

Abstract
The aim of the present work was to prepare and evaluate sublingual fast dissolving films containing metoprolol tartrate-loaded niosomes. Niosomes were utilized to allow for prolonged release of the drug, whereas the films were used to increase the drug’s bioavailability via the sublingual route. Niosomes were prepared using span 60 and cholesterol at different drug to surfactant ratios. The niosomes were characterized for size, zeta-potential, and entrapment efficiency. The selected niosomal formulation was incorporated into polymeric films using hydroxypropyl methyl cellulose E15 and methyl cellulose as film-forming polymers and Avicel as superdisintegrant. The physical characteristics (appearance, texture, pH, uniformity of weight and thickness, disintegration time, and palatability) of the prepared films were studied, in addition to evaluating the in vitro drug release, stability, and in vivo pharmacokinetics in rabbits. The release of the drug from the medicated film was fast (99.9% of the drug was released within 30 minutes), while the drug loaded into the niosomes, either incorporated into the film or not, showed only 22.85% drug release within the same time. The selected sublingual film showed significantly higher rate of drug absorption and higher drug plasma levels compared with that of commercial oral tablet. The plasma levels remained detectable for 24 hours following sublingual administration, compared with only 12 hours after administration of the oral tablet. In addition, the absolute bioavailability of the drug (ie, relative to intravenous administration) following sublingual administration was found to be significantly higher (91.06%±13.28%), as compared with that after oral tablet administration (39.37%±11.4%). These results indicate that the fast dissolving niosomal film could be a promising delivery system to enhance the bioavailability and prolong the therapeutic effect of metoprolol tartrate.
Keywords: anti-hypertensive, β1- antagonist, pharmacokinetics
Introduction
Fast dissolving films have recently attracted increasing interest in the pharmaceutical industry due to the improved patient compliance, accurate dosing, rapid onset of action, pleasant taste, as well as their convenient handling and administration.1–3 These films consist of thin oral strips formulated using hydrophilic polymers that rapidly disintegrate and dissolve when placed in the oral cavity to release the medication, which becomes available for oromucosal absorption, without chewing and intake of water.2,4,5 Hence, it offers a convenient way for patients who cannot be dosed orally like pediatric and geriatric patients6 and also for patients who are unable to swallow large quantity of water, such as those suffering from dysphagia, repeated emesis, motion sickness, and mental disorders.7 However, drugs that require high doses cannot be incorporated into film strip due to its limited surface area.8 A loading of 62.5 mg of simethicone per thin strip was successfully achieved by Novartis Consumer Health’s Gas-X® (East Hanover, NJ, USA).7
For systemic drug delivery, fast dissolving films can be used via sublingual route1,9 due to the high vascularity and permeability of this region, which allows for rapid absorption and quick action of the incorporated drug.10 In addition, sublingual administration avoids first-pass hepatic metabolism.7,9,11,12 Hence, this route can be used to improve oral bioavailability of drugs that undergo extensive first-pass effect.13,14
Metoprolol tartarate (MT) is a synthetic, cardioselective β1-adrenoreceptor antagonist widely used in the treatment of essential hypertension and other cardiac disorders.15–18 After oral administration, MT is almost completely absorbed (95%)19,20 with peak plasma concentrations achieved after 2–3 hours.21 It undergoes extensive first-pass metabolism by the liver that results in low and variable oral bioavailability (40%–50%).19,20,22 The plasma half-life is approximately 3–4 hours,18,23 which needs frequent dosing to maintain the therapeutic blood levels of the drug for a long-term treatment. Several attempts have been attained to enhance its bioavailability by avoidance of first-pass hepatic metabolism such as intravenous,18 transdermal,24 intranasal,25 rectal,15 and buccoadhesive drug delivery systems.16 Other strategies with sustained drug release pattern have been developed to avoid the frequent dosing of MT, including the use of floating tablets,26 mucoadhesive floating beads,27,28 microspheres,29 niosomes,30 and proniosomes.31
Drug delivery systems using colloidal particulate carriers have significant advantages over conventional dosage forms as the particles can act as reservoir for the loaded drug.13 Niosomes are closed bilayer vesicles formed by self-assembly of nonionic surfactants in aqueous media.32 These structures are analogous to liposomes but have the ability to increase the stability of their entrapped drugs.33,34 Due to the flexibility of their structural characteristics (composition, fluidity, and size) and ease of storage and handling,35–37 these lipid vesicles can be tailored for delivery of a wide variety of drugs for drug targeting,38 controlled release,39 and permeation enhancement.40
In the present study, an attempt has been made to prepare and evaluate sublingual fast dissolving film containing MT-loaded niosomes. The sublingual route of administration is expected to enhance the drug bioavailability by avoidance of first-pass hepatic metabolism. In addition, rapid absorption of the drug-loaded niosomes may maintain therapeutic concentrations of the MT for prolonged time period and, thus, avoiding the frequent dosing of the drug.
Materials
MT was supplied by Sid Corporation for Pharmaceutical and Chemical Industry (Cairo, Egypt). Hydroxypropyl methyl cellulose E15 (HPMC) and methyl cellulose (MC) were purchased from Dow Chemical Co. (Midland, MI, USA). Microcrystalline cellulose (Avicel PH105) was purchased from Serva Feinbiochemica (Heidelberg, Germany). Polyethylene Glycol 400 was supplied from BDH Chemicals Ltd (Poole, UK). Cholesterol, span 60, saccharine, and menthol were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Other chemicals and reagents were of analytical grade.
Methods
Formulation of MT-loaded niosomes
Niosomes were prepared by the conventional thin film hydration method41,42 using span 60 as a nonionic surfactant and cholesterol as an enhancer of niosomal membrane rigidity. Four different drug to surfactant ratios were utilized (1:0.5, 1:1, 1:1.5, and 1:2). The drug and cholesterol ratio was similar in all formulations. Drug, nonionic surfactant, and cholesterol were weighed and dissolved in chloroform in a round bottom flask. The solvent was evaporated at a temperature of 60°C under reduced pressure using a rotary evaporator (Buchi 200; BÜCHI Labortechnik AG, Flawil, Switzerland) to form a thin film on the flask wall. The resulting film was hydrated with ultrapure water (deionized water) for 30 minutes at room temperature with gentle shaking. This film was hydrated with 10 mL of deionized water at 60°C. The resulting niosomal suspension was mixed by vortex mixing for 10 minutes and sonicated for 20 minutes at 25°C. The niosomal suspension was left overnight at 4°C and stored at refrigerator temperature (4°C–8°C) for further studies. The compositions of the niosomal formulations are provided in Table 1.
Table 1.
Composition and characterization of MT-loaded niosomal formulations
| Code | Ratio (drug:span 60:cholesterol) | Particle size (nm)* | PI* | Zeta potential (mV)* | EE (%)* |
|---|---|---|---|---|---|
| N1 | 1:0.5:1 | 1,800±67.21 | 0.721±0.48 | −41.8±4.11 | 25.44±9.7 |
| N2 | 1:1:1 | 1,668±114.2 | 0.752±0.42 | −49.6±5.21 | 34.22±7.5 |
| N3 | 1:1.5:1 | 1,254±94.2 | 0.761±0.29 | −56.7±4.95 | 39.34±5.3 |
| N4 | 1:2:1 | 883.7±31.47 | 0.731±0.34 | −64.1±0.95 | 52.64±6.1** |
Notes:
Mean ± SD (n=3).
Significant difference (P<0.05) compared to other EE% from other ratios.
Abbreviations: EE, entrapment efficiency; MT, metoprolol tartarate; PI, polydispersity index; SD, standard deviation.
Characterization of prepared niosomes
Determination of entrapment efficiency
Niosomes containing MT were separated from free drug by cooling centrifugation at 15,000 rpm for 60 minutes at 4°C. The niosomal pellets were suspended in methanol and centrifuged again.32,43 The integrity of vesicles was not affected by centrifugation as reported in literature.32,44 The washing procedure was repeated two times as reported previously.44 The supernatant was separated each time and assayed spectrophotometrically at 275 nm. The amount of entrapped drug was obtained by subtracting the amount of free drug from the total drug.45 The percent of entrapment efficiency (EE%) was then calculated according to Equation 1 (each result is the mean of three separate experiments):
| (1) |
Particle size and zeta potential
The mean particle size (nm) and polydispersity index of the prepared niosomes in both niosomal dispersion and niosomal film were measured by dynamic light scattering laser using a Zetasizer Nano ZS (Malvern Instruments, Worcestershire, UK) equipped with a 4 mW helium/neon laser (λ=633 nm) and thermoelectric temperature controller. The corresponding zeta potentials (mV) were determined by photon correlation spectroscopy using the same Zetasizer Nano instrument.
