

Abstract
Objective:
To critically re-evaluate cases diagnosed as adult neuronal ceroid lipofuscinosis (ANCL) in order to aid clinicopathologic diagnosis as a route to further gene discovery.
Methods:
Through establishment of an international consortium we pooled 47 unsolved cases regarded by referring centers as ANCL. Clinical and neuropathologic experts within the Consortium established diagnostic criteria for ANCL based on the literature to assess each case. A panel of 3 neuropathologists independently reviewed source pathologic data. Cases were given a final clinicopathologic classification of definite ANCL, probable ANCL, possible ANCL, or not ANCL.
Results:
Of the 47 cases, only 16 fulfilled the Consortium's criteria of ANCL (5 definite, 2 probable, 9 possible). Definitive alternate diagnoses were made in 10, including Huntington disease, early-onset Alzheimer disease, Niemann-Pick disease, neuroserpinopathy, prion disease, and neurodegeneration with brain iron accumulation. Six cases had features suggesting an alternate diagnosis, but no specific condition was identified; in 15, the data were inadequate for classification. Misinterpretation of normal lipofuscin as abnormal storage material was the commonest cause of misdiagnosis.
Conclusions:
Diagnosis of ANCL remains challenging; expert pathologic analysis and recent molecular genetic advances revealed misdiagnoses in >1/3 of cases. We now have a refined group of cases that will facilitate identification of new causative genes.
Diagnosis of neurodegenerative diseases in younger adults is challenging due to the relative rarity of the problem, the heterogeneous causes, and the frequent absence of noninvasive diagnostic clues, raising the question of brain biopsy. The neuronal ceroid lipofuscinoses (NCLs) are a group of storage diseases presenting from infancy to adulthood.1 The stored material is largely made up of subunit c of mitochondrial adenosine triphosphate synthase or saposin proteins A and D—that is, of proteins so hydrophobic that they require special mechanisms for their breakdown and disposal.2
Adult-onset NCL (ANCL) is a particularly demanding diagnostic problem. Childhood NCLs have well-characterized clinical patterns typically involving brain and eye, abundant storage in readily accessible peripheral tissues such as skin, and largely solved molecular genetic causes.3–5 In contrast, ANCL can present in a variety of ways with progressive myoclonus epilepsy, dementia, or motor disorders and shows limited storage in peripheral tissue. Stored material is usually only found in a subset of neurons, with the accumulation of age-related lipofuscin often confused with pathologic storage.6–13 Furthermore, molecular genetic characterization of ANCL is at an early stage. Hindered by the small number of cases, and the clinical and pathologic challenges, ANCL is poorly understood.
Kufs disease is the best-known form of ANCL; it differs from most childhood-onset forms because there is no retinal involvement, and the inheritance can be either recessive or dominant. It is widely accepted that the uncommon teenage-onset cases without retinal involvement are grouped with adult-onset cases. The clinical presentation is variable with 2 broad forms identified. Kufs type A presents with progressive myoclonus epilepsy, whereas Kufs type B presents with dementia and motor signs.6,13 Recessive mutations in CLN614–16 and dominant mutations in DNAJC517–19 can cause Kufs type A; Kufs type B can be caused by recessive mutations in CTSF.20,21 Rare cases of ANCL with retinal involvement are described due to mutations in PPT1 (CLN1),22,23 CLN5,24,25 and GRN.26 Finally, some patients with CLN3 mutations present with neurologic manifestations in adult life, on a background of visual failure in childhood.27
Despite these important molecular discoveries, many reported cases that receive a diagnosis of ANCL remain unsolved. As ANCL is rare, we formed a consortium to combine putative unsolved cases from centers around the world and analyzed them as one large cohort. We aimed to refine the clinical spectrum of late-onset NCL by implementing an expert clinical and pathologic review process, followed by consensus diagnosis. Here we report our somewhat surprising results, in particular, the frequent misdiagnosis of a broad spectrum of neurologic disorders as Kufs disease. Our eventual aim is to discover the remaining molecular causes of ANCL.
