

Abstract
ATP-sensitive potassium (KATP) channels play fundamental roles in the regulation of endocrine, neural and cardiovascular function. Small-molecule inhibitors (e.g., sulfonylurea drugs) or activators (e.g., diazoxide) acting on SUR1 or SUR2 have been used clinically for decades to manage the inappropriate secretion of insulin in patients with Type 2 diabetes, hyperinsulinism and intractable hypertension. More recently, the discovery of rare disease-causing mutations in KATP channel-encoding genes has highlighted the need for new therapeutics for the treatment of certain forms of neonatal diabetes mellitus, congenital hyperinsulinism and Cantu syndrome. Here, we provide a high-level overview of the pathophysiology of these diseases and discuss the development of a flexible high-throughput screening platform to enable the development of new classes of KATP channel modulators.
Keywords: : Cantu syndrome, chemical chaperone, diabetes, high-throughput screening, insulin, KATP channels, pancreas, thallium flux, trafficking
ATP-sensitive potassium (KATP) channels are widely expressed in endocrine, muscle and nervous tissues, and serve to couple cellular metabolism, via adenine nucleotides ATP and ADP, to membrane potential and excitability [1]. This characteristic sensitivity to cellular metabolism, which is modulated by multiple cell signaling pathways, enables KATP channels to regulate diverse physiological processes ranging from maintenance of blood glucose homeostasis to control of smooth muscle in vascular and nonvascular tissues, blood pressure and cardiac and skeletal excitation contraction coupling. Pharmacological inhibitors (e.g., sulfonylureas) and activators/openers (e.g., diazoxide) of pancreatic-type KATP channels have been used clinically for decades to treat diabetes mellitus and various forms of hyperinsulinism, respectively. More recently, sulfonylurea inhibitors have been used to treat neonatal diabetes caused by activating mutations in KATP genes [2]. While this represents a victory for translational medicine and the patients involved, the failure to treat other types of neonatal diabetes, as well as certain forms of congenital hyperinsulinism, highlight the need for ‘sharper’ and more diverse pharmacological tools to meet the specific needs of those patients. Furthermore, the recent discovery of activating mutations in cardiovascular KATP channels underlying Cantu syndrome [3] invites the development of cardiovascular-specific KATP channel inhibitors for the treatment of this disease. The goals of this article are: provide a high-level overview of the molecular mechanisms underlying Neonatal Diabetes Mellitus (NDM) and Congenital Hyperinsulinism of Infancy (CHI) and Cantu syndrome, all resulting from gain- or loss-of-function mutations in KATP channel genes; discuss opportunities for developing novel mechanism-based KATP channel therapeutics; describe our recent discovery of a novel-scaffold KATP channel activator using a flexible high-throughput screening assay that could be used to expand the molecular pharmacology for the KATP channel family.
KATP channel structure & regulation by adenine nucleotides
KATP channels are hetero-octameric complexes comprised of four pore-forming inward rectifier potassium channel subunits (Kir6.1 or Kir6.2) and four regulatory sulfonylurea receptor subunits (SUR1 or SUR2) [4–6] (Figure 1). Kir6.1 and Kir6.2 are encoded by the genes KCNJ8 and KCNJ11, whereas SUR1 and SUR2 are encoded by ABCC8 and ABCC9, respectively. Splice variation of ABCC9 gives rise to SUR2A and SUR2B, which differ only in the carboxyl-terminal 42 amino acids. Intriguingly, the SUR2 and Kir6.1 genes are immediately adjacent on human chromosome 12, whereas the SUR1 and Kir6.2 are adjacent genes on chromosome 11. This indicates not only that one pair of genes is a duplication of the other, but additionally suggests that each pair of genes may be co-regulated. Functional channels can be reconstituted by expressing any combination of Kir6.1 or Kir6.2 with SUR1, SUR2A or SUR2B, but as discussed below, the canonical pairing of Kir6.1/SUR2 and Kir6.2/SUR1 may be the key physiological pairings in vivo. Each Kir channel subunit comprises two membrane-spanning domains flanking a re-entrant pore loop that imparts selectivity for K+ over other ions, and a cytoplasmic domain formed from the intracellular amino- and carboxyl-termini of the individual subunits. Four subunits assemble around a water-filled pore through which K+ ions move down their electrochemical gradient and out of the cell. SUR1 and SUR2 are members of the ATP-binding cassette (ABC) superfamily of membrane proteins. Each SUR subunit contains 17 transmembrane domains and two intracellular nucleotide binding domains (NBD), termed NBD1 and NBD2 (Figure 1).
Figure 1. . Molecular composition and membrane topology of KATP channels.

Four subunits of SUR and Kir6.x each co-assemble to form the functional hetero-octameric KATP channel complex. The Kir6.x comprises two membrane-spanning domains that flank a K+ selectivity P-loop, whereas SUR has total 17 transmembrane domains organized in three regions (TM0, TM1 and TM2). The two nucleotide binding domains NBD1 and NBD2 are shown in purple.
The biophysical mechanisms underlying nucleotide-dependent regulation of KATP channels are complex and still not fully understood. Extensive functional studies on mutagenized Kir6.2/SUR1 channels, and in silico docking simulations on structural models, indicate that ATP inhibits the channel directly, binding to sites located at the interface of adjacent Kir6.2 subunits to stabilize channel closure [7–9]. In contrast, activation of the channel by MgADP and MgATP results from interactions with the NBDs of SUR1 that stabilize the open state of the channel pore. Whether or not this involves the hydrolysis of ATP at NBDs to induce channel opening is not yet fully resolved [10–12].
Existing KATP channel pharmacology
KATP channel inhibitors
Kir6.2/SUR1 channels are the molecular targets for inhibitory sulfonylureas and related drugs that are used to treat Type 2 diabetes [13,14]. By inhibiting KATP channels expressed in β cells of the pancreas, these drugs lead to β-cell excitation, induction of insulin secretion and lowering of blood glucose (Figure 2). The discovery that sparked the development of sulfonylureas was made during World War II when French physician Marcel Janbon noted that patients receiving an antibacterial sulfonamide drug developed symptoms of hypoglycemia. Experiments in normal and pancreatectomized dogs led him to conclude that the drugs were stimulating insulin release from the pancreas [15]. Between 1956 and 1966, the first-generation sulfonylureas – tolbutamide, chlorpropamide, acetohexamide and tolazamide – were introduced into clinical practice for the treatment of diabetes mellitus. Lead optimization efforts to improve the hypoglycemic efficacy led to the development of second-generation sulfonylureas glibenclamide (glyburide), gliclazide, glipizide in the late 1960s and early 1970s. The third-generation sulfonylurea glimepiride was introduced in the late 1980s. More recently, a new structural (glinide) class of antagonists typified by metiglinide, nateglinide and repaglinide, has been developed. The potency and selectivity of these drugs toward the major KATP channel subtypes are summarized in Table 1.
Figure 2. . Regulation of pancreatic β-cell insulin secretion by KATP channels comprised of Kir6.2/SUR1.

(Left) Under low-glucose conditions, the elevated ADP/ATP ratio maintains KATP channels in an open state, leading to membrane hyperpolarization, inhibition of L-type calcium (CaL) channels and inhibition of insulin secretion. Elevation of blood glucose leads to Glut2-dependent transport of glucose into β cells and stimulation of oxidative phosphorylation by mitochondria. The elevation in ATP/ADP ration causes KATP channel closure, membrane depolarization, activation of CaL channels and insulin secretion.