Preparation of fast dissolving niosomal films
Fast dissolving films were prepared by solvent casting technique.46 HPMC and MC were used as film-forming polymers. Polyethylene Glycol 400 was used as a plasticizer, saccharine as a sweetener, and menthol as flavoring agent and to give mouth refreshment feeling. Concentrations of plasticizer, sweetener, and flavoring agents were kept constant. Microcrystalline cellulose (Avicel) was used as superdisintegrant at two different concentrations. Specified weight of film-forming polymer was first dissolved in 20 mL of the casting solvent (warm distilled water), and sweetener and flavoring agent were dissolved in the polymeric solution. The calculated amount of superdisintegrant was incorporated into the polymeric solutions after levigation with the required volume of the plasticizer. For the preparation of medicated films (containing free drug), the required amount of MT was directly added and completely dissolved into the polymeric solution before the addition of superdisintegrant. For niosomal film, a specified volume of the selected niosomal dispersion (N4) (corresponding to the required MT dose) was incorporated and gently mixed with the selected polymeric solution. The final volume was adjusted to 25 mL with distilled water, and the beaker was covered with aluminum foil to prevent solvent evaporation. The casting solution was subjected to gentle stirring for 2 hours using magnetic stirrer (L32; Bibby, Staffordshire, UK).
The casting solution (25 mL) was transferred into a previously cleaned and dried Teflon-coated plate (area =28 cm2, each 4 cm2 contains 25 mg of drug). The solvent was allowed to evaporate for 72 hours, and the film was then removed from the Teflon plate and was allowed to dry in a desiccator at least 48 hours before evaluation. The patches were punched into 4 cm2 pieces containing 25 mg of MT, then wrapped in an aluminum foil (to maintain the integrity and elasticity of the films) and were finally stored in a dry place at ambient room temperature. The films were subjected to evaluation within 1 week of their preparation. Composition of various formulations is provided in Table 2.
Table 2.
Composition of different MT fast dissolving films
| Component (mg) | F1 | F2 | F3 | F4 | F5 | F6 |
|---|---|---|---|---|---|---|
| HPMC | 100 | 100 | 100 | 0 | 0 | 0 |
| MC | 0 | 0 | 0 | 200 | 200 | 200 |
| PEG400 | 150 | 150 | 150 | 150 | 150 | 150 |
| Saccharine | 50 | 50 | 50 | 50 | 50 | 50 |
| Menthol | 17 | 17 | 17 | 17 | 17 | 17 |
| MCC | 0 | 7 | 14 | 0 | 7 | 14 |
Note: Contents of each film were casted into PTFE-coated plate (area =28 cm2).
Abbreviations: HPMC, hydroxyl propyl methyl cellulose; MC, methyl cellulose; MCC, microcrystalline cellulose; MT, metoprolol tartarate; PEG, polyethylene glycol.
Evaluation of fast dissolving films
Physical and mechanical properties
The fast dissolving films were evaluated for physical appearance, surface texture, thickness, weight uniformity, folding endurance, surface pH, and drug content uniformity. The physical appearance was checked with visual inspection of films and texture by touch. Thickness was measured by micrometer screw gauge (Mitutoyo, Tokyo, Japan) at different points of each formulation, and the mean values were calculated. Weight variation was studied using electronic Analytical balance (AJ150; Mettler, Greifensee, Switzerland) by taking individual weights of ten randomly selected 4 cm2 patches for each formulation (prepared in different batches).
In vitro disintegration time was determined visually in a Petri dish containing 25 mL of phosphate buffer (pH 6.8) with swirling every 10 seconds. The disintegration time is the time recorded when the film starts to break or disintegrate.11 Folding endurance was determined by repeated folding of the film at the same place till the strip breaks.47 The number of times the film is folded without breaking was computed as the folding endurance value.
For the determination of surface pH, three films of each formulation were allowed to contact with 1 mL of distilled water for 1 hour at room temperature and measured by pH meter (Jenway, Staffordshire, UK). The surface pH was measured by bringing the electrode in contact with the surface of the film and allowing it to equilibrate for 1 minute.7 Finally, to check the uniformity of the drug content in the cast film, 4 cm2 patches were cut from different places in cast film, and each film was dissolved in 100 mL phosphate buffer (pH 6.8). The resulting solution was filtered, and further dilution was made with phosphate buffer and the absorbance was measured spectrophotometrically at 275 nm, and the percentage of the drug content was determined. The same procedure was repeated for at least three patches of each formulation, and the mean values and standard deviations were calculated.7
Fourier transform infrared spectroscopy
The Fourier transform infrared spectroscopy (FTIR) spectra of the selected niosomal film compared to its corresponding physical mixture and the individual solid components were recorded using FTIR spectrophotometer (IR-470; Shimadzu, Kyoto, Japan). Samples were mixed with potassium bromide (spectroscopic grade) and compressed into disks using hydraulic press before scanning from 4,000 to 600 cm−1.
Differential scanning calorimetry
Differential scanning calorimetry (DSC) scans were recorded for the selected niosomal film, its corresponding physical mixture and the individual solid components. The samples (3–5 mg) were hermetically sealed in aluminum pans and heated at a constant rate of 10°C/min, over a temperature range of 25°C–200°C. Thermograms of the samples were obtained using DSC (DSC-60; Shimadzu). Thermal analysis data were recorded using a TA 50I PC system with Shimadzu software programs. Indium standard was used to calibrate the DSC temperature and enthalpy scale. N2 was used as purging gas at rate of 40 mL/min.
In vitro drug release
The previously prepared film was removed from the plate, cut in area of 4 cm2, and weighed on an analytical balance. The release of MT from either the medicated niosomal dispersion or from the prepared films (medicated films or niosomal films) was performed in beakers each containing 100 mL of phosphate buffer at pH 6.8 (beaker method).48 The beakers were placed in a shaking water bath (Gesellschaft für Labortechnik mbH, Burgwedel, Germany) set at 37°C and 100 rpm. Samples were withdrawn (5 mL) at the determined time intervals and were centrifuged, and then the supernatant was filtered and assayed using a UV-visible spectrophotometer (Shimadzu Seisakusho, Ltd., Kyoto, Japan) at 275 nm. Samples were replaced by equal volumes of fresh buffer to maintain the same volume in the flasks. The experiment was conducted in triplicates. The amount of drug released at each time interval was calculated, and the cumulative amount of drug released was calculated as a function of time to construct the drug release profile graphs.49 The release data were kinetically analyzed by curve fitting method to different kinetic models of zero-order, first-order, Higuchi, and Korsemeyer–Peppas models.32,50
Stability study
For the prepared fast dissolving films, stability study was carried out at two different storage conditions, one was normal room conditions and the other was 40°C/75% relative humidity for 8 weeks. Each piece of the conventional medicated film formulation (F6) and the niosomal film (N4F6) were packed in butter paper followed by aluminum foil and plastic tape. After 8 weeks, the films were evaluated for the physical appearance, surface pH, and in vitro drug release.11 Regarding the prepared niosomal dispersion, stability study was carried out at 4°C, room temperature, and elevated temperature for a period of 8 weeks. The samples of selected niosomal dispersion formulation (N4) were sealed in a glass vial and stored at the selected temperature for 4 weeks. Samples from each batch were withdrawn at definite time intervals and evaluated for physical appearance, vesicle size, and zeta potential, as compared to the reconstituted niosomal dispersion after the niosomal film dissolution in phosphate buffer (pH 6.8).
Scanning electron microscope
The surface morphology of the selected fast dissolving niosomal film (N4F6) was observed and compared to that of the selected niosomal dispersion formulation (N4) using scanning electron microscope (JSM-5400 LV; Joel, Japan) operated at an acceleration voltage of 15 kV.
