

Abstract
Importance
Sebaceous neoplasms (SN) define the Muir-Torre syndrome (MTS) variant of Lynch syndrome (LS), which is associated with increased risk for colon and other cancers necessitating earlier and more frequent screening to reduce morbidity and mortality. Immunohistochemical (IHC) staining for mismatch repair (MMR) proteins in SN can be used to screen for LS, but data on subsequent germline genetic testing to confirm LS diagnosis is limited.
Objective
To characterize the utility of IHC screening of SN in identification of germline MMR mutations confirming LS
Design
Retrospective study
Setting
Two academic cancer centers
Participants
86 adult patients referred for clinical genetics evaluation after diagnosis of SN
Main Outcomes and Measures
Results of tumor IHC testing and germline genetic testing were reviewed to determine positive predictive value and sensitivity of IHC in diagnosis of LS. Clinical variables, including age at diagnosis of SN, clinical diagnostic criteria for LS and MTS, and family history characteristics were compared between mutation carriers and non-carriers.
Results
25 (29.1%) of 86 patients with SN had germline MMR mutations confirming LS. Among 77 patients with IHC testing on SN, 38 (49.4%) had loss of staining of one or more MMR proteins, and 14 had germline MMR mutations. IHC correctly identified 13/16 MMR mutation carriers, corresponding to 81.3% sensitivity. Ten of 12 (83.3%) patients with > 1 SN had MMR mutations. 52% of MMR mutation carriers did not meet clinical diagnostic criteria for LS, and 44% did not meet the clinical definition of MTS.
Conclusions and Relevance
IHC screening of SN is effective in identifying patients with germline MMR mutations and can be used as a first line test when LS is suspected. Abnormal IHC, including absence of MSH2, is not diagnostic of LS and should be interpreted cautiously in conjunction with family history and germline genetic testing. Use of family history to select patients for IHC screening has significant limitations, suggesting that universal IHC screening of SN merits further study. Clinical genetics evaluation is warranted for patients with any of the following: abnormal IHC, normal IHC with personal or family history of other LS-associated neoplasms, or multiple SN.
Introduction
Lynch syndrome (LS) is caused by germline mutations in genes involved in the DNA mismatch repair (MMR) pathway (MLH1, MSH2, MSH6, PMS2 and TACSTD1/EPCAM), and is associated with increased risk for several cancers including colorectal (CRC), endometrial, ovarian, gastric, biliary tract, pancreatic, urinary tract, and central nervous system tumors (2). The association of sebaceous neoplasms of the skin (SN), including sebaceous carcinomas (SC) and sebaceous adenomas (SA), with internal malignancy was first described in the dermatology literature in 1967 and referred to as Muir-Torre syndrome (MTS)(3, 4). In 1981, Lynch et al reported SN in three LS kindreds with pathogenic germline mutations in MMR genes, further defining MTS as a clinical variant of LS (5).
Identification of patients with LS is clinically valuable given availability of risk reducing strategies, including earlier and more frequent colonoscopy and prophylactic hysterectomy and bilateral salpingo-oophorectomy, to reduce cancer related morbidity and mortality (6, 7). Routine screening of CRC and endometrial cancers for evidence of MMR deficiency, including presence of microsatellite instability (MSI) and/or absent expression of the MMR proteins by immunohistochemistry (IHC), has shown that 2–4% of CRC and 1–5% of endometrial cancers are associated with LS (8, 9). This universal tumor screening approach has better sensitivity than clinical criteria for identifying patients with LS and has the potential to be cost effective if individuals and their at risk relatives can be identified and screened to reduce morbidity and mortality (10). Given the experience with CRC and endometrial cancers, routine screening of SN for MMR deficiency to identify LS has been proposed (11–13). Several studies have examined the use of MSI and IHC to screen unselected SNs and have shown prevalence of MMR deficiency ranging from 25–60% (12–17). However, most of these studies had limited or no information on germline genetic test results; thus data regarding the prevalence of germline MMR mutations confirming LS among individuals with SN, as well as sensitivity and specificity of SN tumor testing, are limited.
Our objective was to characterize the utility of MSI and IHC screening of SN in identification of germline MMR mutations confirming LS. We analyzed data on all patients with SN evaluated at two large clinical cancer genetics programs to examine outcomes of tumor screening and germline genetic testing.
Methods
Permission for research was approved by the Institutional Review Boards of the University of Michigan Comprehensive Cancer Center (UMCCC) and the Dana Farber Cancer Institute (DFCI). Patients consented to participate in DNA banking registries granting access to de-identified medical and family history and use of this information for publication. Subjects were identified through review of patients enrolled in research registries of the cancer genetics clinics at UMCCC and DFCI from January 2000 through September 2012. Individuals with diagnoses of SC, SA, sebaceoma and sebaceous epithelioma were included in the analysis; sebaceous hyperplasia was excluded. Clinical demographic information including age, SN location and subtype, and other cancer diagnoses were recorded for each subject. Results from clinically-performed tumor testing, including MSI and IHC for DNA MMR proteins, and germline genetic testing for mutations in MLH1, MSH2, MSH6, PMS2, and/or TACSTD1/EPCAM genes were reviewed. All clinical testing was completed over the 12 year study period in laboratories with College of American Pathologists (CAP) accreditation and Clinical Laboratory Improvement Amendments (CLIA) licensure, in accordance with accepted standards. Four generation family histories were evaluated for cancer diagnoses among relatives and categorized according to Amsterdam I/II clinical diagnostic criteria for LS (18, 19). For patients not meeting Amsterdam criteria, PREMM (1,2,6) risk scores (20) were calculated to estimate risk for MMR mutation based on personal and family history of LS related cancers. Colon adenomas were not incorporated. Cancers in relatives were confirmed where records were available.