METHODS
Participants.
We established the ANCL Gene Discovery Consortium encompassing researchers with clinical, neuropathologic, and molecular genetic expertise in ANCL from the United Kingdom, Europe, United States, Canada, and Australia.
Consortium members pooled 47 unrelated, unsolved cases into a shared database. These cases had been referred to individual Consortium researchers for molecular genetic studies over a 20-year period because of diagnosis or putative diagnosis of ANCL. Clinicopathologic support for an ANCL diagnosis varied considerably in these cases that remained unsolved despite study by a variety of molecular genetic and enzymatic assays over time. For familial cases, only the proband was included. No attempt was made to sequence all known NCL genes before entry into the study.
Standard protocol approvals, registrations, and patient consents.
Local institutional review boards at each contributing site approved this research. Participants or their guardians provided informed consent.
Diagnostic criteria.
We developed initial diagnostic criteria based on a combination of clinical and pathologic features. These criteria were based on review of the literature and the clinical experience of Consortium members.6–13
Clinical and pathologic review of cases.
Strenuous attempts were made to source detailed clinical data from treating clinicians. We deliberately used broad clinical criteria (table 1) that allowed for retinal involvement.
Table 1.
Adult-onset neuronal ceroid lipofuscinosis: Clinical and pathologic criteria

Neuropathologic diagnosis remains the gold standard for this group of disorders. A panel of 3 expert neuropathologists (J.F.S., S.C., G.W.A.) developed criteria to classify the pathologic data as definite, probable, or possible NCL (table 1). Images of the pathologic material, where available, were subsequently shared with each of the 3 neuropathologists. They provided independent opinions, which were then discussed, and a consensus on the classification reached.
Overall classification of cases.
Each case was given a final classification of definite ANCL, probable ANCL, possible ANCL, or not ANCL based on clinical and pathologic data. This occurred via regular Consortium teleconferences where the clinical criteria and pathologic classification were integrated to determine an overall classification (table 2).
Table 2.
Adult-onset neuronal ceroid lipofuscinosis (ANCL) classification criteria incorporating both clinical and pathologic criteria (see table 1) for 47 putative cases

Molecular genetic studies.
Screening for known ANCL genes (CLN6, CTSF, DNAJC5, GRN, and PPT1) was performed by both Sanger sequencing and high-throughput sequencing technologies in the majority of the 16 cases (detailed in table e-1 on the Neurology® Web site at Neurology.org). Genomic DNA was extracted from venous blood using standard methods. Briefly, for Sanger sequencing, coding exons and flanking intronic regions including splice sites of the 3 genes were PCR amplified using primers designed for all isoforms with reference human gene transcripts (NCBI Gene; http://www.ncbi.nlm.nih.gov/; primers available on request). Amplification reactions were cycled on a Veriti Thermal Cycler (Applied Biosystems, Carlsbad, CA) and bidirectional sequencing was completed using BigDye v3.1 Terminator Cycle Sequencing Kit (Applied Biosystems). Sequencing products were resolved using a 3730xl DNA Analyzer (Applied Biosystems). Sequence chromatograms were compared to published cDNA sequence with nucleotide changes detected using Codon Code Aligner (CodonCode Corporation, Dedham, MA).
A custom gene panel (CGP), designed to target coding regions (6.88 Mb) of 3,616 OMIM genes with known phenotypes including ANCL, and whole-exome sequencing (WES) was also performed for the majority (table e-1). DNA enrichment was performed according to the manufacturer's protocol using either SeqCap EZ Choice Library (Roche Nimblegen, Madison, WI) for the CGP or Nimblegen SeqCap V3 (Roche Nimblegen) for WES. DNA sequencing was performed on the captured barcoded DNA libraries using either SOLiD 4 System (Applied Biosystems) or Illumina HiSeq 1500. Data analysis was performed as previously described.28 The data were inspected for variations in known ANCL genes and the search for novel genes is ongoing.