In situ disintegration time and palatability studies
The study followed the ethical guidelines of Assiut University and was approved by the Assiut University Medical Ethical Review Board. A taste panel consisting of 14 healthy male volunteers, nine males, and five females (25–45 years old) has tried the selected niosomal film formulation (N4F6). Prior to the study, the volunteers were briefed on the nature, purpose, duration, and risk of the study. After explaining the study, written informed consent was obtained from all volunteers included. The tested film was kept in mouth until disintegration. The volunteers were requested to record the disintegration time, ease of handling, and acceptance of the film and gave a score based on the parameters, namely taste, after-taste, mouth-feel refreshment, as presented in previously reported studies.7,51
In vivo pharmacokinetic study
The protocol of this research was reviewed and approved by the Institutional Animal Ethical Committee of Faculty of Pharmacy, Assiut University, and it adheres to the Guide for the Care and Use of Laboratory Animals, 8th Edition, National Academies Press, Washington, DC.
Animal dosing and sampling scheme
The plasma concentrations of MT were evaluated from healthy rabbits after the sublingual administration of the selected niosomal films (N4F6) compared to the commercial oral tablets, Betaloc® (100 mg MT). Fifteen healthy rabbits (1.5–2.25 kg) were used in the present study. The rabbits were fasted for 24 hours before the administration of the drug and were anesthetized with 0.1 mL thiopentane (0.5 mg/mL). Approximately 7 mg/kg of the drug, corresponding to a 100-mg human dose, was used. This equivalent dose for rabbits was calculated by the aid of surface area ratio, as the therapeutic dose of man was multiplied by a certain mathematical factor obtained from a special table for surface area ratios of some common laboratory species and man.52,53 The following equation was used, Dr = Dh (Wr/Wh),3/4 where Dr is the rabbit dose, Dh is the human dose, Wr is the rabbit weight, and Wh is the human weight.15,16 Rabbits were randomly divided into three groups each of five animals as follows: the first group received MT commercial oral tablets by gastric intubation, the second group was given the selected MT sublingual niosomal film (N4F6), and the third group was given intravenous MT in isotonic saline solutions. Control blood samples were taken from the rabbits immediately before administration of the drug. Multiple blood samples (1–2 mL) were collected in heparinized vacutainer tubes before administration and at 0.25, 0.5, 1, 2, 3, 4, 6, 8, 12, and 24 hours following drug administration. The plasma was then separated after centrifugation and stored frozen at −20°C until analysis.16,54
Analysis of plasma samples
MT plasma concentration was measured using a reported method prescribed in our previous publications15,16 using a sensitive high-performance liquid chromatography assay. The high-performance liquid chromatography system (Knauer K-500 pump, Knauer, K-2500 UV detector, C-R6A, chromatopac integrator; Shimadzu) was used with the reversed-phase mode. Analysis was performed on Aqua RP-C18 packed column (250×4.6 mm internal diameter, 5 mm particle diameters). The 50-mL aliquots were injected and eluted with a mobile phase containing 3.9 g ammonium acetate in 810 mL water, 2 mL triethylamine, 10 mL glacial acetic acid, 3 mL phosphoric acid, and 146 mL acetonitrile.55 The flow rate was set at 1 mL/min, and the eluent was monitored at 275 nm. A calibration curve of MT in the plasma was constructed using blank plasma spiked with standard MT solutions to obtain a concentration range of 0.3–30 mg/mL. The spiked plasma was then subjected to the same extraction procedure of the samples. Triplicate runs were made for each standard sample.
Pharmacokinetics analysis
After measuring MT concentrations in the plasma, MT pharmacokinetics was assessed by fitting the plasma concentration–time data to the suitable model using Win Nonlin standard version 1.5 (Science Consulting, Apex, NC, USA) software. Absorption rate constant (Ka), absorption half-life (t½a), elimination rate constant (Kel), elimination half-life (t½), and the area under the plasma-MT concentration versus time curve (AUC) were calculated. Also, the maximum concentration (Cmax) and the time to reach the maximum concentration (Tmax) were reported. The absolute bioavailability of the drug was calculated by comparing the respective AUC after extravascular and intravenous administration.
Statistical analysis
Experiments were performed in triplicates unless otherwise noted. Statistical assessment of differences between experimental groups was performed by one-way analysis of variance or two-sided Student’s t-test for pairwise comparison (GraphPad Prism 6.0; GraphPad, San Diego, CA, USA). Differences between means were considered statistically nonsignificant (NS) if the P-value was >0.05. The parameters were taken as significantly (S) different for 0.05>P≥0.01 and highly significantly (HS) different for 0.01>P≥0.001.
Results and discussion
Particle size, zeta potential, and EE of the prepared niosomes
MT-loaded niosomes were prepared by thin film hydration technique. Span 60 was selected as nonionic surfactant as it was reported to enhance the entrapment of MT as compared to Tween group surfactants.30 Cholesterol was selected as one of the common additives included in preparing stable niosomes, due to its ability to stabilize niosome bilayers, prevent leakage, and retard permeation of solutes enclosed in the aqueous core of these noisome vesicles.56 Four different drug to surfactant ratios (1:0.5, 1:1, 1:1.5, and 1:2) were used for the preparation of niosomes. Evaluation of the prepared niosomes was carried out by measuring the particle size, zeta potential, and EE.
Table 1 shows the particle size and zeta potential analysis of the freshly prepared niosomes. The size of niosomal formulations decreased with increasing the amount of the added surfactant (span 60) reaching the smallest particle size with drug to surfactant ratio of 1:2 (883.7±31.5 nm) with a polydispersity index of 0.731±0.34. This small vesicle size reflects the stability of the formed niosomal dispersion that is confirmed by the corresponding high negative zeta potential value (−64.1±0.95 mV).
The effect of different mass ratios of span 60 to cholesterol on the EE of the drug in niosomes was also shown in Table 1. Significant (P<0.05) highest EE (52.64%±6.1%) was observed from formulation N4 prepared at span 60 to cholesterol mass ratio of 2:1, which might be attributed to the elevation of phase transition temperature with increasing the span ratio, and thus, forming stable vesicles with higher entrapment of drug. These results were is in agreement with previous studies that reported a maximum EE at this mass ratio of span 60.30,57 Suitable particle size, zeta potential, and EE were obtained from niosomal dispersion formulation N4 with 1:2 drug to surfactant ratio, and thus, it was selected for the preparation of the niosomal film.
Evaluation of fast dissolving films
Physical and mechanical properties
The prepared MT films were elegant enough, transparent, smooth, homogeneous, and flexible. For the niosomal films, opacity was observed due to dispersion of niosomes through the film casting solution. The results of other physical characteristics such as weight and thickness uniformity, film flexibility, surface pH, uniformity of drug content, and in vitro disintegration time are presented in Table 3.
Table 3.
Physical and mechanical properties of MT fast dissolving films
| Formulation code | Thickness (mm)* | Weight (mg)* | Surface pH* | Folding endurance (no of folds)* | Drug content (%)* | Disintegration time (seconds)* |
|---|---|---|---|---|---|---|
| F1 | 0.12±0.003 | 20.53±1.24 | 6.8±0.12 | 230±12 | 97.98±3.21 | 180±12 |
| F2 | 0.14±0.005 | 20.68±2.34 | 7.04±0.14 | 215±20 | 98.6±1.81 | 176±18 |
| F3 | 0.13±0.002 | 21.48±1.44 | 6.9±0.21 | 223±17 | 99.8±2.32 | 175±20 |
| F4 | 0.14±0.003 | 25.43±3.42 | 7.1±0.16 | 219±22 | 97.6±1.22 | 89±5.55 |
| F5 | 0.12±0.004 | 25.7±1.35 | 6.82±0.23 | 235±13 | 98.5±2.11 | 64.33±7.85 |
| F6 | 0.13±0.006 | 25.53±2.18 | 7.1±0.18 | 229±18 | 99.1±1.61 | 38.33±7.78** |
Notes: Evaluated film is 4 cm2 in size, containing 25 mg of the drug.
Mean ± SD (n≥3).
Significant lower disintegration time (P<0.05) compared to other values.
