

Abstract
Objective:
To correlate time to loss of ambulation (LoA) and different truncating DMD gene mutations in a large, prospective natural history study of Duchenne muscular dystrophy (DMD), with particular attention to mutations amenable to emerging molecular treatments.
Methods:
We analyzed data from the Cooperative International Neuromuscular Research Group Duchenne Natural History Study for participants with DMD single- or multi-exon deletions or duplications with defined exon boundaries (n = 186), or small mutations identified by sequencing (n = 26, including 16 nonsense point mutations). We performed a time-to-event analysis of LoA, a strong indicator of overall disease severity, adjusting for glucocorticoid treatment and genetic modifiers.
Results:
Participants with deletions amenable to skipping of exon 44 had later LoA (median 14.8 years, hazard ratio 0.31, 95% confidence interval 0.14–0.69, p = 0.004). Age at LoA did not differ significantly in participants with deletions amenable to exon 45, 51, and 53 skipping, duplications, and small rearrangements. Nonsense mutation DMD also showed a typical median age at LoA (11.1 years), with a few outliers (ambulatory around or after 16 years of age) carrying stop codons within in-frame exons, more often situated in the rod domain.
Conclusions:
As exon 44 skipping–amenable DMD has a later LoA, mutation-specific randomization and selection of placebo groups are essential for the success of clinical trials.
Duchenne muscular dystrophy (DMD) is caused by truncating DMD gene mutations leading to absent dystrophin.1 The typical DMD phenotype encompasses muscle weakness and wasting presenting in early childhood, and loss of independent ambulation (LoA) by age 13 years,2 while the mild allelic form Becker muscular dystrophy (BMD) is defined by nontruncating mutations3 and LoA after 16 years.2 Daily glucocorticoid treatment and improved standards of care,4,5 however, have shifted the median age at LoA in DMD to around 14 years,6–9 with a wide variability attributed to different treatment regimens8,9 and genetic modifiers.10–14
Another potential source of clinical variability is the rescue of small amounts of dystrophin, despite frame-shifting mutations. Possible mechanisms include translation reinitiation downstream of N-terminal domain mutations,15–18 or low-rate splicing out of specific exons.19–21 Even very small quantities of dystrophin, hardly detectable by immunohistochemistry (IHC) or Western blot (WB), might ameliorate the disease and delay LoA by several years. This is particularly relevant for the development of dystrophin-restoring treatments, such as exon skipping by antisense oligonucleotides (AONs)22–30 and stop codon readthrough,31–33 as variation in baseline levels of dystrophin may confound evaluations of efficacy in clinical trials.
We aimed to analyze correlations between different truncating DMD mutations and age at LoA in the Cooperative International Neuromuscular Research Group Duchenne Natural History Study (CINRG-DNHS), a large prospective DMD natural history study,6,7 with particular attention to mutation groups amenable to emerging molecular treatments (i.e., out-of-frame deletions eligible for the skipping of exons 44, 45, 51, and 53, and nonsense mutations).
METHODS
CINRG-DNHS inclusion criteria.
Participants in the parent CINRG-DNHS (distinguished from an added cohort of DMD participants aged 4 to <8 years old, NCT00468832) were recruited between 2006 and 2009, and inclusion criteria have been described.6 Importantly, patients were excluded if they had a proximal (before exon 25) out-of-frame mutation and a BMD phenotype (because of frequent violations to the reading frame rule in this region15). Participants could be included with an in-frame DMD mutation, or no demonstrated DMD mutation but a typical DMD phenotype manifesting by 5 years of age (e.g., progressive proximal weakness, characteristic gait, positive Gowers sign, and calf pseudohypertrophy) and abnormal dystrophin IHC or WB. The average follow-up period for the parent cohort was 4 years.
Additional inclusion criteria.
In order to group participants by DMD mutation type and amenability to molecular therapies, we further selected participants with available evidence of a DMD genetic mutation and the following characteristics: single- or multi-exon out-of-frame deletion with univocally defined exon boundaries (multiplex ligation-dependent probe amplification [MLPA] or multiplex PCR with amplification of immediately flanking exons); single- or multi-exon out-of-frame duplication confirmed by MLPA; small out-of-frame DMD mutation (insertion, deletion); nonsense mutation; splicing mutation.
DMD mutation studies.
Diagnostic genetic testing for causative DMD mutations was performed at local institutions as part of the standard diagnostic workup, and reviewed by a central CINRG genetic counselor, who reviewed rearrangement exon boundaries and unified mutation nomenclature (following HGVS recommendations) and reference sequences (genomic NG_012232.1, transcript NM_004006.2).
Grouping of DMD mutations.
The grouping rationale was to describe the natural history of LoA in groups of participants with typical vs atypical phenotypes or amenability to mutation-specific molecular therapies. We defined the following categories: out-of-frame deletions amenable to skipping of (1) exon 44, (2) exon 45, (3) exon 51, (4) exon 53, (5) deletion of exons 3–7; (6) out-of-frame deletions not amenable to skipping of exons 44, 45, 51, or 53; (7) out-of-frame duplications; (8) nonsense mutations; and (9) out-of-frame small insertions or deletions.
Genetic modifier genotyping.
