

Abstract
The development of an artificial metalloenzyme for CO2 reduction is described. The small-molecule catalyst [NiII (cyclam)]2+ has been incorporated within azurin. Selectivity for CO generation over H+ reduction is enhanced within the protein environment, while the azurin active site metal impacts the electrochemical overpotential and photocatalytic activity. The enhanced catalysis observed for copper azurin suggests an important role for intramolecular electron transfer, analogous to native CO2 reducing enzymes.
Since the industrial age, combustion of fossil fuels has been the primary source of atmospheric greenhouse gases such as carbon dioxide, resulting in marked global effects.1 However, if harnessed appropriately, carbon dioxide presents potential opportunities for utilization as an inexpensive chemical feedstock.2 Currently, progress towards efficient anthropogenic recycling of CO2 has been slow, primarily due to a lack of catalysts,1 while gaseous carbon cycles in nature are balanced using specialized enzymes. Plants assimilate CO2 using RuBisCO, a slow but abundant system, and many microbes rely on highly efficient, redox-active metalloenzymes.1,3,4
One such microbial enzyme is carbon monoxide dehydrogenase (CODH), which uses earth-abundant metals for catalysis.1,5 The anaerobic [NiFe] CODH selectively and reversibly performs the two-electron, proton-coupled interconversion of CO2 to CO with negligible thermodynamic overpotential.3 A functional homodimer, the site of the [NiFe] CODH enzyme is a [4Fe-5S-Ni] C cluster (Fig. 1A).6,7 Supporting [4Fe-4S] clusters, labeled the B and D clusters, serve as electron reservoirs and facilitate rapid electron transfer (ET) between the active site and an external partner, enabling the near-diffusion-limited catalytic rates seen for this enzyme (Fig. 1B). A well-defined secondary coordination sphere is conserved around the C cluster, further enhancing catalytic activity (Fig. 1A).
Figure 1.
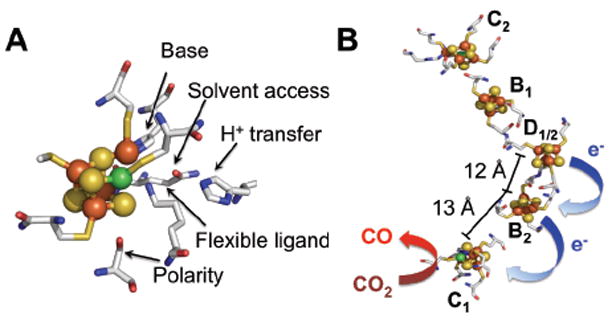
(A) Structure of the CODH/ACS active site C cluster (PDB ID: 1MJG) showing secondary coordination sphere residues with proposed functional roles. (B) CODH cofactor arrangement with center-to-center distances between clusters indicated. Cluster D bridges between two CODH subunits.
To generate improved catalysts for CO2 reduction, a number of synthetic mimics of the [NiFe] CODH active site have been developed.3 However, as is often the case in biomimetic chemistry, purely structural CODH models have not shown any catalytic activity,8 and many of the functional mimics require precious metals or suffer from low turnover numbers.9,10 One model of particular interest is [NiII(cyclam)]2+ ([1], cyclam = 1,4,8,11-tetraazacyclotetradecane), due to its stability and reported selectivity for CO2 reduction over H+ reduction. This selectivity is a challenge for most synthetic catalysts due to the thermodynamic potentials for each reaction; at pH 7, H+ reduction occurs at -413 mV vs. NHE, while CO2 reduction to CO has a potential of -520 mV vs. NHE. Though much of the work characterizing [1] has been carried out using a mercury-based electrode because of its large potential window, recent work by Froehlich and Kubiak has expanded these studies to carbon-based electrodes.11,12 In general, [1] was found to be less active in solution than when adsorbed on the Hg surface. This decreased activity has been attributed to product inhibition and increased conformational flexibility, which may disfavor more active, minority conformations, though interactions between the Hg electrode and [1] may also play a role.12,13
To address some of the aforementioned problems with both natural enzymes and synthetic catalysts, the field of artificial metalloenzymes has been rapidly expanding. Diverse approaches have been employed to generate these engineered systems, including cofactor substitution,14 modification of an existing scaffold, and attachment of an unnatural moiety to a protein,15-17 and there are many successful examples in which new functionality has been introduced into noncatalytic proteins. This approach often provides a straightforward route to tune the reactivity of a system, enabling novel chemical processes or benign reaction conditions that have otherwise been difficult to accomplish.18,19
To develop a system for CO2 reduction with enhanced activity in aqueous media, we have elected to incorporate [1] within a biological scaffold, azurin (Az), as depicted in Figure 2. Az has been used in many protein engineering studies and is robust towards mutation, serving as a tunable platform that imposes a well-defined secondary coordination sphere, which can modulate catalytic efficiency and selectivity.20,21
Figure 2.