Molecular diagnosis of cases initially suspected of having ANCL, where other disorders were later established, were performed at a number of commercial and research laboratories using standard techniques.
RESULTS
Of the 47 Consortium cases, 16 cases fulfilled the Consortium's clinical and pathologic criteria for definite, probable, or possible ANCL disease (table 2). Of the 16 cases, 8 (2 definite) presented with seizures as a main feature, usually with a progressive myoclonus epilepsy (Type A Kufs). The remaining 8 cases (3 definite) presented with dementia and motor signs (Type B Kufs). None of the ANCL cases had retinal involvement. The mean age at onset was 35 years (median 35.5 years). There were 2 clusters; 6 had onset in the second decade of life and 10 in the fourth decade or later. Both Type A and Type B cases were represented in the 2 age clusters. Two cases fell outside of our arbitrary 12- to 60-years range for disease onset (onset at 10 and 62 years); these cases were considered clinically atypical and classified overall as possible ANCL (table 2).
A family history suggestive of dominant inheritance was found in 4/16 ANCL cases; in 12, there was no family history. DNAJC5 mutations were not found in the 4 dominant cases or the 10 other cases that had this gene sequenced. CLN6 and CTSF, known causes of recessive ANCL, were sequenced in 15 cases and 1 case had probable pathogenic compound heterozygous CLN6 variants identified. This patient was classified as definite ANCL with a Type A presentation. This leaves a subset of 15 genetically unsolved ANCL cases (4 definite).
Our criteria for ANCL were not met in 31/47 (66%) cases, including 2 cases previously published in detail as having ANCL.29,30 In 10 of these 31 cases, detailed review with further investigation established definitive alternate diagnoses (table 3). The main reason for misdiagnosis as ANCL was overinterpretation of normal age-related lipopigment as pathologic storage material (figure). Six cases had features suggesting that ANCL was not the correct diagnosis. Two of these had storage material without the characteristics of the lipopigment seen in NCL, but a specific diagnosis could not be made. Two had other neuropathologic abnormalities, again without leading to a specific diagnosis. In 2 cases, biopsy material reported as suggestive of NCL was considered nonspecific on review, and the clinical features were atypical. In the remaining 15 cases, the clinical and pathologic data were inadequate for classification.