The parent CINRG-DNHS cohort showed significant effects of 2 known genetic modifiers of ambulation in DMD14: the single nucleotide polymorphisms (SNPs) rs28357094 in the promoter of the SPP1 gene, encoding Secreted PhosphoProtein 1 or osteopontin,10,11 and rs10880 in the coding portion of the LTBP4 (latent transforming growth factor β binding protein 4) gene.12 Genotyping and grouping by inheritance models was described previously.14
Loss of ambulation.
We defined LoA as participant- or caregiver-reported age at continuous wheelchair use, approximated to the nearest month, and verified by a CINRG-trained clinical evaluator with the inability to perform the 10 m run/walk assessment.
Statistical analysis.
We performed a time-to-event analysis of LoA with age (years) as time variable and LoA as event. Median age at LoA and corresponding 95% confidence intervals (CIs) were estimated by plotting empirical Kaplan-Meier curves for participant groups defined by mutation (as described above), and by GC treatment administered while ambulatory, with the following grouping: untreated (or short treatment of <1 year), prednisone or prednisolone, and deflazacort. Participants switching between prednisone/prednisolone and deflazacort were grouped accordingly to the drug administered for the longest time. We used Cox proportional hazard models to estimate and compare age-related risks of LoA. Covariates included DMD mutations as described above and time-varying GC drug (prednisone/prednisolone or deflazacort) and weight-adjusted dose as previously described.9 Note that GC treatment as a time-varying covariate in the Cox proportional hazard analysis is independent of the grouping of individual participants, used for the empirical Kaplan-Meier estimation of median age at LoA. The same analyses were repeated adding covariates for SPP1 and LTBP4 genotype; this was performed separately, because DNA for genotyping of both SNPs was not available in 41 participants (largely due to regulatory issues in some countries that did not allow DNA shipping). Statistical significance was set at p < 0.05. All analyses were performed with the survival package in R, version 3.2.1.
RESULTS
Selected cohort.
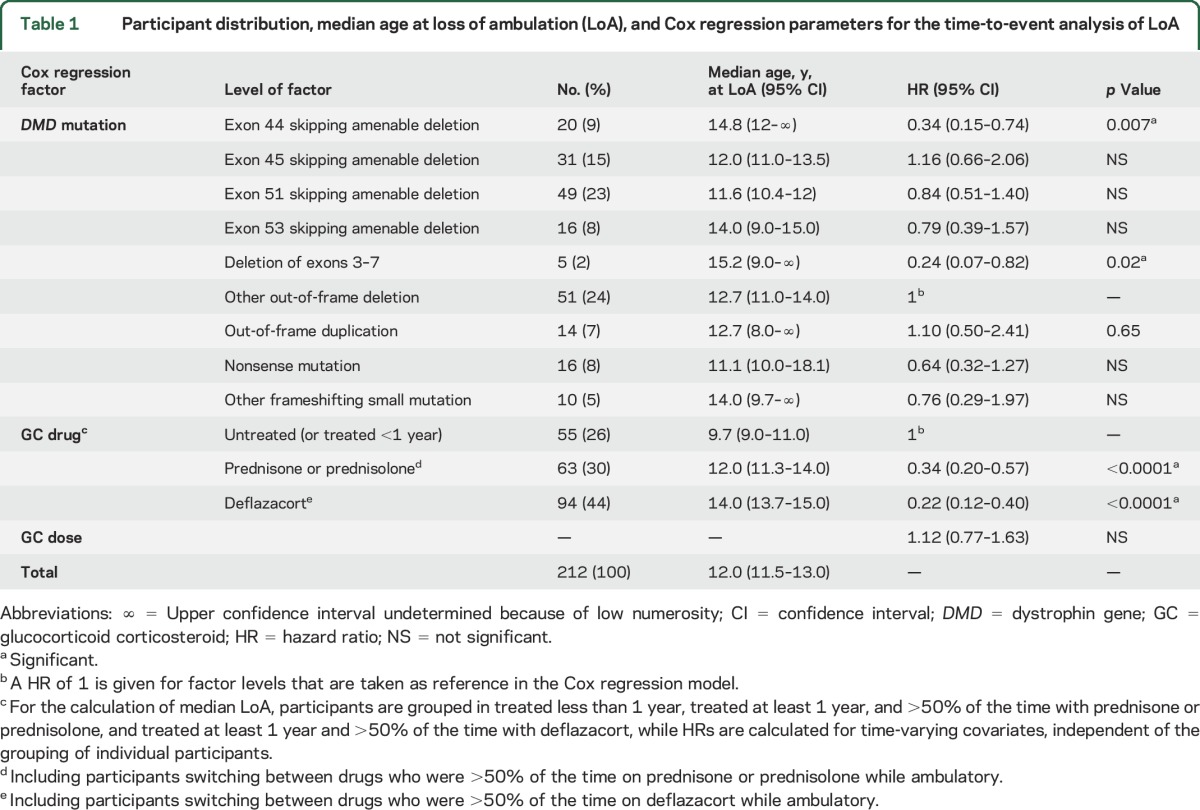
Inclusion criteria described in Methods led to the selection of 212/340 (62.3%) participants to the parent CINRG-DNHS (figure 1). Their distribution by mutation and exon-skipping amenability is described in table 1. Of note, there were 49/212 participants (23.1%) with deletions amenable to skipping of exon 51, 31/212 (14.6%) of exon 45, 20/212 (9.4%) of exon 44, 16/212 (7.5%) of exon 53, and 16/212 (7.5%) with nonsense mutations. Detailed frequencies of single- or multi-exon rearrangements are summarized in table e-1 on the Neurology® Web site at Neurology.org.
