

Abstract
In plants, hybrid vigor is influenced by genetic and epigenetic mechanisms; however, the molecular pathways are poorly understood. We investigated the potential contributions of epigenetic regulators to heterosis in Arabidposis and found that the chromatin remodeler DECREASED DNA METHYLATION 1 (DDM1) affects early seedling growth heterosis in Col/C24 hybrids. ddm1 mutants showed impaired heterosis and increased expression of non-additively expressed genes related to salicylic acid metabolism. Interestingly, our data suggest that salicylic acid is a hormetic regulator of seedling growth heterosis, and that hybrid vigor arises from crosses that produce optimal salicylic acid levels. Although DNA methylation failed to correlate with differential non-additively expressed gene expression, we uncovered DDM1 as an epigenetic link between salicylic acid metabolism and heterosis, and propose that the endogenous salicylic acid levels of parental plants can be used to predict the heterotic outcome. Salicylic acid protects plants from pathogens and abiotic stress. Thus, our findings suggest that stress-induced hormesis, which has been associated with increased longevity in other organisms, may underlie specific hybrid vigor traits.
Keywords: DDM1, DNA methylation, epigenetics, heterosis, non-additive expression
Introduction
Hybrid vigor, or heterosis, refers to the improved performance of hybrid offspring relative to their parents. This is often restricted to a particular trait such as yield, plant height, biomass or defense and can be influenced by parent background and imprinting [1]. Various mechanisms, including dominance, overdominance and epistasis, have been proposed to explain heterosis from a quantitative genetics perspective; however, the molecular mechanisms underlying heterosis are still unclear. Recently, heterosis was found to correlate with circadian-clock-mediated regulation of several biotic and abiotic stress-related genes [2]. An overdominant gene, SINGLE FLOWER TRUSS, has also been identified in tomato, which regulates fruit yield heterosis [3]. In addition, the role of polyploidy and epigenetic interactions between parents, including small RNA, DNA methylation and histone modifications, have also been proposed to influence heterosis [4–8]. Indeed, Arabidopsis accessions C24/Ler, which have very similar genomic sequences, show heterosis. The associated epigenetic modifications, including DNA cytosine methylation, have also shown segregation, which may contribute to a loss of heterosis in the F2 generation [9]. However, the key epigenetic mechanisms regulating heterosis have not been investigated using genetic tests.
RNA-directed DNA methylation (RdDM) is a well-studied epigenetic pathway in plants and has been implicated in heterosis due to the alteration of small RNA profiles after hybridizations [10–13], although there is also evidence that it is not important for heterosis [14, 15]. In the current model of RdDM, de novo DNA methylation is initiated by the production of 24-nt small interfering RNAs (siRNAs), which are generated by a set of factors, including a plant-specific RNA polymerase (Pol IV), RNA-dependent RNA polymerase 2 (RDR2) and Dicer-like 3. These 24-nt siRNAs are then loaded onto AGO4/6 to form an AGO4/6–siRNA complex. Guided by siRNAs, the AGO4/6–siRNA complex binds to nascent noncoding RNAs produced by another plant-specific RNA polymerase (Pol V), through RNA–RNA base pairing. Recent work suggests that the role of siRNAs can be largely replaced by their precursors, which are Pol IV and RDR2 products of 25–50 nt in length [16]. Pol V function also requires the DDR complex (consisting of DEFECTIVE IN RNA-DIRECTED DNA METHYLATION 1, DEFECTIVE IN MERISTEM SILENCING 3 and RNA-DIRECTED DNA METHYLATION 1) and formation of the AGO4/6–siRNA–scaffold RNA complex may require RNA-DIRECTED DNA METHYLATION 3. Eventually, DOMAINS REARRANGED METHYLTRANSFERASE 2 is recruited to this complex and mediates de novo methylation of cytosines in all sequence contexts (CG, CHG and CHH, where H represents A, C or T). This results in transcriptional silencing at genomic loci transcribed by Pol V. Overall, RdDM has been found to occur at a wide range of genomic locations but with preferences for euchromatic regions.
RdDM is excluded to some extent from pericentromeric heterochromatin regions surrounding centromeres [17]. DNA methylation in these regions mostly occurs through an siRNA-independent manner and relies on another SWI2/SNF2-like chromatin remodeler protein called DDM1 (DECREASED DNA METHYLATION 1) [18, 19]. In Arabidopsis, DDM1 preferentially affects DNA methylation in heterochromatin, transposable elements and tandem repeat-rich regions, and primarily targets long terminal repeat retrotransposons and Mutator DNA transposons. In addition, DDM1 can modulate the expression of protein-coding genes by regulating their adjacent transposable elements (TEs) [20]. Recent work also suggested that DDM1 functions separately from RdDM by allowing the DNA methyltransferase CHROMOMETHYLASE 2 to approach H1-containing heterochromatin [17]. In addition, DDM1 can regulate gene expression through histone modifications [21].
Here we used epigenetic mutants to investigate the early seedling biomass heterosis of Arabidopsis Col and C24 accessions. We uncovered DDM1 as a major regulator of heterosis. We found that genes associated with heterosis were generally not regulated by DNA methylation. Further, high-throughput transcriptomics revealed that DDM1 affects salicylic acid (SA)-related genes. SA defends plants from pathogen infection and abiotic stresses, such as drought [22]. Previous findings have suggested that decreased SA contributed to the increased growth of hybrid plants [8, 23]. However, our findings suggest that SA is a hormetic regulator of biomass heterosis and that, depending on the parent, optimal levels of SA can be associated with improved hybrid growth. Thus, our findings indicate that DDM1 connects epigenetics, SA metabolism and hormesis, to the broader context of heterosis regulation in plants.