Table 1. . KATP channel modulators .
| Compound | Structure | Selectivity | IC50/EC50 | Ref. |
|---|---|---|---|---|
|
Inhibitors | ||||
| Tolbutamide |
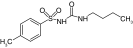 |
SURl >SUR2 |
2–7 μM |
[16,17] |
| Glibenclamide (glyburide) |
 |
SUR1 >SUR2A |
SUR1 ˜4 nM SUR2A ˜27 nM |
[16] |
| Gliclazide |
 |
SURl |
˜50 nM |
[18] |
| Glipizide |
 |
SUR1 |
4 nM |
[19] |
| Mitiglinide |
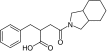 |
SURl ˜ SUR2 |
SUR1 ˜0.3 μM SUR2A ˜0.5 μM |
[20] |
| Nateglinide |
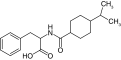 |
SUR1 ˜SUR2B >> SUR2A |
SUR1 ˜SUR2B 100 nM SUR2A >1 μM |
[20] |
|
Activators | ||||
| Diazoxide |
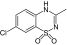 |
SURl > SUR2 |
30–60 μM |
[21,22] |
| Pinacidil |
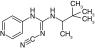 |
SUR2 |
˜1 μM |
[21,23] |
| P1075 |
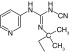 |
SUR2 |
≤50 nM |
[21,24] |
| Levcromakalim |
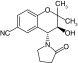 |
SUR2 |
-0.4–3 μM |
[21,25] |
| NN414 |
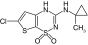 |
SUR1 |
450 nM |
[22] |
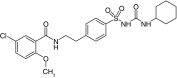
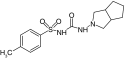
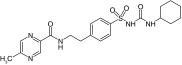
| VU0071063 | 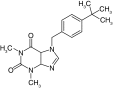 |
SUR1 | ˜7 μM | [26] |
The dose-dependent inhibition of Kir6.2/SUR1 activity with sulfonylureas or glinides follows a biphasic relationship that reflects two inhibitory binding sites: a high-affinity site (i.e., low nanomolar to micromolar) that accounts for typically 50–90% of channel inhibition under physiological conditions, and mediates the clinical efficacy of the drugs; and a low-affinity site (i.e., high micromolar to millimolar) that mediates inhibition of the remaining current and is clinically irrelevant. Results from mutagenesis experiments, binding studies and pharmacophore modeling suggest that the high-affinity site is located on SUR1 [16,27], whereas the low-affinity site is located on Kir6.2 [28,29].
The mechanism of action underlying channel inhibition by sulfonylureas involves complex interactions of the drugs with the activating effect of nucleotides on SUR1 [30]. At submicromolar drug concentrations of gliclazide (i.e. <500 nM), MgATP reduces gliclazide-dependent channel inhibition, possibly through stabilization of the open state of the channel, displacement of gliclazide or both. At therapeutic gliclazide concentrations (e.g. >500 nM), however, MgATP can enhance the relative inhibitory effect of the drug. This has been proposed to result from gliclazide impairing activation of the channel by MgATP, and thereby revealing the full extent of ATP-dependent inhibition at Kir6.2. In contrast, MgADP reduces sulfonylurea inhibition of cardiomyocyte Kir6.2/SUR2A channels across all concentrations, potentially explaining why sulfonylureas exhibit little cardiac side effects [31].
KATP channel openers
Conversely, by activating a K+ conductance, KATP channel openers (KCOs) tend to hyperpolarize the cell membrane potential, dampen cell excitability and inhibit calcium entry into cells. As a consequence, KCOs have been used clinically to inhibit insulin secretion from pancreatic β cells, reduce blood pressure by inducing vascular smooth muscle cell relaxation and hence blood vessel vasodilation, and for the treatment of angina pectoris. Other proposed clinical applications include urinary incontinence, airway hyper-reactivity and cardiac arrhythmias; however, off-target effects due to moderate potency and selectivity have limited their use in these clinical settings.
KCOs are represented by a diverse collection of compounds, such as first-generation benzopyrans, benzothiadiazines, cyanoguanidines, pyridyl nitrates and thioformamides, and second-generation cyclobutenediones, dihydropyridines and tertiarycarbinols (Table 1) (reviewed in [32]). KCO binding sites are localized to the SUR subunits, and some of these have been partially mapped [21,33–34]. As with inhibitory sulfonylureas, the potency and selectivity of some KCOs are flexible and modulated by the interactions of Mg-nucleotides at NBDs [30], making the development of strictly specific modulators a challenging goal. For example, two of the most investigated KCOs, diazoxide and pinacidil, exhibit preferential activity toward SUR1- and SUR2-dependent channels, respectively, but diazoxide exhibits equivalent activity toward SUR2-containing channels in the presence of intracellular MgADP [35]. Whether or not other KCOs exhibit this flexibility has not been systematically explored.
Novel inhibitors of Kir6.2/SUR1 are needed for certain forms of neonatal diabetes
Arguably the best understood role of KATP channels is in the control of insulin secretion in β cells of the pancreas. Comprised primarily of Kir6.2 and SUR1 subunits, high activity of pancreatic KATP channels serves to hyperpolarize the β-cell membrane potential and suppress the electrical activity of the cells, thereby preventing insulin secretion. Following a meal, elevation of blood glucose leads to the stimulation of β-cell mitochondrial oxidative phosphorylation, increased intracellular ATP/ADP ratio [36,37] and closure of KATP channels. The ensuing membrane depolarization leads to activation of voltage-gated calcium channels, influx of calcium and secretion of insulin, which in turn promotes the uptake and utilization of glucose by target organs. Thus, Kir6.2- and SUR1-dependent KATP channels are central regulators of β-cell insulin secretion and blood glucose homeostasis (Figure 2).
Gain-of-function mutations in either KCNJ11 or ABBC9 are the major cause of Neonatal Diabetes Mellitus (NDM; MIM 606176). NDM is typically diagnosed before 6 months of age, patients frequently presenting with low birth weight, symptomatic hyperglycemia and ketoacidosis [38–41]. In extreme cases (DEND syndrome), developmental delay and epilepsy accompany neonatal diabetes. Gain of KATP channel function in pancreatic β cells hyperpolarizes the membrane potential and suppresses excitability, thereby inhibiting calcium-dependent insulin secretion, leading to elevations in blood glucose [42]. How gain of KATP channel function gives rise to generalized epilepsy, muscle weakness and other neurological deficits is less clear, but presumably results from a suppressive effect on neuronal excitability [43]. Transcripts for Kir6.2 and SUR1 are widely expressed in the brain and show overlap in expressed location, consistent with KATP channels being present in the hippocampus, cerebellum and several nuclei of the midbrain and brainstem, among other regions [44]. Using pharmacological and genetic approaches, several groups have linked Kir6.2/SUR1 channels to multiple brain functions, including memory [45], sleep [46], exploratory behavior [47], motor control [39] and central regulation of glucose homeostasis [48,49]. Neuronal Kir6.2/SUR1 channels are regulated in a manner analogous to those of pancreatic β cells, whereby elevations in extracellular glucose lead to channel closure, membrane depolarization and enhanced electrical excitability. It is therefore likely that gain-of-function mutations in Kir6.2 or SUR1 lead to a reduction in neural activity in several brain regions.
Prior to the recognition that KATP channel GOF can cause NDM [39], insulin injections were the only treatment option available for managing blood glucose in these patients. However, it was reported soon after that NDM patients with KATP mutations could be transitioned from insulin injections to oral sulfonylurea drugs [50,51]. Today, sulfonylureas such as glibenclamide are considered first-line therapy choices for KATP-dependent NDM, and in a majority of cases, provide excellent long-term glycemic control [2]. For reasons that are unclear, the therapeutic efficacy of glibenclamide for improving motor and cognitive function in DEND patients is more variable. One possible explanation is that in some patients glibenclamide may fail to accumulate in the brain to concentrations capable of blocking the overactive KATP channels. In support of this idea, studies in rodents and in vitro cell culture systems show that glibenclamide and tolbutamide are efficiently transported out of the brain via P-glycoprotein and other drug efflux pathways present in the blood–brain barrier [52–54]. Another possible explanation is that higher sulfonylurea concentrations could be required to inhibit the activity of mutant KATP channels in some patients. In general, NDM mutations increase KATP channel activity by reducing ATP binding and hence block of the pore-forming subunit, or stabilizing the open-state stability of the channel [55]. As noted earlier, inhibition of KATP channels by sulfonylureas involves interactions between nucleotides and drugs, such that mutations that impair nucleotide-dependent channel inhibition are predicted to reduce sulfonylurea-dependent block. Electrophysiological studies on mutant KATP channels have shown that this is indeed the case; NDM mutations that render KATP channels less sensitive to ATP-dependent inhibition also make the channels less sensitive to sulfonylureas [55,56]. Thus, a combination of poor sulfonylurea brain penetrance/accumulation and sulfonylurea-resistant KATP is a conceivable explanation for the modest improvements in neurological functions observed in some DEND patients. The development of chemically diverse inhibitors with better brain penetrance and novel mechanisms of action would help resolve this issue.
Correctors of KATP channel trafficking are needed for congenital hyperinsulinism
Opposite to NDM, congenital hyperinsulinism (CHI) is a rare disorder characterized by the persistent secretion of insulin and hypoglycemia. Missense mutations in multiple genes have been associated with CHI, but loss-of-function (LOF) mutations in ABCC8 and KCNJ11 are prominent. More than 150 mutations in ABCC8 and 20 in KCNJ11 account for greater than 35% of all CHI cases [57,58]. ‘Type 1’ mutations in either gene lead to impaired trafficking of the KATP channel complex to the plasma membrane, whereas ‘Type 2’ mutations render cell-surface channels less sensitive to activation by Mg nucleotides [57]. Both types of mutations result in net decrease of KATP channel function in β cells, leading to membrane depolarization, constitutive calcium influx and unregulated insulin secretion.