Figure 1. Study flow chart.

Participant selection from the parent (i.e., not including a currently recruiting extension) Cooperative International Neuromuscular Research Group Duchenne Natural History Study (CINRG-DNHS), identifying subgroups analyzed in this article.
Table 1.
Participant distribution, median age at loss of ambulation (LoA), and Cox regression parameters for the time-to-event analysis of LoA
Age at LoA by mutation group.
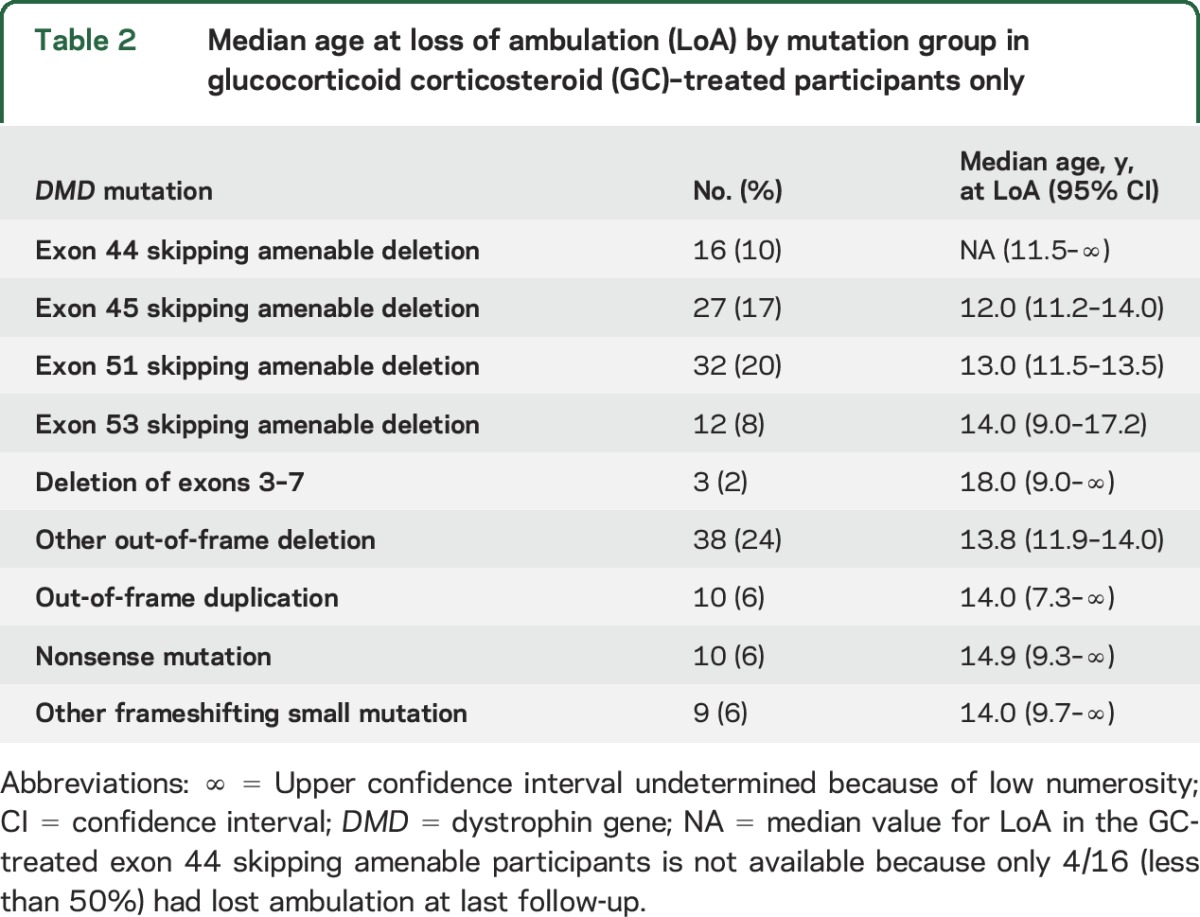
Median age at LoA with 95% CIs by mutation group is shown in table 1 and Kaplan-Meier plots of LoA by age and mutation group are shown in figure 2. The largest group (n = 51, 24%), comprising participants with out-of-frame deletions not amenable to skipping of exons 44, 45, 51, and 53, showed a median age at LoA of 12.7 years (95% CI 11–14 years). Groups with later median LoA included deletion of exons 3–7 (n = 5, median age at LoA 15.2 years, 95% CI 9–undetermined) and deletions amenable to exon 44 skipping (n = 20, median 14.8 years, 95% CI 12–undetermined). Age at LoA for GC-treated participants only within each mutation group is shown in table 2.
Figure 2. Kaplan-Meier plots of loss of ambulation by mutation group.