Results
Heterosis of F1 hybrids was impaired in ddm1 loss-of-function mutants
To test the roles of epigenetic regulators in heterosis, we used a series of homozygous mutants in the C24 background [24–26] and crossed them with their corresponding homozygous alleles in the Col ecotype (for gene accession numbers and SALK IDs of Col and C24, see Supplementary Figure S1A–I and Supplementary Table S1). We made reciprocal crosses between Colmaternal × C24paternal and C24maternal×Colpaternal to obtain F1 and r-F1 hybrids, respectively, and confirmed that they were true hybrids by genotyping (Supplementary Figure S1). We found that F1 seedlings from wild type (WT), rdr2, dms3, drd1, rdm1, nrpd1, nrpe1, ago4, ago6 and rdm3 crosses displayed a significant increase in leaf width compared with their respective parents, indicating a heterosis phenotype (Figure 1). No significant difference in phenotype was observed between reciprocal crosses (Figure 1 and Supplementary Figure S1J).
Figure 1.

Heterotic phenotypes of F1 hybrids from Col and C24 crosses in wild-type and RdDM mutant backgrounds. F1 hybrids were produced from Col and C24 crosses using wild-type and RdDM pathway mutant plants. As a convention, the maternal parent is listed first. The mutant details are described in Supplementary Table S1. Seedlings from all genotypes were gown in pots and then phenotyping was conducted at 18–20 DAS (day after sowed) (scale bar=2 cm.) For quantitative assessment, refer Supplementary Figure S1J. The experiments were performed in triplicate to confirm reproducibility.
To determine whether the chromatin remodeler DDM1 affects heterosis, we generated F1 hybrids of ddm1 loss-of-function mutants. We performed independent reciprocal crosses between three Col (ddm1-1, ddm1-10 and ddm1-16) and three C24 (ddm1-9, ddm1-14 and ddm1-15) ddm1 alleles. The F1 hybrids from different allelic combinations were genotyped to confirm that they are true hybrids (Figure 2 and Supplementary Figure S2). We recently derived ddm1-14, ddm1-15 and ddm1-16, and verified their effects by evaluating the methylation status of 5S-rDNA through methylation-sensitive restriction enzyme PCR (Chop-PCR) (Supplementary Figure S2H) [27]. As ddm1 mutants show developmental defects after a few generations of inbreeding [28], homozygous ddm1 plants were used for only three generations in heterosis tests and then fresh homozygous seeds were obtained from heterozygous parents.
Figure 2.
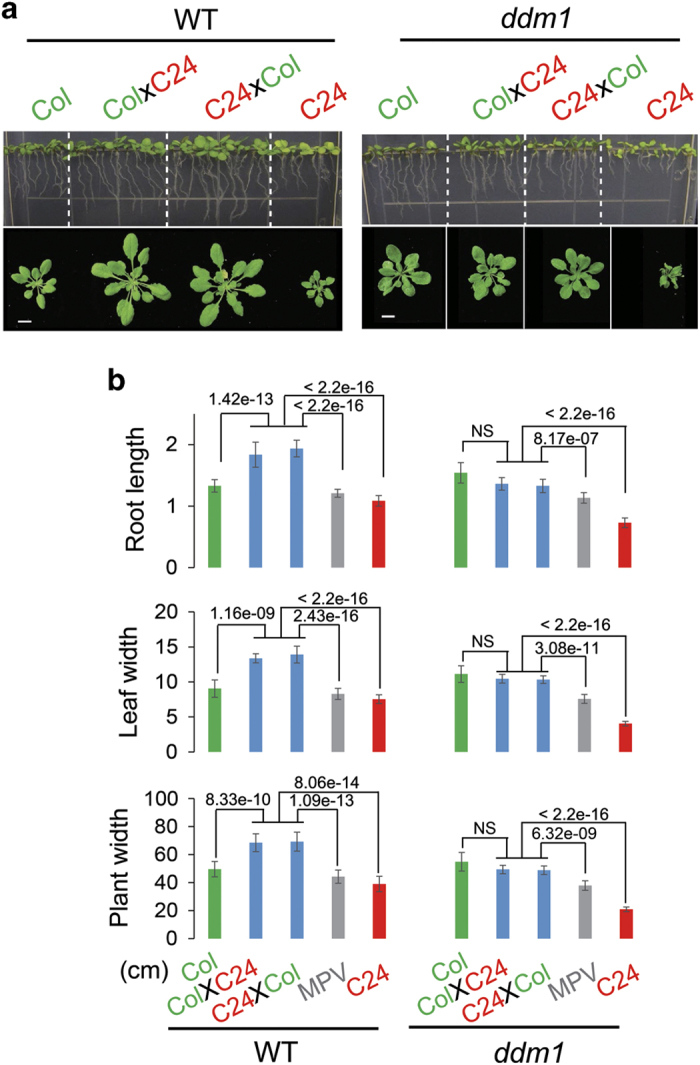
Heterosis was impaired in F1 offspring of Col and C24 in ddm1 mutant background. The phenotyping of F1 wild-type and ddm1 mutant plants was performed by the pot and vertical plate method. WT-F1 showed BPH, whereas ddm1-F1 showed MPH performance (a). The differential phenotype was quantified in terms of root length (at 10 DAS), leaf width and plant width (at 20 DAS) (b). The P-values are shown for corresponding pair-wise comparisons (NS, not significant; P-values>0.05, t-test). The ddm1-10 (Col) and ddm1-15 (C24) were used in this experiment. The experiments were performed in triplicate, to confirm reproducibility. MPV, mid-parent values (scale bar, 1 cm).
We examined the growth pattern at the early seedling stage and found that heterosis was markedly impaired in ddm1-F1 hybrids compared with WT-F1 hybrids (Figure 2a). The key morphological traits of seedlings such as root length, leaf width and rosette width were quantified. The WT-F1 hybrids showed significantly better performance than both parents (P<0.01, t-test), confirming best-parent heterosis (BPH). Unlike WT-F1, ddm1-F1 hybrids showed a dramatic reduction in these morphological traits when compared with WT-F1 (Supplementary Figure S3D). Although ddm1-F1 plants displayed a growth pattern that was more vigorous than the parental ddm1-C24 plants, it was similar to parental ddm1-Col plants. Therefore, ddm1-F1 seedlings lost the BPH that was observed in WT-F1. However, their growth vigor was better than the average performance of the two parents, consistent with mid-parent heterosis (MPH) (Figure 2b). We also evaluated different alleles of ddm1 and did not detect significant differences among the different allelic combinations (Supplementary Figure S3A–D).