To evaluate differences in demographic, clinical, and familial characteristics between groups we conducted t-tests and Wilcoxon-Mann-Whitney tests for continuous variables and Chi-Square and Fisher’s Exact tests for categorical variables. Bonferroni corrections were applied to p-values to adjust for multiple comparisons.
Results
Eighty-six patients with SN from 86 independent families presented for genetic evaluation during the study period (66 UMCCC, 20 DFCI). A total of 107 SNs were diagnosed among the 86 patients, including 52 SA, 45 SC, 5 sebaceomas, 3 sebaceous epitheliomas, and 2 SN without further information. Twelve patients had more than one sebaceous lesion. Pathogenic or suspected pathogenic germline MMR mutations confirming LS were identified in 25 (29.1%) of 86 patients with SN referred for genetic evaluation. Mutations identified included 18 MSH2, 5 MLH1, 1 MSH6, and 1 PMS2. Two variants of uncertain significance (VUS) were identified (1 MSH2, 1 MSH6).
Nine patients presenting on the basis of SN diagnosis were found to carry MMR mutations without any testing of their SN tumors. Reasons testing of SN was not completed included: known MMR mutation in the family (3/9); tumor testing performed on a different LS related tumor (4/9); and family history meeting Amsterdam I/II clinical diagnostic criteria warranting direct germline genetic testing (2/9).
The remaining 77 patients underwent tumor analysis for MMR deficiency on a single SN. MSI testing could not be completed in 37 (40.5%) SN tumors due to insufficient sample (individual MSI results reported in Tables 1 and 2); IHC analysis was completed on all 77 SN tumors. IHC analysis found 38 (49.4%) of 77 SN had absent expression of one or more MMR proteins. Twenty-seven had absent expression of MSH2 or MSH2/MSH6; 9 (33.3%) of these were found to carry germline MSH2 mutations. Nine had absent expression of MLH1 or MLH1/PMS2; 4 (44.4%) of these had germline MLH1 mutations. Two patients had equivocal IHC staining for MSH6 only; one had a pathogenic MSH6 mutation and one had a VUS in MSH6. None of the samples demonstrated isolated absence of PMS2 expression. In total, 14/38 patients with abnormal IHC on SN were confirmed to carry pathogenic germline MMR mutations, for a positive predictive value of 36.8% for IHC (Figure 1, Table 1, Table 2).
Table 1.
Mismatch Repair Mutations Identified Based on Sebaceous Neoplasm
| Gender | Cancer History | Family History of LS Associated Cancers# | PREMM (1,2,6) (%) | Tumor Analysis | Germline Mutation | ||
|---|---|---|---|---|---|---|---|
| Sebaceous Neoplasm (age) | Other (age) | MSI | IHC absent | ||||
| M | SC (67) | UR (64) RCC (65) |
Amsterdam I/II | – | H | MLH1/PMS2 | MLH1 c.1981delGCCA (p.Agg1LfsX3) |
| M | SN (57) | CRC (39) | FA - CRC (60) | 31 | – | MLH1/PMS2 | MLH1 c.1852delAAG (p.K618del) |
| M | SC, SA (53, 53) | – | PGM - CRC (73) | <5 | H | MLH1/PMS2 | MLH1 del exon 16–19 |
| F | SC (72) | EN (59) DCIS (52) | FA - CRC (40, 55) | 39.9 | – | MLH1/PMS2 | MLH1 del exon 15 |
| F | SC, SC (49, 49) | EN(47) | MO – EN(50), CRC (66) | 64 | H | MSH2/MSH6 | MSH2 del exons 3–8 |
| M | SA, SA, SA (47, 47, 47) | – | FA - CRC (43) PU - CRC (38) PU - CRC (49) PU- CRC (52) |
45.6 | H | MSH2/MSH6 | MSH2 c.1408delG (p.V470X) |
| F | SC (49) | ACC (23) FH (38) |
MO - OV (54); MA - CRC (73) | <5 | H | MSH2/MSH6 | MSH2 c.942+3A>T |