Table 3.
Summary of 10 cases with an initial diagnosis of adult-onset neuronal ceroid lipofuscinosis (ANCL) in which an alternate diagnosis was later confirmed
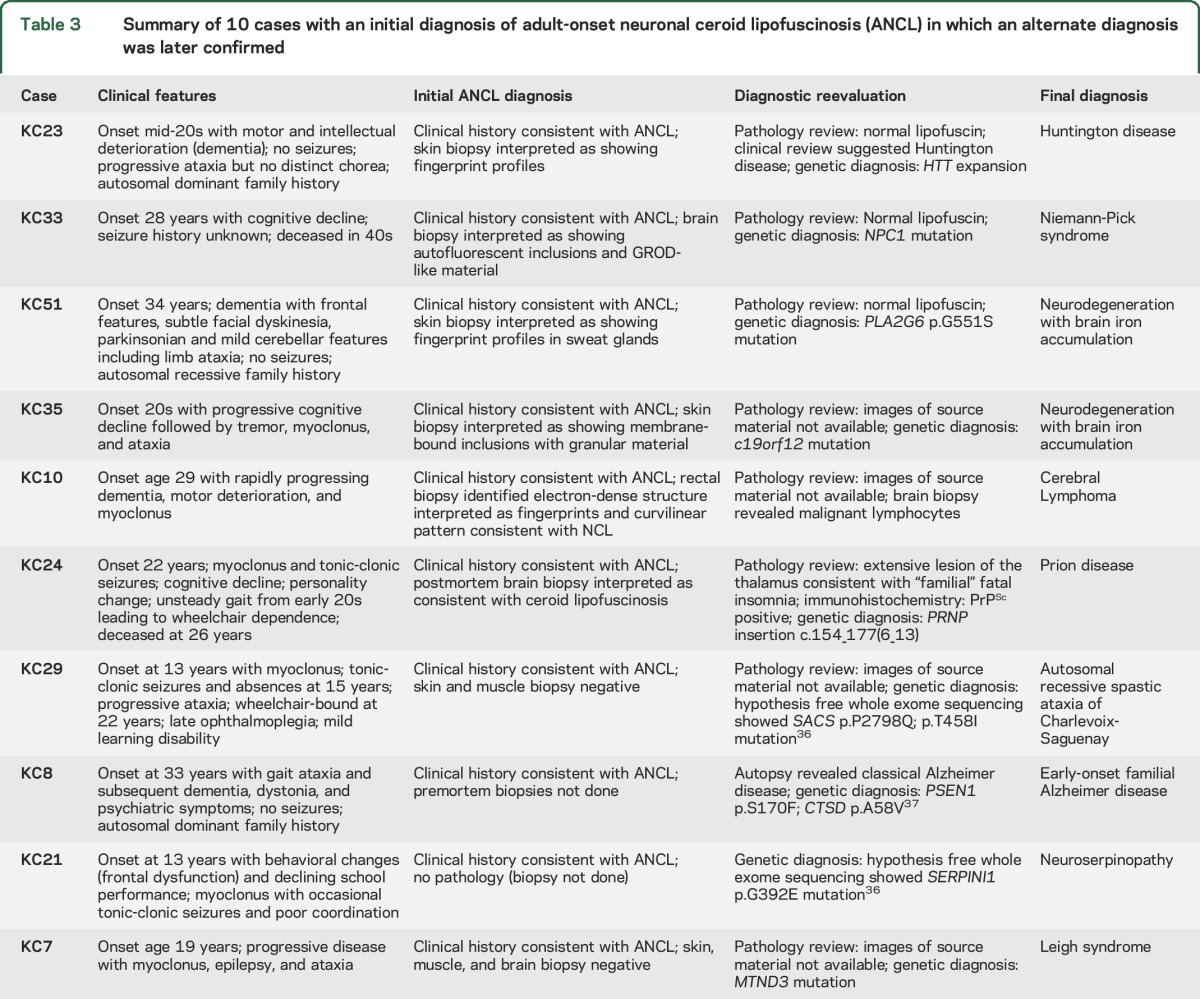
Figure. Pathologic diagnosis and misdiagnosis of adult-onset neuronal ceroid lipofuscinosis (ANCL).

(A) Electron micrograph of KC33 shows one of many deposits of lipofuscin in a cortical neuron. These deposits were originally considered to represent granular osmiophilic deposits (GRODs), leading to a diagnosis of ANCL. Lipid vacuoles, as seen here, are numerous in lipofuscin, but not in GRODs, and the granules in lipofuscin are coarser and less uniform (bar = 5 μm). The patient had cognitive decline beginning at age 28 years; NPC1 mutation was later found (table 3). (B) This electron micrograph of KC15 shows fingerprint profiles from an eccrine secretory cell. The basic paired parallel line pattern even at this magnification tends to appear as a single wide slightly fuzzy line, but the spacing, the wheeling ranks, and the focal crystalline pattern where the ranks intersect (at the left of the asterisk) are very characteristic (bar = 1 μm). Disease onset was at 18 years with stimulus-sensitive and action-induced myoclonus, dementia, and parkinsonism on the background of normal development. This case met out criteria for classification as definite ANCL. The molecular basis remains unknown.
DISCUSSION
In this study of 47 cases considered to have ANCL, we found that the diagnosis could be supported in only 16 cases; of these, 9 were considered possible cases. Moreover, ANCL was confidently excluded in 16 cases, of which 10 had secure alternative diagnoses established. The classification criteria developed by the Consortium were useful in formalizing the approach to problematic cases and has highlighted the challenges and subtleties in diagnosing ANCL.
