

Abstract
Background
The lung is exposed to airborne fungal spores, and fungi that colonize the oral cavity such as Candida albicans, but does not develop disease to opportunistic fungal pathogens unless the immune system is compromised. The Group IVA cytosolic phospholipase A2 (cPLA2α) is activated in response to Candida albicans infection resulting in the release of arachidonic acid for eicosanoid production. Although eicosanoids such as prostaglandins and leukotrienes modulate inflammation and immune responses, the role of cPLA2α and eicosanoids in regulating C. albicans lung infection is not understood.
Methods
The responses of cPLA2α+/+ and cPLA2α−/− Balb/c mice to intratracheal instillation of C. albicans were compared. After challenge, we evaluated weight loss, organ fungal burden, and the recruitment of cells and the levels of cytokines and eicosanoids in bronchoalveolar lavage fluid. The ability of macrophages and neutrophils from cPLA2α+/+ and cPLA2α−/− mice to recognize and kill C. albicans was also compared.
Results
After C. albicans instillation, cPLA2α+/+ mice recovered a modest weight loss by 48 h and completely cleared fungi from the lung by 12 h with no dissemination to the kidneys. In cPLA2α−/− mice, weight loss continued for 72 h, C. albicans was not completely cleared from the lung and disseminated to the kidneys. cPLA2α−/− mice exhibited greater signs of inflammation including higher neutrophil influx, and elevated levels of albumin and pro-inflammatory cytokines/chemokines (IL1α, IL1β, TNFα, IL6, CSF2, CXCL1, CCL20) in bronchoalveolar lavage fluid. The amounts of cysteinyl leukotrienes, thromboxane B2 and prostaglandin E2 were significantly lower in bronchoalveolar lavage fluid from C. albicans-infected cPLA2α−/− mice compared to cPLA2α+/+ mice. Alveolar macrophages and neutrophils from uninfected cPLA2α−/− mice exhibited less killing of C. albicans in vitro than cells from cPLA2α+/+ mice. In addition alveolar macrophages from cPLA2α−/− mice isolated 6 h after instillation of GFP-C. albicans contained fewer internalized fungi than cPLA2α+/+ macrophages.
Conclusions
The results demonstrate that cPLA2α contributes to immune surveillance and host defense in the lung to prevent infection by the commensal fungus C. albicans and to dampen inflammation.
Electronic supplementary material
The online version of this article (doi:10.1186/s12865-016-0165-9) contains supplementary material, which is available to authorized users.
Keywords: Cytosolic phospholipase A2, Prostaglandins, Leukotrienes, Neutrophils, Macrophages, Inflammation, Candida albicans
Background
Group IVA cytosolic phospholipase A2 (cPLA2α) releases arachidonic acid to initiate eicosanoid production [1]. Eicosanoids are secreted and act locally through G-protein coupled receptors, which are expressed in a cell-type specific manner and initiate distinct signaling pathways to promote diverse biological responses [2–4]. Arachidonic acid is metabolized by 5-lipoxygenase (5-LO) to leukotrienes, and by constitutive cyclooxygenase (COX)-1 and inducible COX-2 to prostaglandins and thromboxane [5, 6]. Leukotrienes are pro-inflammatory mediators produced by macrophages, dendritic cells, mast cells, basophils and eosinophils that regulate cell trafficking, cytokine production, vascular permeability and phagocyte function [7]. The cysteinyl leukotrienes including leukotriene C4, leukotriene D4 and leukotriene E4 are bronchoconstrictors involved in asthma and allergic responses [2]. cPLA2α and COXs are widely expressed reflecting the ability of most cells and tissues to produce prostanoids, which have diverse functions [1, 6]. Prostaglandins regulate normal physiological processes such as female reproduction, hemostasis, kidney function and the maintenance of the gastrointestinal tract [1]. Although prostaglandins promote acute and chronic inflammation in response to tissue injury they also play a role in the resolution of inflammation and can be anti-inflammatory and immunosuppressive [8–10]. Therefore cPLA2α mediates the release of arachidonic acid for the production of numerous bioactive lipid mediators that have diverse effects [1]. This makes its role in regulating responses to infection difficult to predict and would be influenced by the specific tissue involved and nature of the microorganism.
Eicosanoids are produced rapidly in response to engagement of pattern recognition receptors by microbial pathogens and modulate immune cell function by affecting phagocytosis, microbial killing, chemotaxis and the transcriptional program [7, 10, 11]. We have used resident tissue macrophages from the peritoneal cavity and the lung to study the mechanisms of cPLA2α activation by the fungal pathogen Candida albicans [12–16]. Resident tissue macrophages are sentinel cells that are first responders to microbial invasion for initiating host defense to infection [17]. In resident peritoneal macrophages, activation of cPLA2α by C. albicans involves engagement of fungal cell wall polysaccharides β-glucan and mannans to C-type lectin receptors dectin-1 and dectin-2, respectively [13, 14]. These receptors act with MyD88-dependent pathways to activate cPLA2α, which involves calcium-induced translocation to membrane and phosphorylation by mitogen-activated protein kinases. In peritoneal macrophages, C. albicans stimulates an autocrine loop involving cPLA2α activation, production of prostaglandins and increases in cAMP that affects expression of genes involved in host defense and to dampen inflammation [15, 16]. In contrast, alveolar macrophages exhibit distinct properties since C. albicans poorly stimulates cPLA2α-mediated arachidonic acid release, however, priming with granulocyte macrophage colony-stimulating factor (GM-CSF) enhances arachidonic acid release by increasing expression of dectin-1 [12].