Plots for participants with (A) deletions amenable to skipping of exon 44, (B) deletions amenable to skipping of exon 45, (C) deletions amenable to skipping of exon 51, (D) deletions amenable to skipping of exon 53, (E) deletion of exons 3–7, (F) single- or multi-exon duplications, (G) nonsense mutations, and (H) small frameshift mutations are compared in each graph with the reference group of participants with single- or multi-exon deletions not amenable to skipping of exons 44, 45, 51, or 53 (other deletions; thin black line). Thin colored lines refer to all the participants in each mutation group, while thick lines refer to glucocorticoid-treated participants only.
Table 2.
Median age at loss of ambulation (LoA) by mutation group in glucocorticoid corticosteroid (GC)–treated participants only
Cox regression analysis of LoA.
Cox regression analysis including GC treatment covariates showed that the delay of LoA was statistically significant in both the group with deletion of exons 3–7 (hazard ratio [HR] 0.24, 95% CI 0.07–0.82, p = 0.02) and the group with deletions amenable to skipping of exon 44 (HR 0.34, 95% CI 0.15–0.74, p = 0.007). Cox regression parameters for all other DMD mutations did not differ significantly from the reference group (table 1). As previously described in the whole CINRG-DNHS population,7 treatment with prednisone/prednisolone or deflazacort were strongly associated with later LoA, with a lower HR (i.e., later LoA) for deflazacort (HR 0.34 and 0.22, respectively, p < 0.0001 for both), while there was no significant independent effect of GC dose in this population. In 171/212 participants with available modifier genotypes, amenability to skipping of exon 44 remained significantly associated with later LoA (HR 0.24, 95% CI 0.10–0.61, p = 0.006), and so was deletion of exons 3–7 (HR 0.21, 95% CI 0.05–0.92, p = 0.01), after adjusting for SPP1 and LTBP4 genotypes as additional covariates. Modifier genotype effects were in the same direction as previously described, but not statistically significant in this multivariate model in this population (table e-2).
DISCUSSION
In the present study, we describe significant differences in age at LoA between DMD participants with different DMD mutations. LoA is a clinically meaningful natural history milestone in DMD and a reliable overall indicator of the severity of disease progression.6,7 It is strongly correlated with the longitudinal changes of functional measures commonly adopted as clinical trial endpoints,34–36 but compared to these, it is less influenced by motivational factors. Furthermore, LoA predicts other major disease milestones such as the need for ventilatory support and survival.37,38
We observed an approximately 2-year delay of median LoA in 20 participants who had mutations amenable to exon 44 skipping. Similar results were observed in a retrospective genotype–phenotype association study carried out in the Netherlands.20 Most exon 44 skippable participants carried the relatively frequent single-exon deletion of exon 45: 60% in our cohort and 66% in the Dutch cohort.20 This mutation has long been known to induce endogenous skipping of the adjacent exon 44, resulting in traces of dystrophin expression.39 Recently, a Japanese research group studied a patient with DMD carrying the exon 45 deletion, and demonstrated a splicing silencer effect of the junction sequence between introns 44 and 45, which promoted the skipping of exon 44.40
Traces of dystrophin were reported in 3/6 exon 44 skipping eligible DNHS participants by IHC and 0/4 by WB. Although these findings were from retrospective laboratory reports and were not performed using some of the quantitation techniques recommended in clinical trials,e1 it can be inferred that limited amounts of dystrophin, only detectable by a sensitive technique such as IHC, may be sufficient to prolong ambulation. This is in keeping with observations in the dystrophin/utrophin double knockout mouse, an animal model of severe dystrophinopathy, in which re-expression of small amounts of dystrophin improves muscle pathology and function.e2
Although most exon 44 skippable participants carry the deletion of exon 45, some carry different deletions upstream of exon 44. Interestingly, we observed an intermediate DMD/BMD phenotype in 2 participants carrying deletions of exons 10–43 and 38–43. These 2 GC-treated participants were still ambulant at ages 21 and 16 years, suggesting a different molecular mechanism for dystrophin rescue than that described for exon 45 deletion.40 One of the 2 participants had traces of dystrophin identified by IHC, but not WB.
A distinct, albeit small group of participants with a milder phenotype in the CINRG-DNHS is represented by 5 participants with DMD carrying the deletion of exons 3–7, previously described as an exception to the reading frame rule.16–18 IHC showed traces of dystrophin in 1/3 participants with this mutation and WB in 0/3 (the 3 participants who underwent IHC and the 3 who underwent WB are not the same). However, the one participant with reported dystrophin traces lost ambulation at age 9 years, despite high-dose weekend prednisone since age 4. A 5' internally deleted dystrophin protein might be less efficient in rescuing the phenotype because of the disruption of the functionally relevant N-terminal actin-binding domain, and other genetic and environmental confounders may influence the clinical outcome. Larger case series are needed to fully understand the phenotypes associated with the deletion of exons 3–7. Notably, some intermediate DMD/BMD cases with this deletion might have been excluded from the CINRG-DNHS due to inclusion criteria (see Methods).
These findings have several potential repercussions for clinical trials. First, some patients eligible for exon 44 skipping might show stabilized function even in the placebo group, especially in short studies, making small drug effects challenging to identify. Second, among patients with DMD with the same exon-skipping eligibility, there might be some who activate endogenous exon-skipping mechanisms, and some who do not. Therefore, accurate quantitation of baseline dystrophin for each patient participating in dystrophin-restoring trials is of paramount importance. Third, we provide data on age at LoA for GC-treated DMD subcohorts with specific exon-skipping eligibility that may be useful as a natural history control cohort to compare to current and future cohorts treated with exon-skipping drugs. For instance, age at LoA for GC-treated participants with exon 51 skipping eligibility does not appear to be significantly different from non-exon-skipping-eligible deletions.