To test whether mutations in both DDM1 alleles are required for the impairment of hybrid vigor, we performed reciprocal crosses with a ddm1 mutant in one ecotype and WT in the other ecotype, and examined the heterosis of F1 hybrids. Heterozygous F1 hybrids from all four crosses, including ddm1-Col×DDM1-C24, ddm1-C24×DDM1-Col, DDM1-Col×ddm1-C24 and DDM1-C24×ddm1-Col, were compared with their corresponding parents and with WT-F1 hybrids as well. No obvious reduction in early seedling growth heterosis was observed in these crosses (Supplementary Figure S3E and F), indicating that loss of BPH requires dysfunction of both DDM1 alleles in F1 hybrids. Further, these results demonstrate that impaired heterosis in ddm1-Col and ddm1-C24 crosses is not simply due to the slower growth rate of ddm1-C24 seedlings.
NEGs related to SA in F1 hybrids
To investigate the molecular basis of BPH-to-MPH conversion in ddm1-F1, we identified differentially expressed genes (DEGs) in different genotypes, but the DEGs failed to provide a clear explanation of the change in heterosis (Supplementary Information and Supplementary Figures S4 and S5).
We then analysed non-additively expressed genes (NEGs) in WT-F1 and ddm1-F1. NEGs are defined by F1 gene expression that is significantly different from the average value of the parental inbred lines (mid-parent value, MPV). We identified 347 NEGs in WT-F1, using t-test (two-sides) and P-value<0.05 as cutoff, of which 35 and 312 genes were above or below the MPV, respectively. Using the same criteria, along with a fold change of >2 cutoff to focus on highly functional NEGs, we identified 529 NEGs in ddm1-F1, of which 93 and 436 were above or below the MPV, respectively. The four classes of NEGs (above or below MPV in WT-F1 and ddm1-F1) were further divided into two groups depending on their upregulation (ddm1-Col>WT-Col) or downregulation (ddm1-Col<WT-Col) in the ddm1 mutant (Figure 3a). This resulted in eight groups of NEGs, which were independently tested for gene ontology (GO)-based functional enrichment analysis. Only two of these groups displayed functional enrichments, which we subsequently refer to as WT-NEGs (140 genes) and ddm1-NEGs (312 genes) (Figure 3a).
Figure 3.

The functional NEGs were enriched in SA metabolism (SA-NEGs). Genes showing differential expression compared with MPVs were defined as NEGs in WT- and ddm1-F1 (a). Venn diagram shows the overlap of WT- and ddm1-NEGs and their functional enrichment in SA metabolism (b). GO-based functional analysis showed that overlapped NEGs between WT and ddm1 are significantly enriched in SA metabolism (c). The NEGs related to SA metabolism were further classified on the basis of their involvement with core SA biosynthesis (d), biosynthesis of pipecolic acid (e), activator of SA biosynthesis (f) and downstream to SA signaling (g). The letter ‘O’ and ‘D’ in brackets before gene IDs indicated that the genes were from ‘overlap’ or ‘ddm1 specifc’ groups in c, respectively. The letter ‘L’ and ‘U’ in brackets after gene names indicated that the gene were from ‘Low-methylation’ or ‘Unmethylated’ groups in Supplementary Figure S7.
Forty-three of the 140 WT-NEGs are related to SA metabolism and response, whereas the remaining WT-NEGs did not show any functional enrichment (Figure 3b). The full set of 312 ddm1-NEGs are statistically enriched in SA, including the 43 WT-NEGs related to SA (Figure 3b and c). For convenience, we refer to these 312 NEGs as SA-NEGs. We further classified the SA-NEGs based on their association with different roles in SA metabolism or response, including core components of SA biosynthesis, activators of SA biosynthesis, downstream of SA signaling and other genes related to SA response (Figure 3d–g, Supplementary Figure S4D and Supplementary Table S6). The broad involvement of SA-NEGs with various aspects of SA metabolism strongly suggest that this process influences heterosis.
Further, to determine whether SA-NEGs are regulated through DNA methylation, we performed bisulfite sequencing (BSseq) analyses of the different genotypes but failed to find any association of SA-NEGs with DNA methylation (Supplementary Information and Supplementary Figures S7 and S8).
Endogenous SA levels determine plant size in WT and ddm1 mutants
To gain further insight into the regulation of SA-NEGs, we examined their expression levels by box plot analysis. As expected, the SA-NEGs were expressed at higher levels in the ddm1 mutant compared with WT in parental and with hybrid backgrounds as well (Figures 3a,d–f and 4a). We noticed that the expression of SA-NEGs in WT-F1 was higher than in Col parents; however, no significant change in the expression of SA-NEGs was observed between ddm1-Col and ddm1-F1 (P>0.05), consistent with the comparable growth phenotypes of these plants (Figure 2a). To evaluate the expression of specific parental alleles in F1 hybrids, we selected 243 SA-NEGs that contain single-nucleotide polymorphisms, which distinguish the Col and C24 parent backgrounds. We evaluated the expression of these SA-NEGs by box plot analysis and found that the expression of the C24 allele, but not the Col allele, was significantly altered and reduced in the hybrids relative to the respective parent. In contrast to WT plants, SA-NEGs in ddm1 mutants are expressed at similar levels from each allele in the Col parent and in the hybrid plants (Figure 4b). The ddm1-Col-like expression of SA-NEGs from both alleles in ddm1-F1 hybrid and Col parent plants may account for their similar growth phenotypes (Figure 2a).
