

Abstract
An innovative approach to the delivery of uncharged peptide nucleic acids (PNA) and phosphorodiamidate morpholino (PMO) oligomers in mammalian cells is described and consists of extending the sequence of those oligomers with a short PNA-polyA or PMO-polyA tail. Recognition of the polyA-tailed PNA or PMO oligomers by an amphipathic trans-acting polythymidylic thiophosphate triester element (dTtaPS) results in efficient internalization of those oligomers in several cell lines. Our findings indicate that cellular uptake of the oligomers occurs through an energy-dependent mechanism and macropinocytosis appears to be the predo-minant endocytic pathway used for internalization. The functionality of the internalized oligomers is demonstrated by alternate splicing of the pre-mRNA encoding luciferase in HeLa pLuc 705 cells. Amphipathic phosphorothioate DNA elements may represent a unique class of cellular transporters for robust delivery of uncharged nucleic acid sequences in live mammalian cells.
Keywords: PNA oligomers, PMO oligomers, nucleic acid-based drug delivery, cellular internalization, alternate splicing of luciferase pre-mRNA
Introduction
Uncharged PNA (Yin et al., 2008) and PMO (Fletcher et al., 2006) oligomers are nuclease-resistant and RNase-H incompetent oligonucleotide analogues. Although these quality attributes are essential in the context of nucleic acid-based drug development, a serious limitation in the use of many types of synthetic oligonucleotides and their analogues, as therapeutic antisense agents, has been poor cellular delivery (Abes et al., 2007). Although cationic lipids are efficient carriers for in vitro cellular transfection of negatively charged DNA/RNA sequences, those carriers could not be successfully used for cellular internalization of uncharged PNA or PMO oligomers (Shiraishi and Nielsen, 2011). The conjugation of cationic cell-penetrating peptides (CPP) to PNAs or PMOs resulted in improved cellular uptake of these oligomers and led to pre-mRNA splicing correction activities in mammalian cells and animal models (Bendifallah et al., 2006; Moulton and Jiang, 2009; Cirak et al., 2011; O'Donovan et al., 2015). However, an inherent limitation of this approach to cellular internalization is the requirement of a PNA-CPP or a PMO-CPP conjugate to be complementary to each mRNA targeted sequence. In addition to convey unpredictable physicochemical and functional properties to uncharged nucleic acids (Tung and Stein, 2000; Prater and Miller, 2004), the preparation and purification of CPP-conjugates are tedious and laborious (Shiraishi and Nielsen, 2011).
The cellular uptake of an uncharged thermosensitive DNA oligonucleotide prodrug in mammalian cells has been achieved though replacement of four neutral thiophosphate triester functions with four positively charged ones (Jain et al., 2013). These findings have led to the development of an innovative method for delivering PNA or PMO oligomers in live mammalian cells (Jain et al., 2015). This method consists of extending a PNA or PMO sequence with a short PNA-polyA or PMO-polyA tail, which provides an affinity recognition site for a positively charged, trans-acting polythymidylic thiophosphate triester element (dTtaPS) through sequence complementarity.
This unit describes the preparation of deoxyribonucleoside phosphoramidites (Basic Protocol 1) that are required for solid-phase synthesis of the trans-acting polythymidylic thiophosphate triester element dTtaPS (Basic Protocol 2). The synthesis of phosphordiamidites needed for the preparation of deoxyribonucleoside building blocks is delineated in Support Protocol 1.
This unit also outlines the: (i) preparation of complexes between PNA/PMO oligomers and dTtaPS (Support Protocol 2); (ii) dTtaPS-mediated cellular internalization of PNA/PMO oligomers in live mammalian cells as measured by flow cytometry (Basic Protocol 3); (iii) production of luciferase activity (Basic Protocol 4) and; (iv) RT-PCR analysis of luciferase pre-mRNA splicing correction (Support Protocol 3).
NOTE: Please see Critical Parameters regarding proper treatment of glassware prior to performing the protocols.
Basic Protocol 1
General Procedure for the Preparation of Deoxyribonucleoside Phosphoramidites (4 and 5)
As shown in Fig. 4.69.1, this protocol provides a general method for the synthesis of deoxyribonucleoside phosphoramidites (4 and 5) from commercial 5′-O-(4,4′-Dimethoxytrityl)-2′-deoxythymidine (1), N,N,N′,N′-tetraisopropyl-O-[3-(N,N-dimethylamino)prop-1-yl]phosphordiamidite (2; see Support Protocol) or N,N,N′,N′-tetraisopropyl-O-(octan-1-yl)phosphordiamidite (3; see Support Protocol) and 1H-tetrazole.
Figure 4.69.1.

Preparation of deoxyribonucleoside phosphoramidites 4 and 5. DMTr, 4,4′-dimethoxytrityl; Thy, thymin-1-yl; i-Pr, isopropyl.
Materials
5′-O-(4,4′-Dimethoxytrityl)-2′-deoxythymidine (1; ChemGenes Corporation)
N,N,N′,N′-tetraisopropyl-O-[3-(N,N-dimethylamino)prop-1-yl]phosphordiamidite (2; see Support Protocol)
N,N,N′,N′-tetraisopropyl-O-(octan-1-yl)phosphordiamidite (3; see Support Protocol)
Anhydrous benzene (Aldrich)
Anhydrous acetonitrile (MeCN, Glen Research)
Argon or Nitrogen source
Dichloromethane (CH2Cl2; Fisher)
Hexane (Fisher)
0.45 M 1H-tetrazole in MeCN (Glen Research)
Triethylamine (Aldrich)
Silica gel (60 Å, 230 to 400 mesh; EMD)
Dry ice/acetone bath
50-, 100-, and 250-mL round-bottom flasks
Rubber septa for 14/20- and 24/40-glass joints
Magnetic stir bars
Magnetic stirrer
Vacuum desiccator
High-vacuum oil pump
1-, 3- and 10-mL Luer-tipped glass syringes
Pasteur pipettes
Syringe needles
5-mm NMR tubes
Buchner funnel setup with vacuum source
Rotary evaporator equipped with a dry ice condenser and connected to an oil pump
2.5 × 20-cm disposable Flex chromatography columns (Kontes)
2.5 × 7.5-cm EMD TLC plates pre-coated with a 250-μm layer of silica gel 60 F254
Hand-held UV254 lamp (UVP)
Lyophilizer
NMR spectrometer (Bruker)
Prepare deoxyribonucleoside phosphoramidites
Place N,N,N′,N′-tetraisopropyl-O-[3-(N,N-dimethylamino)prop-1-yl]phosphordiamidite (2, 2.0 mmol) or N,N,N′,N′-tetraisopropyl-O-[octan-1-yl]phosphordiamidite (3, 2.0 mmol) in a 50-mL round-bottom flask and a magnetic stir bar. Seal the flask with a rubber septum.
Under an argon atmosphere, add 20 mL anhydrous acetonitrile using a 10-mL glass syringe. Stir the solution with a magnetic stirrer.
- While stirring the solution, add vacuum-dried 5′-O-(4,4′-dimethoxytrityl)-thymidine (1, 1.0 mmol) dissolved in 2.2 mL 0.45 M 1H-tetrazole (1.0 mmol) in MeCN using a 3-mL syringe.Other activators including 5-ethylthio-1H-tetrazole, 5-benzylthio-1H-tetrazole or 4,5-dicyanoimidazole could theoretically be used, but have not been tested.
- Carefully transfer ∼0.5 mL of the reaction mixture by syringe to a dry 5-mm NMR tube. Monitor the progress of the reaction in the NMR tube by 31P NMR spectroscopy. Upon completion of the reaction, as seen by the disappearance of 2 or 3, return the NMR sample to the main reaction mixture before proceeding to the next step.The 31P NMR signal corresponding to 2 or 3 (δP 123 ppm) disappears within 4-6 hr at ambient temperature and thus indicates complete phosphitylation of 1.
Add 1 mL triethylamine and immediately concentrate the reaction mixture to a syrup using a rotary evaporator connected to a vacuum pump.
Purify and isolate deoxyribonucleoside phosphoramidites
Suspend the crude product in ∼3 mL 95:5 (v/v) hexane/triethylamine and spread the suspension on the top of a 2.5-cm × 20-cm disposable Flex chromatography column containing ∼25 g silica gel that has been equilibrated in 95:5 (v/v) hexane/triethylamine (APPENDIX 3E).
Elute the column using a gradient of dichoromethane (0 → 95%) in 95:5 (v/v) hexane/triethylamine and collect 6-mL fractions.
- Analyze fractions by TLC (APPENDIX 3D) on a 2.5 × 7.5-cm EMD silica gel 60 F254 TLC plate using 95:5 (v/v) hexane/triethylamine as the eluent. Pool appropriate fractions and rotoevaporate under reduced pressure (∼20 mmHg) until a white amorphous solid is obtained.TLC analysis of diastereomeric 4 or 5 reveals two tight spots; the Rf of each pair of spots is ∼0.55 or ∼0.74, respectively, when 9:1 (v/v) benzene/triethylamine is used as the eluent.