The lung has several mechanisms to clear environmental triggers that are continuously inhaled to prevent excess inflammation and tissue injury that may compromise gas exchange function [18]. Candida is the predominant fungal genus in the oral cavity, and dispersal of microoganisms from this site to the lung is a mechanism for shaping the lung microbiome [19, 20]. Despite potential exposure from the oral cavity, levels of C. albicans in the healthy lung are low indicating mechanisms for efficient clearance to prevent colonization [21, 22]. C. albicans is a commensal of mucosal surfaces that does not cause infection unless the immune system is compromised [23, 24]. Candida lung infection occurs in the critically ill, in patients with cancer and cystic fibrosis, during organ transplantation and in immune compromised individuals [21, 25, 26]. By comparing cPLA2α+/+ and cPLA2α−/− mice, we found that cPLA2α contributes to innate immune defenses in the lung for protection against C. albicans infection.
Methods
Materials
Hank’s Balanced Salts Solution was from Invitrogen (Carlsbad, CA). ELISA kits were from eBioscience (San Diego, CA) (IL1α, IL1β, TNFα, IL6), from Immunology Consultants Laboratory Inc. (Portland, OR) (albumin), from R&D Systems (Minneapolis, MN) (CCL20) and from PeproTech (Rocky Hill, NJ) (CXCL1, CSF2, CSF3). Antibodies for flow cytometry analysis were from eBioscience (San Diego, CA) (anti-mouse CD45 eF450, CD11c PE, CD24 FITC, CD11b APC, MHC-II I-A/E PerCP-eF710, CD103 FITC) and from BD Biosciences (San Jose, CA) (anti-mouse Siglec F-PE and Ly6G (clone 1A8)-PE). QuickIII staining kit for cytospins was obtained from Astral Diagnostics, NJ. Butylated hydroxytoluene and indomethacin were from Fisher Scientific. Percoll, collagenase XI, Trypsin inhibitor, DNase I, RBC lysis solution were from Sigma-Aldrich (St. Louis, MO). Nylon cell strainers (70 μm) were from BD Biosciences (San Jose, CA). Qiasol lysis reagent, RNeasy Mini Kits and Mouse Cytokines & Chemokines RT2 Profiler PCR Array were from Qiagen (Valencia, CA). Paraformaldehyde was from Electron Microscopy Sciences (Hatfield, PA). XTT Cell Viability Kit was from Cell signaling.
Mice
cPLA2α−/− mice were generated as previously described [27], and backcrossed onto a Balb/c background for 10 generations. Balb/c control mice (cPLA2α+/+) were obtained from Charles River (San Diego, CA). Mice were housed under specific pathogen free conditions and used between 8–14 weeks of age. Male mice were used for all experiments with exception as noted in the figure legend. The work with mice was approved by the Institutional Animal Care and Use Committee (IACUC) at National Jewish Health and conducted in accordance with their guidelines.
C. albicans challenge
C. albicans (ATCC SC5314) was grown in YPD medium overnight (30 °C), washed, suspended in endotoxin-free PBS then counted. Counts correlated directly with colony forming units (CFU). C. albicans was administered by intratracheal instillation to cPLA2α+/+ and cPLA2α−/− Balb/c mice under isoflurane anesthesia. The trachea was intubated with a gavage needle to instill (50 μl) C. albicans (106–107 CFU) or endotoxin-free PBS. Mice were euthanized by CO2 asphyxiation or cervical dislocation with similar results. C. albicans expressing green fluorescent protein (GFP) was kindly provided by Dr. Robert Wheeler, The University of Maine. It was generated from the wild type SC5314 strain and exhibits similar virulence as the wild type strain in mice [28].
Bronchoalveolar lavage
Lungs were lavaged 5 times as described [12]. For analysis of eicosanoids in bronchoalveolar lavage fluid (BALF), the lavage solution also contained 5 μM indomethacin and 50 μM butylated hydroxytoluene. Cells in lavage were differentiated on cytospins. Albumin, cytokines and chemokines were measured in BALF by ELISA.
Fungal burden
Blood was drained by cutting the inferior vena cava, and then lungs and kidneys were removed asceptically, weighed and homogenized (Omni Tissue Homogenizer, Omni International) in sterile phenol red-free HBSS. Homogenates were serially diluted, plated on Sabouraud dextrose agar plates containing penicillin and streptomycin, and then C. albicans CFU determined after 48 h incubation at 37 °C.
Histology
Lungs were fixed by inflation (1 ml), immersed in formalin (10 %) then dehydrated and embedded in paraffin. Sections (5 μm) were stained with H & E.
Real-time PCR
Lungs from cPLA2α+/+ and cPLA2α−/− mice were homogenized with an Omni Tissue Homogenizer in Qiasol lysis reagent and RNA isolated using on-column DNase treatment. RNA concentration and purity were determined by UV spectrophotometry, and RNA integrity verified using an Agilent Bioanalyzer 2100. cDNA was synthesized from RNA (200 ng) using RT2 First Strand Kit (Qiagen). Real-time PCR was performed using RT2 qPCR Mastermix and a Mouse Cytokines & Chemokines RT2 Profiler PCR Array according to the manufacturer's protocol using the StepOnePlus Real-Time PCR System (Applied Biosystems). RT2 PCR arrays in a 96-well format were used containing pre-validated primers tested for efficiency (Qiagen). The RT2 Profiler PCR Array System included a reverse transcription control preloaded into the primer buffer of the RT2 First Strand cDNA synthesis kit that measured the relative efficiency of the reverse transcription for all the samples. A genomic DNA control and a positive PCR control were also included in the system. The RT2 Profiler PCR Array data were normalized to the housekeeping gene Gusb and the relative gene expression level (2^(−ΔCt) was calculated using the formula ΔCt = Ct (gene of interest)- Ct (housekeeping gene). The data were analyzed on the PCR array data analysis SA Biosciences web portal (http://pcrdataanalysis.sabiosciences.com/pcr/arrayanalysis.php).