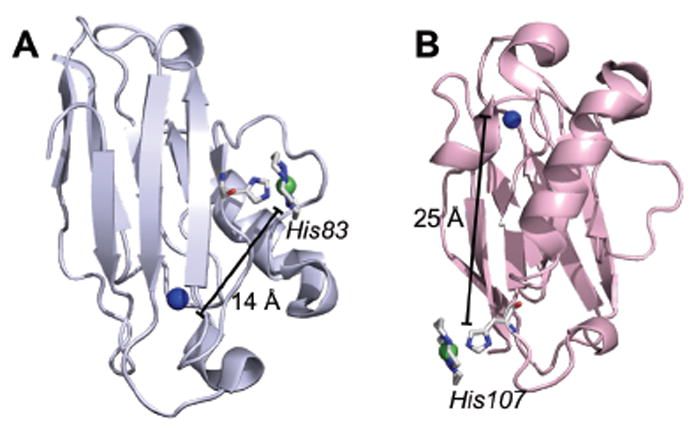
(A) Structure of WT Az (PDB ID: 4AZU) with [1] modeled at the His83 position. (B) Structure of H83Q/Q107H Az (PDB ID: 1R1C) with [1] modeled at the His107 position. Distances between coordinating histidines and native metal-binding site in azurin are shown.
It has been previously established that histidine residues can be used to attach synthetic metal compounds to macromolecules, and [1] is known to bind pyridine,22 which has similar electronic and geometric properties to imidazole. Wild-type (WT) azurin has a single, partially-exposed His residue at position 83 that is available for coordinating [1]; putative secondary sphere interactions around His83 are detailed in Figure S1.23 In addition, a double mutant (H83Q/Q107H) was generated to place [1] in a solvent-exposed environment. Catalyst incorporation within both Cu- and Zn-substituted forms of azurin was accomplished by incubation at a basic pH followed by removal of excess [1] using size-exclusion chromatography. This ensured that all activity seen could be attributed to the bound Az-[1] constructs rather than free [1]. Mutants lacking a surface-exposed His residue did not show coordination of [1] (Fig. S2, ESI).
Due to the weak optical features of [1] (ε ~ 30 M–1 cm–1),24 absorption spectroscopy was only used to quantify labeling efficiencies of the Zn variants (Figs. S3 and S4, ESI). An approximate labeling yield of 40% was achieved for both ZnAz-[1] variants across different batches and conditions; this efficiency was thus also assumed for CuAz-[1] variants due to the similarities in metal charge, ionic radius, and holoprotein structure. Mass spectrometry analysis indicated that the nickel-histidine bond was labile under the experimental measurement conditions, with both [1] and the native metal released using ESI or MALDI ionization techniques.25 Though mass spectrometry proved too harsh to preserve the His-[1] binding, further evidence for the successful labeling of all Az variants is given by the electrochemistry data and activity assays presented below.