Figure 4.

The expression of SA-NEGs correlates with endogenous SA levels. The expression pattern of 312 SA-NEGs were analyzed in different genotypes using boxplots. The differences between the corresponding pairwise comparisons were calculated using t-test. The green and blue lines indicated the differences of WT-Col/ddm1-Col and of WT-F1s/ddm1-F1s, respectively (a). Allele-specific gene expression was also analyzed for WT and ddm1 using publicly available single-nucleotide polymorphism database between Col and C24 accessions (b). The endogenous SA concentration was measured in WT and ddm1 genotypes (both parents and hybrid) at two developmental stages of 14 and 22 DAS (c). The dependence of root length and plant size on SA dose is shown (b, d). The effect of exogenous SA on heterosis performance of WT-F1 with foliar spray (see section Exogenous treatment of SA) was monitored and quantified in terms of plant size and FW (e).
As many SA metabolism-related genes were upregulated in ddm1-F1 compared with WT-F1, we hypothesized that different endogenous SA concentrations might underlie their distinct phenotypes. To test this hypothesis, we quantified SA levels in different genotypes. We found that the levels of SA (Figure 4c) correlated with the expression levels of SA-NEGs (Figure 4a) at both 14 and 22 days after germination. Maximum growth occurred at an optimal endogenous SA concentration observed only with WT-F1 hybrid plants (about 0.11 ng mg−1 fresh weight (FW)). Growth was reduced at sub-optimal endogenous SA concentrations (0.07 ng mg−1 FW, WT-Col) and supra-optimal concentrations (⩾0.3 ng mg−1 FW, WT-C24, all ddm1 parent and hybrid plants) (Figure 4d). The SA level in ddm1-F1 increased by 3.55-fold compared with WT-F1 at 14 day-old seedlings, shifting it from optimal to supra-optimal and from BPH to MPH.
To further validate that loss of the BPH phenotype of ddm1-F1 arises from higher SA concentrations, we treated WT-F1 plants with exogenous SA and monitored growth performance. These treatments abrogated heterosis in WT-F1 hybrids, including heterosis in plant size and FW (Figure 4e). Moreover, low concentrations of SA improved the growth of Col plants but not C24 (Supplementary Figure S6), suggesting that SA is a hormetic regulator of plant growth. Together, these data suggest that heterosis in WT-F1 hybrids reflects an optimal level of SA metabolism, and that sub- and supra-optimal levels of SA negatively influence growth.
Discussion
Col/C24 Arabidopsis hybrids are known to show heterotic phenotypes, including increased biomass and photosynthetic ability [29]. The extent of heterosis changes throughout the growth cycle of plant [30]. therefore, we selected early seedling stages and evaluated the contribution of different epigenetic regulators, such as some RdDM components and DDM1 (chromatin remodeler) in regulating biomass heterosis. The tested RdDM mutants maintained heterosis, at least during the early seedling stage. These results are consistent with recent work in Arabidopsis [14] and also with the observation in maize that loss of MOP1 (modifier of paramutation1), an RDR2 ortholog in the RdDM pathway in maize, did not affect the hybrid vigor of B73 × Mo17 hybrids [15]. Unlike RdDM mutants, F1 hybrids generated in ddm1 mutant backgrounds clearly showed reduced heterosis compared with WT-F1, with the BPH traits in WT-F1 changing to MPH in ddm1-F1.
NEGs, the subset of DEGs that deviate from the MPV, are routinely evaluated in hybrid progeny [1] and it has been proposed that NEGs may explain heterosis [31]. Indeed, NEGs have provided insight into the heterosis observed in Col/C24 hybrids towards abiotic and biotic stimuli [29]. We identified 312 functionally enriched NEGs related to SA (SA-NEGs) in WT/ddm1 F1 hybrids. In general, a relatively higher expression of SA-NEGs correlated negatively with growth, except for WT-Col parental and WT-F1 hybrid plants (Figure 4). Indeed, WT-F1 plants display an optimal level of SA-NEGs expression associated with optimal growth; expression outside this range impairs growth.
We found that the SA-NEGs targeted by DDM1 are enriched for genes related to SA metabolism, including SA biosynthesis and its activation, SA downstream signaling and SA responsive genes. SA is a major phytohormone controlling the defence response, as well as growth and development in plants. Its biosynthesis occurs via two pathways: one from cinnamate, catalyzed by phenylalanine ammonia lyase, and the other from chorismate, catalyzed by isochorismate synthase (ICS) [32, 33]. ICS1 (also known as SID2) is the predominant form of ICS [34] and was an SA-NEGs gene. The SA-NEGs in the SA biosynthesis activation category included genes that function upstream of ICS1 or transcription factors that activate its expression. ALD1 is an aminotransferase mediating the biosynthesis of another immune regulatory metabolite, pipecolic acid, and FMO1 is an essential component of pipecolic acid downstream signaling. Although pipecolic acid positively regulates SA biosynthesis and signaling, there is a significant SA-independent component of ALD1/pipecolic acid/FMO1-mediated defense signaling [35, 36]. Mutations in these genes are known to attenuate SA accumulation in response to pathogen attack [33, 37, 38]. SA-NEGs were also enriched for genes that function downstream of SA, including SA-responsive genes. Interestingly, genes related to SA repression were absent, suggesting that SA-NEGs positively regulate SA signaling.