Real-time PCR was also performed with cDNA synthesized with random hexamer primers (Fermentas Maxima First Strand cDNA Synthesis Kit, Thermo Scientific) using TaqMan fast universal PCR master mix. TaqMan assay probes used were: Clec7a (dectin-1) (Mm01183349_m1), Clec4n (dectin-2) (Mm00490934_m1) and Gusb (Mm01197698_g1). The housekeeping gene Gusb was used for normalization. Threshold cycle values (CT) were determined and used for ∆∆CT analysis of gene expression [29].
Lung digestion and flow cytometry analysis
After performing bronchoalveolar lavage, blood was drained from the lungs by cutting the inferior vena cava. Lungs were removed, cut into small pieces followed by digestion with 5 ml collagenase solution (0.5 mg/ml collagenase XI, 0.2 mg/ml trypsin inhibitor, 5 % FBS in minimum essential medium) for 1 h at 37 °C with occasional mixing. The digested lungs were sheared with an 18-gauge needle, treated with 50 μl of DNase I solution (5 mg/ml) and then incubated for 10 min at 37 °C. Lung digests were filtered through 70-μm nylon cell strainers and the single cell suspension treated with RBC lysis solution. Cells were counted using a Countess cell counter (Invitrogen, Carlsbad, CA) excluding dead cells with trypan blue. Cells were resuspended in flow cytometry (FC) buffer (2 % FBS, 0.1 % BSA, 0.05 % sodium azide in PBS) at 2 × 106 cells/ml. All the steps were done at 4 °C. Cells were dispensed (0.5 x 106 cells in 250 μl) in V-shaped 96 well plates. After centrifugation at 1500 rpm for 5 min, the supernatant was removed and 50 μl of FcBlock (anti-CD16/CD32, clone 2.4G2, 40 μg/ml in FC buffer, eBiosciences) was added followed by incubation on ice for 15 min. Cells were then treated with 50 μl of antibody cocktails, incubated on ice for 30 min followed by addition of 150 μl FC buffer then washed in FC buffer. Cells were fixed with 4 % paraformaldehyde in PBS (100 μl/well), pH 7.4, then transferred to FC tubes in 300 μl FC buffer and stored in the dark at 4 °C until analysis. Data were acquired on a Dako Cyan ADP flow cytometer. Compensation and data analyses were performed using FlowJo software (TreeStar, Ashland, OR). After the exclusion of doublets and debris, immune cells were identified by CD45 positive staining. A sequential gating strategy was used to identify cell populations: alveolar macrophages (CD45+ CD24− CD11b− SiglecF+); tissue macrophages (CD45+ CD24− CD11b+); neutrophils (CD45+ CD11b+ Ly6G+) and CD11b+ dendritic cells (CD11b+ DCs) (CD45+ MHCII+ CD11c+ CD11b+) (Additional file 1) [30].
Eicosanoid measurements
BALF stored at −80 °C was thawed and mixed with an equal volume of cold methanol. Just before analysis, the samples were diluted in water to a final methanol concentration of less than 15 % and then extracted using a solid phase extraction cartridge (Strata Polymeric Reverse Phase 60 mg/ml; Phenomenex, Torrance, CA). The eluate (1 ml of methanol) was dried and reconstituted in 75 μl of high-performance liquid chromatography (HPLC) solvent A (8.3 mM acetic acid buffered to pH 5.7 with NH4OH) and 25 μl of solvent B (acetonitrile/methanol, 65/35, v/v). An aliquot of each sample (30 μl) was injected into an HPLC and metabolites separated on a C18 column (Kinetex EVO C18 100A 50 x 3.0 mm, 5 μm; Phenomenex, Torrance, CA) eluted at a flow rate of 0.25 ml/min with a linear gradient from 25 % to 75 % solvent B in 13 min then increased to 98 % in 2 min and held for 11 min. The HPLC system was directly interfaced into the electrospray ionization source of a triple quadrapole mass spectrometer (Sciex API 5500; PE-Sciex, Thornhill, ON, Canada). Mass spectrometric analyses were performed in the negative ion mode using multiple reaction monitoring of the specific transitions: [d4]PGE2m/z 355 → 275, [d4]PGD2m/z 355➔237, [d4]TXB2m/z 373 → 173, [d4]6-keto-PGF1α m/z 373➔167, [d5]LTC4m/z 629 → 271, [d5]LTD4m/z 500➔177, [d5]LTE4m/z 443➔338, PGE2m/z 351 → 271, PGD2m/z 351➔233, TXB2m/z 369 → 169, 6-Keto-PGF1α m/z 369➔ 163, LTC4m/z 624 → 272, LTD4m/z 495➔177, LTE4m/z 438➔333. Quantitation was performed using a standard isotope dilution curve as described [31].