Dissolve the solid in ∼5 mL anhydrous benzene and add the solution, at the rate of ∼ 1 mL/min, to ∼100 mL vigorously stirred hexane in a 250-mL round-bottom flask placed in a dry ice/acetone bath.
Allow the suspension to settle and carefully decant off most (∼95%) of the supernatant.
Rotoevaporate the wet material to dryness under reduced pressure and then dissolve in ∼10 mL anhydrous benzene.
- Transfer the solution to a 50-mL round-bottom flask, freeze in a dry ice/acetone bath and lyophilize under high vacuum to afford triethylamine-free 4 (0.51 mmol) or 5 (0.70 mmol) as a white powder in a yield of 51% or 70%, respectively.5′-O-(4,4′-dimethoxytrityl)-3′-O-[(N,N-diisopropylamino)(3-[N,N-dimethylamino]prop-1-yl)oxy]phosphinyl-thymidine (4): 31P NMR (121 MHz, C6D6): δ 148.8, 148.3. +HRMS: calcd for C42H57N4O8P [M]+ 777.3986, found 777.3984.5′-O-(4,4′-Dimethoxytrityl)-3′-O-[(N,N-diisopropylamino)(octan-1-yl)oxy]phosphinyl-thymidine (5). 31P NMR (121 MHz, C6D6): δ 147.7, 147.0. +HRMS: calcd for C45H62N3O8P [M + H]+ 804.4353, found 804.4338.
Support Protocol 1
General Procedure for the Preparation of N,N,N′,N′-Tetraisopropylphosphordiamidites (2 and 3)
This protocol delineates the reaction of commercial bis(N,N-diisopropylamino)chlorophosphine with commercial 3-(N,N-dimethylamino)propan-1-ol or 1-octanol in the presence of triethylamine to provide the phosphordiamidites 2 or 3 (see Fig. 4.69.2).
Figure 4.69.2.

Preparation of the phosphordiamidites 2 and 3. i-Pr, isopropyl; Et3N, triethylamine.
Additional Materials (also see Basic Protocol)
Bis(N,N-diisopropylamino)chlorophosphine (Aldrich)
3-(N,N-Dimethylamino)propan-1-ol (Aldrich)
1-Octanol (Aldrich)
Synthesize phosphordiamidites
Place 1.00 g (3.70 mmol) bis(N,N-diisopropylamino)chlorophosphine and a magnetic stir bar in an oven-dried 50-mL round-bottom flask. Seal the flask with a rubber septum.
- Using a 10-mL glass syringe add 20 mL anhydrous benzene under an argon atmosphere. While stirring the suspension with a magnetic stirrer add, consecutively, 436 μL (3.70 mmol) 3-(N,N-dimethylamino)propan-1-ol or 583 μL (3.70 mmol) 1-octanol and 1.00 mL (7.17 mmol) triethylamine using 1-mL syringes. Stir the reaction mixture under argon at ∼25 °C until the reaction is complete.The progress of the reaction is monitored by 31P NMR spectroscopy. The formation of 2 or 3 (singlet, 122.4 ppm, downfield relative to a phosphoric acid external standard) is complete within 2 hr at ∼25°C.
Purify and isolate phosphordiamidites
Using a Pasteur pipette, the entire reaction mixture is loaded on the top of a 2.5-cm × 20-cm disposable Flex chromatography column containing ∼15 g silica gel that has been equilibrated in 9:1 (v/v) benzene/trimethylamine.
Elute the column with 100 mL 9:1 (v/v) benzene/triethylamine. Collect the eluate in a 250-mL round-bottom flask.
Rotoevaporate the volatiles under reduced pressure (∼20 mmHg) to viscous oil.
Dissolve the oily material in a 50-mL round bottom flask with 10-mL anhydrous benzene.
- While swirling the flask in a dry ice-acetone bath, freeze the solution. Lyophilize under high vacuum to give the phosphordiamidite 2 or 3 as viscous oil.N,N,N′,N′-Tetraisopropyl-O-[3-(N,N-dimethylamino)prop-1-yl]phosphordiamidite (2): Yield (1.01 g, 3.03 mmol, 82%). 1H NMR (300 MHz, C6D6): δ 3.70 (dt, J = 7.3, 6.4 Hz, 2H), 3.55 (sept, J = 6.8 Hz, 2H), 3.52 (sept, J = 6.8 Hz, 2H), 2.36 (t, J = 7.1 Hz, 2H), 2.11 (s, 6H), 1.77 (quint, J = 6.8 Hz, 2H), 1.25 (d, J = 6.8 Hz, 12H), 1.20 (d, J = 6.8 Hz, 12H). 13C NMR (75 MHz, C6D6): δ 62.8 (d, 2JCP = 21.8 Hz), 56.9, 45.6, 44.6 (d, 2JCP = 12.6 Hz), 30.4 (d, JCP = 8.6 Hz), 24.8 (d, JCP = 8.6 Hz), 24.1 (d, JCP = 5.6 Hz). 31P NMR (121 MHz, C6D6): δ 123.3.N,N,N′,N′-Tetraisopropyl-O-[octan-1-yl]phosphordiamidite (3): Yield (1.17 g, 3.24mmol, 88%). 1H NMR (300 MHz, C6D6): δ 3.65 (dt, J = 7.1, 6.4 Hz, 2H), 3.57 (sept, J = 6.8 Hz, 2H), 3.53 (sept, J = 6.8 Hz, 2H), 1.65 (dq, J = 6.6, 6.4 Hz, 2H), 1.43 (m, 2H), 1.34-1.18 (br m, 8H), 1.27 (d, J = 6.8 Hz, 12H), 1.22 (d, J = 6.8 Hz, 12H), 0.89 (dd, J = 6.8, 6.4 Hz, 3H). 13C NMR (75 MHz, C6D6): δ 64.7 (d, 2JCP = 21.8 Hz), 44.6 (d, 2JCP =12.6 Hz), 32.2, 32.1 (d, JCP =8.6 Hz), 29.8, 29.7, 26.6, 24.8 (d, JCP = 8.5 Hz), 24.1 (d, JCP =5.6 Hz), 23.1, 14.3. 31P NMR (121 MHz, C6D6): δ 123.2.The phosphordiamidites 2 and 3 are of sufficient purity to be used without further purification in the preparation of deoxyribonucleoside phosphoramidites 4 and 5.
Basic Protocol 2
Solid-Phase Synthesis of the Trans-Acting Polythymidylic Thiophosphate Triester Element dTtaPS (6)
The chemical structure of dTtaPS (6) is presented in Fig. 4.69.3. This protocol describes the solid-phase synthesis and characterization of this amphiphilic phosphorothioate DNA sequence.
Figure 4.69.3.

Chemical structure of the amphipathic polythymidylic thiophosphate triester element dTtaPS shown in its protonated form under physiological conditions.
Materials
Reagents recommended for automated solid-phase oligonucleotide synthesis (Glen Research):
5′-O-(4,4′-dimethoxytrityl)-3′-O-[(N,N-diisopropylamino)(3-[N,N-dimethylamino]prop-1-yl)oxy]phosphinyl-thymidine (4, see Support Protocol 1)
5′-O-(4,4′-Dimethoxytrityl)-3′-O-[(N,N-diisopropylamino)(octan-1-yl)oxy]phosphinyl-thymidine (5, see Support Protocol 1)
Succinylated long chain alkylamine controlled-pore glass support loaded with 0.2 μmol 5′-O-(4,4′-dimethoxytrityl)-thymidine
Deblocking solution: trichloroacetic acid (TCA) in dichloromethane (CH2Cl2)
Activator solution: 1H-tetrazole in acetonitrile
Cap A solution: acetic anhydride in THF/pyridine
Cap B solution: 1-methylimidazole in THF
Sulfuration solution: 0.05 M 3-(dimethylaminomethylidene)amino-3H-1,2,4-dithiazole-3-thione in (2:3 v/v) pyridine/MeCN
Anhydrous acetonitrile (Glen Research)
Low water content acetonitrile (Fisher)
Methylamine gas cylinder (Aldrich)
Triethylamine (Aldrich)
Stainless steel pressure vessel equipped with a valve system (Parr Instrument)
High-vacuum oil pump
DNA/RNA synthesizer (Applied Biosystems)
UV/Vis Spectrophotometer (Agilent Technologies)
MALDI-TOF mass spectrometer
HPLC instrument (Agilent Technologies)
25 cm × 4.6 mm, 300 Å Jupiter® C-4 HPLC Column (5 μm; Phenomenex)
Heating block (VWR)
1.5-mL Microcentrifuge tubes
Pipettor (Corning)
Synthesize oligonucleotide 6 (dTtaPS)
- Perform a 0.2-μmol scale solid-phase synthesis of the phosphorothioate DNA sequence 6 (Fig. 4.69.3) using an Applied Biosystems 394 DNA/RNA synthesizer in “trityl-on” mode according to the manufacturer's recommendations (see also APPENDIX 3C).The deoxyribonucleoside phosphoramidites, 4 and 5 are used as 0.1 M solutions in dry acetonitrile. All ancillary reagents required for the automated preparation of dTtaPS have been purchased from Glen Research and used as recommended by the manufacturer.The synthetic cycle recommended for the preparation of dTtaPS differs from the conventional cycle used for synthesis of unmodified oligonucleotides in that the “capping step” is performed after the oxidative sulfuration reaction (Iyer et al., 1990). The reaction time of each of the following synthesis cycle steps has been extended to ensure optimal production of dTtaPS: (i) 5′-deblocking reaction (3% TCA in CH2Cl2, 60 sec); (ii) 1H-tetrazole-assisted phosphoramidite coupling reaction (600 sec); (iii) oxidation reaction (sulfuration solution, 600 sec); (iv) capping reaction (Cap A and Cap B solutions, 120 sec). Upon complete assembly of dTtaPS, spectrophotometric measurement of the released 5′-DMTr cation at 498 nm revealed an overall yield for dTtaPS (6) in the range of 90 ± 5%.