A majority of mutations that lead to impaired trafficking are located on transmembrane domain 0 (TMD0) of SUR1, which is unique to SUR subunits within the ABC transporter family. Interestingly, Shyng and colleagues discovered that the biogenesis and trafficking defects caused by mutations in TMD0 can be rescued by sulfonylurea drugs [59–61]. An obvious problem, in translating this realization to a potential human therapy, is that the rescued channels will be inhibited, and therefore remain inactive, in the presence of the sulfonylurea. More recently, the same group has shown that the antiepileptic drug carbamazepine, which is structurally unrelated to sulfonylureas, also rescues TMD0 trafficking mutants; but also inhibits KATP channel activity [62]. A future challenge will be identifying small-molecule correctors of KATP channel trafficking that do not inhibit channel activity.
Vascular KATP channel-specific inhibitors are needed for Cantu syndrome, a complex multiorgan disease
Any pair of SURx:Kir6.x tetramers can co-assemble when heterologously expressed [63,64], and even within a single channel more than one SUR isoform or Kir6 isoform can coexist [65–70]. However, it is clear that SUR1/Kir6.2 expression dominates in neuronal and endocrine tissue, explaining the role of Kir6.2/SUR1 in NDM/DEND, but that SUR2 and Kir6.1 expression is predominantly in muscle. All four subunits have been identified in cardiac and skeletal muscle [63,71–73]. Similarly, variable expression of KATP subtypes has been demonstrated in different vascular beds [68,74–80], although low-conductance channels encoded by Kir6.1 may represent the predominant KATP channel subtype in vascular smooth muscle [81].
In mice, knockout of the Kir6.1 or SUR2 genes results in a hypertensive, prinzmetal angina-like phenotype [82,83]. While there is yet no clear human disease resulting from LOF mutations, several independent studies have recently reported multiple coding mutations in both SUR2 and Kir6.1 genes as underlying Cantu syndrome [84–87], a distinctive multiorgan disease. In all cases analyzed [85,87], recombinant expression of mutant channel proteins clearly demonstrated a reduced sensitivity to ATP inhibition which will lead to enhanced KATP channel activity wherever the channels are located.
Cantu syndrome (MIM 239850), or hypertrichosis-osteochondrodysplasia-cardiomegaly syndrome [88] presents with a constellation of features [89–97]. All patients present with congenital hypertrichosis, typically with thick scalp hair and excessive hair growth on the forehead, face, back and extremities. Macrosomia and macrocephaly are typically present at birth, although adult dimensions are probably normal. Various dysmorphic features, including coarse facial appearance and mild skeletal abnormalities are also observed. Extensive cardiovascular complications include enlarged hearts with normal or enhanced contractility, pericardial effusion and pulmonary hypertension secondary to partial pulmonary venous obstruction [91]. Some patients have required pericardiocentesis and ultimately needed pericardial stripping to prevent re-accumulation of the pericardial effusion. A significant number of patients have had patent ductus arteriosus (PDA) requiring surgical closure, as well as bicuspid aortic valves. Lymphedema involving the lower extremities may develop over time, and in one patient, lymphangiogram demonstrated dilated lymphatic vessels with delayed lymphatic drainage in the legs [90]. Interestingly, many of the features of CS have been reported as side effects of the KATP channel openers diazoxide and minoxidil [92,98–100].
Previous studies of Cantu syndrome patients provided no definitive explanation of the underlying cause of the various features, and even now the realization of SUR2 mutations as causal does not immediately provide explanations for all features. Transgenic introduction of Kir6 GOF mutations in vascular muscle cells results in a hypotensive phenotype [101], and the presence of KATP in vascular smooth muscle and potential vasorelaxation effects of KATP GOF may explain persistence of the PDA in Cantu syndrome patients. Altered smooth muscle tone may also underlie lymphedema and pericardial effusion, since the contractile regions of the lymphatic vessels are highly sensitive to KATP activation [102]. Cardiomegaly reported in most cases of Cantu syndrome is due to increased myocardial mass (hypertrophy) with larger cardiac chambers but with no loss of systolic function. This does not fit the diagnostic criteria of dilated or hypertrophic cardiomyopathy [102], but may be a secondary response to reduced vascular tone [92,103], or may also be related the finding that transgenic introduction of Kir6 GOF mutations into cardiac muscle cells results in a counter-intuitive hypercontractile phenotype resulting from a secondarily enhanced L-type Ca channel activity [104]. The mechanism by which minoxidil causes hair growth also remains controversial [105]; while the new realization definitively ties hair growth to KATP channel activation, but does not explain how.
Clearly the realization that GOF in the cardiovascular (Kir6.1/SUR2) ATP isoforms underlie Cantu syndrome presents an opportunity for an isoform-specific channel blocker as a potentially ‘magic-bullet’ therapy, as sulfonylureas have proven to be for NDM. Unfortunately, all commercially approved and available KATP channel blockers are likely to be active on the Kir6.2/SUR1 isoforms as well.
Importantly, the SUR2/Kir6.1 channels exhibit distinct functional as well as pharmacological properties. Unlike classic KATP channels of the heart [63,106] or pancreas [14,107], vascular channels are typically inactive in isolated membrane patches, and require nucleoside diphosphates (ADP, UDP, GDP) in the presence of Mg2+ to open [78–79,108]. Heterologously expressed Kir6.1/SUR2B channels recapitulate these same properties [14,109–113]. Thus the Kir6.1/SUR2B channel may represent the predominant smooth muscle KATP. While high-throughput screening (HTS; see the ‘A flexible HTS platform for KATP channels’ section) may identify novel sulfonylureas with appropriate isoform specificity, the distinct physiological regulatory features provide the possibility of completely new pharmacophores for Kir6.1/SUR2 channels; indeed early studies with the novel compound U37883A [114] may well have already provided a Kir6.1-specific lead.
A flexible HTS platform for KATP channels
Most of the more recent progress toward developing ‘sharper’ KATP channel modulators has come from lead optimization efforts focused on existing scaffolds discovered decades ago. Although this work has led to the generation of more potent and/or selective modulators in some cases, these compounds presumably act via similar mechanisms of actions to their parent compounds and will therefore be of limited use in treating the specific KATP channel defects outlined above. To overcome this barrier, we propose the most direct way of identifying KATP channel ‘correctors’ will be achieved with chemical library screening using functional assays that recapitulate specific disease-causing lesions in KATP channels. These efforts could include: HTS for nanomolar-affinity inhibitors of Kir6.2/SUR1 carrying NDM/DEND mutations that render the channels less sensitive to sulfonylureas; HTS for correctors of mutant Kir6.2/SUR1 biogenesis and trafficking for the treatment of CHI; and HTS for vascular-specific KATP channel inhibitors for the treatment of Cantu syndrome.
We recently developed a cell-based fluorescence assay for Kir6.2/SUR1 [26] that could be adapted to enable the discovery of new classes of subtype-specific KATP channel inhibitors, activators, and even correctors of channel biogenesis and trafficking. The assay reports the flux of the K+ surrogate, thallium (Tl+), through open Kir6.2/SUR1 channels, as an indirect measure of KATP channel activity. Inwardly directed Tl+ flux is reported using the commercially available intracellular dye, Thallos (TEFLabs), which exhibits a dose-dependent increase in fluorescence emission upon Tl+ binding. The assay was developed with a stably transfected HEK-293 cell line that expresses SUR1 constitutively and Kir6.2 from a tetracycline-inducible promoter (Figure 3). The use of a tetracycline-inducible heterologous expression system is intended to minimize cell line degeneration and loss of assay performance associated with constitutive KATP channel overexpression, and enables retesting of screening hits on tetracycline-uninduced cells to rapidly identify and triage compounds acting on Tl+ flux pathways expressed endogenously in HEK-293 cells (e.g., Na-K-ATPase, endogenous K+ channels). Assays can be performed in 384-well plates or other plate formats using a standard kinetic imaging plate reader. Cells are plated in clear-bottomed assay plates and cultured overnight with tetracycline to induce functional KATP channel expression. The next day, the cells are loaded with the acetoxymethylester form of Thallos, washed to remove unincorporated dye and treated with the compounds to be screened. The plate is then moved to the stage of a kinetic imaging plate reader where Tl+ is added simultaneously to each well of the plate to initiate Tl+ flux. Changes in the rate or extent of Tl+ flux are used to identify putative KATP channel modulators in a screen.
Figure 3. . Discovery and characterization of the KATP channel activator VU063.