Our data did not confirm reports that patients with DMD with deletions amenable to exon 53 skipping might present earlier LoA,e3 as these patients appeared to be positioned in the average range for age at LoA in DMD. In order to reproduce the methodology of the cited French study,e3 we calculated mean age at LoA in nonambulatory patients only, which was 11.4 years in 10 nonambulatory CINRG-DNHS participants amenable to exon 53 skipping, vs 8.9 years in 13 patients in the cited study.e3 A higher rate of GC treatment in CINRG-DNHS participants likely plays a relevant role in this difference. Further studies on longitudinal functional measures may confirm the tendency to reduced upper limb function described by the French group.
The DMD population described by the Italian DMD network21 also showed a more rapid deterioration of ambulation-related functional measures, such as 6-minutes walk test and North Star Ambulatory Assessment, in the exon 45 skipping and exon 53 skipping amenable subgroups, which are not confirmed by our LoA data. Again, studies of longitudinal functional measures in the CINRG cohort are warranted to further investigate this correlation.
Ten CINRG-DNHS participants carried a single-exon deletion of exon 52, which could be theoretically amenable to both exon 51 and 53 skipping. Here, we grouped these participants together with deletions amenable to exon 51 skipping, a therapeutic approach that has reached more advanced stages of clinical trials. Interestingly, these exon 52–deleted participants had early LoA, median age being 10.0 vs 11.9 years in all other exon 51 skipping–eligible participants (log-rank p = 0.016). This genotype–phenotype association needs independent validation, but if confirmed, could be relevant for the interpretation of the results of AON clinical trials targeting exons 51 and 53.
A different group of participants amenable to molecular treatment were 16 participants carrying nonsense mutations. This group is important as ambulatory patients with DMD older than 5 years are currently being prescribed Ataluren (Translarna; PTC Therapeutics, South Plainfield, NJ) in several European countries under the provisional approval of the European Medicines Agency (EMA). While the median age at LoA in this group was similar to the reference population carrying non-exon-skipping-eligible mutations, there were 5 outlier cases showing prolonged ambulation (close to or beyond age 16 years), consistent with intermediate DMD/BMD, as shown by a rightward shift of the third and fourth quartiles of the Kaplan-Meier plot (figure 2G). DMD nonsense mutations have previously been described in association with intermediate phenotypes. Furthermore, it has been shown that exons where intermediate nonsense mutations occur are usually in-frame, situated in the functionally dispensable rod domain, and defined by weaker splice signals, explaining easier induction of endogenous exon skipping.19 In fact, of 5 intermediate DMD/BMD participants with nonsense mutations in the CINRG-DNHS, 3 carried stop codons within in-frame exons (14, 29, and 30). On the other hand, 2 carried stop codons in out-of-frame exon 45, again suggesting alternative splicing in this region, and exon 69, suggesting escape from mRNA nonsense-mediated decay with a distally located mutation, which may give rise to a C-terminal truncated protein. Interestingly, another participant lost ambulation at 10 years, despite carrying a proximal nonsense mutation (c.9 G>T, p.Trp3*) with a described founder effect in North America. This is in contrast with the previously described association of this mutation with a mild BMD phenotype,e4 caused probably by dystrophin rescue by downstream translation reinitiation.e5 Patients with a definitely mild BMD phenotype would not have been included in the DNHS because of clinical exclusion criteria; nevertheless, it could be relevant to stratify nonsense mutation patients with DMD by stop codon position (in-frame or out-of-frame exon) in clinical trials. Currently, Ataluren is approved by the EMA for nonsense-mediated DMD only, but cases such as those described here show that the distinction with nonsense-mediated BMD might be blurry and hard to define.
A limitation of this natural history study is the use of DNA mutation data derived from clinical records. Although all CINRG clinical sites actively pursue adherence to the modern standards of care in DMD diagnosis, the identification of the causative mutation (especially those identified by sequencing) was not possible in some cases. As single-exon or multi-exon deletions are easier to identify, the proportions of patients with DMD amenable to skipping of individual exons might be slightly inflated in this report. Future studies including full DMD sequencing at the genomic or RNA level, as well as next-generation sequencing studies of intronic deletion/duplication breakpoints in selected cases, might further refine genotype–phenotype correlations. Another limitation was the lack of an evaluation of other relevant DMD natural history milestones, e.g., scoliosis and survival. Different from LoA, it was not possible for patients who had already developed severe scoliosis (e.g., Cobb angle >40°) to recall the age at which this criterion was met, making a time-to-event analysis challenging to design. As for survival, this endpoint is better analyzed from historical retrospective population studies than in an active follow-up population like the CINRG-DNHS cohort. However, both scoliosis and survival have shown strong correlations with LoA.38 Genotype–phenotype correlations for cardiac and respiratory endpoints in the CINRG-DNHS will be the object of separate studies.