C. albicans recognition and killing assays
Alveolar macrophages were isolated from untreated cPLA2α+/+ and cPLA2α−/− mice by lavage and cultured as previously described [12]. Live opsonized and unopsonized GFP-C. albicans (moi 2) was used for all assays. GFP-C. albicans was opsonized by incubating in DMEM containing 10 % mouse serum for 30 min at 37 °C before incubation with the macrophages. For evaluating binding and internalization (recognition assay), alveolar macrophages (1 × 105) were seeded onto the glass insert of MatTek 35 mm dishes and incubated for 2 h [12]. Cells were washed then incubated with GFP-C. albicans in phenol red-free DMEM containing penicillin, streptomycin and 0.1 % endotoxin-free BSA (stimulation media) for 30 min at 37 °C and 5 % CO2. Macrophages were washed, fixed with 4 % paraformaldehyde for 15 min and then stained with DAPI. Images were captured on a Marianas 200 spinning disk confocal microscope using Intelligent Imaging Innovation Inc. (3I) software (Slidebook 6.0) to determine the number of macrophages containing GFP-C albicans. For killing assays, alveolar macrophages (in 48 well plates) were incubated for 2 h in stimulation media with GFP-C. albicans. Wells containing an equivalent number of GFP-C. albicans (without macrophages) were included as a positive control for determining 100 % viability. Macrophages were lysed with 1 % Triton X-100 and GFP-C. albicans viability was measured using the XTT Cell Viability Kit as described [32].
Bone marrow neutrophils were isolated from untreated cPLA2α+/+ and cPLA2α−/− mice as described previously and purity (>95 %) determined on cytospins [33]. Neutrophils (1 × 105) were plated on polylysine-coated MatTek 35 mm dishes, incubated for 1 h and then incubated with GFP-C. albicans for 30 min. After fixation the cells were incubated for 1 h in PBS containing 10 % FBS and then incubated overnight with anti-Ly6G antibody followed by treatment with anti-rabbit AF594 secondary antibody and with DAPI. For killing assays, GFP-C. albicans was added to neutrophils (5 × 104) in the 96 well plates, centrifuged for 5 min at 300 g to synchronize the infection, and then incubated for 2 h at 37 °C and 5 % CO2. GFP-C. albicans viability was determined as described above for macrophages.
Statistics
The data are presented as mean ± SEM and analyzed using the 2-tailed unpaired t-test or the Mann Whitney method to determine statistical significance (defined as p < 0.05).
Results
C. albicans infection causes greater weight loss in cPLA2α−/− than cPLA2α+/+ mice
The role of cPLA2α in regulating host defense against C. albicans lung infection was investigated by comparing responses of cPLA2α+/+ and cPLA2α−/− Balb/c mice. The LD50 from intratracheal challenge with C. albicans in immune competent mice is approximately 108 CFU [34]. We first determined if concentrations below the LD50 (106 and 107 CFU) induced weight changes (Fig. 1). There was significant weight loss from both cPLA2α+/+ and cPLA2α−/− mice compared to saline controls 24 h after instillation of 107 Candida that continued for 72 h (Fig. 1a). Weight loss was significantly greater from cPLA2α−/− than cPLA2α+/+ mice at 48 and 72 h. Challenging mice with 106C. albicans resulted in a small but significant weight loss in cPLA2α+/+ mice at 12 and 24 h compared to saline controls followed by recovery of normal weight by 48–72 h (Fig. 1b). In cPLA2α−/− mice challenged with 106C. albicans, weight loss continued from 24–72 h and was significantly greater than in cPLA2α+/+ mice. cPLA2α+/+ and cPLA2α−/−mice were challenged with an intermediate amount of C. albicans (5 × 106 CFU) and survival and body weight monitored for 28 days. All cPLA2α+/+ mice survived but 50 % of cPLA2α−/− mice died by 72 h with no further mortality for 28 days (Fig. 1c). cPLA2α−/− mice lost significantly more weight (16.5 % ± 1.1), which was greatest at day 4, than cPLA2α+/+ mice (6.5 % ± 0.4). The surviving cPLA2α−/− mice and all cPLA2α+/+ mice started gaining weight after day 4 that returned to normal by ~18 days. Since the results suggest that cPLA2α regulates early host defense to C. albicans lung infection, we focused on comparing early responses of cPLA2α+/+ and cPLA2α−/− mice up to 72 h after C. albicans challenge.
Fig. 1.
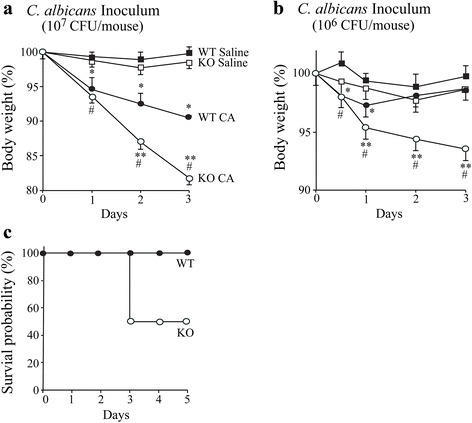
Weight loss is greater in cPLA2α−/− than cPLA2α+/+ mice during C. albicans lung infection. Body weight of cPLA2α−/− (KO, open symbols) and cPLA2α+/+ (WT, closed symbols) mice was monitored after intratracheal instillation of saline (squares) or C. albicans (CA, circles) using an inoculum of (a) 107 or (b) 106 CA. Body weight is expressed as the % of the weight determined just prior to instillation of C. albicans or saline (n = 8–13 mice/group, from 4–6 independent experiments). *P < 0.05 compared to cPLA2α+/+ with saline; #P < 0.05 compared to cPLA2α−/− with saline; **P < 0.05 compared to cPLA2α+/+ with CA. c Survival of female cPLA2α−/− (KO, open symbols) and cPLA2α+/+ (WT, closed symbols) mice was monitored after intratracheal administration of 5 x 106 CFU C. albicans (6 mice/group)
C. albicans is not cleared completely from the lungs of cPLA2α−/− mice and disseminates to the kidney