The enrichment for a wide range of SA-related genes in the set of SA-NEGs suggested that endogenous SA levels may differ between different genotypes and underlie heterosis. SA metabolism has been previously implicated in hybrid vigor in the contexts of the defence response and growth performance in Arabidopsis [8, 23]. Yang et al. [23] found that F1 hybrids with a higher level of SA than parents display heterosis for the defence response but not growth. Groszmann et al. [8] revealed that F1 hybrids with lower endogenous SA levels than MPV displayed heterosis for growth performance, which was compromised by SA treatment. Consistent with these findings, we observed a perfect overlap between SA-NEGs expression and SA concentration at two developmental stages. However, more interestingly, our data suggest hormetic modulation of hybrid vigor by SA. Increased, low endogenous SA concentrations stimulate growth (for example, WT-F1 compared with WT-Col) but, beyond a threshold, SA inhibits growth (for example, WT-C24 and all ddm1 mutant plants). This type of SA dose-dependent growth phenotype with WT-F1 and Col supports the conclusion that SA is hormetic in promoting plant growth, consistent with previous supplementation [39–42] and genetic studies [43]. The endogenous SA concentrations of WT-Col and WT-C24 flank the optimal SA level for growth and the non-additive expression of SA metabolism genes in WT-F1 lead to optimal SA levels and the BPH phenotype. The allele-specific expression data revealed that ddm1 mutants displayed increased expression of SA-NEGs from both alleles compared with WT, in particular from the C24 allele. Thus, both ddm1 mutant parents display supra-optimal endogenous SA levels and SA-NEGs expression, which is maintained in the hybrid progeny. As a result, BPH is converted to MPH in ddm1-F1 due to the hormetic response to SA. Our findings that altered expression of SA metabolism and immune/defence response genes correlates with hybrid vigor suggest that the improved growth of hybrids may be recapitulated in parental plants by optimizing environmental stressors to induce endogenous hormetic responses. Hormesis appears to be induced by many endogenous biological processes, including stress, inflammation and cellular respiration, where a low level of potentially toxic byproducts ultimately improves performance [44–47]. We propose that some improved hybrid traits reflect the optimized, endogenous regulation of stress-induced hormesis.
We found that DDM1 is a major regulator of early stage growth heterosis in Col /C24 accession of Arabidopsis. In order to further dissect how ddm1 drives BPH-to-MPH transformation in ddm1-F1, we performed transcriptome analysis and uncovered SA-NEGs, the differential NEGs enriched in the genes related to SA metabolism. Methylome analysis demonstrated that the SA-NEGs were mostly in the low- and unmethylated categories, which suggested that ddm1 regulates the expression of coding genes by epigenetic mechanisms other than DNA methylation. DDM1-mediated regulation of coding genes has been previously demonstrated through generation of ddm1-induced epigenetic inbred lines, which show flowering time and root length phenotypes [48]. It has been shown that ddm1 mutants display a genome-wide increase in transcription and in histone methylation marks associated with gene activation (H3K4me1/2/3) rather than repression (H3K9me2 and H3K27me3) [21, 49] implying that these histone modifications may be involved in regulation of SA-NEGs; however, other epigenetic marks might also be important, as reported previously [23, 50]. The role of epigenetic modifications in heterosis requires further investigation.
In conclusion, the study revealed that the chromatin remodeler DDM1 is a major regulator of early seedling biomass heterosis in Col/C24 accessions of Arabidopsis thaliana. ddm1 hybrids display elevated expression of NEGs that are functionally enriched for SA metabolism, leading to increased endogenous SA levels. We propose that SA is a hormetic regulator of plant growth, with increased growth associated with increased SA at low concentrations, but inhibition associated with high concentrations. WT-F1 hybrids generated from a cross of WT-Col and WT-C24 produce plants with optimal SA-NEGs expression, endogenous SA concentration and hybrid vigor. Our study links epigenetics to hormone metabolism and hybrid vigor through DDM1. Importantly, we propose that endogenous SA levels and SA-related gene expression can predict whether crosses between two genotypes will show growth heterosis (Figure 5).
Figure 5.

A working model for the proposed action of DDM1-mediated regulation of heterosis through SA metabolism. (a) The promoter status of SA-related genes, SA transcripts abundance, SA concentration and phenotype of parents and F1 in WT and ddm1 mutant background. (b) The effect of dose-dependent SA concentration over plant growth. The F1 hybrids from WT Col and C24 possess SA gene expression/concentration in the optimum zone and, hence, BPH phenotype is seen. As ddm1 increases the ratio of active (green bars) to repressive (red bars) epigenetic marks in promoter elements, SA-related gene expression and SA concentration is increased in ddm1-parents. Thus, F1 hybrids produced from the ddm1 background display supra-optimal SA concentrations and hence MPH phenotype is seen.
Materials and Methods
Plant growth and phenotyping of different genotypes
The Arabidopsis seeds from WT Columbia-0 and C24 background were available in the lab. The epigenetic mutants in Col were procured from Arabidopsis Biological Resource Centre (http://www.biosci.ohio-state.edu/pcmb). The mutants in C24 background were from previous studies [24, 25]. Seeds were surface sterilized and then plated on Murashige and Skoog medium [51]. To ensure homogenous germination, seeds were kept at 4 °C. After 7 days of stratification, seeds were transferred to growth room and then allowed to grow in continuous, cool fluorescent white light (100 μE m−2 s−1) at 22 °C under long day conditions. The 7 day-old seedlings were transplanted on soil pots and at 25 day after sowed, phenotyping was performed in terms of plant width and leaf width. For root phenotyping, 12 day-old seedlings were transplanted on vertical plates and after 7 days increase in root length was measured.
Generation of hybrids and genotyping
The F1 hybrids between Col-0 and C24 under WT and ddm-1 background were generated as described previously [29]. The heterozygotic nature of produced hybrids was confirmed through genotyping according to particular mutant information. At least one natural variant site was selected between Col and C24, to confirm that F1 progenies were really from Col and C24 hybridization. The primers for genotyping and Chop-PCR are listed under Supplementary Table S2.
