholar]
- 30.Micel LN, Tentler JJ, Smith PG, Eckhardt GS. Role of ubiquitin ligases and the proteasome in oncogenesis: novel targets for anticancer therapies. J. Clin. Oncol. 2013;31(9):1231–1238. doi: 10.1200/JCO.2012.44.0958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Da Silva SR, Paiva SL, Lukkarila JL, Gunning PT. Exploring a new frontier in cancer treatment: targeting the ubiquitin and ubiquitin-like activating enzymes. J. Med. Chem. 2013;56(6):2165–2177. doi: 10.1021/jm301420b. [DOI] [PubMed] [Google Scholar]
- 32.Popovic D, Vucic D, Dikic I. Ubiquitination in disease pathogenesis and treatment. Nat. Med. 2014;20(11):1242–1253. doi: 10.1038/nm.3739. [DOI] [PubMed] [Google Scholar]
- 33.Nawrocki ST, Griffin P, Kelly KR, Carew JS. MLN4924: a novel first-in-class inhibitor of NEDD8-activating enzyme for cancer therapy. Expert Opin. Investig. Drugs. 2012;21(10):1563–1573. doi: 10.1517/13543784.2012.707192. [DOI] [PubMed] [Google Scholar]
- 34.Swords RT, Kelly KR, Smith PG, et al. Inhibition of NEDD8-activating enzyme: a novel approach for the treatment of acute myeloid leukemia. Blood. 2010;115(18):3796–3800. doi: 10.1182/blood-2009-11-254862. [DOI] [PubMed] [Google Scholar]
- 35.Deshaies RJ. Proteotoxic crisis, the ubiquitin–proteasome system, and cancer therapy. BMC Biol. 2014;12:94. doi: 10.1186/s12915-014-0094-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morrow JK, Zhang S. Computational prediction of protein hot spot residues. Curr. Pharm. Des. 2012;18(9):1255–1265. doi: 10.2174/138161212799436412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frankland-Searby S, Bhaumik SR. The 26S proteasome complex: an attractive target for cancer therapy. Biochim. Biophys. Acta. 2012;1825(1):64–76. doi: 10.1016/j.bbcan.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kumar SK, Rajkumar SV, Dispenzieri A, et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood. 2008;111(5):2516–2520. doi: 10.1182/blood-2007-10-116129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bonvini P, Zorzi E, Basso G, Rosolen A. Bortezomib-mediated 26S proteasome inhibition causes cell-cycle arrest and induces apoptosis in CD-30+ anaplastic large cell lymphoma. Leukemia. 2007;21(4):838–842. doi: 10.1038/sj.leu.2404528. [DOI] [PubMed] [Google Scholar]
- 40.Groll M, Berkers CR, Ploegh HL, Ovaa H. Crystal structure of the boronic acid-based proteasome inhibitor bortezomib in complex with the yeast 20S proteasome. Structure. 2006;14(3):451–456. doi: 10.1016/j.str.2005.11.019. [DOI] [PubMed] [Google Scholar]
- 41.Hideshima T, Richardson PG, Anderson KC. Mechanism of action of proteasome inhibitors and deacetylase inhibitors and the biological basis of synergy in multiple myeloma. Mol. Cancer Ther. 2011;10(11):2034–2042. doi: 10.1158/1535-7163.MCT-11-0433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gelman JS, Sironi J, Berezniuk I, et al. Alterations of the intracellular peptidome in response to the proteasome inhibitor bortezomib. PLoS ONE. 2013;8(1):e53263. doi: 10.1371/journal.pone.0053263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mattern MR, Wu J, Nicholson B. Ubiquitin-based anticancer therapy: carpet bombing with proteasome inhibitors vs surgical strikes with E1, E2, E3, or DUB inhibitors. Biochim. Biophys. Acta. 2012;1823(11):2014–2021. doi: 10.1016/j.bbamcr.2012.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kubiczkova L, Pour L, Sedlarikova L, Hajek R, Sevcikova S. Proteasome inhibitors – molecular basis and current perspectives in multiple myeloma. J. Cell. Mol. Med. 2014;18(6):947–961. doi: 10.1111/jcmm.12279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Richardson PG, Briemberg H, Jagannath S, et al. Frequency, characteristics, and reversibility of peripheral neuropathy during treatment of advanced multiple myeloma with bortezomib. J. Clin. Oncol. 2006;24(19):3113–3120. doi: 10.1200/JCO.2005.04.7779. [DOI] [PubMed] [Google Scholar]