All cyclic voltammetry experiments were performed using a glassy carbon (GC) electrode in the solution phase, rather than as a protein film, due to the need for potentials beyond those accessible with a pyrolytic graphite electrode. Figure 3A shows the cyclic voltammograms (CVs) of [1], free Az, and the labeled Az-[1] constructs at positive potentials. In addition to the CuII/I couples observed at ~320 mV, a signal around 700 mV is observed in the labeled samples. This signal corresponds to the NiIII/II couple, which is seen at 690 mV vs. NHE for free [1] under these conditions.12 Interestingly, all of the Az-[1] variants displayed a shift in the NiIII/II couple relative to the model compound (Table S1, ESI). Further, the transitions are largely reversible, as reflected in the narrow peak-to-peak separations (Table S1, Fig. S5, ESI).
Figure 3.
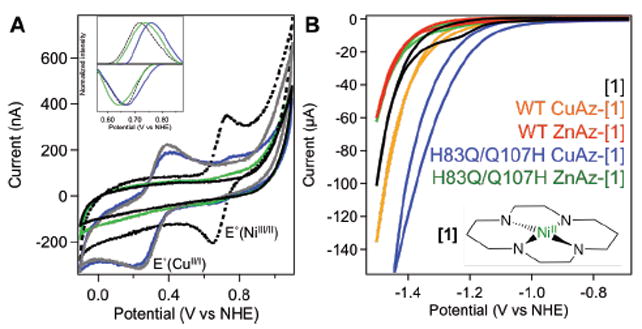
(A) CVs (ν = 10 mV/s) of H83Q/Q107H variants: CuAz (gray), CuAz-[1] (blue), ZnAz (black), and ZnAz-[1] (green), in comparison to free [1] in solution (dotted). (Inset) Baseline-corrected, normalized voltammetric signals of the NiIII/II couple. (B) CVs (ν = 50 mV/s) of Az-[1] constructs as labeled in the figure. (Inset) Molecular structure of [1].
Similar trends in reduction potentials for the hybrid Az-[1] constructs are observed at low potentials. The NiII/I redox couple of [NiII(cyclam)]2+ under an inert atmosphere cannot be observed in aqueous solution due to proton reduction, which masks any non-catalytic signal.12,26 In the presence of CO2, a large catalytic wave suggestive of CO2 reduction is observed for [1] and all labeled proteins (Fig. 3B). Under the experimental conditions, [1] displays a catalytic onset potential of -1.20 V vs. NHE. The onset of catalysis for CO2 reduction by H83Q/Q107H CuAz-[1] occurs approximately +100 mV more positive than free [1] in solution, indicating a decreased overpotential for the reaction. We note that, in contrast to the well-behaved voltammetric response seen for free [1], the azurin-[1] constructs display catalytic currents that do not reach a steady-state limit as the driving force is increased. This type of catalytic wave is commonly seen in solution electrochemistry of proteins,27 as interfacial electron transfer is typically slow relative to protein reorientation and diffusion as well as intrinsic catalytic rates. Because of this non-ideal behavior, the variations in onset potentials for catalysis by the Az-[1] variants relative to [1] were quantified by finding the potential at which the current levels reached those of free [1] at the half-wave potential (Ep = -1.20 V vs. NHE); a detailed description of the process used is given in the Supporting Information (Fig. S6, ESI).