Abbreviations: MT, metoprolol tartarate; SD, standard deviation.
All films had mean weight ranged from 21.48±0.24 to 25.7±0.35 mg and mean thickness in the range of 0.12±0.003 to 0.14±0.005 mm with nonstatistically significant difference (P>0.05) in both weight and thickness among the formulations of the same polymer type.
The mean values of surface pH of all prepared films were close to neutral (6.8±0.12 to 7.1±0.18) with nonsignificant differences obtained (P>0.05). At this pH, the films are less likely to irritate the mucosal lining of the oral cavity, and therefore, they should be fairly comfortable.
Regarding the drug contents, there was nonsignificant difference (P>0.05) in the content of MT among the prepared medicated films (97.6±1.22 to 99.8±2.32). All the formulations were found to contain almost uniform quantity of drug indicating reproducibility of the technique. For the folding and endurance study, high flexibility of all films was demonstrated through their abilities to tolerate folding >200 times without cracking.
The mean in vitro disintegration time for the prepared films containing HPMC polymer was found to have significantly longer (P<0.05) disintegration time (180±12 seconds), as compared with MC polymer (89±2.55 seconds). These results may be attributed to the high viscosity grade of the used HPMC polymer that upon hydration may cause blockade of capillary pores, which prevents the entry of fluid into the film. Similar results were previously reported for the disintegration of montelukast sodium fast dissolving film.58 It was found that neither addition of superdisintegrant nor its concentration had any significant effect on disintegration time of the HPMC films. On the contrary, the mean in vitro disintegration time for the prepared films containing MC polymer was affected by the concentration of superdisintegrant as the disintegration time was significantly decreased (P<0.05) by increasing the concentration of the superdisintegrant (38.33±1.78). These results are in agreement with previously reported data of disintegration time of films prepared with MC.59
Among all the film formulations, the best formulation (F6) was formed from 200 mg MC polymer and 14 mg microcrystalline cellulose, due to its short disintegration time. Hence, it was selected for the preparation of the niosomal film. Niosomal film was prepared by gentle addition of the selected niosomal dispersion formulation (N4) to the polymeric casting solution, followed by gentle stirring until a homogenous mixture was obtained which was transferred into Teflon-coated plate to obtain the dried niosomal film. The resulting niosomal film (N4F6) did not have any significant differences (P>0.05) regarding the surface pH or the in vitro disintegration time compared to the film formulation F6. However, it showed significant increase (P<0.05) in the film weight and thickness after the addition of niosomal dispersion.
Differential scanning calorimetry
DSC provides information on melting, crystallization, decomposition, or a change in heat capacity, and it is useful to assess the physicochemical status of the entrapped drug, as well as the interaction among different components.59 Figure 1 illustrates the DSC thermograms of the selected MT niosomal film (N4F6), its corresponding physical mixture, and their individual solid components. The DSC scan of free MT showed single sharp endothermic melting peak, at 123.7°C with heat fusion of −97.88 mJ, which corresponds to the melting transition of the drug. Data indicate the crystalline nature of the drug and are in good agreement with previously reported melting transitions of thermal analysis of MT.60 Film-forming polymer, MC, did not have any characteristic peaks in the range of studied temperatures, which is similar to the previously reported data for MC.59 Regarding the components of niosomes, it was observed that cholesterol showed sharp peak at 149.6°C with heat of fusion of −54.9 mJ, while span 60 showed two sharp endothermic peaks at 49.57°C (−202.8 mJ) and at 132.12°C (−50.72 mJ). Similar melting transition results were obtained on thermal analysis of both cholesterol and span 60.32,61 Concerning the corresponding physical mixture of MT and MC, cholesterol, or span 60, the characteristic endothermic peak of the drug was still seen with low intensity, only for span 60. These results indicate that the drug is in crystalline form with no degradation and that these components could be considered compatible with MT. From another side, DSC thermogram of MT niosomal film interestingly showed a decreasing intensity of the endothermic peak of span 60 in addition to the disappearance of the other two melting endothermic peaks of MT and cholesterol. These results may be due to the entrapment of the drug in vesicular system within the formed film. These data are in agreement with previously reported data of celecoxib niosomal gel61 and meloxicam niosomal hydrogel.32
Figure 1.
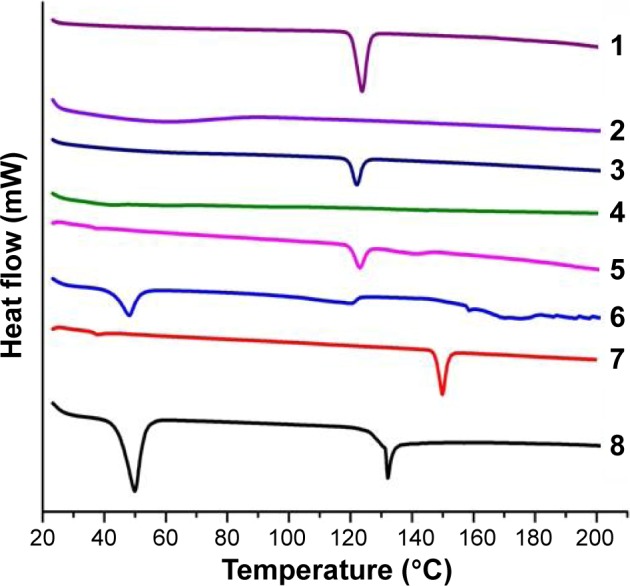
DSC thermograms of (1) pure drug (MT), (2) pure polymer (MC), (3) physical mixture (drug and polymer), (4) MT niosomal film, (5) physical mixture (drug and span 60), (6) pure span 60, (7) pure cholesterol, and (8) physical mixture (drug and cholesterol).
Abbreviations: DSC, differential scanning calorimetry; MC, methyl cellulose; MT, metoprolol tartarate.
Fourier transform infrared spectroscopy
Figure 2 displays the FTIR spectra of the selected MT niosomal film (N4F6), its corresponding physical mixture and their individual solid components. The spectrum of the pure MT drug showed characteristic peaks at 2,981.42, 2,874.36 (OH stretching), 3,462.73 (NH stretching), 1,596.4 (C=O group), 1,249.55 (C–N group), 1,383.48 (OH bending), and 1,180.98 cm−1 (alkyl aryl ether linkage). This pattern is in agreement with previously recorded spectrum for the same drug.31,60,62,63 All these characteristic peaks of the pure drug were also found in the FTIR spectrum of niosomal film and in the spectra of the physical mixtures of the drug with MC, cholesterol, or span 60. Infrared analysis revealed that there was no predominant chemical interaction of MT with ingredients of the selected prepared niosomal film as indicated by the presence of the essential bands and the absence of bands for new functional groups. These findings are in agreement with the previously reported spectra of the same drug with cholesterol, span 60,31 and MC.63
Figure 2.

FTIR spectra of (1) pure drug (MT), (2) pure polymer (MC), (3) physical mixture (drug and polymer), (4) MT niosomal film, (5) physical mixture (drug and cholesterol), (6) physical mixture (drug and span 60), (7) pure cholesterol, and (8) pure span 60.
Abbreviations: FTIR, Fourier transform infrared spectroscopy; MC, methyl cellulose; MT, metoprolol tartarate.
The cumulative percentage
MT release from selected conventional medicated film (F6) in comparison with selected niosomal dispersion (N4) is shown in Figure 3. Conventional fast dissolving film exhibited a significant (P<0.05) faster drug release pattern as compared with the niosomal dispersion. The conventional fast dissolving film, prepared from MC (as a film base) and microcrystalline cellulose (as a superdisintegrant), showed 99.88%±0.89% drug release within 30 minutes, whereas the niosomal drug dispersion showed only 22.85%±0.92% drug release. This fast drug release from the conventional film can be attributed to the hydrophilic nature of the used polymer (MC) that dissolves rapidly and, thus, introducing porosity into the film. The external solvent then diffuses into the film, thereby accelerating the drug dissolution.59,64 In contrast, controlled MT release was achieved from the niosomal dispersion due to the nature of colloidal particulate carrier (niosomes) that have significant advantages over conventional dosage forms of acting as drug reservoirs, which allows for controlled release of the loaded drug.13 In addition, the presence of cholesterol in the niosomal structure reduces the leakage or permeability of the encapsulated drug by decreasing the niosomal membrane fluidity.56
Figure 3.