This study provides mutation-specific natural history data regarding LoA in DMD, carefully adjusting for the effect of other disease-modifying variables. This is relevant for the design and interpretation of clinical trials for innovative therapeutics in DMD. Importantly, patients with DMD with deletions amenable to exon 44 skipping, exon 3–7 deletion, and point mutations within in-frame exons should be excluded from natural history–derived placebo groups for the evaluation of AONs targeting rare deletions, and their distribution in treated/placebo groups should be carefully balanced in clinical trials of non-dystrophin-restoring treatments. Age at LoA in other exon-skipping eligible groups is not significantly different from non-exon-skipping eligible deletions.
Supplementary Material
ACKNOWLEDGMENT
The authors thank all participating patients with DMD and their families and study team members at participating CINRG sites for assistance in collecting and managing data: University of California Davis: M. Cregan, L. Johnson, J. Han, N. Joyce, A. Nicorici, D. Reddy; Sundaram Medical Foundation and Apollo Children's Hospital, Chennai: C. Chidambaranathan, S. Kumar, V. Lakshmi, K. Niroshan, P. Reddappa, V. Reddy, H. Solomon; Holland Bloorview Kids Rehabilitation, Toronto: D. Biggar, M. Dermody, L. Eliasoph, E. Hosaki, V. Harris, G. Lee; AB Children's Hospital, Calgary: A. Chiu, T. Haig, M. Harris, M. Kornelsen, N. Rincon, K. Sanchez, L. Walker; Queen Silvia Children's Hospital: A. Alhander, A. Ekstrom, A. Gustafsson, A. Kroksmark, U. Sterky, L. Wahlgren; Children's National Health System: N. Brody, B. Drogo, M. Leach, R. Leshner, C. Tesi-Rocha, M. Birkmeier, B. Tadese, A. Toles; Royal Children's Hospital: A. Kornberg, K. Carroll, K. DeValle, R. Kennedy, V. Rodriguez, D. Villano; Hadassah Hebrew University Hospital: R. Adani, A. Bar Leve, L. Chen-Joseph, M. Daana, V. Panteleyev-Yitshak, E. Simchovitz, D. Yaffe; Instituto de Neurosciencias Fundacion Favaloro: L. Andreone, F. Bonaudo, J. Corderi, L. Levi, L. Mesa, P. Marco; Children's Hospital of Pittsburgh of UPMC and the University of Pittsburgh: H. Abdel-Hamid, R. Bendixen, C. Bise, A. Craig, K. Karnavas, C. Matthews, G. Niizawa, A. Smith, J. Weimer; Washington University: J. Anger, T. Christenson, J. Florence, R. Gadeken, P. Golumbak, B. Malkus, A. Pestronk, R. Renna, J. Schierbecker, C. Seiner, C. Wulf; Children's Hospital of Richmond at VCU: S. Blair, B. Grillo, E. Monasterio; University of Tennessee: M. Barrett-Adair, C. Benzel, K. Carter, J. Clift, B. Gatlin, R. Henegar, J. Holloway, M. Igarashi, F. Kiphut, A. Parker, A. Phillips, R. Young; Children's Hospital of Westmead: K. Cornett, N. Gabriel, M. Harman, C. Miller, K. North, K. Rose, S. Wicks; University of Alberta: L. Chen, C. Kennedy; Centro Clinico Nemo: M. Beneggi, L. Capone, A. Molteni, V. Morettini; Texas Children's Hospital: A. Gupta, A. Knight, B. Lott, R. McNeil, G. Orozco, R. Schlosser; University of Minnesota: G. Chambers, J. Day, J. Dalton, A. Erickson, M. Margolis, J. Marsh, C. Naughton; Mayo Clinic: K. Coleman-Wood, A. Hoffman, W. Korn-Petersen, N. Kuntz,; University of Puerto Rico: B. Deliz, S. Espada, P. Fuste, C. Luciano, J. Torres; and the CINRG Coordinating Center: A.Cnaan, M. Ahmed, A. Arrieta, N. Bartley, T. Brown-Caines, C. Carty, T. Duong, J. Feng, F. Hu, L. Hunegs, Z. Sund, W. Tang, and A. Zimmerman.
GLOSSARY
- AON
antisense oligonucleotide
- BMD
Becker muscular dystrophy
- CI
confidence interval
- CINRG-DNHS
Cooperative International Neuromuscular Research Group Duchenne Natural History Study
- DMD
Duchenne muscular dystrophy
- EMA
European Medicines Agency
- HR
hazard ratio
- IHC
immunohistochemistry
- LoA
loss of ambulation
- MLPA
multiplex ligation-dependent probe amplification
- SNP
single nucleotide polymorphism
- WB
Western blot
Footnotes
Supplemental data at Neurology.org
Contributor Information
Collaborators: CINRG investigators, A. Cnaan, M. Thangarajh, R.T. Abresch, E. Henricson, V. Viswanathan, L. McAdam, J. Mah, M. Tulinius, M. Ryan, Y. Nevo, A. Dubrovsky, P. Clemens, A. Connolly, J. Teasley, T. Bertorini, R. Webster, H. Kolski, K. Gorni, T. Lotze, P. Karachunski, J. Bodensteiner, and J. Carlo
AUTHOR CONTRIBUTIONS
L.B. contributed to study design, analyzed and interpreted data, and drafted the manuscript. L.P.M. contributed to study design, analyzed and interpreted data, and critically revised the manuscript for intellectual content. H.G.-D. contributed to study design, performed statistical analyses, and critically revised the manuscript for intellectual content. E.P.H. contributed to study design and critically revised the manuscript for intellectual content. C.M.M. contributed to the overall CINRG DMD study design and critically revised the manuscript for intellectual content. S.C. designed the study and critically revised the manuscript for intellectual content.