The shifts of the NiIII/II potentials and the onset potentials for CO2 reduction for the semisynthetic Az-[1] enzymes relative to free [1] indicate perturbations in local environment around [1]. Interestingly, the overpotentials for catalysis are governed by the identity of the azurin metal center, with the Cu-substituted scaffolds displaying more positive onset potentials than the Zn- substituted proteins (Table S1, ESI). The H83Q/Q107H CuAz-[1] construct exhibits the lowest overpotential measured and the highest absolute current, though it is possible that, at lower potentials, the increased current stems from higher levels of CO2 and H+ reduction. Since this engineered His107 is more solvent-exposed than the native His83 (Fig. 2), [1] may be more accessible to substrates at this position. A simple increase in activity for both reduction reactions, however, is not sufficient to explain the differences in electrocatalytic activity between H83Q/Q107H CuAz-[1] and H83Q/Q107H ZnAz-[1], as neither displays particularly high currents in the absence of either [1] or CO2 (Figs. S7-S10, ESI). We offer one possible explanation for these differences below.
To further investigate the activity of the semisynthetic Az-[1] constructs, light-driven assays were conducted and gas chromatography (GC) analyses carried out to quantify the reaction products and assess selectivity. Due to the exceptionally low potentials required for catalysis, it has been challenging to identify a suitable ground-state reducing agent for [1] that is operative in aqueous solutions. However, early reports suggested that CO2 reduction by [1] could be photoinitiated using [RuII(bpy)3]2+ and a sacrificial electron donor.24,28 In the excited state, [*RuII(bpy)3]2+ can act as both a strong oxidant and reductant (E°(Ru *II/I) ~ 0.77 V; E°(RuIII/*II) ~ -0.81 V), able to extract or donate an electron to weak reductants or oxidants.29 On this premise, detailed in Figure 4B, assays to probe the homogeneous, solution-phase activity of the Az-[1] constructs were performed. Excitation of [RuII(bpy)3]2+ in the presence of an ascorbate30 quencher generates the highly reducing RuI state (E°(RuII/I) ~ -1.3 V), which reacts with [1] in a bimolecular fashion to generate the singly-reduced NiI-cyclam moiety. This reduced species is able to bind CO2 and initiate the catalytic cycle. Another electron is provided by a second equivalent of RuI, with protons provided by solvent, and H2O and CO are released.
Figure 4.
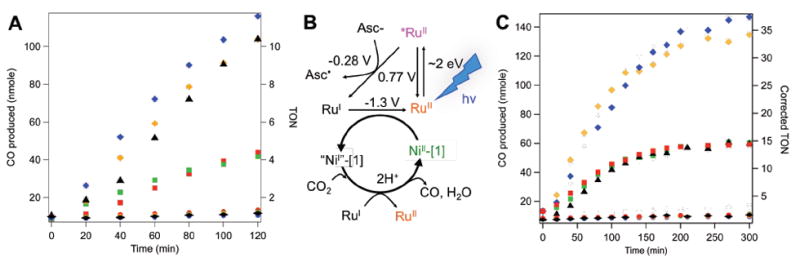
Photoinduced CO2 reduction by [1]-labeled Az variants compared to free [1] in solution. Symbols correspond to labeled CuAz variants (diamonds), labeled ZnAz variants (squares), unlabeled proteins (circles) and free [1] (triangles), with color scheme consistent with that in Figure 3. (A) CO produced (left) and turnover number per protein (TON, right) in the first two hours of the assay. (B) Photocatalytic mechanism for CO2 reduction using [RuII(bpy)3]2+ as a phototrigger. (C) Corrected values for enzyme turnover number over five hours considering the 40% labeling efficiency of Az.
During the assay, the sample headspace was analyzed as a function of time (Fig. 4A). There were no detectable amounts of CO or H2 produced if any of the following components were missing from the assay: [RuII(bpy)3]2+, ascorbate, CO2, or light. CO was not produced when assays were performed with Cu- or Zn-substituted protein controls lacking [1]. In general, photocatalytic experiments reproduced the electrochemical trends. Assuming quantitative labeling efficiencies, H83Q/Q107H CuAz-[1] produced 116 nmol of CO after a two-hour irradiation period, which compares favorably to the 104 nmol produced by free [1] (Figs. 4 and S11, ESI). The ZnAz-[1] variants performed similarly to each other, but showed a significant decrease in CO production and TON when compared to [1] and the analogous CuAz analogs. Rates of CO evolution began to decrease after approximately two hours, and the activity of all samples reached a plateau following five hours of irradiation (Fig. S12, ESI).