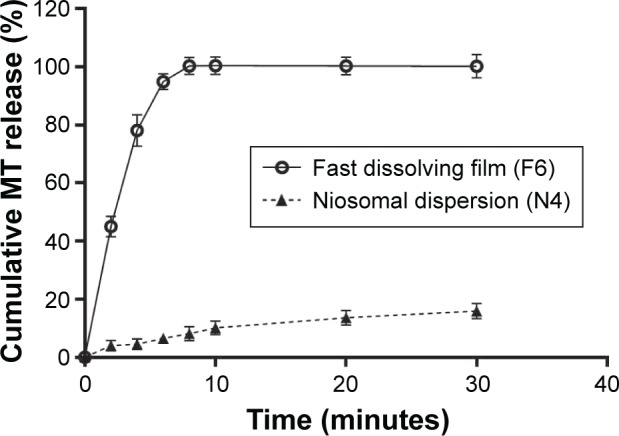
Cumulative MT release from conventional medicated fast dissolving film (F6) in comparison with its niosomal dispersion (N4).
Abbreviation: MT, metoprolol tartarate.
Sublingual administration of MT fast dissolving film (F6) is expected to enhance drug bioavailability through the avoidance of first-pass hepatic metabolism. In addition, inclusion of drug into niosomes (N4), prior to formation of the film, allows delivery of therapeutically significant levels of the drug over prolonged time, which eliminates the need for frequent dosing of the drug that has a short half-life. Figure 4 illustrates the cumulative MT release from the selected niosomal fast dissolving film (N4F6) in comparison with its niosomal dispersion (N4). Incorporation of the medicated niosomes within the fast dissolving film base did not significantly alter (P>0.05) the drug release from the dispersed niosomes. However, the use of film is essential as a suitable dosage form that can be administered to patients. The niosomal dispersion is expected to have very short residence time in the mouth, and hence, there will be no enough time for the absorption of the niosomes/drug through the mucosa. Fast dissolving film can then be used as a convenient dosage form for the delivery of dispersed medicated niosomes that are supposed to continuously deliver therapeutically significant levels of the drug for prolonged time period via sublingual oral mucosa.
Figure 4.
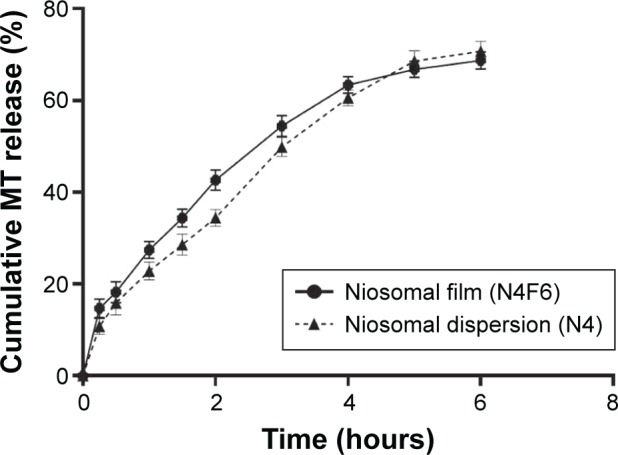
Cumulative MT release from niosomal fast dissolving film (N4F6) in comparison with its niosomal dispersion (N4).
Abbreviation: MT, metoprolol tartarate.
Kinetics of drug release
Table 4 summarizes the release kinetic parameters and correlation coefficients (R2) calculated for the investigated formulations. The in vitro release data indicate that the release of MT from niosomal film as well as niosomal dispersion is most fitted to diffusion-controlled mechanism (Higuchi model) according to the higher correlation coefficient. Korsemeyer–Peppas model was utilized to give more insights on other drug release mechanisms. The diffusion exponent of Peppas model was found to be <0.5, which indicates that a Fickian mechanism is dominant and controls the drug release from the different formulations.32
Table 4.
Kinetics of MT release from niosomal film and dispersion according to different kinetic models
| Formulation | Correlation coefficient (R2)*
|
(n)* Korsmeyer–Peppas equation | |||
|---|---|---|---|---|---|
| Zero order | First order | Higuchi diffusion | Peppas | ||
| Niosomal dispersion (N4) | 0.9798±0.012 | 0.9488±0.019 | 0.9925±0.018 | 0.9735±0.017 | 0.481±0.008 |
| Niosomal film (N4F6) | 0.9526±0.015 | 0.91852±0.021 | 0.9945±0.021 | 0.9793±0.015 | 0.476±0.012 |
Note:
Mean ± SD (n=3).
Abbreviations: MT, metoprolol tartarate; SD, standard deviation.
Stability study
The stability studies were carried out on the selected formulations of fast dissolving film (F6) and fast dissolving niosomal film (N4F6) at room temperature and 40°C for 30 days to assess their long-term stability as summarized in Table 5. After 60 days, it can be noticed that there were nonsignificant differences obtained (P>0.05) in the physicochemical parameters (weight, surface pH, folding endurance, and disintegration time) either at room temperature or at elevated temperature (40°C) compared with initial readings at zero days. Most notably, further assessment of the physicochemical parameters of the selected niosomal dispersion (N4) was hindered due to the fungal contamination observed visually on their surface after only 7±2 days storage at the refrigeration temperature (4°C). These results augment the effective role of the niosomal film preparation on protecting incorporated niosomes from the suspected contamination by keeping these drug-loaded niosomes within the dry form of the forming film. More interestingly, it was found that the drug-loaded niosomes showed a nonsignificant increase (P>0.05) in the vesicle size after their incorporation within the selected film base (953.4±181.3 nm) compared with their original size in the niosomal dispersion (883.7±31.47 nm). This nonsignificant size increase may be attributed to the adsorption of the film-forming polymer (MC) on the surface of the dispersed niosomes.
Table 5.
Physicochemical evaluation of the selected fast dissolving film formulations during stability study
| Formula code
|
Fast dissolving film (F6)
|
Fast dissolving niosomal film (N4F6)
|
||||
|---|---|---|---|---|---|---|
| Time (days) | Zero | 60 (room temperature) | 60 (40°C) | Zero | 60 (room temperature) | 60 (40°C) |
| Weight (mg)* | 25.53±2.18 | 27.53±1.82 | 26.53±2.68 | 39.53±3.5 | 41.23±4.2 | 42.9±5.3 |
| Surface pH* | 7.2±0.15 | 7.1±0.12 | 7.2±0.11 | 6.9±0.2 | 6.8±0.3 | 6.9±0.18 |
| Folding endurance(number of folds)* | 229±18 | 225±22 | 235±16 | 220±21 | 221±17 | 226±23 |
| Disintegration time (seconds)* | 38.33±7.78 | 39.5±5.18 | 34.33±5.22 | 40.21±4.78 | 42.33±3.52 | 39.53±5.18 |
| Particle size (nm)* | – | – | – | 953.4±181.3 | 972.5±150.8 | 986.4±191.6 |
| Zeta potential (mV)* | – | – | – | −51±5.30 | −48±6.30 | −46±4.30 |
Notes:
Mean ± SD (n=3). “–” indicates no data.
Abbreviation: SD, standard deviation.
Scanning electron microscope
The resulting scanning electron micrographs are presented in Figure 5. For the selected niosomal dispersion, Figure 5A clearly demonstrates spherical shape of unilamellar vesicles in nanometer size range with good dispersibility. Additionally, smooth surfaces of the vesicles without any scratches were observed. Previous similar results were reported for the scanning electron micrograph of Montelukast sodium fast dissolving films.58 Similar surface morphology of the niosomes was visualized from the niosomal film micrograph (Figure 5B), which shows spherical vesicles with smooth surfaces without any visible aggregation. The vesicle size was in the nanometer range with good dispersibility. These results confirm that the incorporation of the prepared niosomes within fast dissolving film base did not show any significant changes in the morphology, shape, or dispersibility of the incorporated vesicles.
Figure 5.

Surface morphology of the selected fast dissolving niosomal film and niosomal dispersion.