| F | SA, SC (40, 40) | – | MA - CRC (59); MA - GBM (79) | <5 | H | MSH2/MSH6 | MSH2 c.1861C>T (p.R621X) |
| M | SA,SA, SA (48, 48, 48) | – | MO - OV (42) MGM - OV (57) |
<5 | – | MSH2/MSH6 | MSH2 c.1676ins300b |
| M | SC (66) | – | MO - CRC ×4 | 13.9 | – | MSH2/MSH6 | MSH2 c.942+3A>T |
| M | SN, SN (53, 53) | ACC (53) | Amsterdam I/II | – | – | MSH2 | MSH2 c.2131C>T (p.R711X) |
| F | SA (47) | ACC (42) | PGF - CRC (40s) | <5 | – | MSH2/MSH6 | MSH2 c.792+1G>A |
| M | SC, SC (60, 60) | CRC (50) | Amsterdam I/II | – | – | MSH2/MSH6 | MSH2 c.321delT (p. G108EfsX66) |
| M | SA (55) | – | MO - CRC (56) | 8.3 | – | NORMALˆ | MSH2 c.1511–41G>C (VUS**) |
| F | SA (66) | EN (42) | MO - EN(50);BL(91) | 14.6 | – | NORMAL | MSH2 c.989dupT (p.N331EfsX2) |
| M | SC, SC (57) | – | MU - CRC (81) | <5 | – | MSH6* | MSH6 c.3182T>C (p.L1061P) (VUS**) |
| M | SA (54) | – | S – CRC(53) | 9.5 | – | MSH6* | MSH6 c.3103C>T (p.R1035X) |
| F | SA (51) | – | FA - CRC (71) PA - CRC (60s) |
<5 | – | NORMAL | PMS2 c.137G>A (p.S46N)+ |
LS associated cancers - colorectal, endometrial, ovarian, stomach, small intestine, urinary tract/kidney, bile duct, glioblastoma multiforme, sebaceous gland, pancreas
PMS2 stain read as “weak”; germline testing was completed for all mismatch repair genes
MSH6 stains read as equivocal or uninterpretable; MLH1, MSH2 stains were normal
VUS – variant of uncertain significance
reported as “suspected pathogenic” by clinical laboratory
Legend; LS - Lynch Syndrome; M-male; F-female; SC - sebaceous carcinoma, SA - sebaceous adenoma; SN- sebaceous neoplasm; ACC- adrenocortical carcinoma; FH - fibrous histiocytoma; EN - Endometrial adenocarcinoma; CRC-colorectal cancer; UR – ureteral cancer; RCC - renal cell carcinoma; OV – ovarian cancer; BL – bladder cancer; GBM – glioblastoma multiforme; FA – father; PGM – paternal grandmother; PGM – paternal grandmother; PA – paternal aunt; PU – paternal uncle; MO – mother; MGM – maternal grandmother; MA – maternal aunt; MSI- microsatellite instability; H-high (>30% markers demonstrate instability); L-low (1–30% of markers demonstrate instability); S- stable (0% of markers demonstrate instability) IHC - immunohistochemistry
Amsterdam I/II - 3 or more individuals diagnosed with LS associated tumor, 2 successive generations affected, at least 1 individual diagnosed under age 50
Table 2.
Patients with no germline MMR mutations detected
| Gender | Cancer history | Family History of LS associated cancers#(age) | PREMM,1,2,6 (%) | Tumor analysis | ||
|---|---|---|---|---|---|---|
| Sebaceous Neoplasm (age) | Other (age) | MSI | IHC absent | |||
| M | SC (77) | – | – | 5.1 | H | MLH1/PMS2 |
| F | SC (60) | – | MGF – CRC(88) | <5 | – | MLH1 |
| M | SA (60) | – | MGF – CRC(70s) MU – PAN(73) |
<5 | S | MLH1/PMS2 |
| M | SA (62) | – | MO – CRC(75) | <5 | H | MSH2/MSH6 |
| M | SC (63) | CRC (25) Bladder (61) |
Amsterdam I | – | – | MSH2/MSH6 |
| F | SA (70) | – | MO – RCC(63) MGF – STO(57) |
<5 | – | MSH2/MSH6 |
| F | SA (45) | – | FA – CRC(71) PGM – CRC(92) |
<5 | – | MSH2 |
| M | SN (77) | – | – | 5.1 | H | MSH2/MSH6 |
| M | SA (50) | – | PA – CRC(30s) | 9.6 | – | MSH2/MSH6 |
| M | SC (61) | – | – | 5.1 | H | MSH2/MSH6 |
| M | SA (59) | – | MO – EN(82) | 10.1 | – | MSH2/MSH6 |
| M | SN (77) | CLL/SLL (73) | S – PAN(56) | 6.9 | H | MSH2/MSH6 |
| F | SN (81) | PU – STO(?) | < 5 | – | MSH2/MSH6 | |
