

Abstract
Chronic pain is one of the most ubiquitous diseases in the world, but treatment is difficult with conventional methods, due to undesirable side effects of treatments and unknown mechanisms of pathological pain states. The endogenous peptide, dynorphin A has long been established as a target for the treatment of pain. Interestingly, this unique peptide has both inhibitory (opioid in nature) and excitatory activities (nonopioid) in the CNS. Both of these effects have been found to play a role in pain and much work has been done to develop therapeutics to enhance the inhibitory effects. Here we will review the dynorphin A compounds that have been designed for the modulation of pain and will discuss where the field stands today.
Chronic neuropathic pain
Pain is an unpleasant feeling that has been categorized as acute (lasting a few days or weeks) or chronic (lasting months/years) [2]. Acute pain arises from tissue injury and usually dissipates once the injury is healed, and this pain is useful in alerting the body of damage [2]. On the other hand, chronic pain has no purpose and exists in the absence of tissue injury. Although chronic pain is one of the most prevalent diseases, affecting 100 million Americans [2] (more than heart disease, cancer and diabetes combined), funding from the NIH is miniscule when compared with the other diseases due to rare fatality (Figure 1). Chronic pain greatly limits the patient's quality of life and costs an estimated US$61 billion/year in lost productive time alone [3]. Neuropathic pain, a type of chronic pain, results from the dysfunction of the CNS or the peripheral nervous system (PNS) that can occur in the presence or absence of an initial injury. The hallmarks of this type of pain are hyperalgesia, increased sensitivity to painful stimuli, and allodynia, increased sensitivity to nonpainful stimuli [4].
Figure 1. . Estimates of the number of people suffering from the most common diseases in the USA and estimates of funding from the NIH.

Data taken from [2], American Diabetes Association, American Cancer Society and the American Heart Association.
Therapeutics for chronic neuropathic pain
There are currently many therapeutics for inflammatory acute pain, but unfortunately, there are not many effective treatments for chronic neuropathic pain. Even with quite different mechanisms, acute and chronic pain symptoms are treated with the same therapeutics. Acetaminophen and NSAIDs are generally prescribed for mild-to-moderate pain states and have relatively little side effects and work well for postoperative pain but not for neuropathic pain. For severe pain, opioids are often prescribed and are useful in acute pain states but are not effective in chronic pain states because of serious side effects caused by long-term administration [2]. Anticonvulsants such as pregablin and gabapentin are often prescribed and are effective for patients with postherpetic neuralgia, fibromyalgia and diabetic peripheral neuropathy [5]. For over 40 years, antidepressants such as tricycles, selective serotonin reuptake inhibitors and serotonin norepinephrine reuptake inhibitors have been used for the treatment of neuropathic pain and show efficacy for some patients [6]. Steroids are generally used as an adjunct therapy with opioids and are beneficial to patients with metastatic bone pain, visceral pain and neuropathic pain [7]. Ziconotide, a natural peptide from snail venom, is approved for the treatment of intractable chronic pain [8,9]. Ziconotide administration is limited to intrathecal because of cardiovascular liabilities caused by the intravenous route [10]. There appears to be many treatments for neuropathic pain, but most of them are not effective in some patients showing high numbers needed to treat (NNT) and/or develop serious side effects (Table 1). Therefore, there is still a pressing need for novel therapeutics for the management of chronic neuropathic pain.
Table 1. . Commonly prescribed drugs for chronic neuropathic pain.
| Class of drug/example | Mechanism of action | Structure | Side effects† | NNT | Ref. |
|---|---|---|---|---|---|
| NSAIDs/ibuprofen |
Nonselective inhibitors for COX |
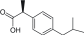 |
Ulcers, stomach bleeding, high blood pressure |
2 |
[11] |
| Acetaminophen |
Inhibition of prostaglandin synthesis. Unknown |
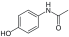 |
Nausea, loss of appetite, jaundice, stomach pain |
4.6 |
[11] |
| Opioids/oxycodone |
Opioid receptors (mainly MOR) |
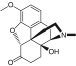 |
Addiction, tolerance, constipation, respiratory depression |
2.6 |
[12] |
| Anticonvulsant/pregablin |
Ca2+ channel α2-δ subunit |
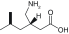 |
Dizziness, somnolence, ataxia, tremor |
4.5 |
[12] |
| Antidepressants/duloxetine |
Selective SNRI |
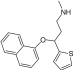 |
Drowsiness, dry mouth, nausea, constipation |
5.0 |
[12] |
| Steroids/dexamethasone |
Inhibits prostaglandin synthesis |
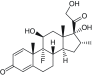 |
Upset stomach, vomiting, headache, increased hair growth. |
N.D. |
|
| Toxin/ziconotide | N-type Ca2+ blocker | Dizziness, drowsiness, headache, nausea | 2‡ | [13] |
†Side effects found at [14].
‡Calculated for postoperation pain.
MOR: μ-opioid receptor; N.D: Not determined; NNT: Numbers needed to treat; SNRI: Serotonin norepinephrine reuptake inhibitor.
Dynorphin A, an extraordinary opioid peptide
In 1979, Goldstein et al. discovered a peptide from the porcine pituitary that was termed Dynorphin A (Dyn A), from the Greek word dynamis (power) [15]. The peptide was found to contain the Leu-enkephalin structure and was 730-fold and threefold more potent than Leu-enkephalin in the guinea pig ileum (GPI) and mouse vas deferens (MVD) contractility assays, respectively. The effects of the peptide in tissues were blocked by an opioid antagonist, naloxone, suggesting the opioid receptors as a target [15]. A few years later, the full structure of this peptide was determined to be a heptadecapeptide, H-Tyr-Gly-Gly-Phe-Leu-Arg-Arg-Ile-Arg-Pro-Lys-Leu-Lys-Trp-Asp-Asn-Gln-OH, with the Dyn A-(1–13) fragment crucial for its potency [16]. Later studies found that Dyn A is derived from prodynorphin [17]. After processing, the inactive precursor protein, prodynorphin, consists of α-neoendorphin and Big Dyn. Cleavage via prohormone convertase 2 affords the bioactive peptides Leu-enkephalin (5 residues) from α-neoendorphin and Dyn A (17 residues) and Dyn B (13 residues) from Big Dyn (Figure 2) [17].
Figure 2. . Cleavage of prodynorphin into the active peptides Dyn A, Dyn B and Leu-enkephalin.

Dyn A, an endogenous ligand for the κ-opioid receptor (KOR)
Although Dyn A has affinity for all three opioid receptors (Table 2), μ-opioid receptor (MOR), KOR and δ-opioid receptor (DOR), it may act primarily at the KOR considering the similar activity with the KOR-selective agonist, ethylketocyclazocine [18]. The KOR is a Gi/o-coupled G-protein coupled receptor (GPCR), in which agonists inhibit adenylate cyclase. There have been some publications suggesting different subtypes of the KOR (subtype 1–3), but to date only the KOR1 has been cloned [19–21]. Work by Devi and colleagues found that KOR, MOR and/or DOR can form heteromeric complexes that produce the pharmacological profiles of the previously reported KOR subtypes [22–24]. For the purpose of this review, we will only discuss ligands designed for the homomeric KOR1. For information involving bivalent ligands designed for the heteromers see the current work done by Portoghese [25,26]. The receptor is widely expressed in the brain, spinal cord and peripheral tissues [27], and the expression of the receptor is found in areas related to pain circuitry; dorsal root ganglia (DRG), dorsal spinal cord, rostral ventromedial medulla and periaqueductal gray [28–30]. Studies have shown that Dyn A interacting with the KOR has inhibitory effects on the brain reward circuit by inhibiting dopamine in brain regions that are associated with drug dependence [31,32]. In terms of pain, there has been some evidence that activation of KOR antagonizes the analgesic effects of the MOR [33]. Other studies have shown that selective KOR agonists have antinociceptive effects as well as being free of the adverse side effects (constipation, respiratory depression, tolerance, addiction) that are mediated by the MOR [34]. These selective KOR agonists have been found to be effective analgesics in visceral and inflammatory pain models [35,36]. Although KOR agonists are free of the side effects from MOR agonists, they have been found to have dysphoria and psychotomimetic effects that limit their use in the clinic [37].
Table 2. . Binding and function of Dynorphin A at the opioid receptors.
| hMOR | hDOR | hKOR | |
|---|---|---|---|
| Ki (nM) |
1.60 ± 0.18 |
1.25 ± 0.12 |
0.05 ± 0.01 |
| EC50 (nM) | 30 ± 5 | 84 ± 11 | 0.43 ± 0.08 |
hDOR; human δ-opioid receptor; hKOR: human κ-opioid receptor hMOR: human μ-opioid receptor.
Data taken from Zhang et al. 1998 [38].