Notes: Scanning electron micrograph of (A) niosomal formulation N4 and (B) niosomal fast dissolving film (N4F6).
In situ disintegration time and palatability evaluation
Volunteers noted that the film formulation N4F6 is flexible and easy to handle. Taste evaluation results showed that 14.28% of response of the volunteers was acceptable, 50% showed good response and 35.71% showed excellent response. Saccharine was added to stimulate salivation, thus masking the undesirable taste of the drug.7 Mouth feel results showed that the response of 21.42% of the volunteers was acceptable, while 42.85% showed good response and 35.71% had an excellent response. Approximately 85.71% of the volunteers exhibited feeling of mouth refreshment. This mouth refreshment feeling is due to the presence of menthol in the film constituents. The mean in situ disintegration time of the selected film formulation was <1 minute (39.74±5.46 seconds), which is in good agreement with previously reported in situ disintegration time of donepezil oral disintegrating film.51 Overall, these results indicate that the prepared selected niosomal film have acceptable palatability.
Plasma concentrations of MT after sublingual and oral administration
The plasma levels of sublingually absorbed MT from niosomal film formulation (N4F6) were assessed in rabbits and compared to the oral MT tablets, Betaloc. This film design was selected due to its short in vitro and in situ disintegration time and acceptable palatability, in addition to small particle size, high zeta potential, high EE, and good dispersibility of the incorporated niosomes. MT plasma concentration was measured using a reported method prescribed in our previous publications using a sensitive high-performance liquid chromatography assay.15,16 MT was completely separated from other constituents giving a sharp MT peak with no interfering peaks, and the retention time was 9.3 minutes. Plasma concentrations of MT after sublingual and oral administration are represented in Figure 6.
Figure 6.
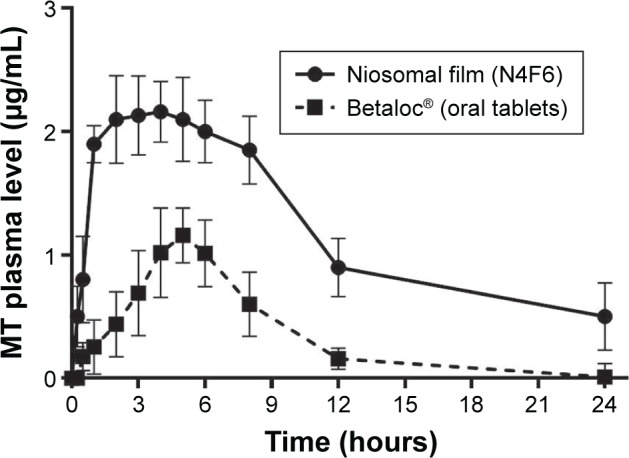
Plasma concentrations of MT after sublingual and oral administration.
Abbreviation: MT, metoprolol tartarate.
The sublingual niosomal film formulation showed a plasma detectable concentration of 0.5±0.245 μg/mL, 15 minutes posttreatment, and the concentration remained detectable for 24 hours. The plasma drug concentration increased progressively to reach the maximum concentration (Cmax) of 2.25±0.086 μg/mL at Tmax of 2.89±0.26 hours, then decreasing to 0.5±0.275 μg/mL at 24 hours after sublingual administration. The oral dose of MT provided a detectable concentration of 0.177±0.113 μg/mL, 30 minutes posttreatment, and the concentration remained detectable for 12 hours. The plasma drug concentration increased progressively to reach the maximum peak concentration (Cmax) of 1.26±0.223 μg/mL at Tmax of 4.22±0.234 hours, then decreasing to 0.16±0.085 μg/mL at 12 hours after oral administration. The observed higher plasma concentrations obtained following the sublingual administration of MT as compared to that of oral tablet reflect the succeeded avoidance of the first-pass metabolism.
Pharmacokinetics and bioavailability of MT after sublingual and oral administration
The one-compartment model without lag time with first-order absorption and elimination represents the best fit model for the MT plasma concentration versus time data. This model was selected after comparison with three other alternative models (one compartment with lag time and two compartments with and without lag time). The fitting of the data did not require incorporation of the lag time. The mean plasma MT pharmacokinetic data based on the plasma concentrations are summarized in Table 6. It could be noticed that the sublingual formulation exhibited faster absorption rate that could be reflected by its shorter Tmax value (2.89±0.26 hours) in comparison with the oral tablet which had a Tmax value of 4.22±0.234 hours. In addition, the peak serum concentration (Cmax) of the drug was markedly higher in the sublingually treated animals 2.25±0.086 μg/mL than that of the oral one (1.26±0.223 μg/mL). This reflects the higher rate of absorption after sublingual delivery of the drug.
Table 6.
Pharmacokinetic parameters of MT in plasma after sublingual administration of fast dissolving niosomal film compared to commercial oral tablets
| Formulation | Cmax* (μg mL−1) | Tmax* (hours) | Ka* (hours−1) | t½a* (hours) | Kel* (hours−1) | t½* (hours) | AUC* (μg h mL−1) | F* (%) |
|---|---|---|---|---|---|---|---|---|
| Betaloc® (MT oral tablet) | 1.26±0.223 | 4.22±0.334 | 0.24±0.024 | 2.93±0.022 | 0.24±0.018 | 2.93±0.019 | 10.73±3.48 | 39.37±11.4 |
| MT niosomal film (N4F6) | 2.25±0.086** | 2.89±0.26** | 0.86±0.16** | 0.80±0.15** | 0.09±0.018** | 7.50±1.47** | 31.88±5.09** | 91.06±7.18** |
Notes:
Mean ± SD (n=5).
Significant differences were obtained compared to that of the oral tablets.
Abbreviations: MT, metoprolol tartarate; SD, standard deviation; Cmax, maximum concentration; Tmax, time of maximum concentration achieved after administration; Ka, absorption rate constant; t½a, absorption half-life; Kel, elimination rate constant; t½, elimination half-life; AUC, area under the MT plasma concentration–time curve; F, the absolute bioavailability.
Moreover, the sublingual administration of the niosomal film showed a significant increase in the area under the plasma concentration time curve (AUC) (31.88±4.09 μg h mL−1) as compared with the AUC obtained after administration of the oral tablet (10.73±3.48 μg h mL−1). This is due to the entrance of MT into the systemic circulation directly to exert its anti-hypertensive action at a controlled rate that is determined by the bioerosion rate of the niosomal bilayers. The narrow AUC obtained after the oral administration of the drug reflects the rapid disappearance of the drug from the plasma due to its short half-life (2.93±0.019 hours) compared with that after sublingual niosomal film administration (7.50±1.47 hours). The absolute bioavailability F after sublingual administration was significantly higher (91.06%±13.28%) than that after oral administration (39.37%±11.4%). The difference between all pharmacokinetic parameters were highly significant at 0.01>P≥0.001. It can be concluded that the systemic bioavailability of MT after sublingual administration was higher than that obtained after oral tablet due to the enhancement of the rate and extent of MT absorption after the sublingual niosomal delivery over prolonged period of time.