(A) Fluorescence heat map of a Tl+ flux assay performed on HEK-293 cells heterologously expressing Kir6.2/SUR1 (see the ‘A flexible HTS platform for KATP channels’ section for details). Fluorescence emission data have been pseudocolored to indicate high Tl+ flux (warm colors – e.g., red, yellow) or low Tl+ flux (cool colors – e.g., blue, cyan). Wells I3-I6 (red box) received different concentrations of VU063, leading to dose-dependent Kir6.2/SUR1 opening and Tl+ flux. (B) Time versus fluorescence emission data recorded from four wells of a 384-well plate that received the indicated doses of VU063. The wells were prodosed with VU063 for 20 min before adding extracellular Tl+ at the ˜15-s time point. (C) Concentration–response curves for VU063, diazoxide and pinacidil from Tl+ flux assays performed on HEK-293-Kir6.2/SUR1 cells.
Reproduced with permission from [26].
Because heterologously expressed KATP channels retain their sensitivity to adenine nucleotides and pharmacological agents, the Kir6.2/SUR1 assay can be readily configured for identifying both inhibitors and activators in an HTS. Under normal cell culture and assay conditions, Kir6.2/SUR1 channels are mostly closed due to the relatively high cytoplasmic ATP/ADP ratio in HEK-293 cells. Therefore, in a screen for Kir6.2/SUR1 antagonists, the channels must first be opened with inhibitors of mitochondrial respiration and glycolysis (e.g., oligomycin, rotenone, 2-deoxyglycose; [raphemot r et al. unpublished observations]), or a KATP channel activator such as diazoxide (Figure 3). Once the channels are opened, the assay can report the dose-dependent inhibition of Tl+ flux by conventional antagonists, such as glibenclamide or tolbutamide, or novel hit antagonists in a compound library. Alternatively, screening for activators can be achieved by simply leaving the channels closed and looking for compounds that induce Tl+ flux. In principle, it should be possible to develop a Kir6.2/SUR1 assay using partially opened channels to screen for both antagonists and openers in a single HTS campaign. Furthermore, because the assay relies on heterologous expression of molecularly defined KATP channel subunits, developing Tl+ flux assays for the other major KATP channel subunits or KATP channels carrying disease-causing mutations should be possible.
Serendipitous discovery of the novel Kir6.2/SUR1 activator VU063
A novel-scaffold KATP channel opener was discovered while evaluating the selectivity of mosquito Kir channel inhibitors against a panel of mammalian inward rectifiers that included Kir6.2/SUR1 [26,115]. Diazoxide, which was being used to open the KATP channels for the inhibitor assay (see the ‘A flexible HTS platform for KATP channels’ section), was inadvertently left out of the compound plate, revealing three wells that received dilutions of a xanthine derivative termed VU063. As shown in Figure 3, VU063 from the VICB library dose dependently activated Tl+ flux (Figure 3) at nominal concentrations of 3, 10 and 30 μM. Given the need for new, chemically diverse KATP modulators, VU063 was re-ordered as a powder, freshly dissolved in DMSO and characterized in Tl+ flux and electrophysiological assays.
VU063 was inactive on tetracycline-uninduced cells lacking Kir6.2 (data not shown), but activated Tl+ flux in induced cells with an EC50 of approximately 10 μM. Diazoxide, in contrast, activated Tl+ flux with a 12-fold higher EC50 of approximately 120 μM. As expected for an assay that reports the activation of Kir6.2/SUR1, the SUR2-specific opener pinacidil had no effect on Tl+ flux. In cells pretreated with VU063 to open Kir6.2/SUR1 channels, the sulfonylurea antagonists glibenclamide and tolbutamide inhibited Tl+ flux with IC50 values expected for SUR1-containing channels. Gold standard voltage clamp electrophysiology experiments demonstrated that VU063 is indeed more potent than diazoxide, activates Kir6.2/SUR1 with faster kinetics than diazoxide, acts directly on Kir6.2/SUR1 to open the channels, and is specific for KATP channels containing SUR1. Importantly, VU063 is active on native KATP channels and inhibits glucose-stimulated calcium entry in primary mouse pancreatic β cells [26]. The discovery of VU063 shows proof-of-concept that novel KATP channel modulators can be readily identified using this HTS platform.
Conclusion
As our understanding of the molecular and biophysical mechanisms underlying KATP channel dysfunction in NDM/DEND, CHI, and Cantu syndrome deepens, so does the realization that the current armament of pharmacological tools available to treat these disorders is inadequate. With few exceptions, most of the KATP channel modulators in use today have origins dating back many decades. We believe that new discovery programs leveraging functional assays based on disease-specific KATP channel mutations will lead to an influx of critical new chemical matter to reinvigorate this field. The HTS platform described here is robust and flexible, and should be readily adaptable for discovering new inhibitors, activators, and correctors of biogenesis and trafficking of molecularly diverse KATP channels. The discovery of the novel-scaffold Kir6.2/SUR1 channel activator VU063 demonstrates that there are indeed chemically diverse KATP modulators just waiting to be discovered.
Future perspective
NDM/DEND, CHI and Cantu syndrome are rare genetic disorders that are unlikely to capture the attention of the pharmaceutical industry; and understandably so. In our view, the best chance of making progress in this area will be through the public support of early stage drug discovery by academic laboratories. The HTS platform described herein should support the discovery of novel-mechanism KATP channel inhibitors, activators and chemical chaperones. Screening libraries of US FDA-approved drugs could dramatically abbreviate the time from hit discovery to testing in existing preclinical animal models and the eventual testing in clinical trials. With adequate support, we speculate that in the next 5–10 years, the scientific community will experience an influx of disease-specific KATP channel modulators that will enable innovative ways to tackle these rare, yet no less devastating, diseases.
Executive summary.
KATP channels regulate blood glucose homeostasis, blood pressure and cardiac function.
KATP channels are validated drug targets for treating diabetes, hyperinsulinism and hypertension.
Neonatal diabetes mellitus (NDM), congenital hyperinsulinism of infancy (CHI) and Cantu syndrome are caused by mutations in KATP channel-encoding genes.
The current pharmaceutical ‘toolkit’ is inadequate to correct all of the molecular defects underlying these diseases.
KATP channels are hetero-octameric proteins containing four pore-forming Kir6x subunits and four regulatory SURx subunits.
Kir6.1/SUR2 and Kir6.2/SUR1 appear to be the major physiological pairings in vivo.
KATP channel inhibitors, including sulfonylureas and glinides, cause membrane depolarization and stimulation of electrical activity.
KATP channel openers, including diazoxide and pinacidil, cause membrane hyperpolarization and inhibition of cell electrical activity.
Both compound classes act on binding sites located on regulatory sulfonylurea receptor (SUR) subunits.
ATP-dependent inhibition of Kir6.2/SUR1 stimulates insulin secretion from pancreatic β cells.
Gain-of-function mutations in genes encoding Kir6.2/SUR1 inhibit insulin secretion, causing NDM. Because Kir6.2/SUR1 channels are also expressed in the brain, severe forms of NDM are associated with developmental delay and epilepsy (DEND).
Some NDM/DEND mutations render Kir6.2/SUR1 channels less sensitive to sulfonylurea inhibitors, making these drugs less efficacious at treating some forms of NDM/DEND.
Development of novel mechanism, brain-penetrant Kir6.1/SUR1 inhibitors would create new treatment options for some NDM/DEND patients.
Some loss-of-function mutations in patients with CHI cause defects in the biosynthesis and trafficking of Kir6.2/SUR1 to the cell surface of pancreatic β cells, resulting in membrane depolarization, unregulated insulin secretion and hypoglycemia.
The biosynthesis and trafficking of KATP channels carrying mutations on transmembrane domain 0 can be rescued with sulfonylureas, but residual channel inhibition by these drugs limits this from being an effective strategy for treating CHI.
Chemical chaperones that rescue KATP channel trafficking and do not block activity at the cell surface are needed for the treatment of CHI.
Gain-of-function mutations in genes encoding Kir6.1 or SUR2 give rise to Cantu syndrome, a complex, multiorgan disorder characterized by cardiovascular complications, hypertrichosis and other clinical features.
Cardiovascular-specific KATP channel inhibitors are needed for the treatment of Cantu syndrome.
A fluorescence based assay that reports thallium flux through heterologously expressed Kir6.2/SUR1 channel expressed in HEK-293 has been developed that enables HTS in 384-well plate format.
The assay should enable HTS for modulators of the major forms of reconstituted KATP channels.
A novel Kir6.2/SUR1-specific activator, termed VU063, was identified using the thallium flux assay.
VU063 is more potent and rapidly acting than diazoxide.
The discovery of VU063 shows proof-of-concept that novel scaffold Kir6.2/SUR1 openers can be readily identified using this thallium flux assay.