Structure–activity relationship of Dyn A at KOR
An early structure–activity relationship (SAR) study by Chavkin and Goldstein examined truncations of Dyn (1–13), since this fragment was found to have the same inhibitory effects as Dyn A [39]. Truncations of the N-terminal tyrosine residue abolished opioid activity in the GPI assay, and therefore, the studies focused on truncations at the C-terminus. From these studies, it was found that a C-terminal modification to an amide retained high affinity [39]. It was also found that Lys13, Lys11 and Arg7 residues were important for the biological activity at the KOR, with Arg7 being the most important residue [39]. From this work, it was suggested that the first four residues of Dyn A contain the message region, whereas residues 5–13 are the address region which is responsible for the potency and specificity at the KOR. Further studies have been performed in many laboratories to discover key residues that imparted selectivity for the KOR over MOR and DOR. Substitution of Gly2 with DAla in Dyn A-(1–11)NH2 to prevent aminopeptidase activity was found to not be a viable option, as this substitution decreased biological activity. Instead, N-terminal methylation, another strategy to prevent aminopeptidase activity, was well tolerated resulting in similar potency (IC50 = 0.42 nM) as parent ligand (IC50 = 0.23 nM) [39]. Our previous studies found that when Gly3 of Dyn-(1–11)NH2 was replaced with DAla, this led to greater selectivity at the KOR over the MOR (350-fold) and DOR (1300-fold) [40]. Our group extensively studied the effects of cyclization between Leu5 and Lys11 in Dyn-(1–11)NH2 and identified a selective KOR ligand [Pen5, Cys11] Dyn-(1–11)NH2 (KOR/MOR/DOR = 1/2.4/165) in binding assays but was inactive in the GPI and MVD assays [41–43]. Similar results were seen with substitutions of Tyr1 with Nα-AcTyr1, DTyr1, Phe1, or Phe(p-Br)1, and Ile8 with DAla8. All the previous compounds were found to be up to 576-fold more selective at KOR than DOR but all had no activity in GPI or MVD assays so were not pursued any further [44]. An enhancement of KOR selectivity was also seen with substitution of DPro in position 10 of Dyn (1–11)NH2 (KOR/MOR/DOR = 1/8/70, Table 3) [45]. Aldrich and colleagues used [DPro10] Dyn A-(1–11) as a template to investigate N-monoalkylated and N,N-dialkylated tyrosine derivatives. They found that all of the N-monoalkylated analogs had greater KOR selectivity when compared with the N,N-dialkylated analogs, with the greatest KOR selectivity shown by N-allyl substitution (KOR/MOR/DOR = 1/220/9200, Table 3) [46]. Many of these analogs did not show efficacy in in vivo models of pain, and one reason may be because of low stability of the analogs. It has been found that degradation of Dyn A occurs at Tyr1-Gly2, Arg6-Arg7 and Pro10-Lys11 [47]. In an effort to increase the stability of the analogs as well as their blood–brain barrier (BBB) penetration, E-2078, was synthesized (H-MeTyr-Gly-Gly-Phe-Leu-Arg-NMeArg-DLeuNHEt). This analog was found to have a half-life (t1/2) of 4 h (cf, Dyn A t1/2 = 0.5 h), and had binding affinities at the opioid receptors comparable to Dyn A (KOR/MOR/DOR = 1/2.4/14, Table 3) [48,49]. E-2078 was found to be analgesic following subcutaneous administration in the formalin and tail flick tests in mice and crossed the BBB [47,50]. Another analog that was designed for an increase in stability was, SK-9709 (H-Tyr-DAla-Phe-Leu-ArgΨ(CH2NH)-Arg-NH2), in which the peptide bond was replaced by a Ψ (CH2NH) bond. SK-9709 showed antinociceptive effects in the acetic acid induced writhing test after subcutaneous, intracerebroventricular and intrathecal injections and was able to cross the BBB [51]. Although E-2078 and SK-9709 both appear to be promising, selective and stable KOR agonists, they were found to be effective in inflammatory pain models, the formalin test and acetic acid-induced writhing test. It is well known that opioids are effective in inflammatory pain states but are less effective in neuropathic pain states [52]. Therefore, tests with these compounds in neuropathic pain states, such as, spinal nerve ligation, diabetes-induced neuropathy or neuropathic bone cancer pain will need to be performed.
Table 3. . Common Dynorphin A agonists at the KOR.
| Compound | Structure | Selectivity (KOR/MOR/DOR) | GPI (nM) | Ref. |
|---|---|---|---|---|
| Dyn A- (1–13) |
H-Y-G-G-F-L-R-R-I-R-P-K-L-K-OH |
1/17/N.D. |
0.63 |
[15,51] |
| Dyn A-(1–11)NH2 |
H-Y-G-G-F-L-R-R-I-R-P-K-NH2 |
1/17/44 |
1.1 ± 0.3 |
[40] |
| [DAla3] DynA-(1–11) NH2 |
H-Y-G-a-F-L-R-R-I-R-P-K-NH2 |
1/350/1300 |
8.1 ± 2.3 |
[40] |
| [DPro] DynA-(1–11) |
H-Y-G-G-F-L-R-R-I-R-p-K-OH |
1/8/70 |
0.22 |
[46] |
| N-allyl [DPro10] DynA-(1–11) |
N-allyl,Y-G-G-F-L-R-R-I-R-p-K-OH |
1/220/920 |
18 |
[46] |
| E-2078 |
H-MeY-G-G-F-L-R-NMeR-l-NHEt |
1/2.4/14 |
0.3 ± 0.03 |
[49] |
| SK-9709 | H-Y-a-F-L-RΨ(CH2NH)-Arg-NH2 | 1/15/350 | N.D. | [51] |
N.D.: Not determined.
Although the gold standard for relief of severe pain is MOR agonists, there are serious side effects caused by long-term administration [53]. KOR agonists were pursued as a pain therapeutic that would not have the adverse side effects as MOR agonists. Unfortunately, it was found that while centrally acting KOR agonists did not have addictive properties, they had unpleasant side effects such as dysphoria, sedation and diuresis [37]. These side effects are mediated through the CNS and limit the use of KOR agonists for treatment of pain at the CNS but may be beneficial in pain at the PNS. Since KOR agonists have effects on mood, they are being examined as a treatment of the manic phase in bipolar disorder [54]. KOR agonists have been found to decrease dopamine levels and are also being examined as a treatment for drug abuse, in particular cocaine [55,56].
KOR antagonists for the treatment of pain
Since Dyn A and the KOR are known to be involved in the body's response to stress, KOR antagonists could have some benefit in anxiety and depression. KOR antagonists have been found to be anxiolytic [57], prevent stress-induced reinstatement of cocaine-seeking behavior [58] and be beneficial in the treatment of opiate addiction [59]. KOR antagonists are also being investigated as a combination therapy with MOR/DOR agonists for the treatment of pain. In an effort to develop KOR antagonists, 1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid (Tic) was replaced for Gly2 in Dyn A-(1–11)NH2 since it is known from dermorphin and Leu-enkephalin that this substitution can convert agonists to antagonists [60]. This substitution did cause antagonism at all three opioid receptors, but the selectivity for KOR was reduced (KOR/MOR/DOR = 1/0.5/0.14, Table 4) [60]. The Schiller group designed (2′,6′-dimethyltyrosine [Dmt]1) Dyn A-(1–11)NH2 analogs with the N-terminal amino group deleted or replaced with a methyl group using 3,4-Dihydro-2H-pyran-2-methanol (Dhp) or (2S)-2-methyl-3-(2′,6′-dimethyl-4′-hydroxyphenyl)-propionic acid (Mdp) to test for KOR antagonism. All of the analogs were potent KOR antagonists with weak MOR and DOR activity, with dynantin ([(2S)-Mdp1] Dyn A-[1–11]NH2) being the most selective for KOR, with subnanomolar binding affinity and antagonist activity [61]. [Pro3] DynA-(1–11)NH2 is one of the most selective KOR analogs (KOR/MOR/DOR =1/2100/3330, Table 4) but is a weak antagonist in both the GPI assay as well as the (35S)GTPγS assay [62]. Aldrich and colleagues identified aromatic dynorphin (arodyn) starting from a novel chimeric peptide, extacet (a MOR-selective analog), substituted at the N-terminus in the message region. Arodyn was found to be selective for KOR (KOR/MOR/DOR = 1/170/580, Table 4) and was able to completely reverse the agonist activity of Dyn A-(1–11)NH2 in the cAMP assay [63]. Aldrich and colleagues also synthesized, cyclodyn, (cycloN,5[Trp3, Trp4, Glu5] DynA-[1–11]NH2) by connecting the N-terminus to the side chain of the Glu5 residue. Cyclodyn was found to be selective for KOR (KOR/MOR/DOR = 1/12/330, Table 4) and was able to block the agonist activity of Dyn A-(1–11)NH2 in the cAMP assay [64]. Another cyclic Dyn A analog is zyklophin, (Nα-benzyl)Tyr1 cyclo(DAsp5, Dap8) DynA-(1–11)NH2, which was selective for KOR (KOR/MOR/DOR = 1/190/330, Table 4) [65]. In the cAMP assay, zyklophin antagonized Dyn A-(1–11)NH2 with Kb= 84 nM, and also antagonized the antinocicpetive actions induced by U50,488 in the warm water tail withdrawal assay following subcutaneous injection [65,66]. Zyklophin was found to be more metabolically stable than other analogs, was able to cross the BBB and had a shorter duration of action (12 h) when compared with other KOR antagonists (e.g., JDTic several weeks) (for a complete review on KOR ligands see [67]).