Conclusion
In this study, sublingual films that contain MT-loaded niosomes were prepared and evaluated for their abilities to enhance systemic delivery of MT, as compared to administration of oral tablets. Several in vitro and in vivo characterizations of both the drug-loaded niosomes and films were performed. MT was first entrapped in different niosomal formulations, and drug-loaded niosomes with small size, low polydispersity, and high EE were selected for incorporation into different fast dissolving films, which were then evaluated for different physical characteristics. The optimal niosomal film showed sustained release of the drug compared to the medicated film containing the free drug. The in vitro release kinetics of drug from the niosomal suspension and niosomal film followed the Higuchi diffusion model. Moreover, the in vivo study in rabbits showed significantly higher rate and extent of MT absorption from sublingual fast dissolving niosomal film compared to that from oral commercial tablets. Consequently, the absolute bioavailability of the drug following sublingual administration was significantly higher than that after oral tablet administration. These results indicated that the prepared sublingual fast dissolving niosomal film could have potential as an efficient delivery system to enhance the bioavailability and prolong the therapeutic effect of MT, thus improving the patient compliance by eliminating the need for frequent dosing of the drug.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Arya A, Chandra A, Sharma V, Pathak K. Fast dissolving oral films: an innovative drug delivery system and dosage form. Int J Chem Tech Res. 2010;2(1):576–583. [Google Scholar]
- 2.Dixit R, Puthli S. Oral strip technology: overview and future potential. J Controlled Release. 2009;139(2):94–107. doi: 10.1016/j.jconrel.2009.06.014. [DOI] [PubMed] [Google Scholar]
- 3.Baghel P, Roy A, Chandrakar S, Bahadur S. Fast dissolving drug delivery systems: a brief review. Res J Pharm Tech. 2013;6(6):597–602. [Google Scholar]
- 4.Bhyan B, Jangra S, Kaur M, Singh H. Orally fast dissolving films: innovations in formulation and technology. Int J Pharm Sci Rev Res. 2011;9(2):50–57. [Google Scholar]
- 5.Brown GL. Formation of films from polymer dispersions. J Polym Sci. 1956;22(102):423–434. [Google Scholar]
- 6.Vishnu YV, Chandrasekhar K, Ramesh G, Rao YM. Development of mucoadhesive patches for buccal administration of carvedilol. Curr Drug Deliv. 2007;4(1):27–39. doi: 10.2174/156720107779314785. [DOI] [PubMed] [Google Scholar]
- 7.Auda SH, Elbadry M, Ibrahim MA. Design, formulation and characterization of fast dissolving films containing dextromethorphan. Digest J Nanomat Biostruc. 2014;9(1):133–141. [Google Scholar]
- 8.Thakur N, Bansal M, Sharma N, Yadav G, Khare P. Overview “a novel approach of fast dissolving films and their patients”. Adv Biol Res. 2013;7(2):50–58. [Google Scholar]
- 9.Dinge A, Nagarsenker M. Formulation and evaluation of fast dissolving films for delivery of triclosan to the oral cavity. AAPS PharmSciTech. 2008;9(2):349–356. doi: 10.1208/s12249-008-9047-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smart JD. Buccal drug delivery. Expert Opin Drug Deliv. 2005;2(3):507–517. doi: 10.1517/17425247.2.3.507. [DOI] [PubMed] [Google Scholar]
- 11.Bhupinder B, Sarita J. Formulation and evaluation of fast dissolving sublingual films of Rizatriptan Benzoate. Int J Drug Dev Res. 2012;4(1):133–143. [Google Scholar]
- 12.Ciper M, Bodmeier R. Preparation and characterization of novel fast disintegrating capsules (Fastcaps) for administration in the oral cavity. Int J Pharm. 2005;303(1):62–71. doi: 10.1016/j.ijpharm.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 13.Zhang H, Zhang J, Streisand JB. Oral mucosal drug delivery. Clin Pharmacokinet. 2002;41(9):661–680. doi: 10.2165/00003088-200241090-00003. [DOI] [PubMed] [Google Scholar]
- 14.Suresh B, Halloran D, James L. Quick dissolving films: a novel approach to drug delivery. Drug Dev Tech. 2006:1–7. [Google Scholar]
- 15.Abou el Ela AESF, Allam AA, Ibrahim EH. Pharmacokinetics and anti-hypertensive effect of metoprolol tartrate rectal delivery system. Drug Deliv. 2016;23(1):69–78. doi: 10.3109/10717544.2014.904021. [DOI] [PubMed] [Google Scholar]
- 16.El Sayeh A, el Ela FA, Ibrahim E, Allam A. Bucco-adhesive tablets containing metoprolol tartarate: formulation, in vitro and in vivo characterization. J Drug Deliv Sci Tech. 2013;23(2):171–179. [Google Scholar]
- 17.Ramana M, Nagda C, Himaja M. Design and evaluation of mucoadhesive buccal drug delivery systems containing metoprolol tartrate. Indian J Pharm Sci. 2007;69(4):515. [Google Scholar]
- 18.Dollery C, Boobis A, Rawlins M, Thomas S, Wilkins M. Therapeutic Drugs. London: Churchill Livingstone; 1999. Metoprolol tartrate. [Google Scholar]
- 19.MERIT-HF Study Group Effect of metoprolol CR/XL in chronic heart failure: metoprolol CR/XL randomised intervention trial in-congestive heart failure (MERIT-HF) Lancet. 1999;353(9169):2001–2007. [PubMed] [Google Scholar]
- 20.Zhang Q, Jin H, Wang L, Chen J, Tang C, Du J. Randomized comparison of metoprolol versus conventional treatment in preventing recurrence of vasovagal syncope in children and adolescents. Med Sci Monit. 2008;14(4):CR199–CR203. [PubMed] [Google Scholar]
- 21.Jin S, Chung H, Chung M, et al. Influence of CYP2D6* 10 on the pharmacokinetics of metoprolol in healthy Korean volunteers. J Clin Pharm Ther. 2008;33(5):567–573. doi: 10.1111/j.1365-2710.2008.00945.x. [DOI] [PubMed] [Google Scholar]
- 22.Luch JR. Metoprolol tartrate. In: Florey K, editor. Analytical Profiles of Drug Substances. Vol. 12. New York: Academic Press; 1983. pp. 325–386. [Google Scholar]
- 23.Fayed MH, Mahrous GM, Ibrahim MA, Sakr A. Influence of Carbopol 71G-NF on the release of dextromethorphan hydrobromide from extended-release matrix tablets. Pharm Dev Tech. 2013;18(5):971–981. doi: 10.3109/10837450.2011.586037. [DOI] [PubMed] [Google Scholar]
- 24.Aqil M, Sultana Y, Ali A, Dubey K, Najmi A, Pillai K. Transdermal drug delivery systems of a beta blocker: design, in vitro, and in vivo characterization. Drug Deliv. 2004;11(1):27–31. doi: 10.1080/10717540490265225. [DOI] [PubMed] [Google Scholar]
- 25.Coucke D, Vervaet C, Foreman P, Adriaensens P, Carleer R, Remon JP. Effect on the nasal bioavailability of co-processing drug and bioadhesive carrier via spray-drying. Int J Pharm. 2009;379(1):67–71. doi: 10.1016/j.ijpharm.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 26.Narendra C, Srinath M, Babu G. Optimization of bilayer floating tablet containing metoprolol tartrate as a model drug for gastric retention. AAPS PharmSciTech. 2006;7(2):E23–E29. doi: 10.1208/pt070234. [DOI] [PubMed] [Google Scholar]
- 27.Jonsson UE, Sandberg JA, Sjogren JA, inventor. Aktiebolaget Hassle, assignee Beads with metoprolol salt, cellulose ether membrane. 4,957,745. United States Patent. 1990 Sep 18;
- 28.Mahayni H, Rekhi G, Uppoor R, et al. Evaluation of “external” predictability of an in vitro – in vivo correlation for an extended-release formulation containing metoprolol tartrate. J Pharm Sci. 2000;89(10):1354–1361. doi: 10.1002/1520-6017(200010)89:10<1354::aid-jps13>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 29.Dahiya S, Gupta ON. Metoprolol tartrate microspheres. Bull Pharm Res. 2011;1(1):31–39. [Google Scholar]