Cleave oligonucleotide 6 from the solid support
Place the dTtaPS synthesis column into a stainless steel pressure vessel. Connect the valve system of the vessel to a high vacuum oil pump. Turn on the valve and evacuate the vessel over a period of 5 min. Turn off the valve system and disconnect it from the vacuum pump.
Connect the pressure vessel to a methylamine gas cylinder through the vessel's valve system. Turn on both the gas cylinder's and the vessel's valves. Fill the pressure vessel to maximum gas pressure (∼2.5 bar, 25 °C).
- Expose the dTtaPS synthesis column to pressurized methylamine gas for 3 min. Turn off both the gas cylinder's and the vessel's valves. Disconnect the gas cylinder from the vessel and open the vessel's valve system to release excess methylamine gas. Connect the vessel's valve system to a high vacuum oil pump. Evacuate the vessel over a period of 2 min and remove the vacuum pump.Do not use an aqueous methylamine or ammonia solution to release dTtaPS from the solid support as it may result in partial internucleotidic chain cleavage of the DNA sequence.
Using a 1-mL glass syringe, elute dTtaPS from the synthesis column with 2 mL of 1:60:39 (v/v/v) triethylamine/acetonitrile/water. Collect the eluate in two 1.5 mL microfuge tubes.
- Evaporate the eluate to dryness using a stream of air. Store the dried DNA sequence 6 at −20 °C.dTtaPS: +MALDI-TOF: calcd for C127H202N19O47P7S7 [M + H]+ 3187, found 3184.
Analyze oligonucleotide 6 by RP-HPLC
- Analyze ∼0.25 OD260 of 6 using a 5-μm Phenomenex Jupiter® C-4 HPLC Column (UNIT 10.5)RP-HPLC analysis is performed employing a 5-μm Phenomenex Jupiter® C-4 HPLC Column (25 cm × 4.6 mm) under the following chromatographic conditions: starting from 20 mM ammonium acetate in 4:6 (v/v) water/acetonitrile, a linear gradient of acetonitrile is pumped at a flow rate of 1 mL/min to 20 mM ammonium acetate in 2:8 (v/v) water/acetonitrile over a period of 15 min and is held, isocratically for 5 min.
Support Protocol 2
General Procedure for the Formation of Complexes Between dTtaPS and PNA or PMO Oligomers
Pre-mixing of dTtaPS with commercial PNA or PMO oligomers (Fig. 4.69.4) results in the formation of complexes facilitating the cellular uptake and bioactivity of the PNA and PMO sequences in live mammalian cells.
Figure 4.69.4.
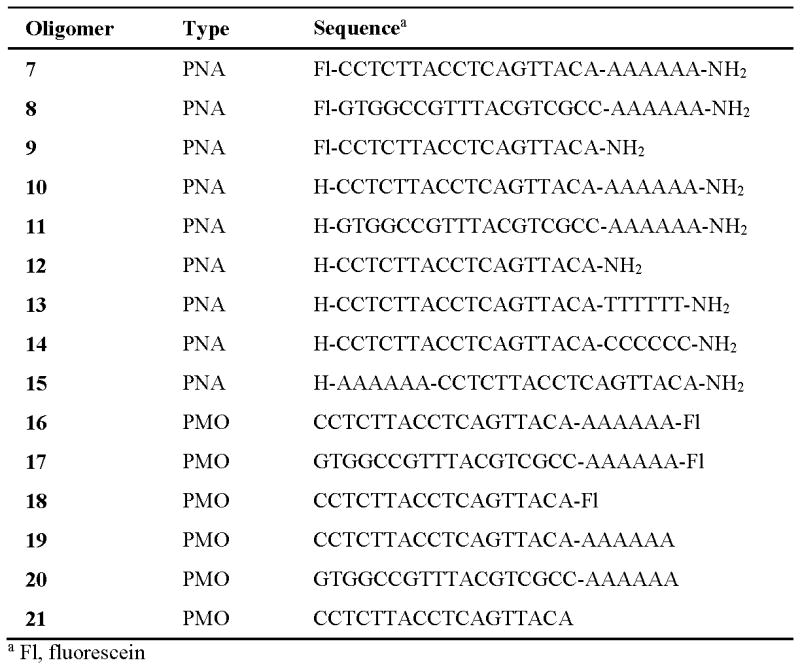
Commercial PNA and PMO sequences needed to demonstrate the dTtaPS-assisted delivery and bioactivity of these oligomers in live mammalian cells.
Additional Materials (see also Basic Protocol)
dTtaPS (6, see Basic Protocol 2)
PNA oligomers 7-15 (PNA Bio, Inc.)
PMO oligomers 16-21 (GeneTools, LLC)
OptiMEM (Life Technologies)
Pipettor (Corning)
1.5-mL Microcentrifuge tubes
Water bath set at 37 °C (Thermo Scientific)
UV/VIS spectrophotometer (Agilent Technologies)
Prepare a 2 × stock solution of PNA or PMO oligomer:dTtaPS complexes
To a 1.5-mL microcentrifuge tube, containing 20 μL OptiMEM, add 1 μL of 100 μM PNA or 100 μM of PMO stock solution using a pipettor.
Pipet 2 μL of 100 μM dTtaPS stock solution and add it to the oligomer solution of step 1 followed by 27 μL of OptiMEM to produce a 2× stock solution of PNA or PMO oligomer:dTtaPS complexes that is 2 μM in PNA or PMO oligomer and 4 μM in dTtaPS in 50 μL OptiMEM.
- Incubate the solution at 37 °C for 30 min. Store the 2 × stock solution of PNA or PMO oligomer:dTtaPS complexes at 4 °C for 15 min prior to immediate use in cellular uptake experiments.When used in cellular uptake experiments, this 2× stock solution of the complexes will provide an extracellular concentration of 1.0 μM in PNA or PMO oligomer and 2.0 μM in dTtaPS (see Basic protocol 3).
Basic Protocol 3
dTtaPS-Mediated Cellular Internalization of PNA or PMO Oligomers in Live Mammalian Cells
This protocol provides the details pertaining to the dTtaPS-mediated cellular internalization of fluorescently-labelled PNA or PMO oligomers in live HEK293, HeLa, HeLa pLuc 705, MCF7 and SK-N-SH cells. Fig. 4.69.5 shows results of the cellular uptake analysis of the dTtaPS:PNA oligomers 7, 8, 9 and dTtaPS:PMO oligomer 16, 17, 18 complexes (Fig. 4.69.4) in the above-mentioned cell lines using a fluorescence-activated cell sorting (FACS) flow cytometer.
Figure 4.69.5.

Internalization of dTtaPS:PNA oligomer complexes in live mammalian cells. The extracellular concentration of dTtaPS and each PNA oligomer (7-9) is 2.0 μM and 1.0 μM, respectively.