STUDY FUNDING
US Department of Education/NIDRR (#H133B031118, #H133B090001); US Department of Defense (#W81XWH-12-1-0417); National Institutes of Health/NIAMS (#R01AR061875); and Italian Ministry of Education PhD grant awarded to L.B. for the 28th Cycle of the Doctorate School of Medical, Clinical, and Experimental, Science at the University of Padova, Italy. S.C. is supported by the German Research Foundation (DFG) and MDA Developmental Grant.
DISCLOSURE
L. Bello, L. Morgenroth, and H. Gordish-Dressman report no disclosures relevant to the manuscript. E. Hoffman reports that he is co-founder and CEO of ReveraGen BioPharma, co-founder and Vice President of AGADA BioSciences, and received grants from National Institutes of Health during the conduct of the study. C. McDonald has served as a consultant for clinical trials for PTC Therapeutics, Prosensa, Sarepta, Eli Lilly, Pfizer, Halo Therapeutics, Cardero, and Mitokyne, outside the submitted work; serves on external advisory boards related to Duchenne muscular dystrophy for PTC Therapeutics and Eli Lilly; and reports grants from US Department of Education/NIDRR, US NIH/NIAMS, US Department of Defense, and Parent Project Muscular Dystrophy US, during the conduct of the study; and grants from Parent Project Muscular Dystrophy US, outside the submitted work. S. Cirak reports a sponsored research agreement with Sarepta Therapeutics. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Hoffman EP, Brown RH Jr, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 1987;51:919–928. [DOI] [PubMed] [Google Scholar]
- 2.Darras BT, Miller DT, Urion DK. Dystrophinopathies. In: Pagon RA, Adam MP, Ardinger HH, et al. eds. GeneReviews®. Seattle: University of Washington; 1993–2016. Available at: http://www.ncbi.nlm.nih.gov/books/NBK1119/. Accessed March 2, 2016. [Google Scholar]
- 3.Hoffman EP, Kunkel LM, Angelini C, Clarke A, Johnson M, Harris JB. Improved diagnosis of Becker muscular dystrophy by dystrophin testing. Neurology 1989;39:1011–1017. [DOI] [PubMed] [Google Scholar]
- 4.Bushby K, Finkel R, Birnkrant DJ, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol 2010;9:77–93. [DOI] [PubMed] [Google Scholar]
- 5.Bushby K, Finkel R, Birnkrant DJ, et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: implementation of multidisciplinary care. Lancet Neurol 2010;9:177–189. [DOI] [PubMed] [Google Scholar]
- 6.McDonald CM, Henricson EK, Abresch RT, et al. The cooperative international neuromuscular research group Duchenne natural history study: a longitudinal investigation in the era of glucocorticoid therapy: design of protocol and the methods used. Muscle Nerve 2013;48:32–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Henricson EK, Abresch RT, Cnaan A, et al. The cooperative international neuromuscular research group Duchenne natural history study: glucocorticoid treatment preserves clinically meaningful functional milestones and reduces rate of disease progression as measured by manual muscle testing and other commonly used clinical trial outcome measures. Muscle Nerve 2013;48:55–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ricotti V, Ridout DA, Scott E, et al. Long-term benefits and adverse effects of intermittent versus daily glucocorticoids in boys with Duchenne muscular dystrophy. J Neurol Neurosurg Psychiatry 2013;84:698–705. [DOI] [PubMed] [Google Scholar]
- 9.Bello L, Gordish-Dressman H, Morgenroth L, et al. Prednisone/prednisolone and deflazacort regimens in the CINRG Duchenne natural history study. Neurology 2015;85:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pegoraro E, Hoffman EP, Piva L, et al. SPP1 genotype is a determinant of disease severity in Duchenne muscular dystrophy. Neurology 2011;76:219–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bello L, Piva L, Barp A, et al. Importance of SPP1 genotype as a covariate in clinical trials in Duchenne muscular dystrophy. Neurology 2012;79:159–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Flanigan KM, Ceco E, Lamar KM, et al. LTBP4 genotype predicts age of ambulatory loss in Duchenne muscular dystrophy. Ann Neurol 2013;73:481–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van den Bergen JC, Hiller M, Bohringer S, et al. Validation of genetic modifiers for Duchenne muscular dystrophy: a multicentre study assessing SPP1 and LTBP4 variants. JNNP 2015;86:1060–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bello L, Kesari A, Gordish-Dressman H, et al. Genetic modifiers of ambulation in the cooperative international neuromuscular research group Duchenne natural history study. Ann Neurol 2015;77:684–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aartsma-Rus A, Van Deutekom JC, Fokkema IF, Van Ommen GJ, Den Dunnen JT. Entries in the Leiden Duchenne muscular dystrophy mutation database: an overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve 2006;34:135–144. [DOI] [PubMed] [Google Scholar]