| M | SN (73) | BL(64) | MO – PAN(70) | 6.9 | – | MSH2/MSH6 |
| M | SA (75) | – | – | 5.1 | H | MSH2/MSH6 |
| F* | SA (71) | – | S – CRC(70), BL(70) | < 5 | – | MSH2/MSH6 |
| M | SN (68) | – | – | 5.1 | – | MSH2/MSH6 |
| M | SC (60) | – | – | 5.1 | H | MSH2/MSH6 |
| F | SA (76) | – | – | 5.1 | – | MSH2/MSH6ˆ |
| M | SC (63) | RCC (52) | MGM – STO(80s) | 5.9 | H | MSH2/MSH6 |
| M | SA(75) | – | MO – BL(80s) | 8.1 | H | MSH2/MSH6 |
| M | SC (69) | – | MU – BL(?) | 5.9 | – | MLH1/PMS2 |
| M | SA (74) | PRO (60) | B – CRC(50) MU – CRC(73) |
< 5 | H | MLH1/PMS2 |
| M | SA (62) | SCC (51) | M – CRC(65) MGM – RCC(70s) |
6.4 | – | Normal |
| F | SA (73) | – | S – CRC(54) MO – CRC(52) |
15.4 | S | Normal |
| M | SC (73) | CRC (69) | – | 6.6 | S | Normal |
| F | SC (85) | CRC (69); EndoSarc (79) | – | 12.3 | S | Normal |
| M | SA (51) | – | PA – OV(50) PGM – PAN(82) |
8.1 | S | Normal |
| F | SN (51) | – | B – CRC(56) | <5 | S | Normal |
| M | SA, SN (73) | – | B – CRC(62), BL(61) PGF – CRC(82) |
<5 | S | Normal |
| M | SC (32) | – | MA – EN(63) | 7.1 | L | Normal |
LS associated cancers - colorectal, endometrial, ovarian, stomach, small intestine, urinary tract/kidney, bile duct, glioblastoma multiforme, sebaceous gland, pancreas
organ transplant recipient
Partial loss of MSH2
Legend; M-male; F-female; SC - sebaceous carcinoma, SA - sebaceous adenoma; SN- sebaceous neoplasm; CRC-colorectal cancer; CLL/SLL- chronic lymphocytic leukemia/small lymphocytic leukemia; BL – bladder cancer; RCC - renal cell carcinoma; PRO – prostate cancer; SCC – squamous cell carcinoma; EndoSarc - endometrial sarcoma; MGF – maternal grandfather; MU – maternal uncle; MO – mother; FA – father; PGM – paternal grandmother; PA – paternal aunt; B – brother; S – sister; PGF – paternal grandfather; MSI- microsatellite instability; H-high (>30% markers demonstrate instability); L-low (1–30% of markers demonstrate instability); S- stable (0% of markers demonstrate instability) IHC - immunohistochemistry
Figure 1.
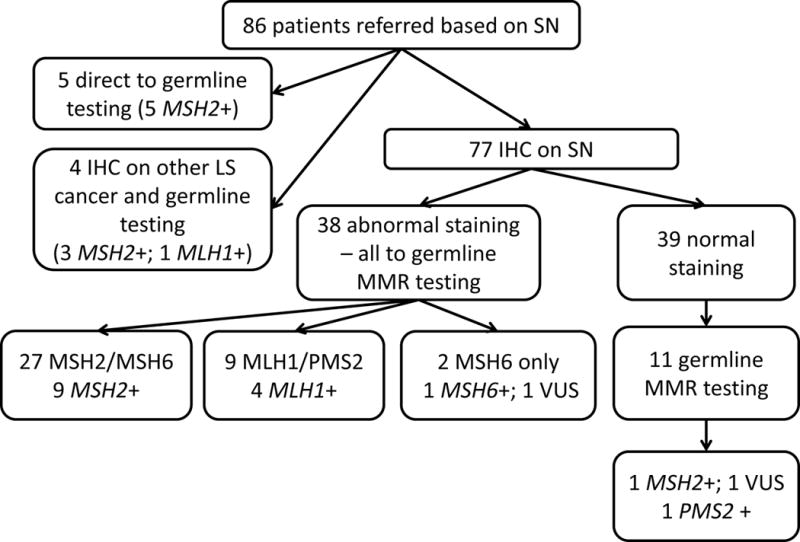
Molecular Testing Outcomes – A flow chart showing results of clinical testing completed (including IHC and germline genetic testing) for patients referred to genetics clinics after diagnosis of sebaceous neoplasm
Several significant differences were noted between those with pathogenic germline MMR mutations (n=25) and those with abnormal IHC expression but without germline MMR mutations (n=23) (Table 3). Mean age at diagnosis of SN among mutation carriers was significantly younger than those without mutations (54.8 vs. 67.2, p=0.0002 t-test). Patients with MMR mutations were significantly more likely to have more than one SN (40.0% vs. 0%, p=0.0007 Fisher’s exact test), and 10/12 patients (83.3%) with more than one SN had pathogenic germline MMR mutations making this a strong predictor of mutation status. One patient with more than one SN had a VUS in MSH6. Family history differences, as measured by adherence to clinical diagnostic family history criteria and risk model scores, were also noted. 48% of patients with MMR mutations met Amsterdam I/II criteria compared to 4.3% in the group without mutations (p=0.0008). Among individuals with family histories not meeting Amsterdam I/II criteria, the mean PREMM (1,2,6) risk score was 31.2% for mutation carriers and 6.4% for the group with no mutations identified (p=0.002).
Table 3.