Footnotes
Financial & competing interests disclosure
This work was supported in part by NIH Grant R01 DK082884 to JS Denton, and by NIH Grant HL45742 to C Nichols. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
- 1.Aguilar-Bryan L, Clement JPT, Gonzalez G, Kunjilwar K, Babenko A, Bryan J. Toward understanding the assembly and structure of KATP channels. Physiol. Rev. 1998;78(1):227–245. doi: 10.1152/physrev.1998.78.1.227. [DOI] [PubMed] [Google Scholar]
- 2.Ashcroft FM. New uses for old drugs: neonatal diabetes and sulphonylureas. Cell Metab. 2010;11(3):179–181. doi: 10.1016/j.cmet.2010.02.004. [DOI] [PubMed] [Google Scholar]
- 3.Nichols CG, Singh GK, Grange DK. KATP channels and cardiovascular disease: suddenly a syndrome. Circ. Res. 2013;112(7):1059–1072. doi: 10.1161/CIRCRESAHA.112.300514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Inagaki N, Gonoi T, Clement JPT, et al. Reconstitution of IKATP: an inward rectifier subunit plus the sulfonylurea receptor. Science. 1995;270(5239):1166–1170. doi: 10.1126/science.270.5239.1166. [DOI] [PubMed] [Google Scholar]
- 5.Shyng S, Nichols CG. Octameric stoichiometry of the KATP channel complex. J. Gen. Physiol. 1997;110(6):655–664. doi: 10.1085/jgp.110.6.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Inagaki N, Gonoi T, Seino S. Subunit stoichiometry of the pancreatic beta-cell ATP-sensitive K+ channel. FEBS Lett. 1997;409(2):232–236. doi: 10.1016/s0014-5793(97)00488-2. [DOI] [PubMed] [Google Scholar]
- 7.Nichols CG. KATP channels as molecular sensors of cellular metabolism. Nature. 2006;440(7083):470–476. doi: 10.1038/nature04711. [DOI] [PubMed] [Google Scholar]
- 8.Antcliff JF, Haider S, Proks P, Sansom MS, Ashcroft FM. Functional analysis of a structural model of the ATP-binding site of the KATP channel Kir6.2 subunit. EMBO J. 2005;24(2):229–239. doi: 10.1038/sj.emboj.7600487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haider S, Antcliff JF, Proks P, Sansom MS, Ashcroft FM. Focus on Kir6.2: a key component of the ATP-sensitive potassium channel. J. Mol. Cell. Cardiol. 2005;38(6):927–936. doi: 10.1016/j.yjmcc.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 10.Matsuo M, Kimura Y, Ueda K. KATP channel interaction with adenine nucleotides. J. Mol. Cell. Cardiol. 2005;38(6):907–916. doi: 10.1016/j.yjmcc.2004.11.021. [DOI] [PubMed] [Google Scholar]
- 11.Proks P, De Wet H, Ashcroft FM. Activation of the K(ATP) channel by Mg-nucleotide interaction with SUR1. J. Gen. Physiol. 2010;136(4):389–405. doi: 10.1085/jgp.201010475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gribble FM, Tucker SJ, Haug T, Ashcroft FM. MgATP activates the beta cell KATP channel by interaction with its SUR1 subunit. Proc. Natl Acad. Sci. USA. 1998;95(12):7185–7190. doi: 10.1073/pnas.95.12.7185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aguilar-Bryan L, Nichols CG, Wechsler SW, et al. Cloning of the beta cell high-affinity sulfonylurea receptor: a regulator of insulin secretion. Science. 1995;268(5209):423–426. doi: 10.1126/science.7716547. [DOI] [PubMed] [Google Scholar]
- 14.Inagaki N, Gonoi T, Clement JP, et al. Reconstitution of I KATP: an inward rectifier subunit plus the sulfonylurea receptor. Science. 1995;270(5239):1166–1170. doi: 10.1126/science.270.5239.1166. [DOI] [PubMed] [Google Scholar]
- 15.Loubatieres-Mariani MM. [The discovery of hypoglycemic sulfonamides] J. Soc. Biol. 2007;201(2):121–125. doi: 10.1051/jbio:2007014. [DOI] [PubMed] [Google Scholar]
- 16.Gribble FM, Tucker SJ, Seino S, Ashcroft FM. Tissue specificity of sulfonylureas: studies on cloned cardiac and beta-cell K(ATP) channels. Diabetes. 1998;47(9):1412–1418. doi: 10.2337/diabetes.47.9.1412. [DOI] [PubMed] [Google Scholar]
- 17.Trube G, Rorsman P, Ohno-Shosaku T. Opposite effects of tolbutamide and diazoxide on the ATP-dependent K+ channel in mouse pancreatic beta-cells. Pflugers Arch. 1986;407(5):493–499. doi: 10.1007/BF00657506. [DOI] [PubMed] [Google Scholar]
- 18.Gribble FM, Ashcroft FM. Differential sensitivity of beta-cell and extrapancreatic K(ATP) channels to gliclazide. Diabetologia. 1999;42(7):845–848. doi: 10.1007/s001250051236. [DOI] [PubMed] [Google Scholar]
- 19.Dorschner H, Brekardin E, Uhde I, Schwanstecher C, Schwanstecher M. Stoichiometry of sulfonylurea-induced ATP-sensitive potassium channel closure. Mol. Pharmacol. 1999;55(6):1060–1066. doi: 10.1124/mol.55.6.1060. [DOI] [PubMed] [Google Scholar]
- 20.Sunaga Y, Gonoi T, Shibasaki T, et al. The effects of mitiglinide (KAD-1229), a new anti-diabetic drug, on ATP-sensitive K+ channels and insulin secretion: comparison with the sulfonylureas and nateglinide. Eur. J. Pharmacol. 2001;431(1):119–125. doi: 10.1016/s0014-2999(01)01412-1. [DOI] [PubMed] [Google Scholar]
- 21.Schwanstecher M, Sieverding C, Dorschner H, et al. Potassium channel openers require ATP to bind to and act through sulfonylurea receptors. EMBO J. 1998;17(19):5529–5535. doi: 10.1093/emboj/17.19.5529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dabrowski M, Larsen T, Ashcroft FM, Bondo Hansen J, Wahl P. Potent and selective activation of the pancreatic beta-cell type K(ATP) channel by two novel diazoxide analogues. Diabetologia. 2003;46(10):1375–1382. doi: 10.1007/s00125-003-1198-1. [DOI] [PubMed] [Google Scholar]
- 23.Lefebvre RA, Horacek J. Relaxant effects of BRL 38227 and pinacidil on the rat gastric fundus. Eur. J. Pharmacol. 1992;214(1):1–6. doi: 10.1016/0014-2999(92)90087-k. [DOI] [PubMed] [Google Scholar]
- 24.Higdon NR, Khan SA, Buchanan LV, Meisheri KD. Tissue and species variation in the vascular receptor binding of 3H-P1075, a potent KATP opener vasodilator. J. Pharmacol. Exp. Ther. 1997;280(1):255–260. [PubMed] [Google Scholar]
- 25.Randall MD, Griffith TM. Modulation of vasodilatation to levcromakalim by hypoxia and EDRF in the rabbit isolated ear: a comparison with pinacidil, sodium nitroprusside and verapamil. Br. J. Pharmacol. 1993;109(2):386–393. doi: 10.1111/j.1476-5381.1993.tb13581.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Raphemot R, Swale DR, Dadi PK, et al. Direct activation of beta-cell KATP channels with a novel xanthine derivative. Mol. Pharmacol. 2014;85(6):858–865. doi: 10.1124/mol.114.091884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ashfield R, Gribble FM, Ashcroft SJ, Ashcroft FM. Identification of the high-affinity tolbutamide site on the SUR1 subunit of the K(ATP) channel. Diabetes. 1999;48(6):1341–1347. doi: 10.2337/diabetes.48.6.1341. [DOI] [PubMed] [Google Scholar]
- 28.Gribble FM, Tucker SJ, Ashcroft FM. The interaction of nucleotides with the tolbutamide block of cloned ATP-sensitive K+ channel currents expressed in Xenopus oocytes: a reinterpretation. J. Physiol. 1997;504(Pt 1):35–45. doi: 10.1111/j.1469-7793.1997.00035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reimann F, Tucker SJ, Proks P, Ashcroft FM. Involvement of the n-terminus of Kir6.2 in coupling to the sulphonylurea receptor. J. Physiol. 1999;518(Pt 2):325–336. doi: 10.1111/j.1469-7793.1999.0325p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koster JC, Sha Q, Nichols CG. Sulfonylurea and K(+)-channel opener sensitivity of K(ATP) channels. Functional coupling of Kir6.2 and SUR1 subunits. J. Gen. Physiol. 1999;114(2):203–213. doi: 10.1085/jgp.114.2.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Proks P, De Wet H, Ashcroft FM. Sulfonylureas suppress the stimulatory action of Mg-nucleotides on Kir6.2/SUR1 but not Kir6.2/SUR2A KATP channels: a mechanistic study. J. Gen. Physiol. 2014;144(5):469–486. doi: 10.1085/jgp.201411222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mannhold R. KATP channel openers: structure-activity relationships and therapeutic potential. Med. Res. Rev. 2004;24(2):213–266. doi: 10.1002/med.10060. [DOI] [PubMed] [Google Scholar]