Materials
HEK293 cells (ATCC)
HeLa cells (ATCC)
HeLa pLuc 705 cells (kindly donated by Prof. Rudolph Juliano, UNC, Chapel Hill)
MCF7 cells (ATCC)
SK-N-SH cells (ATCC)
Dulbecco's Minimum Essential Medium (DMEM) supplemented with 10% heat-inactivated
Fetal Bovine Serum (10% FBS-DMEM, Life Technologies)
20% FBS-DMEM (Life Technologies)
Dulbecco's Phosphate Buffered Saline (pH 7.4) supplemented with 2% Fetal Bovine Serum
(Life Technologies)
MEM medium (MediaTech)
Phenol red-free DMEM medium (MediaTech)
0.4% Trypan Blue (MediaTech)
Monensin (Sigma)
l-glutamine (Life Technologies)
Sodium pyruvate (Life Technologies)
Penicillin (Life Technologies)
Streptomycin (Life Technologies)
Hygromycin (Life Technologies)
Trypsin (MediaTech)
Pipettor (Corning)
T-75 flask (Corning)
Pipette (2 mL, 5 mL and 10 mL; Corning)
1.5-mL microcentrifuge tubes
FACS tubes (Falcon)
Wet-ice bath
Cell counter (Nexcelom)
Flat-bottom tissue culture 96-well plates (BD-Falcon)
37 °C-Humidified incubator under 5% CO2 (Thermo Scientific)
FACScan™ flow cytometer (Becton Dickinson)
Culture cells
An arbitrary number of HEK293, HeLa, HeLa pLuc 705, MCF7 or SK-N-SH cells are allowed to grow at 37 °C in a T-75 flask containing an arbitrary volume of 10% FBS-DMEM until the cells are 70% confluent.
Detach the cells from T-75 flask using Trypsin and count the cells using a cell counter.
Plate cells
In a flat-bottom tissue culture 96-well plates, add 2 × 104 cells/well of HEK293, HeLa, HeLa pLuc 705, MCF7 or SK-N-SH using a pipettor and allow the cells to grow in 100 μL 10% FBS-DMEM for 24 hr at 37 °C.
Internalize the dTtaPS:PNA or PMO complexes in the cells
- Remove the culture medium from all wells using a pipettor. Pipet into each well 50 μL 20% FBS-DMEM followed by 50 μL 2× stock solution of dTtaPS:PNA oligomer 7, 8 or 9 or dTtaPS:PMO oligomer 16, 17 or 18 complexes. Incubate the cell cultures at 37 °C for 18 hr.The final concentration dTtaPS:PNA or dTtaPS:PMO complexes in each well should be 2 μM in dTtaPS and 1.0 μM in PNA or PMO oligomer.
- Remove the medium from each well by vacuum-assisted suction. Pipet into each well 50 μL 0.25% Trypsin.Complete cell detachment occurs within 10 min at 37 °C.
Pipet into each well 100 μL ice-cold Dulbecco's Phosphate Buffered Saline (pH 7.4) supplemented with 2% Fetal Bovine Serum.
Perform FACS analysis
- Pipet the cells of each well into individual FACS tubes. Pipet 50 μL 0.4% Trypan blue and 50 μL of 200 μM monensin; add the solutions to each FACS tube using a pipettor. Place the tubes in an ice-bath and then perform analysis of each tube by flow cytometry using a FACScan™ flow cytometer according to the manufacturer's recommendations.Cell analyses should be performed within 1 h in order to prevent production of misleading results caused by cell death. A total of 5000 events are counted; the results of the FACs analysis are reported as a percentage of the cells that have internalized the fluorescently-labelled oligomers. The extent of dTtaPS-mediated cellular uptake of PNA or PMO oligomers in each of the cell line investigated is presented in Fig. 4.69.5 and 4.69.6, respectively.
Figure 4.69.6.

Internalization of dTtaPS:PMO oligomer complexes in live cells. The extracellular concentration of dTtaPS and each PMO oligomer is 2.0 μM and 1.0 μM, respectively.
Basic Protocol 4
Luciferase Assay for Determining the Bioactivity of PNA and PMO Oligomers in HeLa pLUC 705 Cells
A luciferase assay is described to measure the production of luciferase activity induced by the dTtaPS-mediated internalization of PNA oligomers 10, 11, 12 and PMO oligomers 19, 20, 21 (Fig. 4.69.4) in live HeLa pLuc 705 cells.
Materials
HeLa pLuc 705 cells (kindly donated by Prof. Rudolph Juliano, UNC, Chapel Hill)
Fetal Bovine Serum (FBS, Life Technologies)
DMEM (Life Technologies)
Serum-free Medium (OptiMEM, Life Technologies)
2 × Stock solution of each of the dTtaPS:PNA oligomer 10, 11 or 12 complexes and each of the
dTtaPS:PMO oligomer 19, 20 or 21 complexes (Support Protocol 2)
2 ×Pierce luciferase cell lysis buffer (ThermoFisher)
Bright-Glow reagent (Promega)
Pierce Coomassie (Bradford) protein assay kit (ThermoFisher)
Pipettor (Corning)
1.5-mL Microcentrifuge tubes
Water bath
Mechanical shaker
Black 96-well plate (Corning)
White 96-well plate (Corning)
Luminescence microplate reader (Molecular Devices, FilterMax F3 Multi-Mode Microplate Reader)
Plate HeLa pLuc 705 cells and perform cellular internalization of PNA and PMO oligomers
In a black 96-well plate, allow 2 × 104 cells/well of HeLa pLuc 705 to grow in 100 μL 10% FBS-DMEM at 37 °C for 18 hr.
- Remove the culture medium of each well by vacuum-assisted suction. Pipet 50 μL 20% FBS-OptiMEM in each well.Experiments are performed in triplicates.
- Using a pipettor, add 50 μL 2× stock solution of PNA oligomer 10, 11 or 12:dTtaPS complexes in OptiMEM to the cells of each well. Incubate the plate at 37 °C for 4 hr.PNA oligomers 11 and 12 are negative control sequences (Fig.4.69.4)
- Repeat step 3 using the 2× stock solution of PMO oligomer 19, 20 or 21:dTtaPS complexes.The extracellular concentration of each PNA or PMO oligomer and dTtaPS of steps 3 or 4 is 1.0 μM and 2.0 μM, respectively.PMO oligomers 20 and 21 are negative control sequences (Fig.4.69.4)
Remove the culture medium from each well using a pipettor.
Lyse the cells and perform the luciferase assay
Pipet 50 μL of Pierce luciferase cell lysis buffer to the cells of each well. Using a mechanical shaker, agitate the 96-well plate at 5 °C for 10 min.
- Transfer 30 μL of cell lysate from each well to individual wells of a white 96-well plate using a pipettor. Pipet 20 μL of Bright-Glow reagent in each well. Measure immediately luciferase activity using a luminescence microplate reader.For each well, luminescence is integrated over a period of 1 sec and recorded as relative light units (RLU). Luminescence measurements are reported based on the amount of total protein present in the test sample.Protein concentrations are measured from 5 μL of the cell lysates obtained in step 1 using the Pierce Coomassie (Bradford) protein assay kit as per the manufacturer's instructions.Bright Glow reagent that has been stored frozen at -20 °C or -80 °C should be used to ensure consistent luminescence measurements.The luciferase assay results supporting the bioactivity of dTtaPS:PNA and dTtaPS:PMO oligomer complexes in HeLa pLuc 705 cells are shown in Fig. 4.69.7 and Fig. 4.69.8, respectively.
Figure 4.69.7.

Luciferase activity resulting from the dTtaPS-assisted delivery of PNA oligomers 10, 11 and 12 in HeLa pLuc 705 cells. The concentration of dTtaPS is 2.0 μM in all experiments. Error bars represent the mean ± SD of three independent experiments. Abbreviation: RLU, relative light unit.
Figure 4.69.8.

Luciferase activity resulting from the dTtaPS-assisted delivery of PMO oligomers 19, 20 and 21 in HeLa pLuc 705 cells. The concentration of dTtaPS is 2.0 μM in all experiments. Error bars represent the mean ± SD of three independent experiments. Abbreviations: RLU, relative light unit.
Alternate Protocol 1
Luciferase Pre-mRNA Splice Correction Assay for Determining the Bioactivity of PNA and PMO Oligomers in HeLa pLUC 705 Cells
This protocol provides the details of an orthogonal assay for measuring the bioactivity of PNA oligomer 10 and PMO oligomer 19 upon dTtaPS-mediated delivery in HeLa pLuc 705 cells. The assay is based on an agarose gel analysis of amplified RT-PCR products obtained from incorrectly (268 bp) and correctly (142 bp) spliced luciferase pre-mRNAs.
Materials
HeLa pLuc 705 cells
Fetal Bovine Serum (FBS, Life Technologies)
DMEM (Life Technologies)
Serum-free Medium (Life Technologies)
1× Phosphate buffered saline, pH 7.4 (PBS, MediaTech)
2 × Stock solutions of dTtaPS:PNA oligomer 10 complexes and dTtaPS:PMO oligomer 19 complexes (Support Protocol 2)
HeLa pLuc 705 cell lysates (Basic Protocol 4)
TRIzol (Life Technologies)
20-bp molecular ruler (Bio-Rad)
10× TBE buffer (pH 8.3, Bio-Rad)
5 × Nucleic acid sample buffer (Bio-Rad)
High capacity cDNA Reverse Transcription Kit (Applied Biosystems; Cat# 4368813)
DEPC-treated water (Invitrogen)
0.2 mL PCR 8-strip tubes (Eppendorf)
2 × SYBR green solution (Applied Biosystems)
Forward and reverse primers 5′-TTGATATGTGGATTTCGAGTCGTC and
5′-TGTCAATCAGAGTGCTTTTGGCG, (Integrated DNA Technologies)
PCR Thermocycler (Applied Biosystems® GeneAmp® PCR System 9700)
1.5-mL Microcentrifuge tubes
3% Agarose gels, stained with ethidium bromide (Bio-Rad).