The most notable improvement that the protein scaffold confers towards the activity of [1] is selectivity for CO2 reduction relative to H+ reduction, which was determined by measuring the headspace H2 and CO levels concomitantly (Fig. S13, ESI) and is denoted the selectivity ratio (SR, moles CO/moles H2). At the onset of the photoassay, all Az-[1] constructs display levels of selectivity that far exceed the SR of 0.2 that is observed for free [1] in solution.24,28 Within the first 20 minutes of irradiation, the SRs for WT ZnAz-[1] and WT CuAz-[1] are as large as 3.5 and 1.9, respectively (Fig. S14, Table S1, ESI). The corresponding SRs for H83Q/Q107H ZnAz-[1] and H83Q/Q107H CuAz-[1] are lower than those of WT Az, with values of 0.7 and 0.6, respectively, but are still greater than free [1]. However, by the conclusion of the two-hour irradiation period, all samples displayed increasingly poor selectivity (Fig. S14, ESI). Because low but detectable amounts of H2 were generated in the photoassays by Cu- or Zn-substituted Az lacking [1], all reported measurements of H2 have been corrected for the corresponding background level of H+ reduction.
Developing a catalyst with a bias towards CO2 reduction rather than proton reduction poses significant challenges due to the thermodynamic preference for the latter. Nature is able to supersede this thermodynamic bias by controlling kinetics, an approach that also largely governs the catalytic bias in synthetic systems. Compared to many systems, [1] is a relatively poor catalyst for H2 evolution due to the near-planarity of the ligand; distortion of the compound towards a tetrahedral geometry, which is required for efficient two-electron chemistry and H2 evolution, is energetically disfavored. Constrained within a protein environment, the flexibility of [1] can be further restricted. This constraint would be most pronounced by coordination of [1] to His83, which is partially buried within a hydrophobic patch. Consistent with this hypothesis, the highest levels of selectivity are observed for the WT CuAz-[1] and WT ZnAz-[1] constructs, with SRs exceeding 1. As well as restricting conformational flexibility, this solvent-excluded position may also protect [1] from bimolecular interactions, which would disfavor a homolytic mechanism for H2 evolution.31 The decrease in selectivity of the Az-[1] constructs to the level of free [1] at the conclusion of the assay may be due to deactivation pathways or slow dissociation of [1] from the protein following prolonged exposure at neutral pH.
Considering the 40% labeling efficiency rather than assuming quantitative labeling, the relative activities of the Az-[1] constructs can be adjusted for catalyst concentration (Fig. 4C). Applying this correction, both ZnAz-[1] constructs exhibit comparable CO2 reduction activity to that of free [1] in solution, indicating that any decrease in substrate or reductant accessibility caused by incorporation into the Az protein scaffold is only minor relative to the overall catalyst activity. Notably, both Cu-substituted constructs exhibit significantly higher levels of activity than free [1]. The electrochemistry data are also consistent with these observations, demonstrating higher catalytic currents and lower overpotentials for the CuAz-[1] constructs relative to the ZnAz-[1] variants. Taken together, these results indicate that that the presence of a redox-active center in the Az active site enhances catalytic activity.
The attachment site for [1] is relatively close to the native metal center, only 14 Å from His83 and 25 Å from the His107 variant. These distances are similar to those seen in native CODH, where the nearby B and D clusters serve as electron storage sites for the catalytically active cluster. Like the [4Fe-4S] clusters, which have a higher reduction potential than the [4Fe-5S-Ni] C cluster, the CuII/IAz redox potential is above that of [1]. Thus, at the low potentials used for electrocatalysis or in the presence of ascorbate, the copper center will first be reduced. CO2 binding to [1] occurs after reduction of the Ni center and is likely closely followed by a second, required electron.12,26 Intramolecular ET between CuIAz and [1] during catalysis could provide a means for the additional reducing equivalent to be rapidly shuttled into the active site, occurring on a faster timescale than a bimolecular reduction process.