Table 4. . Common Dynorphin A antagonists at the KOR.
| Compound | Structure | Selectivity (KOR/MOR/DOR) | Ke (nM) | Ref. |
|---|---|---|---|---|
| [Tic2] DynA-(1–11)NH2 |
H-Y-Tic-G-F-L-R-R-I-R-P-K-NH2 |
1/0.5/0.14 |
460 |
[60] |
| Dynantin |
(2S)-Mdp-G-G-F-L-R-R-I-R-P-K-NH2 |
1/260/200 |
3.9 ± 0.7 |
[61] |
| [Pro3] DynA-(1–11)NH2 |
H-Y-G-P-F-L-R-R-I-R-P-K-NH2 |
1/2100/3300 |
240 ± 51 |
[62] |
| Arodyn |
Ac-F-F-F-R-L-R-R-a-R-P-K-NH2 |
1/170/580 |
N.D. |
[63] |
| Cyclodyn |
c[Y-G-W-W-E]-R-R-I-R-P-K-NH2 |
1/12/330 |
N.D. |
[64] |
| Zyklophin | Bz-Y-G-G-F-c[d-R-R-Dap]-R-P-K-NH2 | 1/190/330 | 84† | [65] |
†Kb calculated from cAMP assay.
Ke calculated from GPI assay, see specific citation for further details.
N.D.: not determined
Future directions for Dynorphin A-based KOR ligands
KOR agonists have serious side effects (dysphoria and psychotomimetic effects) that limit their therapeutic use for pain, but have potential to treat bipolar disorder [54]. Since the psychotomimetic side effects are mediated by the CNS, peripherally acting KOR agonists may not have these side effects and thus are currently being investigated as well as mixed MOR/KOR agonists. It has been suggested that the dysphoric effects of KOR agonists occurs through the recruitment of β-arrestin, whereas the analgesic properties do not [68]. Therefore, analogs that are biased and do not recruit β-arrestin may not have the dysphoric effects and this may be another avenue for pain management.
Another potential therapeutic for pain states that has recently been explored in our laboratory is the development of mixed MOR/DOR agonists with KOR antagonism that can be analgesics with limited side effects. One disadvantage of KOR antagonism is the long duration of action (lasting more than 21 days in vivo) [69]. Work is currently being carried out to develop ligands that do not have a long duration of action at the KOR.
Nonopioid effects of Dynorphin A
Although Dyn A has well documented opioid effects that cause analgesia, there are also some effects of Dyn A that cannot be explained by the opioid receptors (see [70] for review on nonopioid effects). Walker and colleagues were one of the first to document the nonopioid effects of the [des-Tyr1]-Dyn A fragments [71]. They observed that when Dyn A-(1–13) was injected into the brain, it produced dramatic motor and behavioral effects that were different than those produced by enkephalin-containing endorphins and these effects could not be blocked by naloxone [72]. These effects of Dyn-(1–13) were similar to the effects of the (des-Tyr1)Dyn A fragment, Dyn A-(2–13), which was found not to interact with the opioid receptors. It has been found that upon release in the synapse, aminopeptidases rapidly degrade Dyn A to the des-tyrosyl fragments [73]. It is well established that Dyn A has serious motor effects when injected into rats, intracerebroventricular injection into the brain (either Dyn A-[1–17] or Dyn A-[1–13]) induced a barrel rotation in rats [74–76] and intrathecal injection at high doses caused paralysis [77–79]. All of these motor effects were not blocked by the opioid antagonist naloxone and are therefore nonopioid in nature. Levels of Dyn A have been shown to increase in the response to stressors, including neuropathic pain [80] and inflammatory pain [81]. An increase in Dyn A was seen in the anterior pituitary, thalamus and spinal cord in a chronic arthritic pain model [82]. Dyn A may also play a role in the inflammatory response, as evident in that Dyn A induced the release of histamine from rat mast cells [83] and induced plasma extravasation [84]. Both of these inflammatory responses were not blocked by naloxone, providing further evidence for the nonopioid effect. An increase in Dyn A has also been found in other pathological pain states such as chronic pancreatitis [85], opioid overuse induced hyperalgesia [86], bone cancer pain [87] and spinal cord trauma [88].
It has been demonstrated that Dyn A is not required for the initiation of pain but instead for the maintenance of pain, which was shown in prodynorphin knockout (KO) mice that had spinal nerve ligation. These KO mice had similar paw withdrawal latency and thresholds when compared with wild-type littermates days 2–6 after injury [89]. After day 10 post-surgery, the KO mice latency and thresholds were reduced to baseline while their wild-type littermates stayed below baseline [89]. Neuropathic pain is characterized by hyperalgesia and allodynia, and it has been demonstrated that intrathecal injection of Dyn A produces long-lasting allodynia (>60 days) that resembles a neuropathic pain state [90]. The administration of a Dyn A antiserum to nerve-injured rats reversed the neuropathic pain state [80]. Similar results were seen in an inflammatory pain model, thus giving more evidence that the higher levels of Dyn A contribute to different pain states [91]. All of these data have led to the hypothesis that during chronic pain states there is an upregulation of Dyn A which results in pronociceptive effects through a nonopioid mechanism to maintain the pain state.
N-Methyl-d-aspartate receptor as the target for nonopioid effects
The N-Methyl-d-aspartate receptor (NMDAR) is a channel that consists of a tetrameric structure of seven subunits [92]. Upon binding of its endogenous agonists, glutamate and glycine, the channel opens and allows the influx of positive ions Na+ and Ca2+. Many studies have found that NMDAR antagonists can prevent Dyn A-induced neurological dysfunctions such as loss of tail-flick reflex [93,94], hindlimb paralysis and mortality [94–96]. Pretreatment of the NMDAR antagonist, MK-801, prevented Dyn A-induced allodynia whereas naloxone did not [90]. Further evidence for the NMDAR as the nonopioid target is that after spinal infusion of Dyn A-(2–13), an increase in prostaglandin E (PGE2) and excitatory amino acids were measured and were blocked by the NMDAR antagonist, amino-5-phosphonovalerate, AP5 [97]. The release of excitatory amino acids was also seen after the addition of Dyn A to the rat hippocampus [98].
Direct evidence for Dyn A interacting at the NMDAR was obtained from competitive binding assays, showing that Dyn A-(1–13) could displace part but not all of [3H] glutamate in rat brain membranes [99]. Further competitive binding of Dyn A with NMDAR antagonists, [3H] MK-801 and [3H] CGP39, 653, showed moderate-to-low affinity [100,101]. Dyn A's affinity at the NMDAR was directly measured using [125I] Dyn A-(2–17) with Kd = 10 nM [102]. In the same study, it was also found that NMDAR antagonists that occupy the closed state of the channel (AP-5, ifenprodil and 7-chlorokynurenic acid) potentiated the affinity of [125I] Dyn A-(2–17) [102]. Therefore, this suggests that Dyn A prefers the closed inactive state of the receptor and may behave as an antagonist. An inhibitory action of Dyn A at the NMDAR does not explain the excitatory effects of Dyn A shown in vivo. Also, it was found that in cortical neurons, Dyn A-(2–17) induced an increase in Ca2+ which cannot be blocked by MK-801, and therefore, suggests a different mechanism [103]. One possible mechanism may be that a different splice variant or combination of splice variants of the NMDAR are responsible for Dyn A's excitatory effects. There are many different splice variants that make up the NMDAR which leads to thousands of possible combinations of receptors that may have Dyn A binding sites. In particular the NR1 subtype has been studied and it was demonstrated that both Dyn A and Dyn A-(2–17) were able to directly bind to the NR1 subunit on the NMDAR [104,105]. Using a decoy peptide that could bind Dyn A-(2–17), Shippenberg et al. were able to prevent the potentiation of NMDA-mediated currents by Dyn A-(2–17) [106]. This decoy was also able to reduce Dyn A-(2–17) evoked cell death in spinal cord neurons in vitro [106]. More interestingly, intrathecal administration of this decoy peptide was able to prevent the motor impairment and tactile allodynia from intrathecal Dyn A-(2–17) [106]. Therefore, peptides that can bind to Dyn A, such as the decoy peptide, would prevent binding to the NMDAR and may be used as a therapeutic.
Non-neuronal cells as the target for the nonopioid effects
There is also some evidence that the neurotoxic effects of Dyn A arise from interaction with microglia. Studies by Mika et al. found that intrathecal minocycline, a microglia inhibitor, prevented Dyn A-induced paralysis in rats. After sciatic nerve injury, minocycline reduced the elevated levels of prodynorphin mRNA [107]. The authors suggest that to control neuropathic pain, drugs that alter the prodynorphin system could be used. More work will need to be carried out to determine if microglia are indeed a target for Dyn A.