- 46.Richardson PG, Barlogie B, Berenson J, et al. A Phase 2 study of bortezomib in relapsed, refractory myeloma. N. Engl. J. Med. 2003;348(26):2609–2617. doi: 10.1056/NEJMoa030288. [DOI] [PubMed] [Google Scholar]
- 47.Suzuki E, Demo S, Deu E, et al. Molecular mechanisms of bortezomib resistant adenocarcinoma cells. PLoS ONE. 2011;6(12):e27996. doi: 10.1371/journal.pone.0027996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vij R, Siegel DS, Jagannath S, et al. An open-label, single-arm, Phase 2 study of single-agent carfilzomib in patients with relapsed and/or refractory multiple myeloma who have been previously treated with bortezomib. Br. J. Haematol. 2012;158(6):739–748. doi: 10.1111/j.1365-2141.2012.09232.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vij R, Wang M, Kaufman JL, et al. An open-label, single-arm, Phase 2 (PX-171–004) study of single-agent carfilzomib in bortezomib-naive patients with relapsed and/or refractory multiple myeloma. Blood. 2012;119(24):5661–5670. doi: 10.1182/blood-2012-03-414359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Meng L, Mohan R, Kwok BH, Elofsson M, Sin N, Crews CM. Epoxomicin, a potent and selective proteasome inhibitor, exhibits in vivo antiinflammatory activity. Proc. Natl Acad. Sci. USA. 1999;96(18):10403–10408. doi: 10.1073/pnas.96.18.10403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Piva R, Ruggeri B, Williams M, et al. CEP-18770: a novel, orally active proteasome inhibitor with a tumor-selective pharmacologic profile competitive with bortezomib. Blood. 2008;111(5):2765–2775. doi: 10.1182/blood-2007-07-100651. [DOI] [PubMed] [Google Scholar]
- 52.Tsukamoto S, Takeuchi T, Rotinsulu H, et al. Leucettamol A: a new inhibitor of Ubc13-Uev1A interaction isolated from a marine sponge, Leucetta aff. microrhaphis. Bioorg. Med. Chem. Lett. 2008;18(24):6319–6320. doi: 10.1016/j.bmcl.2008.10.110. [DOI] [PubMed] [Google Scholar]
- 53.Dalisay DS, Tsukamoto S, Molinski TF. Absolute configuration of the alpha,omega-bifunctionalized sphingolipid leucettamol A from Leucetta microrhaphis by deconvoluted exciton coupled CD. J. Nat. Prod. 2009;72(3):353–359. doi: 10.1021/np800549n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ceccarelli DF, Tang X, Pelletier B, et al. An allosteric inhibitor of the human Cdc34 ubiquitin-conjugating enzyme. Cell. 2011;145(7):1075–1087. doi: 10.1016/j.cell.2011.05.039. [DOI] [PubMed] [Google Scholar]
- 55.Ito T, Ando H, Suzuki T, et al. Identification of a primary target of thalidomide teratogenicity. Science. 2010;327(5971):1345–1350. doi: 10.1126/science.1177319. [DOI] [PubMed] [Google Scholar]
- 56.Kim Y, Schmidt-Wolf IG. Lenalidomide in multiple myeloma. Expert Rev. Anticancer Ther. 2015;15(5):491–497. doi: 10.1586/14737140.2015.1033407. [DOI] [PubMed] [Google Scholar]
- 57.Lacy MQ, Rajkumar SV. Pomalidomide: a new IMiD with remarkable activity in both multiple myeloma and myelofibrosis. Am. J. Hematol. 2010;85(2):95–96. doi: 10.1002/ajh.21610. [DOI] [PubMed] [Google Scholar]
- 58.Vassilev LT, Vu BT, Graves B, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303(5659):844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]; • An original article the described the identification of potent and selective small-molecule antagonists of MDM2 and confirm their mode of action through the crystal structures of complexes and in vitro/in vivo studies.