Surprisingly, the site of attachment of [1] to the protein scaffold seems to confer little advantage in the photocatalytic assays, despite the fact that the His107 position is significantly further from the Cu site than His83. However, even at this distance, ET should occur on the millisecond timescale,32 much faster than the overall turnover frequencies of the different variants. That the same levels of activity are seen for both CuAz-[1] constructs, but not the ZnAz-[1] constructs, suggests that this ET process serves to enhance catalysis overall but is not the rate-determining step. Mechanistic studies to investigate the structures of distinct catalytic intermediates form the basis of ongoing work in our laboratory.
In conclusion, we have developed semisynthetic enzymes for CO2 reduction based on incorporation of a simple inorganic complex, [NiII(cyclam)]2+, into a robust protein scaffold, azurin. The activity of these systems has been characterized electrochemically and photochemically. Relative to free [NiII(cyclam)]2+, increased CO generation was observed for the constructs containing a redox-active ion, copper, in the metal binding site of the scaffold. This suggests that the endogenous metal center can act as a functional mimic for the iron-sulfur clusters in CODH, storing electrons until needed, and highlights the necessity of these electron transfer cofactors for the high levels of activity seen in the natural systems. Further, the initial selectivity of the azurin-[NiII(cyclam)]2+ constructs for CO generation over H+ reduction is significantly enhanced relative to free [NiII(cyclam)]2+ and depends on the site of incorporation within the protein scaffold, indicating an important role for the specific catalyst environment. Within this system, we are now well-poised to identify critical contributions of the secondary coordination sphere towards the selectivity and efficiency of catalytic CO2 reduction by [1], as well as begin to address the catalytic mechanism through trapping and characterization of intermediates. Overall, this work establishes a new, molecular platform upon which to rationally develop increasingly active, robust constructs for CO2 reduction.
Supplementary Material
Acknowledgments
We would like to acknowledge Anastasia Manesis for insightful discussions. This work was supported by the OSU Department of Chemistry and Biochemistry and an OSU IMR Facility Seed Grant. C.R.S. is supported by an NIH Chemistry-Biology Interface Training Program Fellowship (GM08512).
Footnotes
Electronic Supplementary Information (ESI) available: Materials and methods, synthetic and sample preparation protocols and characterization, UV-Vis traces, and further electrochemical and photochemical controls. See DOI: 10.1039/x0xx00000x
References
- 1.Appel AM, et al. Chem Rev. 2013;113(8):6621–6658. doi: 10.1021/cr300463y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aresta M, et al. Reaction Mechanisms in Carbon Dioxide Conversion. Springer Berlin Heidelberg; Berlin, Heidelberg: 2016. [Google Scholar]
- 3.Can M, Armstrong FA, Ragsdale SW. Chem Rev. 2014;114(8):4149–4174. doi: 10.1021/cr400461p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thauer RK, et al. R Nat Rev Microbiol. 2008;6(8):579–591. doi: 10.1038/nrmicro1931. [DOI] [PubMed] [Google Scholar]