The ability of cPLA2α+/+ and cPLA2α−/− mice to clear C. albicans was compared by measuring fungal CFU in lung homogenates. It has previously been reported that immune competent mice are resistant to infection and rapidly eliminate C. albicans from the lung [34, 35]. Analysis of lungs 5 min after intratracheal challenge with 106C. albicans confirmed that greater than 90 % of the inoculum delivered to the lung was recovered in homogenates. By 6 h after instillation most (≥98 %) of the C. albicans was cleared from the lungs of cPLA2α+/+ and cPLA2α−/− mice although significantly more remained in cPLA2α−/− mice than cPLA2α+/+ mice (Fig. 2a). cPLA2α+/+ mice completely cleared C. albicans from the lung with no viable fungi recovered from 12–72 h after instillation, whereas a significant fungal burden persisted in cPLA2α−/− mice during this time period. Using a higher inoculum (107 CFU), a low level of C. albicans was recovered in lungs of cPLA2α+/+ mice (150 ± 14 CFU/g) at 72 h, and 30-fold higher levels in cPLA2α−/− mice (4567 ± 450 CFU/g). We also determined if C. albicans breached the lung and disseminated to the kidney, which is the primary target organ in mice and humans in disseminated candidiasis [36, 37]. C. albicans was recovered from the kidneys of cPLA2α−/− mice challenged with 106 CFU at 12 h that further increased from 24–72 h (Fig. 2b). Using an inoculum of 107 CFU, the kidneys of cPLA2α−/− mice contained considerably more C. albicans than the relatively low level in mice challenged with 106 CFU. In contrast C. albicans was not detected in kidneys of cPLA2α+/+ mice challenged with 106 or 107C. albicans (Fig. 2b). The results demonstrate a critical protective role for cPLA2α in the early stages of Candida clearance and dissemination in vivo. Based on these results we investigated differences in the early host defense responses in cPLA2α+/+ and cPLA2α+/+ mice using the lower inoculum of 106C. albicans.
Fig. 2.
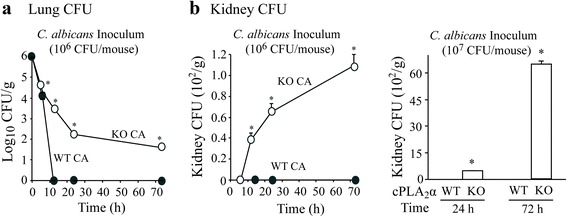
C. albicans is not cleared completely from lungs of cPLA2α−/− mice and disseminates to the kidney. cPLA2α−/− (open circles) and cPLA2α+/+ (closed circles) mice were challenged with either 106 or 107 C. albicans (CA), and CFU were determined at the indicated times in homogenized (a) lung and (b) kidney (n = 3–14 mice/group, from 3–6 independent experiments). *P < 0.05 compared to cPLA2α+/+
cPLA2α−/− mice have higher numbers of neutrophils in BALF and lung tissue than cPLA2α+/+ mice during C. albicans infection
To evaluate the extent of inflammation in cPLA2α+/+ and cPLA2α−/− mice, the recruitment of cells into BALF was compared from 6–72 h after intratracheal instillation of 106C. albicans or saline (Fig. 3). The number of total cells recovered in BALF of untreated (0-time) and saline control mice was significantly higher (~20 %) in cPLA2α−/− than cPLA2α+/+ mice that was due to higher numbers of alveolar macrophages (Fig. 3a and b). Over 95 % of the cells in BALF of cPLA2α+/+ and cPLA2α−/− control mice were alveolar macrophages. The number of total cells in BALF increased 6 h after C. albicans challenge to a slightly greater level in cPLA2α−/− mice, then decreased by 12 h to similar levels in cPLA2α+/+ and cPLA2α−/− mice (Fig. 3a). Between 12 and 24 h after C. albicans instillation the number of total cells in cPLA2α+/+ mice slightly declined but increased in cPLA2α−/− mice due to greater neutrophil influx (Fig. 3a and c). The number of alveolar macrophages in BALF increased at 6 h after C. albicans challenge to similar levels in cPLA2α+/+ and cPLA2α−/− mice followed by a sharp decline in both strains at 12 h that remained low for 72 h (Fig. 3b). The decrease in the number of alveolar macrophages is reminiscent of the macrophage disappearance reaction observed in response to inflammation in the peritoneal cavity that is attributed to macrophage activation resulting in increased adherence or trafficking [17, 38]. Neutrophils significantly increased in BALF from cPLA2α−/− but not cPLA2α+/+ mice 6 h after C. albicans instillation but then increased to similar levels in both strains at 12 h (Fig. 3c). Neutrophil numbers in cPLA2α+/+ mice peaked at 12 h but continued to increase in cPLA2α−/− mice up to 24 h reaching levels 2.3-fold higher than in cPLA2α+/+ mice. Neutrophil numbers in cPLA2α−/− mice remained >2-fold higher than the levels in cPLA2α+/+ mice up to 72 h after infection (Fig. 3c). It has been demonstrated that neutrophil influx into the lung during bacterial pneumonia contributes to alveolar barrier disruption promoting leakage of plasma protein into the alveolar space [39]. To determine if the increased neutrophil influx in C. albicans-infected cPLA2α−/− mice was accompanied by an increase in protein leak into the lung, the amount of albumin in BALF was determined by ELISA (Fig. 3d). Albumin levels increased at 12 h after C. albicans instillation to a similar extent in cPLA2α−/− than cPLA2α+/+ mice. Albumin levels increased by 24 h and were 1.8-fold higher in cPLA2α−/− mice compared to cPLA2α+/+ mice. The results indicate a greater compromise of alveolar barrier function in cPLA2α−/− mice that correlated with a higher level of neutrophil influx in response to C. albicans infection.
Fig. 3.