Bradykinin receptors as the target for nonopioid effects
Our collaborator, Lai, found that in cultured neurons of neonatal rat cortex Dyn A-(2–17) induced an increase in Ca2+ that was not blocked by naloxone or MK-801 [103]. The Lai group later discovered that the nonopioid excitatory effects of Dyn A were mediated by the bradykinin receptors (BRs) [108]. In their study, Dyn A-(2–13) was shown to induce a transient increase in Ca2+ in rat DRG as well as the F11 cell line (hybridoma of rat DRG and mouse neuroblastoma cells). By using Ca2+ channel blockers, they found that this effect was mediated by P/Q and L type Ca2+ channels [108]. In an effort to determine what receptor Dyn A-(2–13) was interacting with to cause the Ca2+ influx, antagonists for each receptor known to be present in the F11 cell line were tested. The bradykinin 2 receptor (B2R) antagonist, HOE140, was the only antagonist that had an ability to abolish Dyn A-(2–13)-induced Ca2+ influx, whereas, the other antagonists including MK-801, an NMDAR antagonist, had no effect [108]. Competitive radioligand binding assays showed that Dyn A-(2–13) displaced [3H] bradykinin (BK) with moderate affinity (IC50 = 4 μM) [108]. Recently, we have found that Dyn A-(2–13) binds with high affinity to BRs in rat brain membranes (IC50 = 170 nM), and an underestimation of the affinity was caused by changes in pH [109]. H89, a PKA inhibitor, was able to abolish Dyn A-(2–13)'s neuroexcitatory effects. On the basis of this, it was proposed that the pronociceptive effects of Dyn A arise from activation of the BRs that couples a PKA pathway to activate L and P/Q type Ca2+ channels [108]. Further study showed that HOE140 was able to block Dyn A-(2–13)'s neuroexcitatory effects in rats [108], supporting the notion that Dyn A's pronociceptive effects are mediated through the BRs.
The mRNA for the B2R was measured in the DRG, and a higher expression of transcript was measured after nerve injury. In the spinal cord of nerve-injured rats, low levels of the precursor to BK, kininogen, was measured. In contrast, high levels of transcript for the precursor of Dyn A, prodynorphin, was measured [108]. Since the BRs are present in the spinal cord but the endogneous ligand, BK, or its precursor are not, it is hypothesized that Dyn A may be the endogenous ligand for the BRs in the spinal cord under pathological pain conditions. Taken together, it is now believed that the development of BRs antagonists to block Dyn A's pronociceptive actions can be an avenue for the treatment of chronic neuropathic pain (Figure 3).
Figure 3. . Dynorphin A in the spinal cord during nerve injury or chronic pain state.

The BRs are Gq-coupled GPCRs, and signal through phospholipase C [110]. The endogenous ligands for the BRs, BK and kallidin, have been found to be mediators of inflammation, and therefore, antagonists have been designed to block the hyperalgesic effects caused by activation of the BRs (in particular the B2R) [111]. In addition to the inflammatory response of BK, the ligand has also been found to be cardioprotective and a regulator of blood pressure [112]. Early studies aimed to develop B2R antagonists based on BK for the treatment of pain had undesirable cardiovascular side effects (see [113] for review on BR antagonists). However, considering the cardiovascular effects are limited to the peripheral B2Rs, centrally acting B2R antagonists can avoid these serious side effects. Since Dyn A's nonopioid effects are mediated through CNS BRs, the inhibition by BR antagonists in the CNS will result in antihyperalgesic effects without cardiovascular liabilities.
Structure–activity relationship of Dynorphin A analogs at the bradykinin receptors
To determine the minimum pharmacophore at the BRs, truncations were made at the N- and C- termini of Dyn A-(2–13) and a distinct SAR was identified. It was found that analogs that had a basic amino acid at the C-terminus such as Dyn A-(3–11) and Dyn A-(4–11) had higher affinity (IC50 = 130 and 140 nM, respectively, Table 5) at the BRs than analogs with hydrophobic residues, Dyn A-(3–8) and Dyn A-(3–10), (IC50 = 2300 and 810 nM, respectively, Table 5) [109]. Truncations at the N-terminus until position 4 did not affect binding, and therefore, the N-terminus is not considered to be important in the binding at the BRs. N-terminal acetylation was well-tolerated (IC50 = 120 nM) which further demonstrates that the N-terminus is not important for BRs recognition. Interestingly, when Arg7, which is known to be an important residue for binding at the KOR was deleted, binding at the BRs was retained [114]. In an effort to improve stability, diverse modifications were performed, in which substitution of the non-natural amino acid, norleucine, for Leu and/or Ile was tolerated. However, C-terminal amidation caused a reduction in affinity at the BRs (IC50 = 6500 nM, Table 5), which suggests that along with a basic amino acid at the C-terminus, the C-terminal carboxylate is also important [109]. The results suggest that amphipathicity, alternating hydrophobic residue, basic residue, is a key component for recognition at the BRs [115]. Nuclear Magnetic Resonance (NMR) studies of selected high-affinity ligands demonstrated a distorted type I β-turn structure at the C-terminus [109]. From the SAR studies, we have identified key structural features for the BRs; basic amino acid at the C-terminus, presence of C-terminal acid, combination of basic and hydrophobic amino acids and presence of Pro residue to make a turn structure. One of the lead ligands, [des-Arg7] Dyn A-(4–11) which retained high affinity (IC50 = 190 nM, Table 5) was tested for analgesic effects in vivo. This ligand blocked Dyn A-(2–13)-induced hyperalgesia providing evidence that this ligand is an antagonist [109]. In nerve-injured rats, intrathecal administration of the ligand reversed thermal hyperalgesia and mechanical hypersensitivity. Importantly, this ligand did not block BK-induced peripheral effects, so it may avoid the cardiovascular liabilities. A main advantage of peptides as therapeutics is that they have high specificity for their target. Further evidence of this was shown in an off-target screening assay of the lead ligand, in which the ligand did not interact with any of the 43 receptors tested [109].
Table 5. . Structure–activity relationship of Dynorphin A analogs at the bradykinin receptors.
| Fragment | Structure | IC50 (nM)† |
|---|---|---|
| Dyn A-(2–13) |
H-G-G-F-L-R-R-I-R-P-K-L-K-OH |
170 |
| Dyn A-(3–11) |
H-G-F-L-R-R-I-R-P-K-OH |
130 |
| Dyn A-(4–11) |
H-F-L-R-R-I-R-P-K-OH |
140 |
| Dyn A-(3–10) |
H-G-F-L-R-R-I-R-P-OH |
810 |
| Dyn A-(3–8) |
H-G-F-L-R-R-I-OH |
2300 |
| [des-Arg7] Dyn A-(4–11) |
H-F-L-R-I-R-P-K-OH |
190 |
| Dyn A-(4–11)NH2 |
H-F-L-R-R-I-R-P-K-NH2 |
6500 |
| [des-Arg7] Ac-Dyn A-(4–11) | Ac-F-L-R-I-R-P-K-OH | 120 |
†Assay carried out according to the conditions in [109].
Future directions for Dynorphin A analogs at bradykinin receptors
Currently, a lead ligand that binds with high affinity to the BRs in the CNS, blocks Dyn A-(2–13)-induced neuroexcitatory effects in naive animals, and reversed hyperalgesia and hypersensitivity in nerve-injured animals has been discovered. Future work will involve modifications of the lead ligand to increase metabolic stability due to the short half-life. Although the lead ligand is effective, it is composed of all natural amino acids and therefore, susceptible to enzymatic degradation. Substitution of unnatural amino acids will be tested in binding as well as stability assays.
Conclusion
Chronic pain, and in particular neuropathic pain, continues to be a disease in which there are no effective treatments. Many of the approved therapeutics for this disease have serious side effects that limit their use. The inhibitory effects of Dyn A were first discovered and were found to be mediated through the opioid receptors. In an effort to obtain a therapeutic with analgesic properties and limited side effects, extensive SAR studies on Dyn A were done for the KOR. Although KOR agonists were found to be analgesics, they had dysphoric side effects that limited their use. KOR antagonists with shorter durations of action are currently being developed and may be used as analgesics in combination with MOR/DOR agonists. Dyn A has been found to be upregulated in many pain states; chronic pancreatitis, bone cancer pain, arthritis, spinal cord trauma, inflammatory pain and neuropathic pain. There has also been evidence about the excitatory effects of Dyn A that cannot be blocked by naloxone and are therefore, nonopioid. The NMDAR has been suggested as the target for the nonopioid effects of Dyn A but the evidence suggests an inhibitory action at the NMDAR which does not explain the neuroexcitatory effects of Dyn A seen in vivo. Instead there is substantial evidence for Dyn A's nonopioid excitatory effects to be mediated through the BRs which through a PKA-mediated pathway activate L and P/Q type calcium channels to cause an influx of Ca2+. Dyn A may be the endogenous ligand for BRs in the spinal cord, as transcripts for the BRs and Dyn A have been measured spinally but the endogenous ligand, BK has not. Extensive SAR studies of Dyn A analogs at the BRs have discovered a lead ligand, (des-Arg7) Dyn A-(4–11) that is able to antagonize Dyn A-(2–13)-induced neuroexcitatory effects and attenuate hyperalgesic effects in a neuropathic pain model. This ligand also does not show antagonist activity at the peripheral BRs that BK has shown and thus, can be a safe drug without the cardiovascular liabilities.
