

Abstract
Colonization resistance refers to the ability of the colonic microbiota to prevent invasion by pathogens including Clostridium difficile. In a recent article, Buffie et al. (2014) have demonstrated that a single metabolic cluster present in the normal, colonic microflora is responsible for preventing C. difficile invasion of healthy hosts.
Infections caused by Clostridium difficile, a Gram-positive, spore-forming, strict anaerobe, have risen dramatically over the past decade. The colonic microflora normally provides protection against invading pathogens. However, treatment of patients with broad-spectrum antibiotics often leads to “collateral damage” to the microflora, thus leaving an opening to C. difficile infection (CDI). Conventional treatment of CDI is with antibiotics such as vancomycin, metronidazole, or fidaxomicin (Koenigsknecht and Young, 2013). While these antibiotics prevent C. difficile growth, they have no effect on spores, and their use prevents full reconstitution of the normal microflora. Thus, due to the continued presence of antibiotic-resistant spores in the colon and the environment, treated patients frequently relapse with CDI (Tannock et al., 2010).
Endospores are dormant forms of bacteria that are resistant to multiple environmental stresses. Upon receiving appropriate environmental signals, these spores germinate and resume active, vegetative growth. C. difficile spore germination is triggered by a mixture of glycine (Sorg and Sonenshein, 2008) and certain bile acids (e.g., taurocholic acid)—small, amphipathic, cholesterol-based compounds that aid in the absorption of fats and cholesterol during digestion.
Upon entry into the colonic environment, bile acids are modified through two processes. First, bile salt hydrolases (BSHs), which are expressed on the surfaces of many colonic commensals, enzymatically cleave the conjugated amino acid from the steroid core (e.g., taurocholic acid is cleaved to taurine + cholic acid) (Ridlon et al., 2006). The second process is performed by the products of a metabolic cluster present in only a small subset of the colonic microbiota. These organisms actively take up deconjugated bile acids and, through a series of enzymatic steps, remove the 7α-hydroxyl group (e.g., cholic acid is converted to deoxycholic acid) (Figure 1) (Ridlon et al., 2006). The actions of these bacteria lead to the complete conversion of the host-derived bile acids (primary bile acids) to secondary bile acids (Ridlon et al., 2006; Theriot et al., 2014).
Figure 1. C. scindens 7α-dehydroxylation Prevents C. difficile Growth.
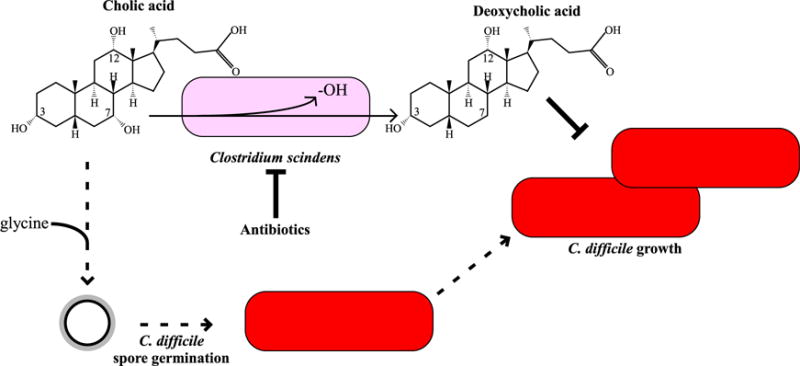
Upon entry into the colonic environment, the 7α-hydroxyl group on cholic acid is removed to produce deoxycholic acid by select members of the colonic microbiota, including C. scindens (Ridlon et al., 2006). Deoxycholic acid inhibits C. difficile vegetative growth (Sorg and Sonenshein, 2008). During antibiotic treatment, the organisms that 7α-dehydroxylate bile acids are killed, leading to an increased abundance of cholic acid derivatives in the colonic environment (Theriot et al., 2014). These compounds, in combination with glycine (Sorg and Sonenshein, 2008), germinate C. difficile spores, which can then grow out as vegetative cells and cause disease.
Previous studies have indicated that secondary bile acids, specifically deoxycholic acid, are inhibitors of C. difficile vegetative cell growth under laboratory conditions, leading to the hypothesis that the microbial conversion of cholic-to-deoxycholic acid in the gut creates an environment that is refractory to C. difficile vegetative growth (Sorg and Sonenshein, 2008; Wilson, 1983). In fact, changes in the colonic microbial population that occur upon antibiotic treatment abolish the ability to 7α-dehydroxylate bile acids and lead to a concomitant increase in the abundance of primary bile acids that induce germination by C. difficile spores (Figure 1) (Theriot et al., 2014). Furthermore, these primary bile acids are not toxic to vegetative C. difficile (Sorg and Sonenshein, 2008).
Now, in a recent article published in Nature, Buffie and colleagues (Buffie et al., 2014) provide evidence that a single metabolic cluster present in healthy microflora correlates with protection against C. difficile invasion. By analyzing those bacteria that are present in the human and mouse colonic environment of C. difficile-sensitive and -resistant individuals, the authors conclude that the presence of a metabolic cluster capable of 7α-dehydroxylation of primary bile acids correlates with protection against CDI. They then show that colonizing mice with Clostridium scindens, a bacterium that is capable of 7α-dehydroxylation of primary bile acids, protects against CDI.