Comparison of characteristics between carriers of MMR mutations, non-carriers, and patients with normal IHC with no germline testing
| MMR Mutation Carriers (N=25) |
Abnormal IHC no germline mutation detected (N=23) |
Normal IHC no genetic testing (N=28) |
|
|---|---|---|---|
|
| |||
| Age at diagnosis (in years) (range in years) |
54.8 (28–76) |
67.2 p=0.0002 (45–81) |
65 (22–93) |
|
| |||
| More than 1 SN (%) | 10 (40.0%) | 0 (0) p=0.0007 |
0 |
|
| |||
| Meets Amsterdam I/II criteria (%) | 12 (48.0%) | 1 (4.3%) p=0.0008 |
0 |
|
| |||
| Not Meet Amsterdam I/II criteria (%) | 13 (52.0%) | 22 (95.7%) | 28 (100%) |
|
| |||
| PREMM <5 (%) | 6 (46.1%) | 8 (36.4%) | 13 (46.4%) |
|
| |||
| PREMM ≥5 (%) | 7 (56.3%) | 14 (63.6%) | 15 (53.6%) |
| Mean PREMM score (all ≥5) | 31.2 | 6.4 p=0.002 |
6.2 |
| Median PREMM score(all ≥5) | 31 | 5.5 p=0.002 |
5.1 |
Thirty-nine (50.6%) of 77 patients had normal expression of all four MMR proteins in their SNs. Eleven (28.2%) went on to have germline genetic testing of all MMR genes due to suspicious personal or family histories. Two patients were found to carry germline mutations: 1 pathogenic mutation in MSH2, 1 suspected pathogenic mutation in PMS2 (Table 1) (2/11; 18.2%). One patient had a VUS in MSH2. Twenty-eight patients did not have germline testing after normal IHC. Fourteen (50%) of these 28 had no personal or family history of any other LS associated cancer. Mean age at diagnosis and strength of family history were not significantly different between the group untested for germline mutations and the group having germline genetic testing with no mutation found (Table 3).
Among the 16 patients with MMR mutations found after tumor screening of SN and germline testing, 9 (56.3%) had personal history of internal malignancy meeting clinical criteria for MTS. Six (37.5%) had PREMM (1,2,6) risk scores of less than 5%. Two of these mutation carriers had personal history of atypical LS cancers not accounted for in risk models or diagnostic criteria (adrenocortical cancer, fibrous histiocytoma), and 1 had more than one SN. Two carriers reported history of classic LS cancers in first cousins, not captured by the PREMM (1,2,6) model which incorporates only diagnoses in first and second degree relatives.
Discussion
In this clinical series of patients with SN selected based on referral for genetic evaluation, IHC was concordant with the MMR gene mutation identified in 13/16 patients (81.3% sensitivity) and had a positive predictive value of for identifying germline MMR mutations of 36.8%. Our series is the first to have germline genetic test results available for all individuals with abnormal IHC in SN. The largest two series to date reported germline data on 5 of 40 patients (12) and 14 of 51 patients (17), with insufficient information to estimate sensitivity and positive predictive value of IHC for diagnosing LS. Prospective study of universal IHC screening in SN would be useful to determine how performance compares to universal IHC screening in CRC, which is gaining acceptance and estimated to be cost effective at a positive predictive value of 23.9% (21, 22). Our results confirm IHC of SN can be used as a first line screening test in patients with suspected LS.
These findings also support previous recommendations for routine IHC screening of all SN (11, 12), regardless of strength of the personal or family history. The majority of mutation carriers identified after IHC of SN and germline testing did not meet Amsterdam I/II clinical diagnostic family history criteria for LS (13/16; 81.3%) and 43.7% (7/16) did not meet the clinical definition of MTS. Presence of two or more relatives with CRC has been suggested as a threshold for offering IHC testing (17) and would have identified 6 (37.5%) of 16 mutation carriers in this series. A PREMM (1,2,6) risk model threshold of >5% has been suggested for consideration of germline testing (23), and would have identified 10 (62.5%) carriers. While a detailed pedigree including third degree relatives and incorporating all cancer diagnoses remains useful to help interpret IHC results, the low sensitivity of family history alone limits its utility as a pre-screen to select patients for IHC.
The majority (60.5%) of patients with abnormal IHC in an SN had no germline mutation identified indicating that MMR phenotype in SN is not diagnostic of LS. This is particularly striking in the sub-group of patients with absent MSH2/MSH6 expression in SN, where only 33.3% had germline MSH2 mutations and no TACSTD1/EPCAM mutations were identified (Figure 1). In CRC and endometrial cancer, absent MSH2/MSH6 staining is widely considered to be diagnostic of LS with 85% of patients having identifiable germline MSH2 or TACSTD1/EPCAM mutations (24, 25). These results suggest that a significant number of patients with abnormal IHC but without germline MMR mutations could have developed SN through somatic, non-heritable molecular events. Caution should be used in interpreting the clinical implications of abnormal IHC in SN in the absence of germline genetic test results.