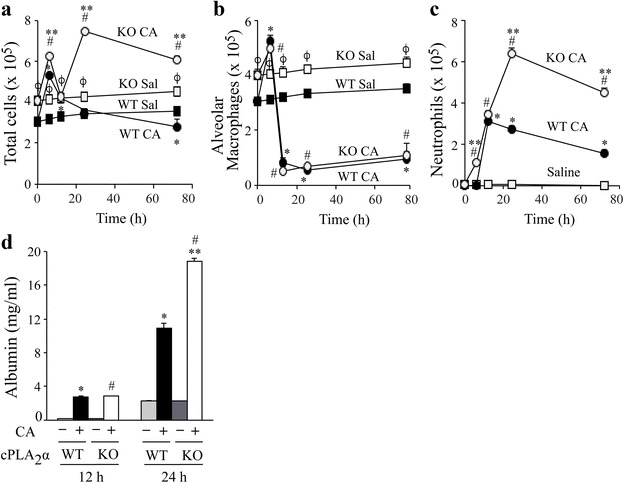
Neutrophils and albumin are higher in BALF of cPLA2α−/− than cPLA2α+/+ mice during C. albicans infection. The number of (a) total cells, (b) alveolar macrophages and (c) neutrophils were determined in BALF (5 lavages) from cPLA2α−/− (KO, open symbols) and cPLA2α+/+ (WT, closed symbols) mice instilled with saline (Sal, squares) or 106 C. albicans (CA, circles). d cPLA2α+/+ (WT) and cPLA2α−/− (KO) mice were lavaged 12 and 24 h after instillation of 106 C. albicans or saline. Albumin levels were determined in BALF by ELISA. (n = 8-13 mice/group, from 4–6 independent experiments). *P < 0.05 compared to cPLA2α+/+ saline control; ϕ P < 0.05 compared to cPLA2α+/+ saline control, # P < 0.05 compared to cPLA2α−/− saline control; **P < 0.05 compared to cPLA2α+/+ with CA
Cell influx into lung tissue of cPLA2α+/+ and cPLA2α−/− mice 24 h after C. albicans infection was also evaluated by flow cytometry (Fig. 4). C. albicans infection stimulated a significant increase in neutrophils (CD45+CD11b+Ly6G+) in lung tissue of both cPLA2α+/+ and cPLA2α−/− mice but numbers were 2.5-fold higher in cPLA2α−/− mice (Fig. 4a). There were similar numbers of alveolar macrophages (CD45+CD24−CD11b−SiglecF+) in lungs of cPLA2α+/+ and cPLA2α−/− mice, and numbers were not affected by C. albicans infection (Fig. 4b). This suggests that the higher number of alveolar macrophages in BALF of uninfected cPLA2α−/− mice (see Fig. 3a) may be due to differences in adherence properties that influence their recovery by lavage. C. albicans infection stimulated an increase in tissue macrophages (CD45+CD24−CD11b+), which were significantly higher in cPLA2α−/− compared to cPLA2α+/+ mice (Fig. 4c). The tissue macrophage population, which includes both interstitial macrophages and monocytes, may increase due to recruitment of monocytes from the blood in response to C. albicans infection. CD11b+ dendritic cells (CD45+MHCII+CD11c+CD11b+) increased in response to C. albicans infection to a greater extent in cPLA2α−/− compared to cPLA2α+/+ mice (Fig. 4d). Representative histograms of the flow cytometry analysis are shown in Additional file 1. Histological examination of lung sections 24 h after C. albicans challenge showed little evidence of inflammation other than an occasional small patch of focal inflammation in cPLA2α+/+ mice. The patches of inflammation were markedly larger and more extensive in cPLA2α−/− than in cPLA2α+/+ mice (Fig. 4e).
Fig. 4.
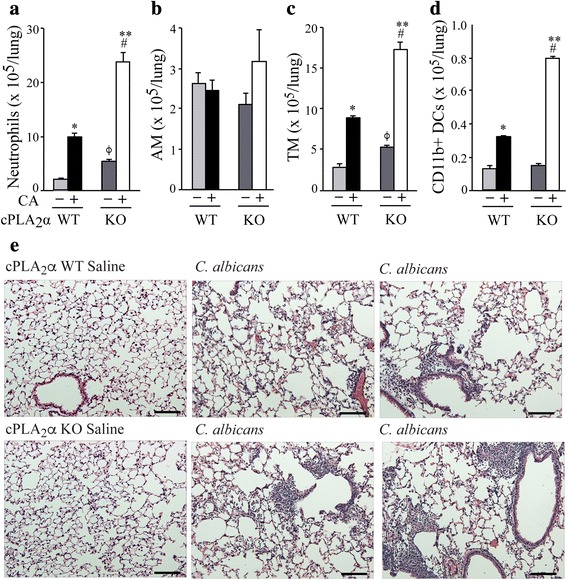
Effect of C. albicans infection on cell composition and histopathology of lung tissue from cPLA2α+/+ and cPLA2α−/− mice. cPLA2α−/− (KO) and cPLA2α+/+ (WT) mice were challenged with saline or 106 CFU C. albicans (CA) for 24 h and then lavaged. Lung tissue was processed for identification of (a) Neutrophils, (b) alveolar macrophages (AM), (c) tissue macrophages (TM) and (d) CD11b+ dendritic cells (DC) by flow cytometry (n = 3 mice/group). *P < 0.05 compared to cPLA2α+/+ saline control; ϕ P < 0.05 compared to cPLA2α+/+ saline control, # P < 0.05 compared to cPLA2α−/− saline control; **P < 0.05 compared to cPLA2α+/+ with CA. e Representative images of H & E stained lung sections from saline controls and from C. albicans-infected cPLA2α WT or cPLA2α KO mice are shown. Scale bar = 100 μm
cPLA2α influences gene expression and cytokine production in lungs of C. albicans infected mice