C. difficile, a member of Clostridium cluster XI, does not 7α-dehydroxylate bile acids. In contrast, C. scindens, belonging to cluster XIVa, does, and its bile acid 7α-dehydroxylation pathway has been studied in detail (Ridlon et al., 2006). Though C. scindens and other bile acid-metabolizing bacteria completely convert the primary bile acids that enter the colonic environment to secondary bile acids, their numbers in the colonic environment are low compared to other organisms. Furthermore, they appear to be sensitive to vancomycin and metronidazole, which are frequently used to treat CDI in patients. Interestingly, fidaxomicin does not inhibit growth of cluster XIVa Clostridia (Tannock et al., 2010), and fidaxomicin-treated patients have a lower recurrence rate than do patients treated with vancomycin or metronidazole.
The present study is a major leap forward in defining the mechanisms of colonization resistance and uses the mouse model of C. difficile infection that has gained popularity for the study of CDI. While mice express similar bile acids as humans, they also synthesize muricholic acids, which are not found in humans. Muricholic acids inhibit germination by C. difficile spores and also inhibit C. difficile vegetative growth with similar MIC values as deoxycholic acid (Francis et al., 2013). Thus, it is unclear why the bacteria capable of 7α-dehydroxylation are needed to protect mice against C. difficile infection when other growth-inhibitory molecules are present in the colonic environment. Additional studies using organisms with bile acid profiles more similar to those of humans will clarify these issues. For example, the Syrian hamster synthesizes a bile acid spectrum that is similar to humans and has been used as a model of C. difficile infection for 35 years.
Bile acids are clearly important for germination by C. difficile spores in the laboratory—and possibly in the host (Giel et al., 2010; Sorg and Sonenshein, 2008; Theriot et al., 2014). However, while it is assumed that cholic acid derivatives are actual in vivo germinants for C. difficile spores, this has not been convincingly demonstrated. Furthermore, deoxycholic acid has not been conclusively shown to be the molecule that protects against CDI. The data supporting the hypotheses that cholic acid is an in vivo spore germinant and that the microbial conversion of cholic acid to deoxycholic acid by C. scindens is the mechanism by which C. difficile growth is inhibited have relied on the use of cholestyramine as a bile acid-binding polymer (Buffie et al., 2014; Giel et al., 2010). Importantly, this polymer functions as a potent anion exchange resin and has also been shown to bind vancomycin (Weiss, 2009). Thus, while cholestyramine has bile acid-binding properties, it cannot be excluded that, in these studies, it also binds other proteins and/or molecules to protect against C. difficile invasion or in vitro growth.
In summary, this study represents a significant finding for C. difficile pathogenesis, mechanisms of colonization resistance, and, possibly, new probiotic therapies to limit recurring infections. Important in this regard, fecal bacteriotherapy has gained popularity as an alternative treatment to antibiotics for recurring CDI and has a nearly 100% cure rate (Koenigsknecht and Young, 2013). However, the microbial diversity in the fecal samples is variable due to the use of different donors. The Buffie study raises the possibility that a single bile acid-metabolizing organism, or a cocktail of bile acid metabolizing organisms, could be used as a probiotic to limit recurring C. difficile disease. Further studies on the metabolic conversion of bile acids, or other compounds, by the colonic microflora will provide a more complete understanding of the mechanisms of resistance against C. difficile colonization.
Acknowledgments
The author thanks Drs. Abraham L. Sonenshein and Matthew Sachs for comments during the preparation of this manuscript. The author acknowledges support by the American Heart Association National Scientist Development grant (No. 11SDG7160013) and support from the National Institute of Allergy and Infectious Diseases (award numbers R21AI07640 and 1R56AI108987). The content is solely the responsibility of the author and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases or the National Institutes of Health.
References
- Buffie CG, Bucci V, Stein RR, McKenney PT, Ling L, Gobourne A, No D, Liu H, Kinnebrew M, Viale A, et al. Nature. 2014 doi: 10.1038/nature13828. Published online October 22, 2014. http://dx.doi.org/10.1016/10.1038/nature13828. [DOI] [PMC free article] [PubMed]
- Francis MB, Allen CA, Sorg JA. PLoS ONE. 2013;8:e73653. doi: 10.1371/journal.pone.0073653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giel JL, Sorg JA, Sonenshein AL, Zhu J. PLoS ONE. 2010;5:e8740. doi: 10.1371/journal.pone.0008740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenigsknecht MJ, Young VB. Curr Opin Gastroenterol. 2013;29:628–632. doi: 10.1097/MOG.0b013e328365d326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridlon JM, Kang DJ, Hylemon PB. J Lipid Res. 2006;47:241–259. doi: 10.1194/jlr.R500013-JLR200. [DOI] [PubMed] [Google Scholar]
- Sorg JA, Sonenshein AL. J Bacteriol. 2008;190:2505–2512. doi: 10.1128/JB.01765-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tannock GW, Munro K, Taylor C, Lawley B, Young W, Byrne B, Emery J, Louie T. Microbiology. 2010;156:3354–3359. doi: 10.1099/mic.0.042010-0. [DOI] [PubMed] [Google Scholar]
- Theriot CM, Koenigsknecht MJ, Carlson PE, Jr, Hatton GE, Nelson AM, Li B, Huffnagle GB, J ZL, Young VB. Nature communications. 2014;5:3114. doi: 10.1038/ncomms4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss K. Int J Antimicrob Agents. 2009;33:4–7. doi: 10.1016/j.ijantimicag.2008.07.011. [DOI] [PubMed] [Google Scholar]
- Wilson KH. J Clin Microbiol. 1983;18:1017–1019. doi: 10.1128/jcm.18.4.1017-1019.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]