We acknowledge our study has certain limitations. Patients were evaluated through 2 different clinics over a 12 year period, with variability in referral practices, and in clinical testing practices and standards. Germline genetic testing was completed in only 11/39 (28%) patients with normal IHC. Without confirmation of normal germline results, specificity cannot be accurately estimated and sensitivity may be overestimated as unidentified mutation carriers could exist in the untested group with normal IHC. However, average age at diagnosis of SN and strength of family history as measured by PREMM (1,2,6) scores among patients with normal IHC who did not have germline testing did not differ significantly from the characteristics of the tested group who did not carry germline mutations (Table 3). It is also possible that some patients with abnormal IHC but no germline mutations may have a germline mutation that could not be identified with currently available testing. Mutation carriers had earlier onset of SN, higher prevalence of multiple SN, and stronger family histories and risk model scores compared with patients with abnormal IHC and no germline mutation found (Table 3). Finally, this series represents a selected population of patients who were identified as having risk for MTS and referred for clinical genetics evaluation at tertiary referral centers, sometimes solely based on SN and sometimes due to additional suggestive personal or family history. There are institutional differences in patient demographics and referral patterns, and this series does not represent a universal screening approach. Defining the true prevalence of LS among all patients with SN will require further prospective study. However, our finding of MMR gene mutations in nearly one in 3 patients presenting based on SNs reinforces the importance of collaboration between dermatology, pathology and clinical genetics to ensure genetic referral for these individuals.
In conclusion, we propose a clinical practice algorithm for patients with SN (Figure 2) beginning with consideration of IHC screening of SN to be ordered by pathologist or clinician, excluding patients with sebaceous hyperplasia or known LS diagnosis. Family history screening for LS-associated cancers in at least first and second degree relatives is also warranted for all patients with SN. Genetics referral should be recommended for patients with SN and any of the following: absent MMR protein expression (abnormal result) on IHC screen; normal MMR protein expression and personal or family history of any LS-associated cancer; more than one SN. Our findings suggest that a combination of routine tumor testing and family history assessment would optimize identification of patients with LS in the dermatology setting.
Figure 2.
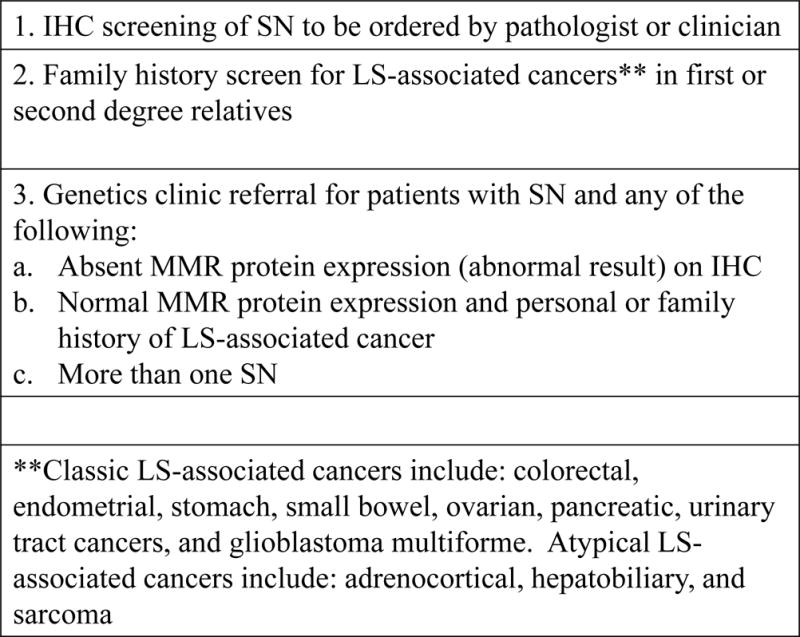
Clinical Practice Algorithm for Sebaceous Neoplasm – Suggested work flow to optimize identification of Lynch syndrome after diagnosis of sebaceous adenoma or carcinoma
Acknowledgments
Funding/Support: This study was supported in part by SR01CA132829-04 (Syngal), K24 – K24CA113433-08 (Syngal), P30 CA014089 (Gruber), P30 CA046592 (Gruber), T32-DK007425 (Else), K07CA120448-5 (Stoffel), and P30 CA042014 (Kohlmann)
Funding/Sponsor was involved?
Design and conduct of the study Yes__ NoX
Collection, management, analysis and interpretation of data Yes_ NoX
Preparation, review, or approval of the manuscript Yes__ NoX
Decision to submit the manuscript for publication Yes__ NoX
Footnotes
Presented at the 16th Annual Meeting of the Collaborative Group of the Americas on Inherited Colon Cancer, Boston, MA, October 27–29 2012(1)
Author Contributions: Ms. Everett and Dr. Stoffel had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Study concept and design: Everett, Raymond, Dandapani, Kohlmann, Syngal, Gruber, Stoffel. Analysis and interpretation of data: Everett, Raymond, Koeppe, Stoffel; Drafting of the manuscript: Everett, Raymond; Critical revision of the manuscript for important intellectual content: Dandapani, Marvin, Kohlmann, Chittenden, Gustafson, Else, Fullen, Johnson, Syngal, Gruber, Stoffel. Statistical analysis: Everett, Raymond, Koeppe. Obtained funding: Gruber, Syngal; Administrative, technical, or material support: Everett, Raymond, Syngal, Gruber.