The pathologic diagnosis is challenging and is compounded by the relative rarity of these disorders such that most neuropathologists have very limited experience with the condition. In general, misdiagnosis occurred by overinterpretation of ultrastructural findings in biopsy samples, with failure to distinguish normal lipofuscin that accumulates with age, from abnormal lipopigment seen in NCL, which may be very limited in ANCL.6–13 In children, this distinction is easier, contrasting abundant storage in disease with minimal normal age-related pigment. Rather than relying on the amount of lipopigment, accurate diagnosis depends on analysis of the particular ultrastructural features.7,31 In all late-onset neuronal storage diseases, only a subset of neurons may be involved. This becomes important when it comes to electron microscopy on a brain biopsy. If the blocks have not been trimmed to include involved neurons, the study may be limited to normal neurons that inevitably contain lipofuscin, and confusion with ANCL may result.
Importantly, the removal of these cases from the cohort helps simplify the apparent clinical and pathologic heterogeneity of ANCL. The clinical profile of our ANCL cases fit broadly into the previously described categories of presentation as progressive myoclonus epilepsy (Type A) and dementia with motor disturbances (Type B)6; the single case that was solved molecularly with a CLN6 mutation had a Type A presentation, as has been previously described.14
The degree of genetic heterogeneity of ANCL remains unclear. It is already known that contrary to the childhood forms, which are essentially all recessive disorders, the adult forms can have either recessive or dominant inheritance.6,32,33 While it is now possible to survey the whole exome (or genome) for genetic variation with high-throughput sequencing technology, determining which of the many thousand variants identified are pathogenic remains challenging. However, this is still a good option for treating clinicians and may avoid the need for brain biopsy should a plausible variant be found in one of the known ANCL genes or in a gene known to cause an alternative late-onset neurologic disease. The efficacy of genetic testing for diagnostic purposes will only improve as further ANCL genes are discovered. At this point in time, pathologic diagnosis remains the gold standard practice.
Future research into the underlying genetic etiology of the unsolved ANCL will be supported by the removal of misdiagnosed cases that would hinder these important efforts. By pooling together unrelated ANCL cases that putatively may share the same causative gene, the genomic search space can be considerably narrowed. This ongoing work, resulting in a deeper understanding of the molecular genetic basis, will provide improved guidance for the accurate and early diagnosis of ANCL. We anticipate that the recently accepted nomenclature for the NCL34 will be expanded as new genes are identified.
Supplementary Material
GLOSSARY
- ANCL
adult-onset neuronal ceroid lipofuscinosis
- CGP
custom gene panel
- NCL
neuronal ceroid lipofuscinosis
- WES
whole-exome sequencing
Footnotes
Supplemental data at Neurology.org
Contributor Information
Collaborators: ANCL Gene Discovery Consortium, Umberto Aguglia, Danielle M Andrade, Francesca Bisulli, Sylvia Boesch, Laura Canafoglia, Hans-Henrik M Dahl, Rainer Ehling, Silvana Franceschetti, Antonio Gambardella, Michael F Gonzales, Renate Kalnins, Anthony Lang, Eliza Lewandowska, Laura Licchetta, Tiago A Mestre, Michela Morbin, Chantal F Morel, Klary E Niezen-Koning, Filippo M Santorelli, Alessandro Simonati, and Paolo Tinuper
AUTHOR CONTRIBUTIONS