RNAseq and data analysis
All the different genotypes were grown on plates in triplicates. At 14 day after sowed, entire seedling was collected, RNA was isolated and RNA sequencing (RNAseq) was performed on Illumina HiSeq 2000 platform (San Diego, CA, USA). The sequencing statistics of different RNAseq libraries can be found as Supplementary Table S4. The raw sequencing reads from all the runs were first trimmed for lo- quality regions and adapter sequences. Clean reads were aligned to TAIR10 Arabidopsis reference genome using TopHat2 software [52]. The resulted BAM alignment files were used as input to summarize the gene expression levels using HTSeq-count [53]. The low expressed genes were removed and only genes with an expression level of at least 1 read per million in at least three samples were retained for further analysis. The R package edgeR, which uses counts per gene from different samples as input and performs data normalization using trimmed mean of M-values method, was used to identify DEGs [54]. To identify NEGs, we compared the average expression value of two parents as MPV, with the expression values of F1 using the same algorithm and criteria as used for DEGs. The log2 transformed values of normalized gene expression (reads per million) was used to build an expression matrix. The matrix from different replicates from each genotypes was independently transposed (Supplementary Table S7), then prcomp function in R [55] was used to perform principal component analysis. The R package pheatmap (Kolde, 2011) was used to draw the heat map for gene expression of NEGs. The bedtools intersect was used to find the abundance of transposons and Fisher’s exact test was used to test for statistical significance. The single-nucleotide polymorphisms information was collected from publicly available database for Col and C24 (http://1001genomes.org/data/MPI/MPISchneeberger2011/releases/current/C24/Marker/C24.SNPs.TAIR9.txt) and allele-specific mapping was extracted from bam alignment file of F1 hybrids using asSeq [56], which was used as input for HTSeq-count to calculate the allelic expression level of NEGs.
The RNAseq data were validated using quantitative real-time PCR analysis, as described previously [57]. The primer details can be seen under Supplementary Table S3.
GO analysis
To analysis the functional enrichments of NEGs, we performed the GO analysis using GOEAST (http://omicslab.genetics.ac.cn/GOEAST/). After we obtained the GO terms lists, the P-value was then adjusted by Benjamini and Hochberg method, to control the false discovery rate with<0.01 cutoff. The top 10 GO terms were showed in the figures as P-value bar plots.
Measurement of SA contents in plants
The endogenous SA contents were measured using liquid chromatography–mass spectrometry. Approximately 25 mg fresh plant sample was homogenized with 500 μl acetone:50 mM citric acid extraction solvent (70:30, vol/vol). The content was centrifuged at 1 000 r.p.m. at 4 °C in dark for 3 h. The supernatant was separated and then evaporated using in a concentrator (Labconco) at −4 °C under vacuum for 1 h. The residual was added with 300 μl of diethyl ether and then centrifuged at 5 000 r.p.m. for 5 min. The upper organic phase was collected thrice. The pooled organic phase was dried at −4 °C under vacuum for 1 h and then dissolved in 200 μl of 50% acetonitrile. After centrifuging at 10 000 r.p.m. for 5 min, the supernatant was collected and used for liquid chromatography–mass spectrometry analysis. During extraction, SA-d4 was also added, which serve as internal standard.
Exogenous treatment of SA
The exogenous supply to SA was given in the form of foliar spray or medium supplement. For foliar spray, SA was applied at two different developmental stages of the plant. Two different doses of SA such as 1 and 5 mM were used for plants at 15 and 22 day after sowed, respectively. The SA was prepared along with 0.01% Tween-20. The plants sprayed with Tween-20 alone served as control. For the medium supplement, Arabidopsis seedlings were grown on 1/2 Murashige and Skoog (MS) medium with different concentrations of SA. At 12 days after growth, seedlings were transplanted to soil for further phenotyping in the next development stages.
BSseq analysis
All the genotypes were grown on plates for 14 days and then collected for BSseq analysis. The sequencing statistics of different BSseq libraries can be found as Supplementary Table S5. The raw reads were mapped to Col-0 TAIR10 A. thaliana genome with BSMAP aligner allowing up to four mismatches [58]. Only uniquely mapped reads were used for subsequent calculation. Only cytosines with a depth of at least four were kept for the determination of cytosine methylation levels [59]. We used the DNA methylation levels of gene body, upstream (2 kb) and downstream for each annotated, to build the DNA methylation matrix (Supplementary Table S8), then used the prcomp function in R [55] for principal component analysis.
Statistical analysis and sequencing data deposition
The experiments during the entire course of the study were carried out in a completely randomized design. All the experiments were repeated at least twice to check reproducibility. Student’s t-test was performed to determine the significant difference between treatments using R statistical computing package [55]. Genome-wide DNA methylation profile was visualized in Integrative Genomics Viewer [60] or Integrated Genome Browser [61].
Acknowledgments
We thank Drs Craig S Pikaard, Eric Richards, Marjori Matzke, Xin-Jian He and Ortrun Mittelsten Scheid for providing various mutant seeds. We thank Dr Yi-Min She, Dr Jingjing Fang and Mr Jun Ma, at Core Facility of Plant Proteomics and Metabolomics, for measurements of endogenous SA in Arabidopsis. We thank Angela Andersen (Life Science Editors) for scientific input and editorial assistance. This work was supported by the Chinese Academy of Sciences and by US National Institutes of Health Grants R01GM070795 and R01GM059138 (to J-KZ). We acknowledge Dr Gaurav Zinta for discussion during manuscript preparation.
NGS accesion numbers
The data generated for this work have been deposited in the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) and are accessible through accession number GSE72993. Genome sequencing data from previous study have been deposited in the NCBI under accession number GSE74551 and GSE72993.
The authors declare no conflict of interest.
Footnotes
(Supplementary information is linked to the online version of the paper on the Cell Discovery website.)