- 30.Bharkatiya M, Nema R, Bhatnagar M. Formulation and evaluation of metoprolol tartrate entrapped niosomes. Int J Chem Sci. 2010;8(4):2055–2062. [Google Scholar]
- 31.Abbas HK, Hussein IFAH, Al-yousuf MD, Shaheed DQ. Preparation and in-vitro evaluation of metoprolol tartrate proniosomal gel. Kerbala J Pharm Sci. 2013;6:153–163. [Google Scholar]
- 32.El-Badry M, Fetih G, Fathalla D, Shakeel F. Transdermal delivery of meloxicam using niosomal hydrogels: in vitro and pharmacodynamic evaluation. Pharm Dev Technol. 2015;20(7):820–826. doi: 10.3109/10837450.2014.926919. [DOI] [PubMed] [Google Scholar]
- 33.Manconi M, Valenti D, Sinico C, Lai F, Loy G, Fadda AM. Niosomes as carriers for tretinoin: II. Influence of vesicular incorporation on tretinoin photostability. Int J Pharm. 2003;260(2):261–272. doi: 10.1016/s0378-5173(03)00268-0. [DOI] [PubMed] [Google Scholar]
- 34.Nishimura M, Matsuura K, Tsukioka T, et al. In vitro and in vivo characteristics of prochlorperazine oral disintegrating film. Int J Pharm. 2009;368(1):98–102. doi: 10.1016/j.ijpharm.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 35.Khan R, Irchhaiya R. Niosomes: a potential tool for novel drug delivery. J Pharm Investig. 2016;46(3):195–204. [Google Scholar]
- 36.Marianecci C, Di Marzio L, Rinaldi F, et al. Niosomes from 80s to present: the state of the art. Adv Colloid Interface Sci. 2014;205:187–206. doi: 10.1016/j.cis.2013.11.018. [DOI] [PubMed] [Google Scholar]
- 37.Moghassemi S, Hadjizadeh A. Nano-niosomes as nanoscale drug delivery systems: an illustrated review. J Control Release. 2014;185:22–36. doi: 10.1016/j.jconrel.2014.04.015. [DOI] [PubMed] [Google Scholar]
- 38.Tortella FC, Pellicano M, Bowery NG. Dextromethorphan and neuromodulation: old drug coughs up new activities. Trends Pharmacol Sci. 1989;10(12):501–507. doi: 10.1016/0165-6147(89)90050-3. [DOI] [PubMed] [Google Scholar]
- 39.Abdelkader H, Ismail S, Kamal A, Alany RG. Design and evaluation of controlled-release niosomes and discomes for naltrexone hydrochloride ocular delivery. J Pharm Sci. 2011;100(5):1833–1846. doi: 10.1002/jps.22422. [DOI] [PubMed] [Google Scholar]
- 40.Kamboj S, Jhawat V, Saini V, Bala S. Recent advances in permeation enhancement techniques for transdermal drug delivery systems: a review. Curr Drug Ther. 2016;8(3):181–188. [Google Scholar]
- 41.Shahiwala A, Misra A. Studies in topical application of niosomally entrapped nimesulide. J Pharm Pharm Sci. 2002;5(3):220–225. [PubMed] [Google Scholar]
- 42.Bayindir ZS, Yuksel N. Characterization of niosomes prepared with various nonionic surfactants for paclitaxel oral delivery. J Pharm Sci. 2010;99(4):2049–2060. doi: 10.1002/jps.21944. [DOI] [PubMed] [Google Scholar]
- 43.El-Badry M, Fetih G, Shakeel F. Comparative topical delivery of anti-fungal drug croconazole using liposome and micro-emulsion-based gel formulations. Drug Deliv. 2014;21(1):34–43. doi: 10.3109/10717544.2013.843610. [DOI] [PubMed] [Google Scholar]
- 44.Mokhtar M, Sammour OA, Hammad MA, Megrab NA. Effect of some formulation parameters on flurbiprofen encapsulation and release rates of niosomes prepared from proniosomes. Int J Pharm. 2008;361(1):104–111. doi: 10.1016/j.ijpharm.2008.05.031. [DOI] [PubMed] [Google Scholar]
- 45.Aggarwal D, Pal D, Mitra AK, Kaur IP. Study of the extent of ocular absorption of acetazolamide from a developed niosomal formulation, by microdialysis sampling of aqueous humor. Int J Pharm. 2007;338(1):21–26. doi: 10.1016/j.ijpharm.2007.01.019. [DOI] [PubMed] [Google Scholar]
- 46.Rowe R, Sheskey P, Owen S. Handbook of Pharmaceutical Excipients. 5th ed. London: Pharmaceutical Press; 2006. [Google Scholar]
- 47.Singh S, Gangwar S, Garg G, Garg V, Sharma P. Formulation and evaluation of rapidly disintegrating film of levocetrizine hydrochloride. Der Pharmacia Lettre. 2010;2(2):434–439. [Google Scholar]
- 48.Acartürk F, Altug N. In-vitro and in-vivo evaluation of a matrix-controlled bromocriptine mesilate-releasing vaginal ring. J Pharm Pharmacol. 2001;53(12):1721–1726. doi: 10.1211/0022357011778124. [DOI] [PubMed] [Google Scholar]
- 49.Hiremath J, Sarfaraz M, Hiremath D, Sarudkar S. Preparation and physicochemical characterization of simvastatin loaded mucoadhesive bilayered tablet. Indian J Nov Drug Deliv. 2009;1(1):18–24. [Google Scholar]
- 50.Korsmeyer RW, Gurny R, Doelker E, Buri P, Peppas NA. Mechanisms of solute release from porous hydrophilic polymers. Int J Pharm. 1983;15(1):25–35. doi: 10.1002/jps.2600721021. [DOI] [PubMed] [Google Scholar]
- 51.Liew KB, Tan YTF, Peh KK. Characterization of oral disintegrating film containing donepezil for Alzheimer disease. AAPS PharmSciTech. 2012;13(1):134–142. doi: 10.1208/s12249-011-9729-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weibel ER, Bacigalupe LD, Schmitt B, Hoppeler H. Allometric scaling of maximal metabolic rate in mammals: muscle aerobic capacity as determinant factor. Respir Physiol Neurobiol. 2004;140(2):115–132. doi: 10.1016/j.resp.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 53.West GB, Brown JH. The origin of allometric scaling laws in biology from genomes to ecosystems: towards a quantitative unifying theory of biological structure and organization. J Exp Biol. 2005;208(9):1575–1592. doi: 10.1242/jeb.01589. [DOI] [PubMed] [Google Scholar]
- 54.Sastry MS, Satyanarayana N, Diwan P, Krishna D. Formulation, pharmacokinetic and pharmacodynamic evaluation of fast releasing compressed propranolol. HCL suppositories. Drug Dev Ind Pharm. 1993;19(9):1089–1096. [Google Scholar]
- 55.Chmielowiec D, Schuster D, Gengo F. Determination of pindolol in human serum by HPLC. J Chromatogr Sci. 1991;29(1):37–39. doi: 10.1093/chromsci/29.1.37. [DOI] [PubMed] [Google Scholar]
- 56.Abdelbary G, El-gendy N. Niosome-encapsulated gentamicin for ophthalmic controlled delivery. AAPS PharmSciTech. 2008;9(3):740–747. doi: 10.1208/s12249-008-9105-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.El-Menshawe SF, Hussein AK. Formulation and evaluation of meloxicam niosomes as vesicular carriers for enhanced skin delivery. Pharm Dev Technol. 2013;18(4):779–786. doi: 10.3109/10837450.2011.598166. [DOI] [PubMed] [Google Scholar]
- 58.Ghorwade V, Patil A, Patil S, Srikonda K, Kotagiri R, Patel P. Development and evaluation of fast-dissolving film of montelukast sodium. World J Med Pharm Biol Sci. 2011;1(1):6–12. [Google Scholar]
- 59.Auda SH, Mahrous GM, El-Badry M, Fathalla D. Development, preparation and evaluation of oral dissolving films containing metoclopramide. Lat Am J Pharm. 2014;33(6):1027–1033. [Google Scholar]
- 60.Raju KN, Velmurugan S, Deepika B, Vinushitha S. Formulation and in-vitro evaluation of buccal tablets of metoprolol tartrate. Int J Pharm Pharm Sci. 2011;3(2):239–246. [Google Scholar]
- 61.Auda SH, Fathalla D, Fetih G, El-Badry M, Shakeel F. Niosomes as transdermal drug delivery system for celecoxib: in vitro and in vivo studies. Polym Bull. 2016;73(2):1229–1245. [Google Scholar]
- 62.Nagendrakumar D, Shirsand S, Para M, Makadia C. Design and evaluation of metoprolol tartrate containing buccal tablets. Dhaka Univ J Pharm Sci. 2012;10(2):101–108. [Google Scholar]
- 63.Ranganathan VSY, Chetty MC, Sasikala C, Varma MM, Kumar MK. Buccal drug delivery from metoprolol tartrate polymeric muco adhesive film. J Pharm Res. 2011;4(11):3892–3896. [Google Scholar]
- 64.Harini C, Sowjanya CH, Kumar KS. Formulation and evaluation of mouth dissolving film containing cetirizine hydrochloride. IJARMPS. 2016;1(1):20–25. [Google Scholar]