Dr. Berkovic drafted/revised the manuscript for content, contributed to study concept/design, contributed to acquisition of the data, performed analysis/interpretation of the data, and provided study supervision and coordination. Dr. Staropoli revised the manuscript for content, contributed to study concept/design, contributed to acquisition of the data, performed analysis/interpretation of the data, and provided study coordination. Dr. Carpenter revised the manuscript for content and performed analysis/interpretation of the data. K.L. Oliver drafted/revised the manuscript for content, contributed to acquisition of the data, performed analysis/interpretation of the data, and provided study coordination. Dr. Kmoch revised the manuscript for content, contributed to study concept/design, contributed to acquisition of the data, and performed analysis/interpretation of the data. G.W. Anderson revised the manuscript for content and performed analysis/interpretation of the data. J.A. Damiano revised the manuscript for content and performed analysis/interpretation of the data. Dr. Hildebrand revised the manuscript for content and performed analysis/interpretation of the data. Dr. Sims revised the manuscript for content, contributed to acquisition of the data, and performed analysis/interpretation of the data. Dr. Cotman revised the manuscript for content, contributed to acquisition of the data, and performed analysis/interpretation of the data. Dr. Bahlo revised the manuscript for content and performed analysis/interpretation of the data. Dr. Smith revised the manuscript for content and performed analysis/interpretation of the data. M. Cadieux-Dion revised the manuscript for content and performed analysis/interpretation of the data. Dr. Cossette revised the manuscript for content and contributed to acquisition of the data. I. Jedličková revised the manuscript for content and performed analysis/interpretation of the data. A. Přistoupilová revised the manuscript for content and performed analysis/interpretation of the data. Dr. Mole revised the manuscript for content, contributed to study concept/design, contributed to acquisition of the data, performed analysis/interpretation of the data, and provided study coordination.
STUDY FUNDING
S.F.B. was supported by a National Health and Medical Research Council Program Grant (ID: 628952). S.K., I.J., and A.P. are funded by Charles University institutional programs PRVOUK-P24/LF1/3, UNCE 204011, and SVV2016/260148 and by the project LQ1604 NPU II from the Ministry of Education of the Czech Republic. This work was specifically supported by grant 15-28208A from the Ministry of Health of the Czech Republic and GA UK No. 269615 (I.J.) and No. 1402213 (A.P.). Instrumental support for exome and gene panel sequencing was provided by the Genomic facility in Motol University Hospital in Prague (OPPK.CZ.2.16/3.100/24022). S.E.M. received specific funding towards the work of the Rare NCL Gene Consortium and Kufs disease from the USA Batten Disease Support and Research Association. S.L.C. is funded by the National Institutes of Health: National Institute of Neurologic Disorders & Stroke (R01NS073813). M.C.-D. is supported by the Fonds de Recherche du Québec–Santé (FRQS).
DISCLOSURE
S. Berkovic has served on scientific advisory boards for UCB and Janssen-Cilag; serves on the editorial boards of Lancet Neurology and Epileptic Disorders and the Advisory Board of Brain; may accrue future revenue on pending patent WO61/010176: Therapeutic Compound that relates to discovery of PCDH19 gene as the cause of familial epilepsy with mental retardation limited to females; is one of the inventors listed on a patent held by Bionomics Inc. on diagnostic testing using the SCN1A gene, WO2006/133508; has received speaker honoraria from UCB; has received unrestricted educational grants from UCB, Janssen-Cilag, and Sanofi-Aventis; and receives/has received research support from the National Health and Medical Research Council of Australia and NINDS. J. Staropoli, S. Carpenter, K. Oliver, S. Kmoch, G. Anderson, J. Damiano, M. Hildebrand, K. Sims, S. Cotman, M. Bahlo, K. Smith, M. Cadieux-Dion, P. Cossette, I. Jedličková, A. Přistoupilová, and S. Mole report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Mole SE, Williams RE, Goebel HH. The Neuronal Ceroid Lipofuscinoses (Batten Disease). 2nd ed. Oxford: Oxford University Press; 2011. [Google Scholar]