We previously reported that activation of cPLA2α in C. albicans-infected macrophages influences gene expression through an autocrine loop involving the production of prostaglandins and increases in cAMP [15, 16]. We first screened differences in gene expression in total lung tissue of cPLA2α+/+ and cPLA2α−/− mice at 12 and 24 h after instillation of C. albicans or saline by using a cytokine/chemokine PCR array (Additional file 2). C. albicans infection stimulated an increase in expression of several pro-inflammatory cytokines (Il1α, Il1β, Tnfα, Il6), and the immune mediators Csf2 and Ccl20, in lungs of cPLA2α+/+ and cPLA2α−/− mice. The level of these cytokines was significantly higher in cPLA2α−/− compared to cPLA2α+/+mice particularly 12 h after C. albicans challenge. The chemokines Ccl2, Ccl7 and Cxcl1 were also expressed at higher levels in cPLA2α−/− compared to cPLA2α+/+mice 12 h after infection, but at 24 h they decreased to a greater extent in cPLA2α−/− than cPLA2α+/+mice. Cxcl10 and Ccl12 increased during C. albicans infection to the same extent in cPLA2α+/+ and cPLA2α−/− mice at 12 h but were significantly lower in cPLA2α−/− than cPLA2α+/+mice at 24 h. The results evaluating gene expression in the total lung suggested that cPLA2α activation suppresses the expression of several pro-inflammatory cytokines but also influences the duration of gene expression particularly for certain chemokines (Ccl2, Ccl7, Ccl12, Cxcl1, Cxcl10).
Since C. albicans first encounters cells lining the airways and in the alveoli, cytokines and chemokines were measured in BALF from cPLA2α+/+ and cPLA2α−/− mice collected 6–24 h after C. albicans infection (Fig. 5). The pro-inflammatory cytokines IL1α (Fig. 5a), IL1β (Fig. 5b), TNFα (Fig. 5c) and IL6 (Fig. 5d) were significantly higher in BALF of cPLA2α−/− compared to cPLA2α+/+ mice at all time points but the time of peak production differed. IL1α production was transient and peaked at 12 h in cPLA2α−/− mice reaching levels that were 8-fold higher than in cPLA2α+/+ mice (Fig. 5a). TNFα continued to increase in cPLA2α−/− mice for 24 h (Fig. 5c). IL6 was 10-fold higher in cPLA2α−/− compared to cPLA2α+/+ mice at 6 and 12 h after infection then decreased by 24 h (Fig. 5d). There was early production of IL1β in cPLA2α−/− mice that continued to increase up to 24 h after C. albicans infection (Fig. 5b). CSF2 (Fig. 5e) and CCL20 (Fig. 5f) were significantly higher in cPLA2α−/− than cPLA2α+/+mice at 12 and 24 h after infection. The neutrophilic chemokine CXCL1 was higher in BALF of cPLA2α−/− than cPLA2α+/+ mice particularly 24 h after C. albicans infection (Fig. 5g). Although levels of Csf3 mRNA were similar in lungs of C. albicans infected cPLA2α−/− and cPLA2α+/+mice, analysis of BALF showed that CSF3 was higher in cPLA2α−/− than cPLA2α+/+ mice at 24 h (Fig. 5h). The results demonstrate that cPLA2α−/− mice have higher levels of pro-inflammatory cytokines and chemokines consistent with increased neutrophil recruitment.
Fig. 5.
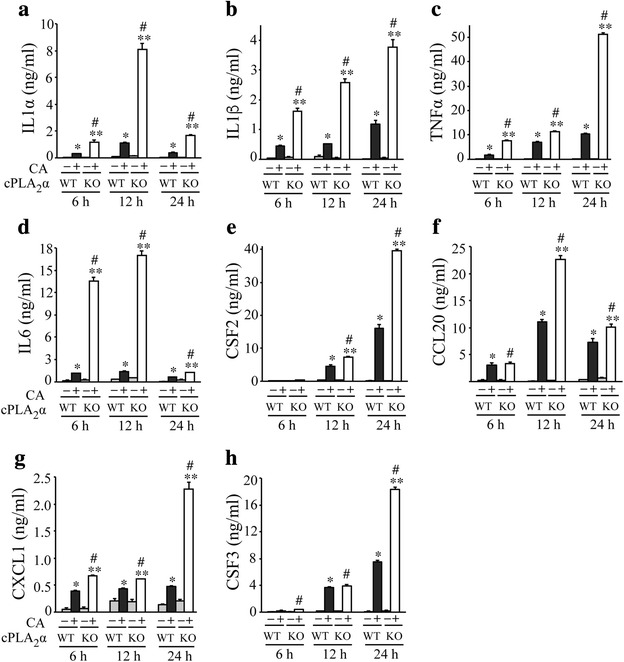
Pro-inflammatory cytokines and chemokines are higher in BALF from cPLA2α−/− than cPLA2α+/+ mice during C. albicans infection. cPLA2α−/− (KO) and cPLA2α+/+ (WT) mice were challenged with saline or 106 C. albicans (CA) for 6, 12 and 24 h. The levels of (a) IL1α, (b) IL1β, (c) TNFα, (d) IL6, (e) CSF2, (f) CCL20, (g) CXCL1 and (h) CSF3 were determined by ELISA (n = 6–10 mice/group in 3–5 experiments). *P < 0.05 compared to cPLA2α+/+ saline control; # P < 0.05 compared to cPLA2α−/− saline control; **P < 0.05 compared to cPLA2α+/+ with CA
Levels of eicosanoids in BALF from cPLA2α+/+ and cPLA2α−/− mice during C. albicans infection
cPLA2α releases arachidonic acid for production of eicosanoids, which play diverse roles in regulating inflammation and innate immunity. Eicosanoids were analyzed by mass spectrometry in BALF collected 24 h after C. albicans infection from cPLA2α+/+ and cPLA2α−/− mice (Fig. 6). Since cyclooxygenase metabolites and oxidation products can be generated during tissue processing from available free arachidonic acid, the cyclooxygenase inhibitor indomethacin and antioxidant butylated hydroxytoluene were added to the lavage solution before administration. By including D8-arachidonic acid in the lavage solution along with