Future perspective
Future advances in this field would greatly benefit from determining the mechanisms of neuropathic pain. These mechanisms are still unknown but it is known that it is different than acute pain and therefore, the treatment for the two pain states should not be the same. One potential mechanism of neuropathic pain is the upregulation of Dyn A which has been found to contribute to the maintenance of a neuropathic pain state. Developing antagonists that block this interaction may provide novel therapeutics that do not have the liabilities of opioids.
Executive summary.
Neuropathic pain is a debilitating disease that results from the dysfunction of the CNS or peripheral nervous system. There are no effective treatments and a better understanding of the underlying mechanisms is needed.
Dynorphin A (Dyn A) shows neuroinhibitory effects through the opioid receptors, specifically the κ-opioid receptor (KOR). The peptide also has neuroexcitatory effects that are thought to be mediated through the N-Methyl-d-aspartate receptor (NMDAR) and/or bradykinin receptors (BRs).
Extensive structure–activity relationship studies have been completed on Dyn A and key pharmacophores have been found to be important for agonist and antagonist function at the KOR.
KOR agonists based on Dyn A are effective analgesics that do not have μ-opioid receptor induced side effects but have dysphoric side effects that limit their use.
KOR antagonists based on Dyn A structure that have a short duration of action are currently being pursued in combination with μ-opioid receptor/δ-opioid receptor agonists for analgesics with limited side effects.
Blockade of the neuroexcitatory effects of nonopioid Dyn A by the NMDAR antagonist, MK-801, suggest that these effects are mediated through the NMDAR. Evidence of a direct interaction of nonopioid Dyn A with the NMDAR suggests an inhibitory effect at the receptor, which does not account for the excitatory effects seen in vivo.
Recent studies discovered that the BRs are the nonopioid target for Dyn A. Dyn A-(2–13) has been found to bind with high affinity at the BRs and cause an increase in Ca2+.
An antagonist that blocked Dyn A's excitatory actions at the BRs has been identified and was effective in a neuropathic pain model.
Footnotes
Financial & competing interests disclosure
This work has been supported by U.S. Public Health Services, NIH, and NIDA P01DA006284 and R01DA013449. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
- 1.Laupacis A, Sackett DL, Roberts RS. An assessment of clinically useful measures of the consequences of treatment. N. Engl. J. Med. 1988;318(26):1728–1733. doi: 10.1056/NEJM198806303182605. [DOI] [PubMed] [Google Scholar]
- 2.Relieving Pain in America: A Blueprint for Transforming Prevention, Care, Education, and Research. The National Academies Press; Washington DC, USA: 2011. Committee on Advancing Pain Research, Care, Education; Board on Health Sciences Policy; Institute of Medicine; pp. 1–19. [PubMed] [Google Scholar]
- 3.Stewart WF, Ricci JA, Chee E, Morganstein D, Lipton R. Lost productive time and cost due to common pain conditions in the US workforce. JAMA. 2003;290(18):2443–2454. doi: 10.1001/jama.290.18.2443. [DOI] [PubMed] [Google Scholar]
- 4.Baron R. Mechanisms of disease: neuropathic pain – a clinical perspective. Nat. Clin. Pract. Neurol. 2006;2(2):95–106. doi: 10.1038/ncpneuro0113. [DOI] [PubMed] [Google Scholar]; • A good review on the mechanisms of neuropathic pain.
- 5.Moore RA, Straube S, Wiffen PJ, Derry S, Mcquay HJ. Pregabalin for acute and chronic pain in adults. Cochrane Database Syst. Rev. 2009;2(3) doi: 10.1002/14651858.CD007076.pub2. CD007076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sindrup SH, Otto M, Finnerup NB, Jensen TS. Antidepressants in the treatment of neuropathic pain. Basic Clin. Pharmacol. Toxicol. 2005;96(6):399–409. doi: 10.1111/j.1742-7843.2005.pto_96696601.x. [DOI] [PubMed] [Google Scholar]
- 7.Watanabe S, Bruera E. Corticosteroids as adjuvant analgesics. J. Pain Symptom Manag. 1994;9(7):442–445. doi: 10.1016/0885-3924(94)90200-3. [DOI] [PubMed] [Google Scholar]
- 8.Mcintosh M, Cruz LJ, Hunkapiller MW, Gray WR, Olivera BM. Isolation and structure of a peptide toxin from the marine snail Conus magus . Arch. Biochem. Biophys. 1982;218(1):329–334. doi: 10.1016/0003-9861(82)90351-4. [DOI] [PubMed] [Google Scholar]
- 9.Lynch SS, Cheng CM, Yee JL. Intrathecal ziconotide for refractory chronic pain. Ann. Pharmacother. 2006;40(7–8):1293–1300. doi: 10.1345/aph.1G584. [DOI] [PubMed] [Google Scholar]
- 10.Mcgivern JG. Ziconotide: a review of its pharmacology and use in the treatment of pain. Neuropsychiatr. Dis. Treat. 2007;3(1):69–85. doi: 10.2147/nedt.2007.3.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sachs CJ. Oral analgesics for acute nonspecific pain. Am. Fam. Physician. 2005;71(5):913–918. [PubMed] [Google Scholar]
- 12.Finnerup NB, Sindrup SH, Jensen TS, et al. The evidence for pharmacological treatment of neuropathic pain. Pain. 2010;150(3):573–581. doi: 10.1016/j.pain.2010.06.019. [DOI] [PubMed] [Google Scholar]
- 13.Mohammed SI, Eldabe S, Simpson KH, et al. Bolus intrathecal injection of ziconotide (Prialt®) to evaluate the option of continuous administration via an implanted intrathecal drug delivery (ITDD) system: a pilot study. Neuromodulation. 2013;16(6):576–581. doi: 10.1111/ner.12003. discussion 582. [DOI] [PubMed] [Google Scholar]
- 14.Medline Plus. Drugs, herbs and supplements. http://www.nlm.nih.gov/medlineplus/druginfo/meds
- 15.Goldstein A, Tachibana S, Lowney LI, Hunkapiller M, Hood L. Dynorphin-(1–13), an extraordinarily potent opioid peptide. Proc. Natl Acad. Sci. USA. 1979;76(12):6666–6670. doi: 10.1073/pnas.76.12.6666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goldstein A, Fischli W, Lowney LI, Hunkapiller M, Hood L. Porcine pituitary dynorphin: complete amino acid sequence of the biologically active heptadecapeptide. Proc. Natl Acad. Sci. USA. 1981;78(11):7219–7223. doi: 10.1073/pnas.78.11.7219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schwarzer V. 30 years of dynorphins – new insights on their functions in neuropsychiatric diseases. Pharmacol. Ther. 2009;123:353–370. doi: 10.1016/j.pharmthera.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chavkin C, James IF, Goldstein A. Dynorphin is a specific endogenous ligand of the kappa opioid receptor. Science. 1982;215(22):413–415. doi: 10.1126/science.6120570. [DOI] [PubMed] [Google Scholar]
- 19.Zukin RS, Eghbali M, Olive D, Unterwald EM, Tempel A. Characterization and visualization of rat and guinea pig brain kappa opioid receptors: evidence for kappa 1 and kappa 2 opioid receptors. Proc. Natl Acad. Sci. USA. 1988;85(11):4061–4065. doi: 10.1073/pnas.85.11.4061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clark JA, Liu L, Price M, Hersh B, Edelson M, Pasternak GW. Kappa opiate receptor multiplicity: evidence for two U50,488-sensitive kappa 1 subtypes and a novel kappa 3 subtype. J. Pharmacol. Exp. Ther. 1989;251(2):461–468. [PubMed] [Google Scholar]
- 21.Simonin F, Gaveriaux-Ruff C, Befort K, et al. Kappa-opioid receptor in humans: cDNA and genomic cloning, chromosomal assignment, functional expression, pharmacology, and expression pattern in the central nervous system. Proc. Natl Acad. Sci. USA. 1995;92(15):7006–7010. doi: 10.1073/pnas.92.15.7006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cvejic S, Devi LA. Dimerization of the delta opioid receptor: implication for a role in receptor internalization. J. Biol. Chem. 1997;272(43):26959–26964. doi: 10.1074/jbc.272.43.26959. [DOI] [PubMed] [Google Scholar]