- 33.Uhde I, Toman A, Gross I, Schwanstecher C, Schwanstecher M. Identification of the potassium channel opener site on sulfonylurea receptors. J. Biol. Chem. 1999;274(40):28079–28082. doi: 10.1074/jbc.274.40.28079. [DOI] [PubMed] [Google Scholar]
- 34.Matsuoka T, Matsushita K, Katayama Y, et al. C-terminal tails of sulfonylurea receptors control ADP-induced activation and diazoxide modulation of ATP-sensitive K(+) channels. Circ. Res. 2000;87(10):873–880. doi: 10.1161/01.res.87.10.873. [DOI] [PubMed] [Google Scholar]
- 35.D'hahan N, Moreau C, Prost AL, et al. Pharmacological plasticity of cardiac ATP-sensitive potassium channels toward diazoxide revealed by ADP. Proc. Natl Acad. Sci. USA. 1999;96(21):12162–12167. doi: 10.1073/pnas.96.21.12162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nilsson T, Schultz V, Berggren PO, Corkey BE, Tornheim K. Temporal patterns of changes in ATP/ADP ratio, glucose 6-phosphate and cytoplasmic free Ca2+ in glucose-stimulated pancreatic beta-cells. Biochem. J. 1996;314(Pt 1):91–94. doi: 10.1042/bj3140091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Detimary P, Dejonghe S, Ling Z, Pipeleers D, Schuit F, Henquin JC. The changes in adenine nucleotides measured in glucose-stimulated rodent islets occur in beta cells but not in alpha cells and are also observed in human islets. J. Biol. Chem. 1998;273(51):33905–33908. doi: 10.1074/jbc.273.51.33905. [DOI] [PubMed] [Google Scholar]
- 38.Hattersley AT, Ashcroft FM. Activating mutations in Kir6.2 and neonatal diabetes: new clinical syndromes, new scientific insights, and new therapy. Diabetes. 2005;54(9):2503–2513. doi: 10.2337/diabetes.54.9.2503. [DOI] [PubMed] [Google Scholar]
- 39.Gloyn AL, Pearson ER, Antcliff JF, et al. Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. N. Engl. J. Med. 2004;350(18):1838–1849. doi: 10.1056/NEJMoa032922. [DOI] [PubMed] [Google Scholar]
- 40.Proks P, Arnold AL, Bruining J, et al. A heterozygous activating mutation in the sulphonylurea receptor SUR1 (ABCC8) causes neonatal diabetes. Hum. Mol. Genet. 2006;15(11):1793–1800. doi: 10.1093/hmg/ddl101. [DOI] [PubMed] [Google Scholar]
- 41.Babenko AP, Polak M, Cave H, et al. Activating mutations in the ABCC8 gene in neonatal diabetes mellitus. N. Engl. J. Med. 2006;355(5):456–466. doi: 10.1056/NEJMoa055068. [DOI] [PubMed] [Google Scholar]
- 42.Koster JC, Marshall BA, Ensor N, Corbett JA, Nichols CG. Targeted overactivity of beta cell K(ATP) channels induces profound neonatal diabetes. Cell. 2000;100(6):645–654. doi: 10.1016/s0092-8674(00)80701-1. [DOI] [PubMed] [Google Scholar]
- 43.Clark RH, Mctaggart JS, Webster R, et al. Muscle dysfunction caused by a KATP channel mutation in neonatal diabetes is neuronal in origin. Science. 2010;329(5990):458–461. doi: 10.1126/science.1186146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Karschin C, Ecke C, Ashcroft FM, Karschin A. Overlapping distribution of K(ATP) channel-forming Kir6.2 subunit and the sulfonylurea receptor SUR1 in rodent brain. FEBS Lett. 1997;401(1):59–64. doi: 10.1016/s0014-5793(96)01438-x. [DOI] [PubMed] [Google Scholar]
- 45.Betourne A, Bertholet AM, Labroue E, et al. Involvement of hippocampal CA3 K(ATP) channels in contextual memory. Neuropharmacology. 2009;56(3):615–625. doi: 10.1016/j.neuropharm.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 46.Varin C, Rancillac A, Geoffroy H, Arthaud S, Fort P, Gallopin T. Glucose induces slow-wave sleep by exciting the sleep-promoting neurons in the ventrolateral preoptic nucleus: a new link between sleep and metabolism. J. Neurosci. 2015;35(27):9900–9911. doi: 10.1523/JNEUROSCI.0609-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schiemann J, Schlaudraff F, Klose V, et al. K-ATP channels in dopamine substantia nigra neurons control bursting and novelty-induced exploration. Nat. Neurosci. 2012;15(9):1272–1280. doi: 10.1038/nn.3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miki T, Liss B, Minami K, et al. ATP-sensitive K+ channels in the hypothalamus are essential for the maintenance of glucose homeostasis. Nat. Neurosci. 2001;4(5):507–512. doi: 10.1038/87455. [DOI] [PubMed] [Google Scholar]
- 49.Hussain S, Richardson E, Ma Y, et al. Glucokinase activity in the arcuate nucleus regulates glucose intake. J. Clin. Invest. 2015;125(1):337–349. doi: 10.1172/JCI77172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sagen JV, Raeder H, Hathout E, et al. Permanent neonatal diabetes due to mutations in KCNJ11 encoding Kir6.2: patient characteristics and initial response to sulfonylurea therapy. Diabetes. 2004;53(10):2713–2718. doi: 10.2337/diabetes.53.10.2713. [DOI] [PubMed] [Google Scholar]
- 51.Pearson ER, Flechtner I, Njolstad PR, et al. Switching from insulin to oral sulfonylureas in patients with diabetes due to Kir6.2 mutations. N. Engl. J. Med. 2006;355(5):467–477. doi: 10.1056/NEJMoa061759. [DOI] [PubMed] [Google Scholar]
- 52.Lahmann C, Kramer HB, Ashcroft FM. Systemic administration of glibenclamide fails to achieve therapeutic levels in the brain and cerebrospinal fluid of rodents. PLoS ONE. 2015;10(7):e0134476. doi: 10.1371/journal.pone.0134476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Takanaga H, Murakami H, Koyabu N, et al. Efflux transport of tolbutamide across the blood-brain barrier. J. Pharm. Pharmacol. 1998;50(9):1027–1033. doi: 10.1111/j.2042-7158.1998.tb06918.x. [DOI] [PubMed] [Google Scholar]
- 54.Bessadok A, Garcia E, Jacquet H, et al. Recognition of sulfonylurea receptor (ABCC8/9) ligands by the multidrug resistance transporter P-glycoprotein (ABCB1): functional similarities based on common structural features between two multispecific ABC proteins. J. Biol. Chem. 2011;286(5):3552–3569. doi: 10.1074/jbc.M110.155200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Koster JC, Remedi MS, Dao C, Nichols CG. ATP and sulfonylurea sensitivity of mutant ATP-sensitive K+ channels in neonatal diabetes: implications for pharmacogenomic therapy. Diabetes. 2005;54(9):2645–2654. doi: 10.2337/diabetes.54.9.2645. [DOI] [PubMed] [Google Scholar]
- 56.Proks P, De Wet H, Ashcroft FM. Molecular mechanism of sulphonylurea block of K(ATP) channels carrying mutations that impair ATP inhibition and cause neonatal diabetes. Diabetes. 2013;62(11):3909–3919. doi: 10.2337/db13-0531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Flanagan SE, Clauin S, Bellanne-Chantelot C, et al. Update of mutations in the genes encoding the pancreatic beta-cell K(ATP) channel subunits Kir6.2 (KCNJ11) and sulfonylurea receptor 1 (ABCC8) in diabetes mellitus and hyperinsulinism. Hum. Mutat. 2009;30(2):170–180. doi: 10.1002/humu.20838. [DOI] [PubMed] [Google Scholar]