DNA Sub Cell electrophoresis horizontal gel chamber (Bio-Rad)
Power supply (Pharmacia)
GE ImageQuant LAS 4000 scanner (GE Healthcare)
Prepare HeLa pLuc 705 cell lysates
- Repeat steps 1 through 5 and step 1 under the section “Lyse the cells and perform the luciferase assay” of Basic Protocol 4.Step 3 of Basic Protocol 4 is modified as follows: Using a pipettor, add 50 μL of 2× stock solution of PNA oligomer 10:dTtaPS complexes in OptiMEM to the cells of each well. Incubate the plate at 37 °C for 4 hr.Step 4 of Basic Protocol 4 is modified as follows: Repeat step 3 using the 2× stock solution of PMO oligomer 19:dTtaPS complexes.
Prepare Total RNA
- To each well of the 96-well plate add 100μL of TRIzol using a pipettor and isolate total RNA as per the manufacturer's recommendations.Caution: Exposure to TRIzol can be a serious health hazard as it can lead to serious chemical burns and permanent scarring. The use of a lab coat, gloves and plastic apron is recommended when handling this reagent.
Prepare single-stranded cDNA products
Place 1 μg of RNA in a PCR 8 strip tube using a pipettor. Pipet 10 μL of high capacity cDNA reverse transcription master mix into the tube and proceed as per the manufacturer's recommendations.
Prepare double-stranded cDNA products
Add 12.5 μL 2× SYBR® Green PCR Master Mix, 2.5 μL of 10 μM forward primer in PBS, 2.5 μL 10 μM reverse primer in PBS, 5 μL of the cDNA produced from step 3 and 2.5 μL DEPC-treated water in a PCR tube using a pipettor.
Amplify the cDNA products (30 cycles) using a PCR thermocycler.
Analyze the cDNA products by electrophoresis on agarose gels
Mix 8 μL of the amplified cDNA products with 2 μL 5 × nucleic acid sample buffer using a pipettor.
- Pipet the cDNA solution obtained from step 6 into a well of a 3% agarose gel placed in a horizontal electrophoresis chamber filled with 1× TBE buffer (pH 8.3). Electrophorese at 75 V until the bromophenol blue marker travels the full length of the gel.The electropherogram presented in Fig. 4.69.9 demonstrates the correct splicing of lucifererase pre-mRNA as a result of the dTtaPS-mediated delivery of PNA oligomer 10 and PMO oligomer 19 in HeLa pLuc 705 cells.
Figure 4.69.9.
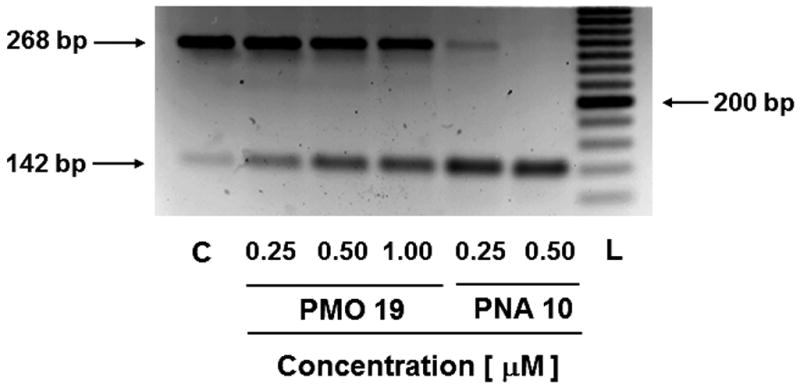
Agarose gel analysis of amplified RT-PCR products obtained from incorrectly (268 bp) and correctly (142 bp) spliced luciferase pre-mRNA induced by the dTtaPS-mediated delivery of PMO oligomer 19 and PNA oligomer 10 into HeLa pLuc 705 cells. The extracellular concentration of dTtaPS is kept at 2.0 μM in all experiments. Abbreviations: C, untreated cell control; L, 20 bp molecular ruler.
Commentary
Background Information
Most human genes undergo alternative splicing events, which is a highly regulated machinery requiring the sequence-specific binding of several proteins to nuclear pre-mRNAs (Pan et al., 2008). Steric interference imparted by RNase H-incompetent oligonucleotide analogues, complementary to specific pre-mRNA splice sites, has been shown to be efficient at redirecting the splicing machinery during assembly of mature mRNAs (Aartsma-Rus and Van Ommen, 2007; Järver et al., 2014). Indeed, skipping the mutated exons in dystrophin pre-mRNA as an approach to the clinical treatment of Duchenne muscular dystrophy underscores the biomedical significance of alternative splicing events (Lu et al., 2005; Harding et al., 2007; Moulton and Moulton, 2010). Negatively charged 2′-O-methyl RNA phosphorothioate sequences (Mann et al., 2002; Lu et al., 2003) and uncharged peptide nucleic acid (PNA) (Shiraishi et al., 2005; Yin et al., 2008) or phosphorodiamidate morpholino (PMO) (Moulton et al., 2003, 2004; Abes et al., 2006; Fletcher et al., 2006, Cirak et al., 2011) oligomers are nuclease-resistant and RNase-H incompetent oligonucleotide analogues that have been found potent at inducing splice redirecting events. While cationic lipids are efficient carriers for in vitro cellular transfection of negatively charged DNA/RNA sequences and their analogues, these carriers could not be successfully used for cellular internalization of uncharged PNA or PMO oligomers (Shiraishi and Nielsen, 2011). The conjugation of cationic cell-penetrating peptides (CPP) to PNAs or PMOs has improved cellular uptake of those oligomers and has led to pre-mRNA splicing correction activities in mammalian cells and animal models (Zatsepin et al., 2005; Bendifallah et al., 2006; Abes et al., 2007; O'Donovan et al., 2015). Interestingly, the use of PNA-CPP conjugates leads to internalization of PNA oligomers in HeLa pLuc 705 cells by virtue of sequence complementarity (Shiraishi and Nielsen, 2011). An inherent limitation of this approach to cellular internalization is the requirement for a PNA-CPP conjugate to be complementary to each PNA targeting a specific mRNA sequence.
During the course of our studies on the cellular uptake of an uncharged thermosensitive DNA oligonucleotide prodrug (15-mer) in mammalian cells, we had discovered that replacement of four neutral thiophosphate triester functions with four positively charged ones had greatly improved uptake of the modified DNA prodrug in Vero, GC-2 and HeLa cells (Jain et al., 2013). We have then hypothesized that extending PNA or PMO oligomers with a short PNA-polyA or PMO-polyA tail could provide an affinity recognition site for a positively charged, trans-acting polythymidylic thiophosphate triester element (dTtaPS) through, perhaps, sequence complementarity. In principle, the affinity recognition of polyA-tailed PNA or PMO oligomers by dTtaPS could facilitate the uptake of these oligomers in live cells while averting the shortcomings inherent to cationic CPP conjugates. With the intent of validating our hypothesis, the synthesis of dTtaPS, a polythymidylic acid (8-mer) with three N,N-dimethylaminopropyl and four octyl thiophosphate triester functions (Fig. 4.69.3) has been performed. At physiological pH, the N,N-dimethylaminopropyl functions are positively charged while the hydrophobic octyl groups are necessary to impart amphiphilicity, which has been reported (Zhi et al., 2010; Maslov et al., 2012) to facilitate in vitro cellular delivery of nucleic acids.
The solid-phase synthesis of dTtaPS begins with the preparation of the phosphordiamidites 2 and 3, which are required for the preparation of deoxyribonuleoside phosphoramidites 4 and 5 (Fig. 4.69.1). Thus, the reaction of equimolar amounts of commercial bis(N,N-diisopropylamino)chlorophosphine with either 3-(N,N-dimethylamino)propan-1-ol or 1-octanol in the presence of excess triethylamine in anhydrous benzene, provides the phosphordiamidite 2 or 3 in yields exceeding 85% after purification on silica gel (Jain et al., 2015).
When the 5′-O-protected deoxythymidine 1, phosphordiamidite 2 or 3 and 1H-tetrazole are mixed together in anhydrous MeCN the deoxyribonucleoside phosphoramidites 4 or 5 are formed and isolated as white powders in yields of 75-85% after purification on silica gel and lyophilization from dry benzene. The identity of phosphoramidites 4 and 5 has been confirmed by 31P-NMR spectroscopy and high-resolution mass spectrometry (Basic Protocol 1).