- 59.Vassilev LT. MDM2 inhibitors for cancer therapy. Trends Mol. Med. 2007;13(1):23–31. doi: 10.1016/j.molmed.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 60.Vu B, Wovkulich P, Pizzolato G, et al. Discovery of RG7112: a small-molecule MDM2 inhibitor in clinical development. ACS Med. Chem. Lett. 2013;4(5):466–469. doi: 10.1021/ml4000657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gembarska A, Luciani F, Fedele C, et al. MDM4 is a key therapeutic target in cutaneous melanoma. Nat. Med. 2012;18(8):1239–1247. doi: 10.1038/nm.2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chang YS, Graves B, Guerlavais V, et al. Stapled alpha-helical peptide drug development: a potent dual inhibitor of MDM2 and MDMX for p53-dependent cancer therapy. Proc. Natl Acad. Sci. USA. 2013;110(36):E3445–E3454. doi: 10.1073/pnas.1303002110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang H, Ma X, Ren S, Buolamwini JK, Yan C. A small-molecule inhibitor of MDMX activates p53 and induces apoptosis. Mol. Cancer Ther. 2011;10(1):69–79. doi: 10.1158/1535-7163.MCT-10-0581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ungermannova D, Lee J, Zhang G, Dallmann HG, Mchenry CS, Liu X. High-throughput screening AlphaScreen assay for identification of small-molecule inhibitors of ubiquitin E3 ligase SCFSkp2-Cks1. J. Biomol. Screen. 2013;18(8):910–920. doi: 10.1177/1087057113485789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Orlicky S, Tang X, Neduva V, et al. An allosteric inhibitor of substrate recognition by the SCF(Cdc4) ubiquitin ligase. Nat. Biotechnol. 2010;28(7):733–737. doi: 10.1038/nbt.1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wu L, Grigoryan AV, Li Y, Hao B, Pagano M, Cardozo TJ. Specific small molecule inhibitors of Skp2-mediated p27 degradation. Chem. Biol. 2012;19(12):1515–1524. doi: 10.1016/j.chembiol.2012.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen Q, Xie W, Kuhn DJ, et al. Targeting the p27 E3 ligase SCF(Skp2) results in p27- and Skp2-mediated cell-cycle arrest and activation of autophagy. Blood. 2008;111(9):4690–4699. doi: 10.1182/blood-2007-09-112904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chan CH, Morrow JK, Li CF, et al. Pharmacological inactivation of Skp2 SCF ubiquitin ligase restricts cancer stem cell traits and cancer progression. Cell. 2013;154(3):556–568. doi: 10.1016/j.cell.2013.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• An original paper that developed first-in-class inhibitors of the SCF Skp2 E3 ubiquitin ligase which directly bind to Skp2 and disrupt Skp2–Skp2 interactions.