- 58.Kapoor RR, Flanagan SE, Arya VB, Shield JP, Ellard S, Hussain K. Clinical and molecular characterisation of 300 patients with congenital hyperinsulinism. Eur. J. Endocrinol. 2013;168(4):557–564. doi: 10.1530/EJE-12-0673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yan FF, Lin YW, Macmullen C, Ganguly A, Stanley CA, Shyng SL. Congenital hyperinsulinism associated ABCC8 mutations that cause defective trafficking of ATP-sensitive K+ channels: identification and rescue. Diabetes. 2007;56(9):2339–2348. doi: 10.2337/db07-0150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yan FF, Casey J, Shyng SL. Sulfonylureas correct trafficking defects of disease-causing ATP-sensitive potassium channels by binding to the channel complex. J. Biol. Chem. 2006;281(44):33403–33413. doi: 10.1074/jbc.M605195200. [DOI] [PubMed] [Google Scholar]
- 61.Yan F, Lin CW, Weisiger E, Cartier EA, Taschenberger G, Shyng SL. Sulfonylureas correct trafficking defects of ATP-sensitive potassium channels caused by mutations in the sulfonylurea receptor. J. Biol. Chem. 2004;279(12):11096–11105. doi: 10.1074/jbc.M312810200. [DOI] [PubMed] [Google Scholar]
- 62.Chen PC, Olson EM, Zhou Q, et al. Carbamazepine as a novel small molecule corrector of trafficking-impaired ATP-sensitive potassium channels identified in congenital hyperinsulinism. J. Biol. Chem. 2013;288(29):20942–20954. doi: 10.1074/jbc.M113.470948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Inagaki N, Gonoi T, Clement JP, et al. A family of sulfonylurea receptors determines the pharmacological properties of ATP-sensitive K+ channels. Neuron. 1996;16(5):1011–1017. doi: 10.1016/s0896-6273(00)80124-5. [DOI] [PubMed] [Google Scholar]
- 64.Inagaki N, Gonoi T, Clement JP, et al. Reconstitution of IKATP: an inward rectifier subunit plus the sulfonylurea receptor. Science. 1995;270(5239):1166–1170. doi: 10.1126/science.270.5239.1166. [DOI] [PubMed] [Google Scholar]
- 65.Cheng WW, Tong A, Flagg TP, Nichols CG. Random assembly of SUR subunits in K(ATP) channel complexes. Channels (Austin) 2008;2(1):34–38. doi: 10.4161/chan.2.1.6046. [DOI] [PubMed] [Google Scholar]
- 66.Chan KW, Wheeler A, Csanady L. Sulfonylurea receptors type 1 and 2A randomly assemble to form heteromeric KATP channels of mixed subunit composition. J. Gen. Physiol. 2008;131(1):43–58. doi: 10.1085/jgp.200709894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wheeler A, Wang C, Yang K, et al. Coassembly of different sulfonylurea receptor subtypes extends the phenotypic diversity of ATP-sensitive potassium (KATP) channels. Mol. Pharmacol. 2008;74(5):1333–1344. doi: 10.1124/mol.108.048355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cui Y, Tran S, Tinker A, Clapp LH. The molecular composition of K(ATP) channels in human pulmonary artery smooth muscle cells and their modulation by growth. Am. J. Respir. Cell Mol. Biol. 2002;26(1):135–143. doi: 10.1165/ajrcmb.26.1.4622. [DOI] [PubMed] [Google Scholar]
- 69.Pountney DJ, Sun ZQ, Porter LM, et al. Is the molecular composition of K(ATP) channels more complex than originally thought? J. Mol. Cell. Cardiol. 2001;33(8):1541–1546. doi: 10.1006/jmcc.2001.1407. [DOI] [PubMed] [Google Scholar]
- 70.Kono Y, Horie M, Takano M, et al. The properties of the Kir6.1–6.2 tandem channel co-expressed with SUR2A. Pflugers Archiv. - European J. Physiol. 2000;440(5):692–698. doi: 10.1007/s004240000315. [DOI] [PubMed] [Google Scholar]
- 71.Morrissey A, Parachuru L, Leung M, et al. Expression of ATP-sensitive K+ channel subunits during perinatal maturation in the mouse heart. Pediatr. Res. 2005;58:185–192. doi: 10.1203/01.PDR.0000169967.83576.CB. [DOI] [PubMed] [Google Scholar]
- 72.Glukhov AV, Flagg TP, Fedorov VV, Efimov IR, Nichols CG. Differential K(ATP) channel pharmacology in intact mouse heart. J. Mol. Cell. Cardiol. 2009;48(1):152–160. doi: 10.1016/j.yjmcc.2009.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Flagg TP, Kurata HT, Masia R, et al. Differential structure of atrial and ventricular KATP: atrial KATP channels require SUR1. Circ. Res. 2008;103(12):1458–1465. doi: 10.1161/CIRCRESAHA.108.178186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Blanco-Rivero J, Gamallo C, Aras-Lopez R, et al. Decreased expression of aortic KIR6.1 and SUR2B in hypertension does not correlate with changes in the functional role of K(ATP) channels. Eur. J. Pharmacol. 2008;587(1–3):204–208. doi: 10.1016/j.ejphar.2008.03.039. [DOI] [PubMed] [Google Scholar]
- 75.Standen NB, Quayle JM, Davies NW, Brayden JE, Huang Y, Nelson MT. Hyperpolarizing vasodilators activate ATP-sensitive K+ channels in arterial smooth muscle. Science. 1989;245(4914):177–180. doi: 10.1126/science.2501869. [DOI] [PubMed] [Google Scholar]
- 76.Miyoshi Y, Nakaya Y, Wakatsuki T, et al. Endothelin blocks ATP-sensitive K+ channels and depolarizes smooth muscle cells of porcine coronary artery. Circ. Res. 1992;70(3):612–616. doi: 10.1161/01.res.70.3.612. [DOI] [PubMed] [Google Scholar]
- 77.Ottolia M, Toro L. Reconstitution in lipid bilayers of an ATP-sensitive K+ channel from pig coronary smooth muscle. J. Membr. Biol. 1996;153(3):203–209. doi: 10.1007/s002329900123. [DOI] [PubMed] [Google Scholar]
- 78.Beech DJ, Zhang H, Nakao K, Bolton TB. K channel activation by nucleotide diphosphates and its inhibition by glibenclamide in vascular smooth muscle cells. Br. J. Pharmacol. 1993;110(2):573–582. doi: 10.1111/j.1476-5381.1993.tb13849.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kajioka S, Kitamura K, Kuriyama H. Guanosine diphosphate activates an adenosine 5′-triphosphate-sensitive K+ channel in the rabbit portal vein. J. Physiol. 1991;444:397–418. doi: 10.1113/jphysiol.1991.sp018885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kamouchi M, Kitamura K. Regulation of ATP-sensitive K+ channels by ATP and nucleotide diphosphate in rabbit portal vein. Am. J. Physiol. 1994;266(5 Pt 2):H1687–H1698. doi: 10.1152/ajpheart.1994.266.5.H1687. [DOI] [PubMed] [Google Scholar]
- 81.Cole WC, Clement-Chomienne O. ATP-sensitive K+ channels of vascular smooth muscle cells. J. Cardiovasc. Electrophysiol. 2003;14(1):94–103. doi: 10.1046/j.1540-8167.2003.02376.x. [DOI] [PubMed] [Google Scholar]
- 82.Chutkow WA, Pu J, Wheeler MT, et al. Episodic coronary artery vasospasm and hypertension develop in the absence of Sur2 K(ATP) channels. J. Clin. Invest. 2002;110(2):203–208. doi: 10.1172/JCI15672. see comment. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Miki T, Suzuki M, Shibasaki T, et al. Mouse model of Prinzmetal angina by disruption of the inward rectifier Kir6.1. Nat. Med. 2002;8(5):466–472. doi: 10.1038/nm0502-466. [DOI] [PubMed] [Google Scholar]
- 84.Van Bon BW, Gilissen C, Grange DK, et al. Cantu syndrome is caused by mutations in ABCC9. Am. J. Hum. Genet. 2012;90(6):1094–1101. doi: 10.1016/j.ajhg.2012.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Harakalova M, Van Harssel JJ, Terhal PA, et al. Dominant missense mutations in ABCC9 cause Cantu syndrome. Nat. Genet. 2012;44(7):793–796. doi: 10.1038/ng.2324. [DOI] [PubMed] [Google Scholar]