- 2.Palmer DN, Barry LA, Tyynela J, Cooper JD. NCL disease mechanisms. Biochim Biophys Acta 2013;1832:1882–1893. [DOI] [PubMed] [Google Scholar]
- 3.Kousi M, Lehesjoki AE, Mole SE. Update of the mutation spectrum and clinical correlations of over 360 mutations in eight genes that underlie the neuronal ceroid lipofuscinoses. Hum Mutat 2012;33:42–63. [DOI] [PubMed] [Google Scholar]
- 4.Mole SE. NCL Mutation and Patient Database [online]. Available at: http://www.ucl.ac.uk/ncl/mutation.shtml. Accessed November 1, 2015. [Google Scholar]
- 5.Kmoch S, Stranecky V, Emes RD, Mitchison HM. Bioinformatic perspectives in the neuronal ceroid lipofuscinoses. Biochim Biophys Acta 2013;1832:1831–1841. [DOI] [PubMed] [Google Scholar]
- 6.Berkovic SF, Carpenter S, Andermann F, et al. Kufs' disease: a critical reappraisal. Brain 1988;111:27–62. [DOI] [PubMed] [Google Scholar]
- 7.Anderson G, Elleder M, Goebel HH. Morphological diagnostic and pathological considerations. In: Mole SE, Williams RE, Goebel HH, eds. The Neuronal Ceroid Lipofuscinoses (Batten Disease). 2nd ed. Oxford: Oxford University Press; 2011:35–49. [Google Scholar]
- 8.Anderson GW, Goebel HH, Simonati A. Human pathology in NCL. Biochim Biophys Acta 2013;1832:1807–1826. [DOI] [PubMed] [Google Scholar]
- 9.Carpenter S. Morphological diagnosis and misdiagnosis in Batten-Kufs disease. Am J Med Genet Suppl 1988;5:85–91. [DOI] [PubMed] [Google Scholar]
- 10.Goebel HH, Braak H. Adult neuronal ceroid-lipofuscinosis. Clin Neuropathol 1989;8:109–119. [PubMed] [Google Scholar]
- 11.Goebel HH, Sharp JD. The neuronal ceroid-lipofuscinoses: recent advances. Brain Pathol 1998;8:151–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pasquinelli G, Cenacchi G, Piane EL, et al. The problematic issue of Kufs disease diagnosis as performed on rectal biopsies: a case report. Ultrastruct Pathol 2004;28:43–48. [PubMed] [Google Scholar]
- 13.Sadzot B, Reznik M, Arrese-Estrada JE, Franck G. Familial Kufs' disease presenting as a progressive myoclonic epilepsy. J Neurol 2000;247:447–454. [DOI] [PubMed] [Google Scholar]
- 14.Arsov T, Smith KR, Damiano J, et al. Kufs disease, the major adult form of neuronal ceroid lipofuscinosis, caused by mutations in CLN6. Am J Hum Genet 2011;88:566–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Andrade DM, Paton T, Turnbull J, et al. Mutation of the CLN6 gene in teenage-onset progressive myoclonus epilepsy. Pediatr Neurol 2012;47:205–208. [DOI] [PubMed] [Google Scholar]
- 16.Canafoglia L, Gilioli I, Invernizzi F, et al. Electroclinical spectrum of the neuronal ceroid lipofuscinoses associated with CLN6 mutations. Neurology 2015;85:316–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cadieux-Dion M, Andermann E, Lachance-Touchette P, et al. Recurrent mutations in DNAJC5 cause autosomal dominant Kufs disease. Clin Genet 2013;83:571–575. [DOI] [PubMed] [Google Scholar]