References
- Schnable PS, Springer NM. Progress toward understanding heterosis in crop plants. Annu Rev Plant Biol 2013; 64: 71–88. [DOI] [PubMed] [Google Scholar]
- Miller M, Song Q, Shi X, Juenger TE, Chen ZJ. Natural variation in timing of stress-responsive gene expression predicts heterosis in intraspecific hybrids of Arabidopsis. Nat Commun 2015; 6: 7453. [DOI] [PubMed] [Google Scholar]
- Krieger U, Lippman ZB, Zamir D. The flowering gene SINGLE FLOWER TRUSS drives heterosis for yield in tomato. Nat Genet 2010; 42: 459–463. [DOI] [PubMed] [Google Scholar]
- Groszmann M, Greaves IK, Fujimoto R, Peacock WJ, Dennis ES. The role of epigenetics in hybrid vigour. Trends Genet 2013; 29: 684–690. [DOI] [PubMed] [Google Scholar]
- Chen ZJ. Genomic and epigenetic insights into the molecular bases of heterosis. Nat Rev Genet 2013; 14: 471–482. [DOI] [PubMed] [Google Scholar]
- Kirkbride RC, Yu HH, Nah G, Zhang C, Shi X, Chen ZJ. An epigenetic role for disrupted paternal gene expression in postzygotic seed abortion in Arabidopsis interspecific hybrids. Mol Plant 8: 1766–17752015. [DOI] [PubMed] [Google Scholar]
- Fort A, Ryder P, McKeown PC et al. Disaggregating polyploidy, parental genome dosage and hybridity contributions to heterosis in Arabidopsis thaliana. New Phytol 209: 590–5992015. [DOI] [PubMed] [Google Scholar]
- Groszmann M, Gonzalez-Bayon R, Lyons RL et al. Hormone-regulated defense and stress response networks contribute to heterosis in Arabidopsis F1 hybrids. Proc Natl Acad Sci USA 2015; 112: E6397–E6406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaves IK, Groszmann M, Wang A, Peacock WJ, Dennis ES. Inheritance of trans chromosomal methylation patterns from Arabidopsis F1 hybrids. Proc Natl Acad Sci USA 2014; 111: 2017–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holoch D, Moazed D. RNA-mediated epigenetic regulation of gene expression. Nat Rev Genet 2015; 16: 71–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzke MA, Kanno T, Matzke AJ. RNA-directed DNA methylation: the evolution of a complex epigenetic pathway in flowering plants. Annu Rev Plant Biol 2015; 66: 243–267. [DOI] [PubMed] [Google Scholar]
- Matzke MA, Mosher RA. RNA-directed DNA methylation: an epigenetic pathway of increasing complexity. Nat Rev Genet 2014; 15: 394–408. [DOI] [PubMed] [Google Scholar]
- Zhang H, Zhu J-K. RNA-directed DNA methylation. Curr Opin Plant Biol 2011; 14: 142–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Wang D, Lang Z et al. Methylation interactions in Arabidopsis hybrids require RNA-directed DNA methylation and are influenced by genetic variation. Proc Natl Acad Sci USA (e-pub ahead of print 5 July 2016; doi:10.1073/pnas.1607851113). [DOI] [PMC free article] [PubMed]
- Barber WT, Zhang W, Win H et al. Repeat associated small RNAs vary among parents and following hybridization in maize. Proc Natl Acad Sci USA 2012; 109: 10444–10449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang DL, Zhang G, Tang K et al. Dicer-independent RNA-directed DNA methylation in Arabidopsis. Cell Res 2016; 26: 66–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemach A, Kim MY, Hsieh PH et al. The Arabidopsis nucleosome remodeler DDM1 allows DNA methyltransferases to access H1-containing heterochromatin. Cell 2013; 153: 193–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeddeloh JA, Bender J, Richards EJ. The DNA methylation locusDDM1 is required for maintenance of gene silencing in Arabidopsis. Genes Dev 1998; 12: 1714–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vongs A, Kakutani T, Martienssen R, Richards E. Arabidopsis thaliana DNA methylation mutants. Science 1993; 260: 1926–1928. [DOI] [PubMed] [Google Scholar]
- Lippman Z, Gendrel A-V, Black M et al. Role of transposable elements in heterochromatin and epigenetic control. Nature 2004; 430: 471–476. [DOI] [PubMed] [Google Scholar]
- Baubec T, Dinh HQ, Pecinka A et al. Cooperation of multiple chromatin modifications can generate unanticipated stability of epigenetic states in Arabidopsis. Plant Cell 2010; 22: 34–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malamy J, Carr JP, Klessig DF, Raskin I. Salicylic acid: a likely endogenous signal in the resistance response of tobacco to viral infection. Science 1990; 250: 1002–1004. [DOI] [PubMed] [Google Scholar]
- Yang L, Li B, Zheng X-Y et al. Salicylic acid biosynthesis is enhanced and contributes to increased biotrophic pathogen resistance in Arabidopsis hybrids. Nat Commun 2015; 6: 7309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Z, Morales-Ruiz T, Ariza RR, Roldán-Arjona T, David L, Zhu J-K. ROS1, a repressor of transcriptional gene silencing in Arabidopsis, encodes a DNA glycosylase/lyase. Cell 2002; 111: 803–814. [DOI] [PubMed] [Google Scholar]
- He X-J, Hsu Y-F, Pontes O et al. NRPD4, a protein related to the RPB4 subunit of RNA polymerase II, is a component of RNA polymerases IV and V and is required for RNA-directed DNA methylation. Genes Dev 2009; 23: 318–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinders J, Wulff BBH, Mirouze M et al. Compromised stability of DNA methylation and transposon immobilization in mosaic Arabidopsis epigenomes. Genes Dev 2009; 23: 939–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieu O, Jasencakova Z, Vaillant I et al. Changes in 5S rDNA chromatin organization and transcription during heterochromatin establishment in Arabidopsis. Plant Cell 2003; 15: 2929–2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakutani T, Jeddeloh JA, Flowers SK, Munakata K, Richards EJ. Developmental abnormalities and epimutations associated with DNA hypomethylation mutations. Proc Natl Acad Sci USA 1996; 93: 12406–12411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto R, Taylor JM, Shirasawa S, Peacock WJ, Dennis E. Heterosis of Arabidopsis hybrids between C24 and Col is associated with increased photosynthesis capacity. Proc Natl Acad Sci USA 2012; 109: 7109–7114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groszmann M, Gonzalez-Bayon R, Greaves IK et al. Intraspecific Arabidopsis hybrids show different patterns of heterosis despite the close relatedness of the parental genomes. Plant Physiol 2014; 166: 265–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZJ. Molecular mechanisms of polyploidy and hybrid vigor. Trends Plant Sci 2010; 15: 57–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlot AC, Dempsey DA, Klessig DF. Salicylic Acid, a multifaceted hormone to combat disease. Annu Rev Phytopathol 2009; 47: 177–206. [DOI] [PubMed] [Google Scholar]