- 16.Muntoni F, Gobbi P, Sewry C, et al. Deletions in the 5' region of dystrophin and resulting phenotypes. J Med Genet 1994;31:843–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Winnard AV, Mendell JR, Prior TW, Florence J, Burghes AH. Frameshift deletions of exons 3-7 and revertant fibers in Duchenne muscular dystrophy: mechanisms of dystrophin production. Am J Hum Genet 1995;56:158–166. [PMC free article] [PubMed] [Google Scholar]
- 18.Gualandi F, Rimessi P, Trabanelli C, et al. Intronic breakpoint definition and transcription analysis in DMD/BMD patients with deletion/duplication at the 5' mutation hot spot of the dystrophin gene. Gene 2006;370:26–33. [DOI] [PubMed] [Google Scholar]
- 19.Flanigan KM, Dunn DM, von Niederhausern A, et al. Nonsense mutation-associated Becker muscular dystrophy: interplay between exon definition and splicing regulatory elements within the DMD gene. Hum Mutat 2011;32:299–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van den Bergen JC, Ginjaarb HB, Niksa EH, Aartsma-Rus A, Verschuuren JJGM. Prolonged ambulation in Duchenne patients with a mutation amenable to exon 44 skipping. J Neuromuscul Dis 2014;1:91–94. [PubMed] [Google Scholar]
- 21.Pane M, Mazzone ES, Sormani MP, et al. 6-Minute walk test in Duchenne MD patients with different mutations: 12 month changes. PLoS One 2014;9:e83400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Deutekom JC, Janson AA, Ginjaar IB, et al. Local dystrophin restoration with antisense oligonucleotide PRO051. N Engl J Med 2007;357:2677–2686. [DOI] [PubMed] [Google Scholar]
- 23.Goemans NM, Tulinius M, van den Akker JT, et al. Systemic administration of PRO051 in Duchenne's muscular dystrophy. N Engl J Med 2011;364:1513–1522. [DOI] [PubMed] [Google Scholar]
- 24.Flanigan KM, Voit T, Rosales XQ, et al. Pharmacokinetics and safety of single doses of drisapersen in non-ambulant subjects with Duchenne muscular dystrophy: results of a double-blind randomized clinical trial. Neuromuscul Disord 2014;24:16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Voit T, Topaloglu H, Straub V, et al. Safety and efficacy of drisapersen for the treatment of Duchenne muscular dystrophy (DEMAND II): an exploratory, randomised, placebo-controlled phase 2 study. Lancet Neurol 2014;13:987–996. [DOI] [PubMed] [Google Scholar]
- 26.Kinali M, Arechavala-Gomeza V, Feng L, et al. Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: a single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol 2009;8:918–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cirak S, Arechavala-Gomeza V, Guglieri M, et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet 2011;378:595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mendell JR, Rodino-Klapac LR, Sahenk Z, et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann Neurol 2013;74:637–647. [DOI] [PubMed] [Google Scholar]
- 29.Lu QL, Cirak S, Partridge T. What can we learn from clinical trials of exon skipping for DMD? Mol Ther Nucleic Acids 2014;3:e152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hoffman EP, McNally EM. Exon-skipping therapy: a roadblock, detour, or bump in the road? Sci Transl Med 2014;6:230fs14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mendell JR, Goemans N, Lowes LP, et al. Longitudinal effect of eteplirsen vs. historical control on ambulation in DMD. Ann Neurol 2016;79:257–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bushby K, Finkel R, Wong B, et al. Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve 2014;50:477–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Haas M, Vlcek V, Balabanov P, et al. European Medicines Agency review of ataluren for the treatment of ambulant patients aged 5 years and older with Duchenne muscular dystrophy resulting from a nonsense mutation in the dystrophin gene. Neuromuscul Disord 2015;25:5–13. [DOI] [PubMed] [Google Scholar]
- 34.Mazzone E, Vasco G, Sormani MP, et al. Functional changes in Duchenne muscular dystrophy: a 12-month longitudinal cohort study. Neurology 2011;77:250–256. [DOI] [PubMed] [Google Scholar]
- 35.Mazzone ES, Pane M, Sormani MP, et al. 24 month longitudinal data in ambulant boys with Duchenne muscular dystrophy. PLoS One 2013;8:e52512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pane M, Mazzone ES, Sivo S, et al. Long term natural history data in ambulant boys with Duchenne muscular dystrophy: 36-month changes. PLoS One 2014;9:e108205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Humbertclaude V, Hamroun D, Bezzou K, et al. Motor and respiratory heterogeneity in Duchenne patients: implication for clinical trials. Eur J Paediatr Neurol 2012;16:149–160. [DOI] [PubMed] [Google Scholar]
- 38.Jimenez C, Moreno, Eagle M, et al. Impact of three decades of improvement in standards of care for Duchenne muscular dystrophy. Neuromuscul Dis 2015;25:S201–S202. [Google Scholar]
- 39.Prior TW, Bartolo C, Papp AC, et al. Dystrophin expression in a Duchenne muscular dystrophy patient with a frame shift deletion. Neurology 1997;48:486–488. [DOI] [PubMed] [Google Scholar]
- 40.Dwianingsih EK, Malueka RG, Nishida A, et al. A novel splicing silencer generated by DMD exon 45 deletion junction could explain upstream exon 44 skipping that modifies dystrophinopathy. J Hum Genet 2014;59:423–429. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.