- 18.Noskova L, Stranecky V, Hartmannova H, et al. Mutations in DNAJC5, encoding cysteine-string protein alpha, cause autosomal-dominant adult-onset neuronal ceroid lipofuscinosis. Am J Hum Genet 2011;89:241–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Velinov M, Dolzhanskaya N, Gonzalez M, et al. Mutations in the gene DNAJC5 cause autosomal dominant Kufs disease in a proportion of cases: study of the Parry family and 8 other families. PLoS One 2012;7:e29729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith KR, Dahl HH, Canafoglia L, et al. Cathepsin F mutations cause type B Kufs disease, an adult-onset neuronal ceroid lipofuscinosis. Hum Mol Genet 2013;22:1417–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Di Fabio R, Moro F, Pestillo L, et al. Pseudo-dominant inheritance of a novel CTSF mutation associated with type B Kufs disease. Neurology 2014;83:1769–1770. [DOI] [PubMed] [Google Scholar]
- 22.van Diggelen OP, Thobois S, Tilikete C, et al. Adult neuronal ceroid lipofuscinosis with palmitoyl-protein thioesterase deficiency: first adult-onset patients of a childhood disease. Ann Neurol 2001;50:269–272. [DOI] [PubMed] [Google Scholar]
- 23.Ramadan H, Al-Din AS, Ismail A, et al. Adult neuronal ceroid lipofuscinosis caused by deficiency in palmitoyl protein thioesterase 1. Neurology 2007;68:387–388. [DOI] [PubMed] [Google Scholar]
- 24.Sleat DE, Ding L, Wang S, et al. Mass spectrometry-based protein profiling to determine the cause of lysosomal storage diseases of unknown etiology. Mol Cell Proteomics 2009;8:1708–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xin W, Mullen TE, Kiely R, et al. CLN5 mutations are frequent in juvenile and late-onset non-Finnish patients with NCL. Neurology 2010;74:565–571. [DOI] [PubMed] [Google Scholar]
- 26.Smith KR, Damiano J, Franceschetti S, et al. Strikingly different clinicopathological phenotypes determined by progranulin-mutation dosage. Am J Hum Genet 2012;90:1102–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lauronen L, Munroe PB, Jarvela I, et al. Delayed classic and protracted phenotypes of compound heterozygous juvenile neuronal ceroid lipofuscinosis. Neurology 1999;52:360–365. [DOI] [PubMed] [Google Scholar]
- 28.Park EJ, Grabinska KA, Guan Z, et al. Mutation of Nogo-B receptor, a subunit of cis-prenyltransferase, causes a congenital disorder of glycosylation. Cell Metab 2014;20:448–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reif A, Schneider MF, Hoyer A, et al. Neuroleptic malignant syndrome in Kufs' disease. J Neurol Neurosurg Psychiatry 2003;74:385–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zini A, Cenacchi G, Nichelli P, et al. Early-onset dementia with prolonged occipital seizures: an atypical case of Kufs disease. Neurology 2008;71:1709–1712. [DOI] [PubMed] [Google Scholar]
- 31.Carpenter S, Karpati G, Andermann F, et al. The ultrastructural characteristics of the abnormal cytosomes in Batten-Kufs' disease. Brain 1977;100:137–156. [DOI] [PubMed] [Google Scholar]
- 32.Boehme DH, Cottrell JC, Leonberg SC, Zeman W. A dominant form of neuronal ceroid-lipofuscinosis. Brain 1971;94:745–760. [DOI] [PubMed] [Google Scholar]
- 33.Burneo JG, Arnold T, Palmer CA, Kuzniecky RI, Oh SJ, Faught E. Adult-onset neuronal ceroid lipofuscinosis (Kufs disease) with autosomal dominant inheritance in Alabama. Epilepsia 2003;44:841–846. [DOI] [PubMed] [Google Scholar]
- 34.Williams RE, Mole SE. New nomenclature and classification scheme for the neuronal ceroid lipofuscinoses. Neurology 2012;79:183–191. [DOI] [PubMed] [Google Scholar]
- 35.Ferlazzo E, Gasparini S, Pasquinelli G, et al. Usefulness of rectal biopsy for the diagnosis of Kufs disease: a controlled study and review of the literature. Eur J Neurol 2012;19:1331–1336. [DOI] [PubMed] [Google Scholar]
- 36.Muona M, Berkovic SF, Dibbens LM, et al. A recurrent de novo mutation in KCNC1 causes progressive myoclonus epilepsy. Nat Genet 2015;47:39–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ehling R, Nosková L, Stranecký V, et al. Cerebellar dysfunction in a family harboring the PSEN1 mutation co-segregating with a cathepsin D variant p.A58V. J Neurol Sci 2013;326:75–82. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.