- 23.Gomes I, Jordan BA, Gupta A, Rios C, Trapaidze N, Devi LA. G protein coupled receptor dimerization: implications in modulating receptor function. J. Mol. Med. 2001;79(5–6):226–242. doi: 10.1007/s001090100219. [DOI] [PubMed] [Google Scholar]
- 24.Jordan BA, Devi LA. G-protein-coupled receptor heterodimerization modulates receptor function. Nature. 1999;399(6737):697–700. doi: 10.1038/21441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berg KA, Rowan MP, Gupta A, et al. Allosteric interactions between delta and kappa opioid receptors in peripheral sensory neurons. Mol. Pharmacol. 2012;81(2):264–272. doi: 10.1124/mol.111.072702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Le Naour M, Lunzer MM, Powers MD, et al. Putative kappa opioid heteromers as targets for developing analgesics free of adverse effects. J. Med. Chem. 2014;57(15):6383–6392. doi: 10.1021/jm500159d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bruchas MR, Land BB, Chavkin C. The dynorphin/kappa opioid system as a modulator of stress-induced and pro-addictive behaviors. Brain Res. 2010;1314:44–55. doi: 10.1016/j.brainres.2009.08.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Winkler CW, Hermes SM, Chavkin CI, Drake CT, Morrison SF, Aicher SA. Kappa opioid receptor (KOR) and GAD67 immunoreactivity are found in OFF and NEUTRAL cells in the rostral ventromedial medulla. J. Neurophysiol. 2006;96(6):3465–3473. doi: 10.1152/jn.00676.2006. [DOI] [PubMed] [Google Scholar]
- 29.Neugebauer V, Li W, Bird GC, Han JS. The amygdala and persistent pain. Neuroscientist. 2004;10(3):221–234. doi: 10.1177/1073858403261077. [DOI] [PubMed] [Google Scholar]
- 30.Gutstein HB, Mansour A, Watson SJ, Akil H, Fields HL. Mu and kappa opioid receptors in periaqueductal gray and rostral ventromedial medulla. Neuroreport. 1998;9(8):1777–1781. doi: 10.1097/00001756-199806010-00019. [DOI] [PubMed] [Google Scholar]
- 31.Bruijnzeel AW. Kappa-opioid receptor signaling and brain reward function. Brain Res. Rev. 2009;62(1):127–146. doi: 10.1016/j.brainresrev.2009.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mysels D, Sullivan MA. The kappa-opiate receptor impacts the pathophysiology and behavior of substance use. Am. J. Addict. 2009;18(4):272–276. doi: 10.1080/10550490902925862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pan ZZ, Tershner SA, Fields HL. Cellular mechanism for anti-analgesic action of agonists of the kappa-opioid receptor. Nature. 1997;389(6649):382–385. doi: 10.1038/38730. [DOI] [PubMed] [Google Scholar]
- 34.Millan MJ. Kappa-opioid receptors and analgesia. Trends Pharmacol. Sci. 1990;11(2):70–76. doi: 10.1016/0165-6147(90)90321-x. [DOI] [PubMed] [Google Scholar]
- 35.Ko MC, Butelman ER, Woods JH. Activation of peripheral kappa opioid receptors inhibits capsaicin-induced thermal nociception in rhesus monkeys. J. Pharmacol. Exp. Ther. 1999;289(1):378–385. [PMC free article] [PubMed] [Google Scholar]
- 36.Walker JS. Anti-inflammatory effects of opioids. Adv. Exp. Med. Biol. 2003;521:148–160. [PubMed] [Google Scholar]
- 37.Pfeiffer A, Brantl V, Herz A, Emrich HM. Psychotomimesis mediated by kappa opiate receptors. Science. 1986;233(4765):774–776. doi: 10.1126/science.3016896. [DOI] [PubMed] [Google Scholar]
- 38.Zhang S, Tong Y, Tian M, et al. Dynorphin A as a potential endogenous ligand for four members of the opioid receptor gene family. J. Pharmacol. Exp. Ther. 1998;286(1):136–141. [PubMed] [Google Scholar]
- 39.Chavkin C, Goldstein A. Specific receptor for the opioid peptide dynorphin: structure–activity relationships. Proc. Natl Acad. Sci. USA. 1981;78(10):6543–6547. doi: 10.1073/pnas.78.10.6543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lung FD, Meyer JP, Li G, et al. Highly kappa receptor-selective dynorphin A analogues with modifications in position 3 of dynorphin A(1–11)-NH2. J. Med. Chem. 1995;38(4):585–586. doi: 10.1021/jm00004a002. [DOI] [PubMed] [Google Scholar]
- 41.Kawasaki AM, Knapp RJ, Kramer TH, et al. Design and synthesis of highly potent and selective cyclic dynorphin A analogues. J. Med. Chem. 1990;33(7):1874–1879. doi: 10.1021/jm00169a007. [DOI] [PubMed] [Google Scholar]
- 42.Kawasaki AM, Knapp RJ, Kramer TH, et al. Design and synthesis of highly potent and selective cyclic dynorphin A analogs. 2. New analogs. J. Med. Chem. 1993;36(6):750–757. doi: 10.1021/jm00058a012. [DOI] [PubMed] [Google Scholar]
- 43.Meyer JP, Collins N, Lung FD, et al. Design, synthesis, and biological properties of highly potent cyclic dynorphin A analogues. Analogues cyclized between positions 5 and 11. J. Med. Chem. 1994;37(23):3910–3917. doi: 10.1021/jm00049a010. [DOI] [PubMed] [Google Scholar]
- 44.Kawasaki AM, Knapp RJ, Walton A, et al. Syntheses, opioid binding affinities, and potencies of dynorphin A analogues substituted in positions, 1, 6, 7, 8 and 10. Int. J. Pept. Protein Res. 1993;42(5):411–419. doi: 10.1111/j.1399-3011.1993.tb00148.x. [DOI] [PubMed] [Google Scholar]
- 45.Gairin JE, Gouarderes C, Mazarguil H, Alvinerie P, Cros J. [D-Pro10]Dynorphin-(1–11) is a highly potent and selective ligand for k opioid receptors. Eur. J. Pharmacol. 1985;106:457–458. doi: 10.1016/0014-2999(84)90741-6. [DOI] [PubMed] [Google Scholar]
- 46.Choi H, Murray TF, Delander GE, Schmidt WK, Aldrich JV. Synthesis and opioid activity of [D-Pro(10)]dynorphin A-(1–11) analogues with N-terminal alkyl substitution. J. Med. Chem. 1997;40(17):2733–2739. doi: 10.1021/jm960747t. [DOI] [PubMed] [Google Scholar]
- 47.Nakazawa T, Furuya Y, Kaneko T, Yamatsu K, Yoshino H, Tachibana S. Analgesia produced by E-2078, a systemically active dynorphin analog, in mice. J. Pharmacol. Exp. Ther. 1990;252(3):1247–1254. [PubMed] [Google Scholar]
- 48.Tachibana S, Yoshino H, Arakawa Y, et al. Biowarning System in the Brain. University of Tokyo Press; Tokyo, Japan: 1988. [Google Scholar]
- 49.Yoshino H, Nakazawa T, Arakawa Y, et al. Synthesis and structure–activity relationships of dynorphin A-(1–8) amide analogues. J. Med. Chem. 1990;33(1):206–212. doi: 10.1021/jm00163a034. [DOI] [PubMed] [Google Scholar]
- 50.Yu J, Butelman ER, Woods JH, Chait BT, Kreek MJ. Dynorphin A (1–8) analog, E-2078, crosses the blood–brain barrier in rhesus monkeys. J. Pharmacol. Exp. Ther. 1997;282(2):633–638. [PubMed] [Google Scholar]
- 51.Hiramatsu M, Inoue K, Ambo A, Sasaki Y, Kameyama T. Long-lasting antinociceptive effects of a novel dynorphin analogue, Tyr-D-Ala-Phe-Leu-Arg psi (CH(2)NH) Arg-NH(2), in mice. Br. J. Pharmacol. 2001;132(8):1948–1956. doi: 10.1038/sj.bjp.0703982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Obara I, Parkitna JR, Korostynski M, et al. Local peripheral opioid effects and expression of opioid genes in the spinal cord and dorsal root ganglia in neuropathic and inflammatory pain. Pain. 2009;141(3):283–291. doi: 10.1016/j.pain.2008.12.006. [DOI] [PubMed] [Google Scholar]
- 53.Klepstad P, Kaasa S, Cherny N, Hanks G, De Conno F Research Steering Committee of the EAPC. Pain and pain treatments in European palliative care units. A cross sectional survey from the European Association for Palliative Care Research Network. Palliat. Med. 2005;19(6):477–484. doi: 10.1191/0269216305pm1054oa. [DOI] [PubMed] [Google Scholar]
- 54.Cohen BM, Murphy B. The effects of pentazocine, a kappa agonist, in patients with mania. Int. J. Neuropsychopharmacol. 2008;11(2):243–247. doi: 10.1017/S1461145707008073. [DOI] [PubMed] [Google Scholar]