The synthesis of the polythymidylic thiophosphate triester 6 (dTtaPS, Fig. 4.69.3) on an appropriately functionalized controlled-pore glass (CPG) support is easily achieved using the deoxyribonucleoside phosphoramidites 4 and 5 as 0.1 M solutions in anhydrous acetonitrile. With the objective of optimizing the stepwise coupling efficiencies of 1H-tetrazole-activated phosphoramidites, the coupling time of 4 and 5 has been extended to 10 min. This coupling time extension has led to stepwise coupling efficiencies in the range of 98-99%, which have been measured using the 4,4′-dimethoxytrityl cation assay (Ellington and Pollard, 2000). The purity of diastereomeric dTtaPS has been assessed by C-4 RP-HPLC analysis after release from the CPG support. As shown in Fig. 4.69.10 the C-4 RP-HPLC chromatogram of dTtaPS shows a broad product peak accounting for 87% of all peak areas. The broad retention time (9.5-16.5 min) and shape of the product peak are consistent with the diastereomeric and amphiphilic nature of the nucleic acid sequence. The purity of dTtaPS is nonetheless deemed adequate for its intended purpose.
Fig. 4.69.10.
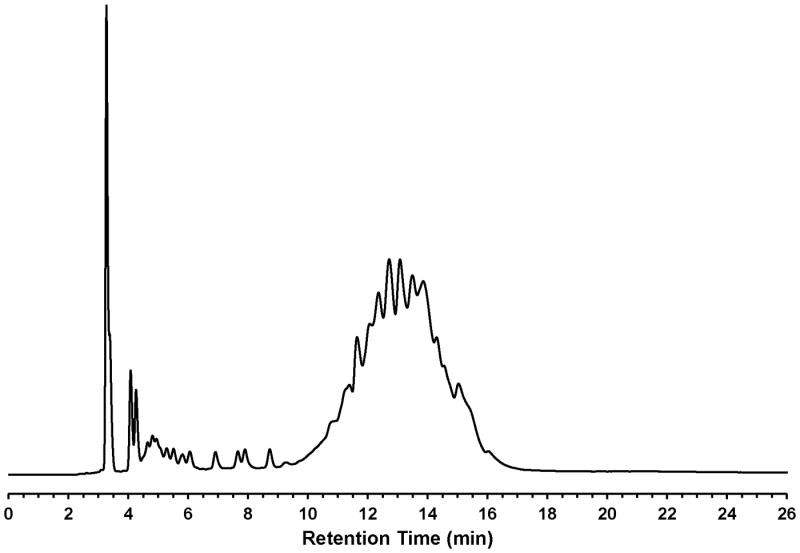
RP-HPLC profile of unpurified of dTtaPS (6). RP-HPLC analysis is performed employing a 5-μm Phenomenex Jupiter® C-4 HPLC Column (25 cm × 4.6 mm) under the following chromatographic conditions: starting from 20 mM ammonium acetate in 4:6 (v/v) water/acetonitrile, a linear gradient of acetonitrile is pumped at a flow rate of 1 mL/min to 20 mM ammonium acetate in 2:8 (v/v) water/acetonitrile over a period of 15 min and is held, isocratically for 5 min.
With the aim of demonstrating that the extension of PNA or PMO sequences with a short PNA-polyA or PMO-polyA stretch is necessary and sufficient for dTtaPS-mediated internalization and bioactivity of PNA oligomers in live mammalian cells, the dTtaPS-mediated cellular uptake of commercial fluorescently-labelled PNA and PMO oligomers (Fig. 4.69.4) in HEK 293, HeLa, HeLa pLuc 705, MCF7 and SK-N-SH live cells has been evaluated by FACS analysis. Figs. 4.69.5 and 4.69.6 show that polyA-tailed PNA and PMO oligomers 7, 8, 16 and 17 are efficiently internalized in the presence of dTtaPS in all cell lines. In contrast to these findings, both PNA and PMO oligomers 9 and 18 (Fig. 4.69.4) lacking the polyA stretch are not substantially internalized by dTtaPS in any of the cell lines under similar conditions (Figs. 4.69.5 and 4.69.6), thereby suggesting that PNA and PMO oligomers require a polyA tail for dTtaPS-assisted cellular internalization.
The efficiency of dTtaPS in mediating the cellular uptake of polyA-tailed PNA and PMO oligomers has been further assessed through a splice correction assay measuring restoration of luciferase production in HeLa pLuc 705 cells. Thus, dTtaPS-mediated internalization of the polyA-tailed PNA and PMO positive control sequences 10 and 19 (Fig. 4.69.4) in HeLa pLuc 705 cells has been shown to efficiently restore the production of luciferase (Figs. 4.69.7 and Fig. 4.69.8). As expected, the polyA-tailed PNA and PMO negative control sequences 11 and 20 (Fig. 4.69.4) have failed to restore significant luciferase activity production under similar conditions (Figs. 4.69.7 and Fig. 4.69.8). Both PNA and PMO positive control sequences 12 and 21 (Fig. 4.69.4) lacking the polyA stretch have also failed to produce luciferase activity under near identical conditions (Figs. 4.69.7 and Fig. 4.69.8). Although the results obtained from the luciferase assays of dTtaPS-assisted delivery of PNA and PMO oligomers 10 and 19 provide evidence that the uncharged PNA and PMO sequences have reached the nuclei of HeLa pLuc 705 cells and induced splice correction of the luciferase pre-mRNA, those results have not provided information on the extent splice correction had occurred. Reverse transcription of the aberrantly and correctly spliced luciferase pre-mRNAs and PCR amplification of the corresponding cDNAs have revealed, after separation of the PCR products by agarose electrophoresis, the presence of the 142 bp cDNA fragment corresponding to the correctly spliced luciferase pre-mRNA along with the 268 bp cDNA fragment corresponding to the aberrantly spliced pre-mRNA (Fig. 4.69.9). This assessment is supported by the basal expression of luciferase in the untreated Hela pLuc 705 cell control experiment (Bendifallah et al., 2006), which shows the presence of both cDNA fragments. The dTtaPS-mediated delivery of the PNA oligomer 10 leads to a high level of splicing correction (∼90%) at an oligomer concentration of 250 nM, whereas the delivery of PMO oligomer 19 by dTtaPS induces splicing redirection to the extent of ∼50% at an oligomer concentration of 1.0 μM (Fig. 4.69.9). These results correlate quite well with luciferase activity measurements presented in Figs. 4.69.7 and Fig. 4.69.8.
The recognition specificity of dTtaPS for the polyA tail of PNA oligomer 10 has been convincingly confirmed by replacement of the polyA tail of 10 with a polyT or a polyC tail (Jain et al., 2015). Fig. 4.69.11 clearly shows that dTtaPS fails to deliver PNA oligomers 13 and 14 (Fig. 4.69.4) in HeLa pLuc 705 cells based on the lack of luciferase activity production. However, when the polyA tail of PNA oligomer 10 is relocated from its 3′-terminus to its 5′-terminus, the dTtaPS-assisted internalization of PNA oligomer 15 (Fig. 4.69.4) in HeLa pLuc 705 cells results in full restoration of luciferase activity production (Fig. 4.69.11).
Figure 4.69.11.

Recognition of the polyA-stretch of PNA oligomers 10 and 15 by dTtaPS is specific, necessary and sufficient for internalization of these oligomers in HeLa pLuc 705 cells and restoration of luciferase activity.
The results of all experiments presented above strongly suggest that recognition of the polyA tail of PNA and PMO oligomers 10 and 19 by dTtaPS is specific and appears to occur through weak Watson-Crick base-pairing interactions given that thermal denaturation of dTtaPS:10 complexes in PBS buffer (pH 7.4) does not produce a typical sigmoidal denaturation profile from which a melting temperature could be determined (Jain et al., 2015). The formation of weak Watson-Crick base-pairing interactions between dTtaPS and the poly A-tailed PNA or PMO oligomer 10 or 19 has been further substantiated through experiments conducted in the presence of urea. As a denaturant, urea is expected to interfere with the ability of dTtaPS to recognize the polyA tail of 10 or 19 and inhibit the cellular internalization of these sequences, thereby leading to poorer production of luciferase activity. The results of those experiments, performed in the presence of urea at an extracellular concentration of 67 mM, indicate that the dTtaPS-mediated internalization of PNA sequence 10 or PMO sequence 19 is decreased leading to a reduction of luciferase activity production by more than 80% or 65%, respectively, relative to that of a control experiment performed in the absence of urea (Jain et al., 2015). These findings strongly support our contention that the recognition of the polyA tail of PNA or PMO oligomers proceeds through weak Watson-Crick base-pairing interactions.