- 69.Nakajima H, Fujiwara H, Furuichi Y, Tanaka K, Shimbara N. A novel small-molecule inhibitor of NF-kappaB signaling. Biochem. Biophys. Res. Commun. 2008;368(4):1007–1013. doi: 10.1016/j.bbrc.2008.01.166. [DOI] [PubMed] [Google Scholar]
- 70.Blees JS, Bokesch HR, Rubsamen D, et al. Erioflorin stabilizes the tumor suppressor Pdcd4 by inhibiting its interaction with the E3-ligase beta-TrCP1. PLoS ONE. 2012;7(10):e46567. doi: 10.1371/journal.pone.0046567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Khoury K, Domling A. P53 mdm2 inhibitors. Curr. Pharm. Des. 2012;18(30):4668–4678. doi: 10.2174/138161212802651580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tabernero J, Dirix L, Schoffski P, et al. A Phase I first-in-human pharmacokinetic and pharmacodynamic study of serdemetan in patients with advanced solid tumors. Clin. Cancer Res. 2011;17(19):6313–6321. doi: 10.1158/1078-0432.CCR-11-1101. [DOI] [PubMed] [Google Scholar]
- 73.Kapuria V, Peterson LF, Fang D, Bornmann WG, Talpaz M, Donato NJ. Deubiquitinase inhibition by small-molecule WP1130 triggers aggresome formation and tumor cell apoptosis. Cancer Res. 2010;70(22):9265–9276. doi: 10.1158/0008-5472.CAN-10-1530. [DOI] [PubMed] [Google Scholar]
- 74.Lee BH, Lee MJ, Park S, et al. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature. 2010;467(7312):179–184. doi: 10.1038/nature09299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Aleo E, Henderson CJ, Fontanini A, Solazzo B, Brancolini C. Identification of new compounds that trigger apoptosome-independent caspase activation and apoptosis. Cancer Res. 2006;66(18):9235–9244. doi: 10.1158/0008-5472.CAN-06-0702. [DOI] [PubMed] [Google Scholar]
- 76.Altun M, Kramer HB, Willems LI, et al. Activity-based chemical proteomics accelerates inhibitor development for deubiquitylating enzymes. Chem. Biol. 2011;18(11):1401–1412. doi: 10.1016/j.chembiol.2011.08.018. [DOI] [PubMed] [Google Scholar]
- 77.Chauhan D, Catley L, Li G, et al. A novel orally active proteasome inhibitor induces apoptosis in multiple myeloma cells with mechanisms distinct from Bortezomib. Cancer Cell. 2005;8(5):407–419. doi: 10.1016/j.ccr.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 78.Millward M, Price T, Townsend A, et al. Phase 1 clinical trial of the novel proteasome inhibitor marizomib with the histone deacetylase inhibitor vorinostat in patients with melanoma, pancreatic and lung cancer based on in vitro assessments of the combination. Invest. New Drugs. 2012;30(6):2303–2317. doi: 10.1007/s10637-011-9766-6. [DOI] [PubMed] [Google Scholar]
- 79.Kupperman E, Lee EC, Cao Y, et al. Evaluation of the proteasome inhibitor MLN9708 in preclinical models of human cancer. Cancer Res. 2010;70(5):1970–1980. doi: 10.1158/0008-5472.CAN-09-2766. [DOI] [PubMed] [Google Scholar]
- 80.Ushiyama S, Umaoka H, Kato H, et al. Manadosterols A and B, sulfonated sterol dimers inhibiting the Ubc13–Uev1A interaction, isolated from the marine sponge Lissodendryx fibrosa . J. Nat. Prod. 2012;75(8):1495–1499. doi: 10.1021/np300352u. [DOI] [PubMed] [Google Scholar]
- 81.Metzger MB, Hristova VA, Weissman AM. HECT and RING finger families of E3 ubiquitin ligases at a glance. J. Cell Sci. 2012;125(3):531–537. doi: 10.1242/jcs.091777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nijman SM, Luna-Vargas MP, Velds A, et al. A genomic and functional inventory of deubiquitinating enzymes. Cell. 2005;123(5):773–786. doi: 10.1016/j.cell.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 83.Skaar JR, Pagan JK, Pagano M. Mechanisms and function of substrate recruitment by F-box proteins. Nat. Rev. Mol. Cell Biol. 2013;14(6):369–381. doi: 10.1038/nrm3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lu G, Middleton RE, Sun H, et al. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science. 2014;343(6168):305–309. doi: 10.1126/science.1244917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fischer ES, Bohm K, Lydeard JR, et al. Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature. 2014;512(7512):49–53. doi: 10.1038/nature13527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lopez-Girona A, Mendy D, Ito T, et al. Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide. Leukemia. 2012;26(11):2326–2335. doi: 10.1038/leu.2012.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhu YX, Braggio E, Shi CX, et al. Cereblon expression is required for the antimyeloma activity of lenalidomide and pomalidomide. Blood. 2011;118(18):4771–4779. doi: 10.1182/blood-2011-05-356063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lee JT, Gu W. The multiple levels of regulation by p53 ubiquitination. Cell Death Differ. 2010;17(1):86–92. doi: 10.1038/cdd.2009.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387(6630):296–299. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- 90.Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997;420(1):25–27. doi: 10.1016/s0014-5793(97)01480-4. [DOI] [PubMed] [Google Scholar]
- 91.Fang S, Jensen JP, Ludwig RL, Vousden KH, Weissman AM. Mdm2 is a RING finger-dependent ubiquitin protein ligase for itself and p53. J. Biol. Chem. 2000;275(12):8945–8951. doi: 10.1074/jbc.275.12.8945. [DOI] [PubMed] [Google Scholar]
- 92.Chargari C, Leteur C, Angevin E, et al. Preclinical assessment of JNJ-26854165 (Serdemetan), a novel tryptamine compound with radiosensitizing activity in vitro and in tumor xenografts. Cancer Lett. 2011;312(2):209–218. doi: 10.1016/j.canlet.2011.08.011. [DOI] [PubMed] [Google Scholar]
- 93.Van Maerken T, Rihani A, Van Goethem A, De Paepe A, Speleman F, Vandesompele J. Pharmacologic activation of wild-type p53 by nutlin therapy in childhood cancer. Cancer Lett. 2014;344(2):157–165. doi: 10.1016/j.canlet.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 94.Joseph TL, Madhumalar A, Brown CJ, Lane DP, Verma CS. Differential binding of p53 and nutlin to MDM2 and MDMX: computational studies. Cell Cycle. 2010;9(6):1167–1181. doi: 10.4161/cc.9.6.11067. [DOI] [PubMed] [Google Scholar]
- 95.Deshaies RJ, Joazeiro CA. RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 2009;78:399–434. doi: 10.1146/annurev.biochem.78.101807.093809. [DOI] [PubMed] [Google Scholar]
- 96.Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 2005;6(1):9–20. doi: 10.1038/nrm1547. [DOI] [PubMed] [Google Scholar]
- 97.Aghajan M, Jonai N, Flick K, et al. Chemical genetics screen for enhancers of rapamycin identifies a specific inhibitor of an SCF family E3 ubiquitin ligase. Nat. Biotechnol. 2010;28(7):738–742. doi: 10.1038/nbt.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chen BB, Coon TA, Glasser JR, et al. A combinatorial F box protein directed pathway controls TRAF adaptor stability to regulate inflammation. Nat. Immunol. 2013;14(5):470–479. doi: 10.1038/ni.2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Frescas D, Pagano M. Deregulated proteolysis by the F-box proteins SKP2 and beta-TrCP: tipping the scales of cancer. Nat. Rev. Cancer. 2008;8(6):438–449. doi: 10.1038/nrc2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bielskiene K, Bagdoniene L, Mozuraitiene J, Kazbariene B, Janulionis E. E3 ubiquitin ligases as drug targets and prognostic biomarkers in melanoma. Medicina (Kaunas) 2015;51(1):1–9. doi: 10.1016/j.medici.2015.01.007. [DOI] [PubMed] [Google Scholar]
- 101.Skaar JR, Pagan JK, Pagano M. SCF ubiquitin ligase-targeted therapies. Nat. Rev. Drug Discov. 2014;13(12):889–903. doi: 10.1038/nrd4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.http://www.onenucleus.com/media/Events/ONC%20Seminar/Oncology%20seminar%20UB%20Pharma%20111011.pdf Ub Pharma overview.
- 103.Nakayama K, Nagahama H, Minamishima YA, et al. Skp2-mediated degradation of p27 regulates progression into mitosis. Dev. Cell. 2004;6(5):661–672. doi: 10.1016/s1534-5807(04)00131-5. [DOI] [PubMed] [Google Scholar]
- 104.Chan CH, Li CF, Yang WL, et al. The Skp2-SCF E3 ligase regulates Akt ubiquitination, glycolysis, herceptin sensitivity, and tumorigenesis. Cell. 2012;149(5):1098–1111. doi: 10.1016/j.cell.2012.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]; • An original report describing the nonproteolytic ubiquitination of Akt by Skp2 E3 ligase to promote Akt translocation and activation. This has been implicated in tumorigenesis, cancer metabolism and drug resistance.