- 5.Hille R. Chem Rev. 1996;96(7):2757–2816. doi: 10.1021/cr950061t. [DOI] [PubMed] [Google Scholar]
- 6.Drennan C. Curr Opin Struct Biol. 2003;13(2):220–226. doi: 10.1016/s0959-440x(03)00038-1. [DOI] [PubMed] [Google Scholar]
- 7.Dobbek H, et al. Science. 2001;293(5533):1281–1285. doi: 10.1126/science.1061500. [DOI] [PubMed] [Google Scholar]
- 8.Tan GO, et al. Proc Natl Acad Sci. 1992;89(10):4427–4431. doi: 10.1073/pnas.89.10.4427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fujita E. Coord Chem Rev. 1999;185:373–384. [Google Scholar]
- 10.Majumdar A. Dalton Trans. 2014;43(32):12135. doi: 10.1039/c4dt00729h. [DOI] [PubMed] [Google Scholar]
- 11.Beley M, et al. J Am Chem Soc. 1986;108(24):7461–7467. doi: 10.1021/ja00284a003. [DOI] [PubMed] [Google Scholar]
- 12.Froehlich JD, Kubiak CP. Inorg Chem. 2012;51(7):3932–3934. doi: 10.1021/ic3001619. [DOI] [PubMed] [Google Scholar]
- 13.Song J, et al. Inorg Chem. 2014;53(14):7500–7507. doi: 10.1021/ic500829p. [DOI] [PubMed] [Google Scholar]
- 14.Slater JW, Shafaat HS. J Phys Chem Lett. 2015;6(18):3731–3736. doi: 10.1021/acs.jpclett.5b01750. [DOI] [PubMed] [Google Scholar]
- 15.Petrik ID, et al. Curr Opin Chem Biol. 2014;19:67–75. doi: 10.1016/j.cbpa.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ward TR. Acc Chem Res. 2011;44(1):47–57. doi: 10.1021/ar100099u. [DOI] [PubMed] [Google Scholar]
- 17.Lewis JC. Curr Opin Chem Biol. 2015;25:27–35. doi: 10.1016/j.cbpa.2014.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pàmies O, et al. Adv Synth Catal. 2015;357(8):1567–1586. [Google Scholar]
- 19.Palomo JM, Filice M. Biotechnol Adv. 2015;33(5):605–613. doi: 10.1016/j.biotechadv.2014.12.010. [DOI] [PubMed] [Google Scholar]
- 20.McLaughlin, et al. J Am Chem Soc. 2012;134(48):19746–19757. doi: 10.1021/ja308346b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Manesis AC, Shafaat HS. Inorg Chem. 2015;54(16):7959–7967. doi: 10.1021/acs.inorgchem.5b01103. [DOI] [PubMed] [Google Scholar]
- 22.Saravanakumar D, et al. ChemSusChem. 2012;5(4):634–636. doi: 10.1002/cssc.201100481. [DOI] [PubMed] [Google Scholar]
- 23.Bjerrum MJ, et al. J Bioenerg Biomembr. 1995;27(3):295–302. doi: 10.1007/BF02110099. [DOI] [PubMed] [Google Scholar]
- 24.Craig CA, et al. J Phys Chem. 1990;94(20):7957–7960. [Google Scholar]
- 25.Lay JO. Mass Spectrom Rev. 2001;20(4):172–194. doi: 10.1002/mas.10003. [DOI] [PubMed] [Google Scholar]
- 26.Froehlich JD, Kubiak CP. J Am Chem Soc. 2015;137(10):3565–3573. doi: 10.1021/ja512575v. [DOI] [PubMed] [Google Scholar]
- 27.Sommer DJ, et al. Chem Commun. 2014;50(100):15852–15855. doi: 10.1039/c4cc06700b. [DOI] [PubMed] [Google Scholar]
- 28.Grant JL, et al. J Chem Soc Dalton Trans. 1987;(9):2105–2109. [Google Scholar]
- 29.Prier CK, et al. Chem Rev. 2013;113(7):5322–5363. doi: 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakada A, et al. Inorg Chem. 2015;54(4):1800–1807. doi: 10.1021/ic502707t. [DOI] [PubMed] [Google Scholar]
- 31.Collin JP, et al. Inorg Chem. 1988;27(11):1986–1990. [Google Scholar]
- 32.Gray HB, Winkler JR. Chem Phys Lett. 2009;483(1-3):1–9. doi: 10.1016/j.cplett.2009.10.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.