- 86.Brownstein CA, Towne MC, Luquette LJ, et al. Mutation of KCNJ8 in a patient with Cantu syndrome with unique vascular abnormalities – support for the role of K(ATP) channels in this condition. Eur. J. Med. Genet. 2013;56(12):678–682. doi: 10.1016/j.ejmg.2013.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cooper PE, Reutter H, Woelfle J, et al. Cantu syndrome resulting from activating mutation in the KCNJ8 gene. Hum. Mutat. 2014;35(7):809–813. doi: 10.1002/humu.22555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cantu JM, Garcia-Cruz D, Sanchez-Corona J, Hernandez A, Nazara Z. A distinct osteochondrodysplasia with hypertrichosis- Individualization of a probable autosomal recessive entity. Hum. Genet. 1982;60(1):36–41. doi: 10.1007/BF00281261. [DOI] [PubMed] [Google Scholar]
- 89.Scurr I, Wilson L, Lees M, et al. Cantu syndrome: report of nine new cases and expansion of the clinical phenotype. Am. J. Med. Genet. A. 2011;155A(3):508–518. doi: 10.1002/ajmg.a.33885. [DOI] [PubMed] [Google Scholar]
- 90.Garcia-Cruz D, Mampel A, Echeverria MI, et al. Cantu syndrome and lymphoedema. Clin. Dysmorphol. 2011;20(1):32–37. doi: 10.1097/MCD.0b013e32833d015c. [DOI] [PubMed] [Google Scholar]
- 91.Kobayashi D, Cook AL, Williams DA. Pulmonary hypertension secondary to partial pulmonary venous obstruction in a child with Cantu syndrome. Pediatr. Pulmonol. 2010;45(7):727–729. doi: 10.1002/ppul.21215. [DOI] [PubMed] [Google Scholar]
- 92.Grange DK, Lorch SM, Cole PL, Singh GK. Cantu syndrome in a woman and her two daughters: further confirmation of autosomal dominant inheritance and review of the cardiac manifestations. Am. J. Med. Genet. A. 2006;140(15):1673–1680. doi: 10.1002/ajmg.a.31348. [DOI] [PubMed] [Google Scholar]
- 93.Engels H, Bosse K, Ehrbrecht A, et al. Further case of Cantu syndrome: exclusion of cryptic subtelomeric chromosome aberrations. Am. J. Med. Genet. 2002;111(2):205–209. doi: 10.1002/ajmg.10560. [DOI] [PubMed] [Google Scholar]
- 94.Lazalde B, Sanchez-Urbina R, Nuno-Arana I, Bitar WE, De Lourdes Ramirez-Duenas M. Autosomal dominant inheritance in Cantu syndrome (congenital hypertrichosis, osteochondrodysplasia, and cardiomegaly) Am. J. Med. Genet. 2000;94(5):421–427. doi: 10.1002/1096-8628(20001023)94:5<421::aid-ajmg15>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 95.Concolino D, Formicola S, Camera G, Strisciuglio P. Congenital hypertrichosis, cardiomegaly, and osteochondrodysplasia (Cantu syndrome): a new case with unusual radiological findings. Am. J. Med. Genet. 2000;92(3):191–194. [PubMed] [Google Scholar]
- 96.Robertson SP, Kirk E, Bernier F, Brereton J, Turner A, Bankier A. Congenital hypertrichosis, osteochondrodysplasia, and cardiomegaly: Cantu syndrome. Am. J. Med. Genet. 1999;85(4):395–402. [PubMed] [Google Scholar]
- 97.Rosser EM, Kaariainen H, Hurst JA, et al. Three patients with the osteochondrodysplasia and hypertrichosis syndrome--Cantu syndrome. Clin. Dysmorphol. 1998;7(2):79–85. doi: 10.1097/00019605-199804000-00001. [DOI] [PubMed] [Google Scholar]
- 98.Mehta PK, Mamdani B, Shansky RM, Mahurkar SD, Dunea G. Severe hypertension. Treatment with minoxidil. JAMA. 1975;233(3):249–252. [PubMed] [Google Scholar]
- 99.Kaler SG, Patrinos ME, Lambert GH, Myers TF, Karlman R, Anderson CL. Hypertrichosis and congenital anomalies associated with maternal use of minoxidil. Pediatrics. 1987;79(3):434–436. [PubMed] [Google Scholar]
- 100.Rosa FW, Idanpaan-Heikkila J, Asanti R. Fetal minoxidil exposure. Pediatrics. 1987;80(1):120. [PubMed] [Google Scholar]
- 101.Li A, Knutsen RH, Zhang H, et al. Hypotension due to Kir6.1 gain-of-function in vascular smooth muscle. J. Am. Heart Assoc. 2013;2(4):e000365. doi: 10.1161/JAHA.113.000365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Richardson P, Mckenna W, Bristow M, et al. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of cardiomyopathies. Circulation. 1996;93(5):841–842. doi: 10.1161/01.cir.93.5.841. [DOI] [PubMed] [Google Scholar]
- 103.Mehta PA, Dubrey SW. High output heart failure. QJM. 2009;102(4):235–241. doi: 10.1093/qjmed/hcn147. [DOI] [PubMed] [Google Scholar]
- 104.Flagg TP, Charpentier F, Manning-Fox J, et al. Remodeling of excitation-contraction coupling in transgenic mice expressing ATP-insensitive sarcolemmal KATP channels. Am. J. Physiol. Heart Circ. Physiol. 2004;286(4):H1361–H1369. doi: 10.1152/ajpheart.00676.2003. [DOI] [PubMed] [Google Scholar]
- 105.Rossi A, Cantisani C, Melis L, Iorio A, Scali E, Calvieri S. Minoxidil use in dermatology, side effects and recent patents. Recent Pat. Inflamm. Allergy Drug Discov. 2012;6(2):130–136. doi: 10.2174/187221312800166859. [DOI] [PubMed] [Google Scholar]
- 106.Aguilar-Bryan L, Nichols CG, Rajan AS, Parker C, Bryan J. Co-expression of sulfonylurea receptors and KATP channels in hamster insulinoma tumor (HIT) cells. Evidence for direct association of the receptor with the channel. J. Biol. Chem. 1992;267(21):14934–14940. [PubMed] [Google Scholar]
- 107.Ashcroft FM, Harrison DE, Ashcroft SJ. Glucose induces closure of single potassium channels in isolated rat pancreatic beta-cells. Nature. 1984;312(5993):446–448. doi: 10.1038/312446a0. [DOI] [PubMed] [Google Scholar]
- 108.Zhang HL, Bolton TB. Two types of ATP-sensitive potassium channels in rat portal vein smooth muscle cells. Br. J. Pharmacol. 1996;118(1):105–114. doi: 10.1111/j.1476-5381.1996.tb15372.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Farzaneh T, Tinker A. Differences in the mechanism of metabolic regulation of ATP-sensitive K+ channels containing Kir6.1 and Kir6.2 subunits. Cardiovasc. Res. 2008;79(4):621–631. doi: 10.1093/cvr/cvn138. [DOI] [PubMed] [Google Scholar]
- 110.Isomoto S, Kondo C, Yamada M, et al. A novel sulfonylurea receptor forms with BIR (Kir6.2) a smooth muscle type ATP-sensitive K+ channel. J. Biol. Chem. 1996;271(40):24321–24324. doi: 10.1074/jbc.271.40.24321. [DOI] [PubMed] [Google Scholar]
- 111.Satoh E, Yamada M, Kondo C, et al. Intracellular nucleotide-mediated gating of SUR/Kir6.0 complex potassium channels expressed in a mammalian cell line and its modification by pinacidil. J. Physiol. 1998;511(Pt 3):663–674. doi: 10.1111/j.1469-7793.1998.663bg.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yamada M, Isomoto S, Matsumoto S, et al. Sulphonylurea receptor 2B and Kir6.1 form a sulphonylurea-sensitive but ATP-insensitive K+ channel. J. Physiol. 1997;499(Pt 3):715–720. doi: 10.1113/jphysiol.1997.sp021963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Babenko AP, Bryan J. A conserved inhibitory and differential stimulatory action of nucleotides on K(IR)6.0/SUR complexes is essential for excitation-metabolism coupling by K(ATP) channels. J. Biol. Chem. 2001;276(52):49083–49092. doi: 10.1074/jbc.M108763200. [DOI] [PubMed] [Google Scholar]
- 114.Smith MP, Humphrey SJ, Jackson WF. Selective in vivo antagonism of pinacidil-induced hypotension by the guanidine U37883A in anesthetized rats. Pharmacology. 1994;49(6):363–375. doi: 10.1159/000139255. [DOI] [PubMed] [Google Scholar]
- 115.Raphemot R, Rouhier MF, Swale DR, et al. Discovery and characterization of a potent and selective inhibitor of Aedes aegypti inward rectifier potassium channels. PLoS ONE. 2014;9(11):e110772. doi: 10.1371/journal.pone.0110772. [DOI] [PMC free article] [PubMed] [Google Scholar]