- Dempsey DA, Vlot AC, Wildermuth MC, Klessig DF. Salicylic acid biosynthesis and metabolism. Arabidopsis Book 2011; 9: e0156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wildermuth MC, Dewdney J, Wu G, Ausubel FM. Isochorismate synthase is required to synthesize salicylic acid for plant defence. Nature 2001; 414: 562–565. [DOI] [PubMed] [Google Scholar]
- Bernsdorff F, Döring A-C, Gruner K, Schuck S, Bräutigam A, Zeier J. Pipecolic acid orchestrates plant systemic acquired resistance and defense priming via salicylic acid-dependent and -independent pathways. Plant Cell 2016; 28: 102–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarova H, Bernsdorff F, Doring AC, Zeier J. Pipecolic acid, an endogenous mediator of defense amplification and priming, is a critical regulator of inducible plant immunity. Plant Cell 2012; 24: 5123–5141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Xu S, Ding P et al. Control of salicylic acid synthesis and systemic acquired resistance by two members of a plant-specific family of transcription factors. Proc Natl Acad Sci USA 2010; 107: 18220–18225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawrath C, Metraux JP. Salicylic acid induction-deficient mutants of Arabidopsis express PR-2 and PR-5 and accumulate high levels of camalexin after pathogen inoculation. Plant Cell 1999; 11: 1393–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javaheri M, Mashayekhi K, Dadkhah AR, Tavallaee FZ. Effects of salicylic acid on yield and quality characters of tomato fruit (Lycopersicum esculentum Mill.). Int J Agri Crop Sci 2012; 4: 1184–1187. [Google Scholar]
- Fariduddin Q, Hayat S, Ahmad A. Salicylic acid influences net photosynthetic rate, carboxylation efficiency, nitrate reductase activity, and seed yield in Brassica juncea. Photosynthetica 2003; 41: 281–284. [Google Scholar]
- Khandaker L, Akond AM, Oba S. Foliar application of salicylic acid improved the growth, yield and leaf's bioactive compounds in red amaranth (amaranthus tricolor l.). Vegetable Crops Res Bull 2011; 74: 77–86. [Google Scholar]
- Fariduddin Q, Hayat S, Ahmad A. Salicylic acid influences net photosynthetic rate, carboxylation efficiency, nitrate reductase activity, and seed yield in Brassica juncea. Photosynthetica 2003; 41: 281–284. [Google Scholar]
- Sasek V, Janda M, Delage E et al. Constitutive salicylic acid accumulation in pi4kIIIbeta1beta2 Arabidopsis plants stunts rosette but not root growth. New Phytol 2014; 203: 805–816. [DOI] [PubMed] [Google Scholar]
- Gems D, Partridge L. Stress-response hormesis and aging: ‘that which does not kill us makes us stronger’. Cell Metab 2008; 7: 200–203. [DOI] [PubMed] [Google Scholar]
- Mirisola MG, Longo VD. A radical signal activates the epigenetic regulation of longevity. Cell Metab 2013; 17: 812–813. [DOI] [PubMed] [Google Scholar]
- Schroeder EA, Raimundo N, Shadel GS. Epigenetic silencing mediates mitochondria stress-induced longevity. Cell Metab 2013; 17: 954–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz TJ, Zarse K, Voigt A, Urban N, Birringer M, Ristow M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab 2007; 6: 280–293. [DOI] [PubMed] [Google Scholar]
- Cortijo S, Wardenaar R, Colomé-Tatché M et al. Mapping the epigenetic basis of complex traits. Science 2014; 343: 1145–1148. [DOI] [PubMed] [Google Scholar]
- Lippman Z, Gendrel AV, Black M et al. Role of transposable elements in heterochromatin and epigenetic control. Nature 2004; 430: 471–476. [DOI] [PubMed] [Google Scholar]
- Ni Z, Kim E-D, Ha M et al. Altered circadian rhythms regulate growth vigour in hybrids and allopolyploids. Nature 2009; 457: 327–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murashige T, Skoog F. A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiol Plantarum 1962; 15: 473–497. [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 2013; 14: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 2015; 31: 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010; 26: 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R-Core-Team R. A Language and Environment for Statistical Computing. R Foundation for Statistical Computing: Vienna, Austria, 2015. [Google Scholar]
- Sun W, Zhabotynsky V. asSeq: A Set Of Tools for the Study of Allele-Specific RNA-seq Data. 2013. http://www.bios.unc.edu/~weisun/software/asSeq.pdf.
- Shen H, He H, Li J et al. Genome-wide analysis of DNA methylation and gene expression changes in two Arabidopsis ecotypes and their reciprocal hybrids. Plant Cell 2012; 24: 875–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi Y, Li W. BSMAP: whole genome bisulfite sequence MAPping program. BMC Bioinformatics 2009; 10: 232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, O'Malley RC, Tonti-Filippini J et al. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 2008; 133: 523–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W et al. Integrative genomics viewer. Nat Biotechnol 2011; 29: 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicol JW, Helt GA, Blanchard SG Jr, Raja A, Loraine AE. The Integrated Genome Browser: free software for distribution and exploration of genome-scale datasets. Bioinformatics 2009; 25: 2730–2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.