- 105.Hershko DD. Oncogenic properties and prognostic implications of the ubiquitin ligase Skp2 in cancer. Cancer. 2008;112(7):1415–1424. doi: 10.1002/cncr.23317. [DOI] [PubMed] [Google Scholar]
- 106.Lin HK, Wang G, Chen Z, et al. Phosphorylation-dependent regulation of cytosolic localization and oncogenic function of Skp2 by Akt/PKB. Nat. Cell Biol. 2009;11(4):420–432. doi: 10.1038/ncb1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chan CH, Lee SW, Li CF, et al. Deciphering the transcriptional complex critical for RhoA gene expression and cancer metastasis. Nat. Cell Biol. 2010;12(5):457–467. doi: 10.1038/ncb2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lee SW, Li CF, Jin G, et al. Skp2-dependent ubiquitination and activation of LKB1 is essential for cancer cell survival under energy stress. Mol. Cell. 2015;57(6):1022–1033. doi: 10.1016/j.molcel.2015.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zheng N, Schulman BA, Song L, et al. Structure of the Cul1-Rbx1-Skp1-F boxSkp2 SCF ubiquitin ligase complex. Nature. 2002;416(6882):703–709. doi: 10.1038/416703a. [DOI] [PubMed] [Google Scholar]; • One of the earliest and most comprehensive crystal structure studies of the SCF complex that can be used for Skp2 functional studies and structure-based design of SCF Skp2 inhibitors.
- 110.Hao B, Zheng N, Schulman BA, et al. Structural basis of the Cks1-dependent recognition of p27(Kip1) by the SCF(Skp2) ubiquitin ligase. Mol. Cell. 2005;20(1):9–19. doi: 10.1016/j.molcel.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 111.Sakai T, Sakaue H, Nakamura T, et al. Skp2 controls adipocyte proliferation during the development of obesity. J. Biol. Chem. 2007;282(3):2038–2046. doi: 10.1074/jbc.M608144200. [DOI] [PubMed] [Google Scholar]
- 112.Chan CH, Morrow JK, Zhang S, Lin HK. Skp2: a dream target in the coming age of cancer therapy. Cell Cycle. 2014;13(5):679–680. doi: 10.4161/cc.27853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ganoth D, Bornstein G, Ko TK, et al. The cell-cycle regulatory protein Cks1 is required for SCF(Skp2)-mediated ubiquitinylation of p27. Nat. Cell Biol. 2001;3(3):321–324. doi: 10.1038/35060126. [DOI] [PubMed] [Google Scholar]
- 114.Montagnoli A, Fiore F, Eytan E, et al. Ubiquitination of p27 is regulated by Cdk-dependent phosphorylation and trimeric complex formation. Genes Dev. 1999;13(9):1181–1189. doi: 10.1101/gad.13.9.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Tsvetkov LM, Yeh KH, Lee SJ, Sun H, Zhang H. p27(Kip1) ubiquitination and degradation is regulated by the SCF(Skp2) complex through phosphorylated Thr187 in p27. Curr. Biol. 1999;9(12):661–664. doi: 10.1016/s0960-9822(99)80290-5. [DOI] [PubMed] [Google Scholar]
- 116.Komander D, Clague MJ, Urbe S. Breaking the chains: structure and function of the deubiquitinases. Nat. Rev. Mol. Cell Biol. 2009;10(8):550–563. doi: 10.1038/nrm2731. [DOI] [PubMed] [Google Scholar]
- 117.Yang Y, Kitagaki J, Wang H, Hou DX, Perantoni AO. Targeting the ubiquitin–proteasome system for cancer therapy. Cancer Sci. 2009;100(1):24–28. doi: 10.1111/j.1349-7006.2008.01013.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Daviet L, Colland F. Targeting ubiquitin specific proteases for drug discovery. Biochimie. 2008;90(2):270–283. doi: 10.1016/j.biochi.2007.09.013. [DOI] [PubMed] [Google Scholar]