- 55.Shippenberg TS, Zapata A, Chefer VI. Dynorphin and the pathophysiology of drug addiction. Pharmacol. Ther. 2007;116(2):306–321. doi: 10.1016/j.pharmthera.2007.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mello NK, Negus SS. Interactions between kappa opioid agonists and cocaine. Preclinical studies. Ann. NY Acad. Sci. 2000;909:104–132. doi: 10.1111/j.1749-6632.2000.tb06678.x. [DOI] [PubMed] [Google Scholar]
- 57.Knoll AT, Meloni EG, Thomas JB, Carroll FI, Carlezon WA., Jr Anxiolytic-like effects of kappa-opioid receptor antagonists in models of unlearned and learned fear in rats. J. Pharmacol. Exp. Ther. 2007;323(3):838–845. doi: 10.1124/jpet.107.127415. [DOI] [PubMed] [Google Scholar]
- 58.Carey AN, Borozny K, Aldrich JV, Mclaughlin JP. Reinstatement of cocaine place-conditioning prevented by the peptide kappa-opioid receptor antagonist arodyn. Eur. J. Pharmacol. 2007;569(1–2):84–89. doi: 10.1016/j.ejphar.2007.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rothman RB, Gorelick DA, Heishman SJ, et al. An open-label study of a functional opioid kappa antagonist in the treatment of opioid dependence. J. Subst. Abuse Treat. 2000;18(3):277–281. doi: 10.1016/s0740-5472(99)00074-4. [DOI] [PubMed] [Google Scholar]
- 60.Guerrini R, Capasso A, Marastoni M, et al. Rational design of dynorphin A analogues with delta-receptor selectivity and antagonism for delta- and kappa-receptors. Bioorg. Med. Chem. 1998;6(1):57–62. doi: 10.1016/s0968-0896(97)10008-6. [DOI] [PubMed] [Google Scholar]
- 61.Lu Y, Nguyen TM, Weltrowska G, et al. [2′,6′-Dimethyltyrosine]dynorphin A(1–11)-NH2 analogues lacking an N-terminal amino group: potent and selective kappa opioid antagonists. J. Med. Chem. 2001;44(19):3048–3053. doi: 10.1021/jm0101186. [DOI] [PubMed] [Google Scholar]
- 62.Schlechtingen G, Zhang L, Maycock A, et al. [Pro(3)]Dyn A(1–11)-NH(2): a dynorphin analogue with high selectivity for the kappa opioid receptor. J. Med. Chem. 2000;43(14):2698–2702. doi: 10.1021/jm990442p. [DOI] [PubMed] [Google Scholar]
- 63.Bennett MA, Murray TF, Aldrich JV. Identification of arodyn, a novel acetylated dynorphin A-(1–11) analogue, as a kappa opioid receptor antagonist. J. Med. Chem. 2002;45(26):5617–5619. doi: 10.1021/jm025575g. [DOI] [PubMed] [Google Scholar]
- 64.Vig BS, Murray TF, Aldrich JV. A novel N-terminal cyclic dynorphin A analogue cyclo(N,5)[Trp(3),Trp(4),Glu(5)] dynorphin A-(1–11)NH(2) that lacks the basic N-terminus. J. Med. Chem. 2003;46(8):1279–1282. doi: 10.1021/jm0256023. [DOI] [PubMed] [Google Scholar]
- 65.Patkar KA, Yan X, Murray TF, Aldrich JV. [Nalpha-benzylTyr1,cyclo(D-Asp5,Dap8)]- dynorphin A-(1–11)NH2 cyclized in the “address” domain is a novel kappa-opioid receptor antagonist. J. Med. Chem. 2005;48(14):4500–4503. doi: 10.1021/jm050105i. [DOI] [PubMed] [Google Scholar]
- 66.Aldrich JV, Patkar KA, Mclaughlin JP. Zyklophin, a systemically active selective kappa opioid receptor peptide antagonist with short duration of action. Proc. Natl Acad. Sci. USA. 2009;106(43):18396–18401. doi: 10.1073/pnas.0910180106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Aldrich JV, Mclaughlin JP. Peptide kappa opioid receptor ligands: potential for drug development. AAPS J. 2009;11(2):312–322. doi: 10.1208/s12248-009-9105-4. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• A good review on peptide κ-opioid receptor (KOR) ligands and their potentials as drugs.
- 68.Bruchas MR, Chavkin C. Kinase cascades and ligand-directed signaling at the kappa opioid receptor. Psychopharmacology (Berl.) 2010;210(2):137–147. doi: 10.1007/s00213-010-1806-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Carroll I, Thomas JB, Dykstra LA, et al. Pharmacological properties of JDTic: a novel kappa-opioid receptor antagonist. Eur. J. Pharmacol. 2004;501(1–3):111–119. doi: 10.1016/j.ejphar.2004.08.028. [DOI] [PubMed] [Google Scholar]
- 70.Wollemann M, Benyhe S. Non-opioid actions of opioid peptides. Life Sci. 2004;75(3):257–270. doi: 10.1016/j.lfs.2003.12.005. [DOI] [PubMed] [Google Scholar]; • A good review examining the nonopioid actions of various opioid peptides.
- 71.Walker JM, Moises HC, Coy DH, Baldrighi G, Akil H. Nonopiate effects of dynorphin and des-Tyr-dynorphin. Science. 1982;218(4577):1136–1138. doi: 10.1126/science.6128791. [DOI] [PubMed] [Google Scholar]; • First reference of the nonopioid effects of Dynorphin A (Dyn A).
- 72.Walker JM, Katz RJ, Akil H. Behavioral effects of dynorphin 1–13 in the mouse and rat: initial observations. Peptides. 1980;1(4):341–345. doi: 10.1016/0196-9781(80)90012-1. [DOI] [PubMed] [Google Scholar]
- 73.Young EA, Walker JM, Houghten R, Akil H. The degradation of dynorphin A in brain tissue in vivo and in vitro . Peptides. 1987;8(4):701–707. doi: 10.1016/0196-9781(87)90046-5. [DOI] [PubMed] [Google Scholar]
- 74.Walker JM, Moises HC, Coy DH, Young EA, Watson SJ, Akil H. Dynorphin (1–17): lack of analgesia but evidence for non-opiate electrophysiological and motor effects. Life Sci. 1982;31(16–17):1821–1824. doi: 10.1016/0024-3205(82)90219-3. [DOI] [PubMed] [Google Scholar]
- 75.Kaneko T, Nakazawa T, Ikeda M, et al. Sites of analgesic action of dynorphin. Life Sci. 1983;33(Suppl. 1):661–664. doi: 10.1016/0024-3205(83)90589-1. [DOI] [PubMed] [Google Scholar]
- 76.Herrera-Marschitz M, Hokfelt T, Ungerstedt U, Terenius L, Goldstein M. Effect of intranigral injections of dynorphin, dynorphin fragments and alpha-neoendorphin on rotational behaviour in the rat. Eur. J. Pharmacol. 1984;102(2):213–227. doi: 10.1016/0014-2999(84)90253-x. [DOI] [PubMed] [Google Scholar]
- 77.Faden AI, Jacobs TP. Dynorphin-related peptides cause motor dysfunction in the rat through a non-opiate action. Br. J. Pharmacol. 1984;81(2):271–276. doi: 10.1111/j.1476-5381.1984.tb10074.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Herman BH, Goldstein A. Antinociception and paralysis induced by intrathecal dynorphin A. J. Pharmacol. Exp. Ther. 1985;232(1):27–32. [PubMed] [Google Scholar]
- 79.Przewlocki R, Shearman GT, Herz A. Mixed opioid/nonopioid effects of dynorphin and dynorphin related peptides after their intrathecal injection in rats. Neuropeptides. 1983;3(3):233–240. doi: 10.1016/0143-4179(83)90019-7. [DOI] [PubMed] [Google Scholar]
- 80.Malan TP, Ossipov MH, Gardell LR, et al. Extraterritorial neuropathic pain correlates with multisegmental elevation of spinal dynorphin in nerve-injured rats. Pain. 2000;86(1–2):185–194. doi: 10.1016/s0304-3959(00)00243-8. [DOI] [PubMed] [Google Scholar]
- 81.Dubner R, Ruda MA. Activity-dependent neuronal plasticity following tissue injury and inflammation. Trends Neurosci. 1992;15(3):96–103. doi: 10.1016/0166-2236(92)90019-5. [DOI] [PubMed] [Google Scholar]
- 82.Millan MJ, Millan MH, Pilcher CW, Colpaert FC, Herz A. Chronic pain in the rat: selective alterations in CNS and pituitary pools of dynorphin as compared with vasopressin. Neuropeptides. 1985;5(4–6):423–424. doi: 10.1016/0143-4179(85)90044-7. [DOI] [PubMed] [Google Scholar]
- 83.Sydbom A, Terenius L. The histamine-releasing effect of dynorphin and other peptides possessing Arg–Pro sequences. Agents Actions. 1985;16(3–4):269–272. doi: 10.1007/BF01983157. [DOI] [PubMed] [Google Scholar]
- 84.Chahl LA, Chahl JS. Plasma extravasation induced by dynorphin-(1–13) in rat skin. Eur. J. Pharmacol. 1986;124(3):343–347. doi: 10.1016/0014-2999(86)90237-2. [DOI] [PubMed] [Google Scholar]
- 85.Vera-Portocarrero LP, Xie JY, Kowal J, Ossipov MH, King T, Porreca F. Descending facilitation from the rostral ventromedial medulla maintains visceral pain in rats with experimental pancreatitis. Gastroenterology. 2006;130(7):2155–2164. doi: 10.1053/j.gastro.2006.03.025. [DOI] [PubMed] [Google Scholar]