Financial Conflict of Interest Disclosure: None reported
All Financial Interests: Dr. Gruber, Ms. Everett, and Ms. Gustafson have served as paid consultants to Myriad Genetic Laboratories, Inc.
References
- 1.The Proceedings of the Collaborative Group of the Americas on Inherited Colorectal Cancer Sheraton Boston Hotel, Boston, Massachusetts, October 27–29, 2012. Fam Cancer. 2013 doi: 10.1007/s10689-012-9592-9. Epub 2013/05/03. [DOI] [Google Scholar]
- 2.Aarnio M, Sankila R, Pukkala E, Salovaara R, Aaltonen LA, de la Chapelle A, et al. Cancer risk in mutation carriers of DNA-mismatch-repair genes. Int J Cancer. 1999;81(2):214–8. doi: 10.1002/(sici)1097-0215(19990412)81:2<214::aid-ijc8>3.0.co;2-l. Epub 1999/04/03. doi: 10.1002/(SICI)1097-0215(19990412)81:2<214::AID-IJC8>3.0.CO;2-L [pii] [DOI] [PubMed] [Google Scholar]
- 3.Torre D. Multiple sebaceous tumors. Arch Dermatol. 1968;98(5):549–51. doi: 10.1001/archderm.98.5.549. Epub 1968/11/01. [DOI] [PubMed] [Google Scholar]
- 4.Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363(9):809–19. doi: 10.1056/NEJMoa1002011. Epub 2010/09/08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lynch HT, Lynch PM, Pester J, Fusaro RM. The cancer family syndrome. Rare cutaneous phenotypic linkage of Torre’s syndrome. Arch Intern Med. 1981;141(5):607–11. Epub 1981/04/01. [PubMed] [Google Scholar]
- 6.Jarvinen HJ, Aarnio M, Mustonen H, Aktan-Collan K, Aaltonen LA, Peltomaki P, et al. Controlled 15-year trial on screening for colorectal cancer in families with hereditary nonpolyposis colorectal cancer. Gastroenterology. 2000;118(5):829–34. doi: 10.1016/s0016-5085(00)70168-5. Epub 2000/04/28. doi: S0016508500404555 [pii] [DOI] [PubMed] [Google Scholar]
- 7.Lindor NM, Petersen GM, Hadley DW, Kinney AY, Miesfeldt S, Lu KH, et al. Recommendations for the care of individuals with an inherited predisposition to Lynch syndrome: a systematic review. JAMA. 2006;296(12):1507–17. doi: 10.1001/jama.296.12.1507. Epub 2006/09/28. doi: 296/12/1507 [pii] [DOI] [PubMed] [Google Scholar]
- 8.Hampel H, Frankel WL, Martin E, Arnold M, Khanduja K, Kuebler P, et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer) N Engl J Med. 2005;352(18):1851–60. doi: 10.1056/NEJMoa043146. Epub 2005/05/06. doi: 352/18/1851 [pii] 10.1056/NEJMoa043146. [DOI] [PubMed] [Google Scholar]
- 9.Hampel H, Frankel W, Panescu J, Lockman J, Sotamaa K, Fix D, et al. Screening for Lynch syndrome (hereditary nonpolyposis colorectal cancer) among endometrial cancer patients. Cancer Res. 2006;66(15):7810–7. doi: 10.1158/0008-5472.CAN-06-1114. Epub 2006/08/04. doi: 66/15/7810 [pii] 10.1158/0008-5472.CAN-06-1114. [DOI] [PubMed] [Google Scholar]
- 10.Recommendations from the EGAPP Working Group: genetic testing strategies in newly diagnosed individuals with colorectal cancer aimed at reducing morbidity and mortality from Lynch syndrome in relatives. Genet Med. 2009;11(1):35–41. doi: 10.1097/GIM.0b013e31818fa2ff. Epub 2009/01/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mathiak M, Rutten A, Mangold E, Fischer HP, Ruzicka T, Friedl W, et al. Loss of DNA mismatch repair proteins in skin tumors from patients with Muir-Torre syndrome and MSH2 or MLH1 germline mutations: establishment of immunohistochemical analysis as a screening test. Am J Surg Pathol. 2002;26(3):338–43. doi: 10.1097/00000478-200203000-00007. Epub 2002/02/23. [DOI] [PubMed] [Google Scholar]
- 12.Plocharczyk EF, Frankel WL, Hampel H, Peters SB. Mismatch Repair Protein Deficiency is Common in Sebaceous Neoplasms and Suggests the Importance of Screening for Lynch Syndrome. Am J Dermatopathol. 2012 doi: 10.1097/DAD.0b013e31825f7efe. Epub 2012/06/23. [DOI] [PubMed] [Google Scholar]
- 13.Orta L, Klimstra DS, Qin J, Mecca P, Tang LH, Busam KJ, et al. Towards identification of hereditary DNA mismatch repair deficiency: sebaceous neoplasm warrants routine immunohistochemical screening regardless of patient’s age or other clinical characteristics. Am J Surg Pathol. 2009;33(6):934–44. doi: 10.1097/PAS.0b013e318199edca. Epub 2009/04/04. [DOI] [PubMed] [Google Scholar]