- 119.D’arcy P, Wang X, Linder S. Deubiquitinase inhibition as a cancer therapeutic strategy. Pharmacol. Ther. 2015;147C:32–54. doi: 10.1016/j.pharmthera.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 120.Shinji S, Naito Z, Ishiwata S, et al. Ubiquitin-specific protease 14 expression in colorectal cancer is associated with liver and lymph node metastases. Oncol. Rep. 2006;15(3):539–543. [PubMed] [Google Scholar]
- 121.Wu N, Liu C, Bai C, Han YP, Cho WC, Li Q. Over-Expression of deubiquitinating enzyme USP14 in lung adenocarcinoma promotes proliferation through the accumulation of beta-catenin. Int. J. Mol. Sci. 2013;14(6):10749–10760. doi: 10.3390/ijms140610749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Peterson LF, Sun H, Liu Y, et al. Targeting deubiquitinase activity with a novel small-molecule inhibitor as therapy for B-cell malignancies. Blood. 2015;125(23):3588–3597. doi: 10.1182/blood-2014-10-605584. [DOI] [PubMed] [Google Scholar]
- 123.Bartholomeusz GA, Talpaz M, Kapuria V, et al. Activation of a novel Bcr/Abl destruction pathway by WP1130 induces apoptosis of chronic myelogenous leukemia cells. Blood. 2007;109(8):3470–3478. doi: 10.1182/blood-2006-02-005579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bartholomeusz G, Talpaz M, Bornmann W, Kong LY, Donato NJ. Degrasyn activates proteasomal-dependent degradation of c-Myc. Cancer Res. 2007;67(8):3912–3918. doi: 10.1158/0008-5472.CAN-06-4464. [DOI] [PubMed] [Google Scholar]
- 125.Seiberlich V, Borchert J, Zhukareva V, Richter-Landsberg C. Inhibition of protein deubiquitination by PR-619 activates the autophagic pathway in OLN-t40 oligodendroglial cells. Cell Biochem. Biophys. 2013;67(1):149–160. doi: 10.1007/s12013-013-9622-8. [DOI] [PubMed] [Google Scholar]
- 126.Seiberlich V, Goldbaum O, Zhukareva V, Richter-Landsberg C. The small molecule inhibitor PR-619 of deubiquitinating enzymes affects the microtubule network and causes protein aggregate formation in neural cells: implications for neurodegenerative diseases. Biochim. Biophys. Acta. 2012;1823(11):2057–2068. doi: 10.1016/j.bbamcr.2012.04.011. [DOI] [PubMed] [Google Scholar]
- 127.Eichhorn PJ, Rodon L, Gonzalez-Junca A, et al. USP15 stabilizes TGF-beta receptor I and promotes oncogenesis through the activation of TGF-beta signaling in glioblastoma. Nat. Med. 2012;18(3):429–435. doi: 10.1038/nm.2619. [DOI] [PubMed] [Google Scholar]
- 128.Inui M, Manfrin A, Mamidi A, et al. USP15 is a deubiquitylating enzyme for receptor-activated SMADs. Nat. Cell Biol. 2011;13(11):1368–1375. doi: 10.1038/ncb2346. [DOI] [PubMed] [Google Scholar]
- 129.Zou Q, Jin J, Hu H, et al. USP15 stabilizes MDM2 to mediate cancer-cell survival and inhibit antitumor T cell responses. Nat. Immunol. 2014;15(6):562–570. doi: 10.1038/ni.2885. [DOI] [PMC free article] [PubMed] [Google Scholar]; • An excellent report demonstrating how USP15 regulates T-cell activation and can be targeted for cancer immunotherapy.