- 86.Vanderah TW, Suenaga NMH, Ossipov MH, Malan TP, Lai J, Porreca F. Tonic descending facilitation from the rostral ventromedial medulla mediates opioid-induced abnormal pain and antinociceptive tolerance. J. Neurosci. 2001;21(1):279–286. doi: 10.1523/JNEUROSCI.21-01-00279.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Peters CM, Lindsay TH, Pomonis JD, et al. Endothelin and the tumorigenic component of bone cancer pain. Neuroscience. 2004;126(4):1043–1052. doi: 10.1016/j.neuroscience.2004.04.027. [DOI] [PubMed] [Google Scholar]
- 88.Faden AI, Molineaux CJ, Rosenberger JG, Jacobs TP, Cox BM. Increased dynorphin immunoreactivity in spinal cord after traumatic injury. Regul. Pept. 1985;11(1):35–41. doi: 10.1016/0167-0115(85)90029-1. [DOI] [PubMed] [Google Scholar]
- 89.Wang ZJ, Gardell LR, Ossipov MH, et al. Pronociceptive actions of dynorphin maintain chronic neuropathic pain. J. Neurosci. 2001;21(5):1779–1786. doi: 10.1523/JNEUROSCI.21-05-01779.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Vanderah TW, Laughlin T, Lashbrook JM, et al. Single intrathecal injections of dynorphin A or des-Tyr-dynorphins produce long-lasting allodynia in rats: blockade by MK-801 but not naloxone. Pain. 1996;68(2–3):275–281. doi: 10.1016/s0304-3959(96)03225-3. [DOI] [PubMed] [Google Scholar]
- 91.Luo MC, Chen QM, Ossipov MH, Rankin DR, Porreca F, Lai J. Spinal dynorphin and bradykinin receptors maintain inflammatory hyperalgesia. J. Pain. 2008;9(12):1096–1105. doi: 10.1016/j.jpain.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ghasemi M, Schachter SC. The NMDA receptor complex as a therapeutic target in epilepsy: a review. Epilepsy Behav. 2011;22(4):617–640. doi: 10.1016/j.yebeh.2011.07.024. [DOI] [PubMed] [Google Scholar]
- 93.Caudle RM, Isaac L. A novel interaction between dynorphin(1–13) and an N-methyl-D-aspartate site. Brain Res. 1988;443(1–2):329–332. doi: 10.1016/0006-8993(88)91628-9. [DOI] [PubMed] [Google Scholar]
- 94.Stewart P, Isaac L. A strychnine-sensitive site is involved in dynorphin-induced paralysis and loss of the tail-flick reflex. Brain Res. 1991;543(2):322–326. doi: 10.1016/0006-8993(91)90044-v. [DOI] [PubMed] [Google Scholar]
- 95.Bakshi R, Faden AI. Blockade of the glycine modulatory site of NMDA receptors modifies dynorphin-induced behavioral effects. Neurosci. Lett. 1990;110(1–2):113–117. doi: 10.1016/0304-3940(90)90797-d. [DOI] [PubMed] [Google Scholar]
- 96.Long JB, Rigamonti DD, Oleshansky MA, Wingfield CP, Martinez-Arizala A. Dynorphin A-induced rat spinal cord injury: evidence for excitatory amino acid involvement in a pharmacological model of ischemic spinal cord injury. J. Pharmacol. Exp. Ther. 1994;269(1):358–366. [PubMed] [Google Scholar]
- 97.Koetzner L, Hua XY, Lai J, Porreca F, Yaksh T. Nonopioid actions of intrathecal dynorphin evoke spinal excitatory amino acid and prostaglandin E-2 release mediated by cyclooxygenase-1 and-2. J. Neurosci. 2004;24(6):1451–1458. doi: 10.1523/JNEUROSCI.1517-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Faden AI. Dynorphin increases extracellular levels of excitatory amino acids in the brain through a non-opioid mechanism. J. Neurosci. 1992;12(2):425–429. doi: 10.1523/JNEUROSCI.12-02-00425.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Examines the excitatory effects of nonopioid Dyn A.
- 99.Massardier D, Hunt PF. A direct non-opiate interaction of dynorphin-(1–13) with the N-methyl-D-aspartate (NMDA) receptor. Eur. J. Pharmacol. 1989;170(1–2):125–126. doi: 10.1016/0014-2999(89)90149-0. [DOI] [PubMed] [Google Scholar]
- 100.Dumont M, Lemaire S. Dynorphin potentiation of [3H]CGP-39653 binding to rat brain membranes. Eur. J. Pharmacol. 1994;271(1):241–244. doi: 10.1016/0014-2999(94)90288-7. [DOI] [PubMed] [Google Scholar]
- 101.Shukla VK, Bansinath M, Dumont M, Lemaire S. Selective involvement of kappa opioid and phencyclidine receptors in the analgesic and motor effects of dynorphin-A-(1–13)-Tyr-Leu-Phe-Asn-Gly-Pro. Brain Res. 1992;591(1):176–180. doi: 10.1016/0006-8993(92)90994-k. [DOI] [PubMed] [Google Scholar]
- 102.Tang QB, Gandhoke R, Burritt A, Hruby VJ, Porreca F, Lai J. High-affinity interaction of (des-tyrosyl) dynorphin A(2–17) with NMDA receptors. J. Pharmacol. Exp. Ther. 1999;291(2):760–765. [PubMed] [Google Scholar]
- 103.Tang QB, Lynch RM, Porreca F, Lai J. Dynorphin A elicits an increase in intracellular calcium in cultured neurons via a non-opioid, non-NMDA mechanism. J. Neurophysiol. 2000;83(5):2610–2615. doi: 10.1152/jn.2000.83.5.2610. [DOI] [PubMed] [Google Scholar]
- 104.Woods AS, Huestis MA. A study of peptide–peptide interaction by matrix-assisted laser desorption/ionization. J. Am. Soc. Mass Spectrom. 2001;12(1):88–96. doi: 10.1016/S1044-0305(00)00197-5. [DOI] [PubMed] [Google Scholar]
- 105.Jackson SN, Wang HY, Yergey A, Woods AS. Phosphate stabilization of intermolecular interactions. J. Proteome Res. 2006;5(1):122–126. doi: 10.1021/pr0503578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Woods AS, Kaminski R, Oz M, et al. Decoy peptides that bind dynorphin noncovalently prevent NMDA receptor-mediated neurotoxicity. J. Proteome Res. 2006;5(4):1017–1023. doi: 10.1021/pr060016+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Rojewska E, Makuch W, Przewlocka B, Mika J. Minocycline prevents dynorphin-induced neurotoxicity during neuropathic pain in rats. Neuropharmacology. 2014;86:301–310. doi: 10.1016/j.neuropharm.2014.08.001. [DOI] [PubMed] [Google Scholar]
- 108.Lai J, Luo MC, Chen QM, et al. Dynorphin A activates bradykinin receptors to maintain neuropathic pain. Nat. Neurosci. 2006;9(12):1534–1540. doi: 10.1038/nn1804. [DOI] [PubMed] [Google Scholar]; •• Reference demonstrates the evidence for the nonopioid effects of Dyn A being mediated through the bradykinin receptors.
- 109.Lee YS, Muthu D, Hall SM, et al. Discovery of amphipathic dynorphin A analogues to inhibit the neuroexcitatory effects of dynorphin A through bradykinin receptors in the spinal cord. J. Am. Chem. Soc. 2014;136(18):6608–6616. doi: 10.1021/ja501677q. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Structure–activity relationship of Dyn A analogs at the bradykinin receptors.
- 110.Hess JF, Borkowski JA, Young GS, Strader CD, Ransom RW. Cloning and pharmacological characterization of a human bradykinin (BK-2) receptor. Biochem. Biophys. Res. Comm. 1992;184(1):260–268. doi: 10.1016/0006-291x(92)91187-u. [DOI] [PubMed] [Google Scholar]
- 111.Hess JF, Borkowski JA, Macneil T, et al. Differential pharmacology of cloned human and mouse B2 bradykinin receptors. Mol. Pharmacol. 1994;45(1):1–8. [PubMed] [Google Scholar]
- 112.Lopes P, Regoli D, Couture R. Cardiovascular effects of intrathecally administered bradykinin in the rat: characterization of receptors with antagonists. Br. J. Pharmacol. 1993;110(4):1369–1374. doi: 10.1111/j.1476-5381.1993.tb13971.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bathon JM, Proud D. Bradykinin antagonists. Annu. Rev. Pharmacol. Toxicol. 1991;31:129–162. doi: 10.1146/annurev.pa.31.040191.001021. [DOI] [PubMed] [Google Scholar]
- 114.Lee YS, Rankin D, Hall SM, et al. Structure-activity relationships of non-opioid [des-Arg(7)]-dynorphin A analogues for bradykinin receptors. Bioorg. Med. Chem. Lett. 2014;24(21):4976–4979. doi: 10.1016/j.bmcl.2014.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lee YS, Hall SM, Ramos-Colon C, et al. Modification of amphipathic non-opioid dynorphin A analogues for rat brain bradykinin receptors. Bioorg. Med. Chem. Lett. 2015;25(1):30–33. doi: 10.1016/j.bmcl.2014.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]