- 14.Chhibber V, Dresser K, Mahalingam M. MSH-6: extending the reliability of immunohistochemistry as a screening tool in Muir-Torre syndrome. Mod Pathol. 2008;21(2):159–64. doi: 10.1038/modpathol.3800997. Epub 2007/12/11. doi: 3800997 [pii] 10.1038/modpathol.3800997. [DOI] [PubMed] [Google Scholar]
- 15.Cesinaro AM, Ubiali A, Sighinolfi P, Trentini GP, Gentili F, Facchetti F. Mismatch repair proteins expression and microsatellite instability in skin lesions with sebaceous differentiation: a study in different clinical subgroups with and without extracutaneous cancer. Am J Dermatopathol. 2007;29(4):351–8. doi: 10.1097/DAD.0b013e318057713c. Epub 2007/08/02. doi: 10.1097/DAD.0b013e318057713c 00000372-200708000-00004 [pii] [DOI] [PubMed] [Google Scholar]
- 16.Morales-Burgos A, Sanchez JL, Figueroa LD, De Jesus-Monge WE, Cruz-Correa MR, Gonzalez-Keelan C, et al. MSH-2 and MLH-1 protein expression in Muir Torre syndrome-related and sporadic sebaceous neoplasms. P R Health Sci J. 2008;27(4):322–7. Epub 2008/12/17. [PMC free article] [PubMed] [Google Scholar]
- 17.Roberts ME, Riegert-Johnson DL, Thomas BC, Thomas CS, Heckman MG, Krishna M, et al. Screening for Muir-Torre Syndrome Using Mismatch Repair Protein Immunohistochemistry of Sebaceous Neoplasms. J Genet Couns. 2012 doi: 10.1007/s10897-012-9552-4. Epub 2012/12/06. [DOI] [PubMed] [Google Scholar]
- 18.Vasen HF, Mecklin JP, Khan PM, Lynch HT. The International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer (ICG-HNPCC) Dis Colon Rectum. 1991;34(5):424–5. doi: 10.1007/BF02053699. Epub 1991/05/01. [DOI] [PubMed] [Google Scholar]
- 19.Vasen HF, Watson P, Mecklin JP, Lynch HT. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology. 1999;116(6):1453–6. doi: 10.1016/s0016-5085(99)70510-x. Epub 1999/05/29. doi: S0016508599005715 [pii] [DOI] [PubMed] [Google Scholar]
- 20.Kastrinos F, Steyerberg EW, Mercado R, Balmana J, Holter S, Gallinger S, et al. The PREMM(1,2,6) model predicts risk of MLH1, MSH2, and MSH6 germline mutations based on cancer history. Gastroenterology. 140(1):73–81. doi: 10.1053/j.gastro.2010.08.021. Epub 2010/08/24. doi: S0016-5085(10)01239-4 [pii] 10.1053/j.gastro.2010.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hampel H, Frankel WL, Martin E, Arnold M, Khanduja K, Kuebler P, et al. Feasibility of screening for Lynch syndrome among patients with colorectal cancer. J Clin Oncol. 2008;26(35):5783–8. doi: 10.1200/JCO.2008.17.5950. Epub 2008/09/24. doi: JCO.2008.17.5950 [pii] 10.1200/JCO.2008.17.5950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gudgeon JM, Williams JL, Burt RW, Samowitz WS, Snow GL, Williams MS. Lynch syndrome screening implementation: business analysis by a healthcare system. Am J Manag Care. 2011;17(8):e288–300. Epub 2011/08/20. doi: 50524 [pii] [PubMed] [Google Scholar]
- 23.Dinh TA, Rosner BI, Atwood JC, Boland CR, Syngal S, Vasen HF, et al. Health benefits and cost-effectiveness of primary genetic screening for Lynch syndrome in the general population. Cancer Prev Res (Phila) 2011;4(1):9–22. doi: 10.1158/1940-6207.CAPR-10-0262. Epub 2010/11/23. doi: 1940-6207.CAPR-10-0262 [pii] 10.1158/1940-6207.CAPR-10-0262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rumilla K, Schowalter KV, Lindor NM, Thomas BC, Mensink KA, Gallinger S, et al. Frequency of deletions of EPCAM (TACSTD1) in MSH2-associated Lynch syndrome cases. J Mol Diagn. 13(1):93–9. doi: 10.1016/j.jmoldx.2010.11.011. Epub 2011/01/14. doi: S1525-1578(10)00025-5 [pii] 10.1016/j.jmoldx.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ligtenberg MJ, Kuiper RP, Chan TL, Goossens M, Hebeda KM, Voorendt M, et al. Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3′ exons of TACSTD1. Nat Genet. 2009;41(1):112–7. doi: 10.1038/ng.283. Epub 2008/12/23. doi: ng.283 [pii] 10.1038/ng.283. [DOI] [PubMed] [Google Scholar]