Although dTtaPS has been comparably efficient at internalizing the fluoresceinated polyA-tailed PNA and PMO oligomers 7 and 16 in HeLa pLuc 705 cells in terms of percentage of transfected cells (see Figs. 4.69.5 and 4.69.6), dTtaPS did not deliver comparable amounts of 7 and 16 to the transfected cells. Indeed, FACS analysis of the dTtaPS-mediated uptake of PNA and PMO oligomers 7 and 16, when performed in the mean fluorescence intensity mode, reveals that the internalization of PNA oligomer 7 is ∼10-fold greater than that of the PMO oligomer 16 (Jain et al., 2015). These results are in agreement with those obtained from the dTtaPS-assisted internalization of PMO oligomer 19 in HeLa pLuc 705 cells (Fig. 4.69.8), indicating that the restoration of luciferase activity is about 5- to 8-fold less than that obtained with the PNA oligomer 10 under conditions of similar oligomer concentrations (Fig. 4.69.7).
A number of experiments have been carried out to provide insights into the mechanism by which the dTtaPS-mediated internalization of PNA oligomer 10 or PMO oligomer 21 in HeLa pLuc 705 cells is occurring. When the dTtaPS-assisted internalization of PNA oligomer 10 is performed at either 37°C or at 4°C, production of luciferase activity is reduced by at least 50% at 4°C (Jain et al., 2015). The outcome of this experiment suggests that the dTtaPS-assisted internalization of PNA oligomer 10 in HeLa pLuc 705 cells proceeds through an energy-dependent process consistent with an endocytic uptake mechanism (Khalil et al., 2006). Several experiments have also been performed to identify the most probable endocytic pathway leading to the dTtaPS-mediated uptake of PNA oligomer 10 and PMO oligomer 19 in HeLa pLuc 705 cells. Well-known endocytic pathway inhibitors including chlorpromazine for inhibition of clathrin-coated pits-mediated endocytosis, nystatin for inhibition of caveolae-mediated endocytosis and 5-(N-ethyl-N-isopropyl)amiloride (EIPA) for inhibition of macropinocytosis have been employed for this purpose (Khalil et al., 2006; Ivanova et al., 2008).
Nystatin does not inhibit luciferase expression, whereas chlorpromazine and EIPA inhibit restoration of luciferase activity to the extent of ∼35% and ∼95%, respectively. Although, the clathrin-coated pits-mediated endocytosis pathway is used to some extent for dTtaPS-assisted internalization of 10 and 21 in HeLa pLuc 705 cells, macropinocytosis clearly appears to be the prevailing endocytic pathway used for this internalization process (Jain et al., 2015).
The cytotoxicity of PNA oligomer 10:dTtaPS and PMO oligomer 19:dTtaPS complexes in HeLa pLuc 705 cells has been evaluated over a period of 18 hr using a commercial cell-counting kit. Increasing the concentration of 10 or 19 from 1.0 μM to 2.5 μM while keeping the concentration of dTtaPS at 2.0 μM does not induce significant cytotoxicity relative to that of the medium or in the absence of dTtaPS (Jain et al., 2015). When taken all together the data provide evidence that amphipathic phosphorothioate DNA elements may represent a unique class of cellular transporters for efficient delivery of uncharged nucleic acid sequences in live mammalian cells.
Critical Parameters and Troubleshooting
All glassware is flame-dried and allowed to cool in a desiccator over phosphorus pentoxide prior to use. Dimethyl sulfoxide and dichloromethane are distilled over calcium hydride and stored in amber glass bottle over activated 4 Å (8-12 mesh) molecular sieves.
The deoxyribonucleoside phosphoramidite 4 and 5 (Basic Protocol 1) must also be prepared under strictly anhydrous conditions and must not be exposed to any acid, which would render these compounds prone to hydrolysis. With the intent of preventing this undesirable outcome, readers are referred to the Critical Parameters and Troubleshooting sections of UNITS 2.7 & 3.17, which address critical issues associated with preparation and use of nucleoside phosphoramidites. The precipitation of 4 and 5 from cold hexane after silica-gel purification facilitates the removal of H-phosphonate impurities that often co-elute with the desired phosphoramidite monomers. Also removed during the course of the precipitation step is residual triethylamine, which if not removed, can neutralize 1H-tetrazole and reduce the coupling efficiency of phosphoramidites during automated solid-phase DNA synthesis. The presence of moisture can also compromise the coupling efficiency of phosphoramidites and should be taken into consideration; this concern has also been addressed in UNIT 2.7.
When preparing the 2 × stock solution of PNA or PMO oligomer:dTtaPS complexes it is critically important to use freshly made dTtaPS solution from solid dTtaPS stored at -20 °C. It is also imperative to add the dTtaPS solution to the PNA or PMO oligomer solution, as stated in step 2 of Support Protocol 2. The use of dTtaPS:PNA or PMO complexes immediately after the 15-min storage at 4°C is strongly recommended for optimal cellular uptake experiments.
In order to ensure consistent measurements of luciferase activity production, it is critical to employ a Bright Glow reagent (Basic Protocol 4, step 7) that has been stored frozen at -20 °C or -80 °C and measure luminescence immediately after addition of the reagent to cell lysates.
Anticipated Results
The synthesis of phosphordiamidites is straightforward and consistently provides post-purification yields exceeding 80%. Similarly, the protocol for the preparation of dexyribonucleoside phosphoramidites that are required for solid-phase synthesis of dTtaPS is uncomplicated and proceeds in high yields (> 90%) based on 31P NMR spectroscopy. However, the isolated yield of each deoxyribonucleoside phosphoramidite is variable and depends on the solubility of the phosphoramidite precipitate in cold hexane.
When every step of the solid-phase synthesis of dTtaPS is performed as delineated in Basic Protocol 2, an optimal yield of 6 is obtained, that is 90 ± 5%, based on spectrophotometric measurement of the DMTr cation released upon completion of the last coupling step of the synthesis cycle. Alternatively, the yield of dTtaPS is 87% when based on RP-HPLC integration of the peak area corresponding to 6 relative to the total area of all chromatographic peaks.
In order to obtain optimal cellular uptake of PNA or PMO oligomer:dTtaPS complexes in live mammalian cells, Support Protocol 2 must be carefully executed. Indeed, when preparing 2 × stock solutions of PNA or PMO oligomer:dTtaPS complexes, fresh dTtaPS solutions made from solid dTtaPS stored at -20 °C should be used. The addition of the dTtaPS solution to the PNA or PMO oligomer solution, as stated in step 2 of Support protocol 2 should be performed. Also recommended for optimal cellular uptake experiments is the use of dTtaPS:PNA or PMO complexes immediately after the mandatory15-min storage at 4 °C.
As mentioned above, careful execution of the steps delineated in Support Protocol 2 will ensure consistent cellular uptake results by FACS analysis or luciferase activity measurements, where appropriate.
Careful implementation of Alternate Protocol 1 and the use of 30 cycles of PCR amplification of the cDNA products should produce, as shown in Fig. 4.69.9, consistent luciferase pre-mRNA splicing correction results
Time Considerations
The preparation and purification of either phosphordiamidite 2 or 3 needed for the synthesis of deoxyribonucleoside phosphoramidites takes approximately 20 hr; lyophilization of silica gel-purified 2 or 3 is complete within 14 hr. The synthesis, purification and hexane precipitation of deoxyribonucleoside phosphoramidite 4 or 5 is achieved within 11 hr. The lyophilization of each phosphoramidite precipitate is accomplished within 14 hr.
The automated solid-phase synthesis of the amphipathic trans-acting phosphorothioate DNA element (dTtaPS, 6) including its release and elution from the solid support, followed by complete evaporation of the eluate, require 8 hr to complete.
Prior to performing cellular uptake experiments, formation of complexes between dTtaPS and PNA or PMO oligomers is achieved within 1 hr. Culture of HEK293, HeLa, HeLa pLuc 705, MCF7 or SK-N-SH cells to 70% confluency takes 3 days. Cell plating and performing the dTtaPS-assisted internalization of fluorescently labelled PNA or PMO oligomers in the selected cell lines can be done within 43 hr. Measurements of the number of cells that have internalized PNA or PMO oligomers by FACS take 2 hr. Luciferase activity production resulting from the dTtaPS-assisted internalization of PNA or PMO oligomers in HeLa pLuc 705 cells can be measured within 42 h. Evaluation of the bioactivity of internalized PNA and PMO oligomers in HeLa pLuc 705 cells is achieved through analysis of luciferase pre-mRNA splice correction. This analysis involves the preparation and extraction of total cellular RNA, reverse transcription of the RNA to cDNA products and PCR amplification of the cDNAs; these steps require 30 hr to complete. Qualitative measurements of the amplified cDNA products obtained from incorrectly and correctly spliced luciferase pre-mRNA are obtained, within 3 hr, via agarose gel electrophoresis.
Acknowledgments
This work was supported through FDA intramural funds.