- 130.Ernst A, Avvakumov G, Tong J, et al. A strategy for modulation of enzymes in the ubiquitin system. Science. 2013;339(6119):590–595. doi: 10.1126/science.1230161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Nalepa G, Rolfe M, Harper JW. Drug discovery in the ubiquitin–proteasome system. Nat. Rev. Drug Discov. 2006;5(7):596–613. doi: 10.1038/nrd2056. [DOI] [PubMed] [Google Scholar]
- 132.Welcker M, Clurman BE. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat. Rev. Cancer. 2008;8(2):83–93. doi: 10.1038/nrc2290. [DOI] [PubMed] [Google Scholar]
- 133.Yen HC, Elledge SJ. Identification of SCF ubiquitin ligase substrates by global protein stability profiling. Science. 2008;322(5903):923–929. doi: 10.1126/science.1160462. [DOI] [PubMed] [Google Scholar]
- 134.Yen HC, Xu Q, Chou DM, Zhao Z, Elledge SJ. Global protein stability profiling in mammalian cells. Science. 2008;322(5903):918–923. doi: 10.1126/science.1160489. [DOI] [PubMed] [Google Scholar]
- 135.Maspero E, Valentini E, Mari S, et al. Structure of a ubiquitin-loaded HECT ligase reveals the molecular basis for catalytic priming. Nat. Struct. Mol. Biol. 2013;20(6):696–701. doi: 10.1038/nsmb.2566. [DOI] [PubMed] [Google Scholar]
- 136.Huang Z, Choi BK, Mujoo K, et al. The E3 ubiquitin ligase NEDD4 negatively regulates HER3/ErbB3 level and signaling. Oncogene. 2014;34(9):1105–1115. doi: 10.1038/onc.2014.56. [DOI] [PubMed] [Google Scholar]
- 137.Zeng F, Xu J, Harris RC. Nedd4 mediates ErbB4 JM-a/CYT-1 ICD ubiquitination and degradation in MDCK II cells. FASEB J. 2009;23(6):1935–1945. doi: 10.1096/fj.08-121947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Persaud A, Alberts P, Hayes M, et al. Nedd4–1 binds and ubiquitylates activated FGFR1 to control its endocytosis and function. EMBO J. 2011;30(16):3259–3273. doi: 10.1038/emboj.2011.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Rotin D, Kumar S. Physiological functions of the HECT family of ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 2009;10(6):398–409. doi: 10.1038/nrm2690. [DOI] [PubMed] [Google Scholar]
- 140.Maspero E, Mari S, Valentini E, et al. Structure of the HECT:ubiquitin complex and its role in ubiquitin chain elongation. EMBO Rep. 2011;12(4):342–349. doi: 10.1038/embor.2011.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Bouyain S, Leahy DJ. Structure-based mutagenesis of the substrate-recognition domain of Nrdp1/FLRF identifies the binding site for the receptor tyrosine kinase ErbB3. Protein Sci. 2007;16(4):654–661. doi: 10.1110/ps.062700307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Liu Q, Zhou H, Langdon WY, Zhang J. E3 ubiquitin ligase Cbl-b in innate and adaptive immunity. Cell Cycle. 2014;13(12):1875–1884. doi: 10.4161/cc.29213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Lutz-Nicoladoni C, Wolf D, Sopper S. Modulation of Immune Cell Functions by the E3 Ligase Cbl-b. Front. Oncol. 2015;5:58. doi: 10.3389/fonc.2015.00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Loeser S, Penninger JM. The ubiquitin E3 ligase Cbl-b in T cells tolerance and tumor immunity. Cell Cycle. 2007;6(20):2478–2485. doi: 10.4161/cc.6.20.4797. [DOI] [PubMed] [Google Scholar]
- 145.Udeshi ND, Svinkina T, Mertins P, et al. Refined preparation and use of anti-diglycine remnant (K-epsilon-GG) antibody enables routine quantification of 10,000s of ubiquitination sites in single proteomics experiments. Mol. Cell. Proteomics. 2013;12(3):825–831. doi: 10.1074/mcp.O112.027094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Nguyen VN, Huang KY, Huang CH, et al. Characterization and identification of ubiquitin conjugation sites with E3 ligase recognition specificities. BMC Bioinform. 2015;16(Suppl. 1):S1. doi: 10.1186/1471-2105-16-S1-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]