Literature Cited
- Aartsma-Rus A, Van Ommen GJB. Antisense-mediated exon skipping: A versatile tool with therapeutic and research applications. RNA. 2007;13:1609–1624. doi: 10.1261/rna.653607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abes S, Moulton HM, Clair P, Prevot P, Youngblood DS, Wu RP, Iversen PL, Lebleu B. Vectorization of morpholino oligomers by the (R-Ahx-R)4 peptide allows efficient splicing correction in the absence of endosomolytic agents. J Control Release. 2006;116:304–313. doi: 10.1016/j.jconrel.2006.09.011. [DOI] [PubMed] [Google Scholar]
- Abes S, Turner JJ, Ivanova GD, Owen D, Williams D, Arzumanov A, Clair P, Gait MJ, Lebleu B. Efficient splicing correction by PNA conjugation to an R6-Penetratin delivery peptide. Nucleic Acids Res. 2007;35:4495–4502. doi: 10.1093/nar/gkm418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendifallah N, Rasmussen FW, Zachar V, Ebbesen P, Nielsen PE, Koppelhus U. Evaluation of cell-penetrating peptides (CPPs) as vehicles for intracellular delivery of antisense peptide nucleic acid (PNA) Bioconjugate Chem. 2006;17:750–758. doi: 10.1021/bc050283q. [DOI] [PubMed] [Google Scholar]
- Cirak S, Arechavala-Gomeza V, Guglieri M, Feng L, Torelli S, Anthony K, Abbs S, Garralda MA, Bourke J, Wells DJ, Dickson G, Wood MJA, Wilton SD, Straub V, Kole R, Shrewsbury SB, Sewry C, Morgan JE, Bushby K, Muntoni F. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet. 2011;378:595–605. doi: 10.1016/S0140-6736(11)60756-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellington A, Pollard JD., Jr . Introduction to the synthesis and purification of oligonucleotides. In: Beaucage SL, Bergstrom DE, Glick GD, Jones RA, editors. Current Protocols in Nucleic Acid Chemistry. I. John Wiley & Sons, Inc; Hoboken: 2000. pp. A.3C.1–A.3C.22. [DOI] [PubMed] [Google Scholar]
- Fletcher S, Honeyman K, Fall AM, Harding PL, Johnsen RD, Wilton SD. Dystrophin expression in the mdx mouse after localised and systemic administration of a morpholino antisense oligonucleotide. J Gene Med. 2006;8:207–216. doi: 10.1002/jgm.838. [DOI] [PubMed] [Google Scholar]
- Harding PL, Fall AM, Honeyman K, Fletcher S, Wilton SD. The influence of antisense oligonucleotide length on dystrophin exon skipping. Mol Ther. 2007;15:157–166. doi: 10.1038/sj.mt.6300006. [DOI] [PubMed] [Google Scholar]
- Ivanova GD, Arzumanov A, Abes R, Yin H, Wood MJA, Lebleu B, Gait MJ. Improved cell-penetrating peptide-PNA conjugates for splicing redirection in HeLa cells and exon skipping in mdx mouse muscle. Nucleic Acids Res. 2008;36:6418–6428. doi: 10.1093/nar/gkn671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer RP, Phillips LR, Egan W, Regan JB, Beaucage SL. The automated synthesis of sulfur-containing oligodeoxyribonucleotides using 3H-1,2-benzodithiol-3-one 1,1-dioxide as a sulfur-transfer reagent. J Org Chem. 1990;55:4693–4699. [Google Scholar]
- Jain HV, Takeda K, Tami C, Verthelyi D, Beaucage SL. Assessment of the cellular internalization of thermolytic phosphorothioate DNA oligonucleotide prodrugs. Bioorg Med Chem. 2013;21:6224–6232. doi: 10.1016/j.bmc.2013.04.071. [DOI] [PubMed] [Google Scholar]
- Jain HV, Verthelyi D, Beaucage SL. Amphipathic trans-acting phosphorothioate DNA elements mediate the delivery of uncharged nucleic acid sequences in mammalian cells. RSC Adv. 2015;5:65245–65254. [Google Scholar]
- Järver P, O'Donovan L, Gait MJ. A chemical view of oligonucleotides for exon skipping and related drug applications. Nucleic Acid Ther. 2014;24:37–47. doi: 10.1089/nat.2013.0454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil IA, Kogure K, Akita H, Harashima H. Uptake pathways and subsequent intracellular trafficking in nonviral gene delivery. Pharmacol Rev. 2006;58:32–45. doi: 10.1124/pr.58.1.8. [DOI] [PubMed] [Google Scholar]
- Lu QL, Mann CJ, Lou F, Bou-Gharios G, Morris GE, Xue SA, Fletcher S, Partridge TA, Wilton SD. Functional amounts of dystrophin produced by skipping the mutated exon in the mdx dystrophic mouse. Nat Med. 2003;9:1009–1014. doi: 10.1038/nm897. [DOI] [PubMed] [Google Scholar]
- Lu QL, Rabinowitz A, Chen YC, Yokota T, Yin H, Alter J, Jadoon A, Bou-Gharios G, Partridge T. Systemic delivery of antisense oligoribonucleotide restores dystrophin expression in body-wide skeletal muscles. Proc Natl Acad Sci USA. 2005;102:198–203. doi: 10.1073/pnas.0406700102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann CJ, Honeyman K, McClorey G, Fletcher S, Wilton SD. Improved antisense oligonucleotide induced exon skipping in the mdx mouse model of muscular dystrophy. J Gene Med. 2002;4:644–654. doi: 10.1002/jgm.295. [DOI] [PubMed] [Google Scholar]
- Maslov MA, Kabilova TO, Petukhov IA, Morozova NG, Serebrennikova GA, Vlassov VV, Zenkova MA. Novel cholesterol spermine conjugates provide efficient cellular delivery of plasmid DNA and small interfering RNA. J Control Release. 2012;160:182–193. doi: 10.1016/j.jconrel.2011.11.023. [DOI] [PubMed] [Google Scholar]
- Moulton HM, Hase MC, Smith KM, Iversen PL. HIV Tat peptide enhances cellular delivery of antisense morpholino oligomers. Antisense Nucleic Acid Drug Dev. 2003;13:31–43. doi: 10.1089/108729003764097322. [DOI] [PubMed] [Google Scholar]
- Moulton HM, Nelson MH, Hatlevig SA, Reddy MT, Iversen PL. Cellular uptake of antisense morpholino oligomers conjugated to arginine-rich peptides. Bioconjugate Chem. 2004;15:290–299. doi: 10.1021/bc034221g. [DOI] [PubMed] [Google Scholar]
- Moulton JD, Jiang S. Gene knockdowns in adult animals: PPMOs and Vivo-Morpholinos. Molecules. 2009;14:1304–1323. doi: 10.3390/molecules14031304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulton HM, Moulton JD. Morpholinos and their peptide conjugates: Therapeutic promise and challenge for Duchenne muscular dystrophy. Biochim Biophys Acta. 2010;1798:2296–2303. doi: 10.1016/j.bbamem.2010.02.012. [DOI] [PubMed] [Google Scholar]
- O'Donovan L, Okamoto I, Arzumanov AA, Williams DL, Deuss P, Gait MJ. Parallel synthesis of cell-penetrating peptide conjugates of PMO toward exon skipping enhancement in Duchenne Muscular Dystrophy. Nucleic Acid Ther. 2015;25:1–10. doi: 10.1089/nat.2014.0512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Q, Shai1 O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet. 2008;40:1413–1415. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- Prater CE, Miller PS. 3′-Methylphosphonate-modified oligo-2′-O-methylribonucleotides and their Tat peptide conjugates: Uptake and stability in mouse fibroblasts in culture. Bioconjugate Chem. 2004;15:498–507. doi: 10.1021/bc049977+. [DOI] [PubMed] [Google Scholar]
- Shiraishi T, Nielsen PE. Peptide nucleic acid (PNA) cell penetrating peptide (CPP) conjugates as carriers for cellular delivery of antisense oligomers. Artificial DNA: PNA & XNA. 2011;2:90–99. doi: 10.4161/adna.18739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tung CH, Stein S. Preparation and applications of peptide−oligonucleotide conjugates. Bioconjugate Chem. 2000;11:605–618. doi: 10.1021/bc0000334. [DOI] [PubMed] [Google Scholar]
- Yin H, Lu Q, Wood M. Effective exon skipping and restoration of dystrophin expression by peptide nucleic acid antisense oligonucleotides in mdx mice. Mol Ther. 2008;16:38–45. doi: 10.1038/sj.mt.6300329. [DOI] [PubMed] [Google Scholar]
- Zatsepin TS, Turner JJ, Oretskaya TS, Gait MJ. Conjugates of oligonucleotides and analogues with cell penetrating peptides as gene silencing agents. Curr Pharm Des. 2005;11:3639–3654. doi: 10.2174/138161205774580769. [DOI] [PubMed] [Google Scholar]
- Zhi D, Zhang S, Wang B, Zhao Y, Yang B, Yu S. Transfection efficiency of cationic lipids with different hydrophobic domains in gene delivery. Bioconjugate Chem. 2010;21:563–577. doi: 10.1021/bc900393r. [DOI] [PubMed] [Google Scholar]