indomethacin, preliminary experiments showed that cyclooxygenase products were not generated during the lavage procedure since D8 metabolites were not found. In addition we found that it was necessary to include butylated hydroxytoluene during lavage to prevent the formation of isoprostanes. As shown in Fig. 6, C. albicans stimulated an increase in cysteinyl leukotriene production in cPLA2α+/+ mice with the stable metabolite leukotriene E4 being the most abundant followed by leukotriene C4 and leukotriene D4 (Fig. 6a). Cysteinyl leukotrienes were at very low or undetectable levels in saline controls and in BALF from C. albicans infected cPLA2α−/− mice indicating that cPLA2α initiates their production. There was no significant production of leukotriene B4 in saline controls or in response to C. albicans infection in either cPLA2α+/+ or cPLA2α−/− mice. The cyclooxygenase metabolites thromboxane B2 (the stable metabolite of thromboxane A2), prostaglandin E2 and prostaglandin D2 were detected at the highest levels in BALF of C. albicans infected cPLA2α+/+ mice (Fig. 6b). Thromboxane B2 and Prostaglandin E2 were significantly lower in C. albicans-infected cPLA2α−/− than cPLA2α+/+ mice. Although prostaglandin D2 levels were lower in C. albicans-infected cPLA2α−/− mice than cPLA2α+/+ mice this did not reach statistical significance. The stable metabolite of prostaglandin I2 (6-keto-prostaglandin F1α) was detected in BALF at relatively high endogenous levels but was not increased by C. albicans and not significantly different in cPLA2α+/+ and cPLA2α−/− mice (Fig. 6b). Therefore the higher levels of cysteinyl leukotrienes, thromboxane A2 or prostaglandin E2 in cPLA2α+/+ than cPLA2α−/− mice may be important for protecting the lung against C. albicans infection.
Fig. 6.
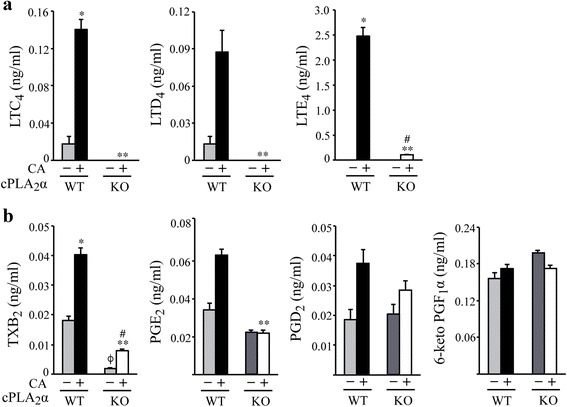
Eicosanoids are lower in BALF from cPLA2α−/− than cPLA2α+/+ mice during C. albicans infection. Levels of (a) cysteinyl leukotrienes, leukotriene C4 (LTC4), leukotriene D4 (LTD4) and leukotriene E4 (LTE4) and (b) cyclooxygenase metabolites thromboxane B2 (TXB2), prostaglandin E2 (PGE2), prostaglandin D2 (PGD2) and 6-keto prostaglandin F1α (6-keto PGF1α) were determined by mass spectrometry in BALF from cPLA2α−/− (KO) and cPLA2α+/+ (WT) mice instilled with saline or 106 C. albicans (CA) for 24 h. Results are the mean ± SEM (n = 6 mice/group). Statistical differences were determined by the Mann Whitney protocol. *P < 0.05 compared to WT saline control; ϕ P < 0.05 compared to WT saline control, # P < 0.05 compared to KO saline control; **P < 0.05 compared to WT with CA
Functional differences in alveolar macrophages and neutrophils from cPLA2α+/+ and cPLA2α−/− mice
Neutrophils and alveolar macrophages play an important role in host defense against C. albicans, however, despite the increase in neutrophil influx in cPLA2α−/− mice the fungus was not completely cleared from these mice. Therefore the ability of these cells to recognize and kill C. albicans was determined. Alveolar macrophages and neutrophils were isolated from uninfected cPLA2α+/+ and cPLA2α−/− mice, and their ability to bind and internalize GFP-C. albicans (recognition assay) and to kill the fungus was examined in vitro. The binding/internalization of GFP-C. albicans (unopsonized and opsonized) by cPLA2α+/+ and cPLA2α−/− alveolar macrophages was similar but killing of GFP-C. albicans was ~25–30 % lower in cPLA2α−/− macrophages (Fig. 7a). Alveolar macrophages from cPLA2α+/+ and cPLA2α−/− mice expressed similar levels of the lectin receptors dectin-1 (Clec7a) and dectin-2 (Clec4n). Neutrophils from cPLA2α−/− mice exhibited ~20–30 % less recognition and killing of GFP-C. albicans than cPLA2α+/+ neutrophils (Fig. 7b). Neutrophils from cPLA2α+/+ and cPLA2α−/− mice expressed similar levels of dectin-1 but the levels of dectin-2 (Clec4n) were significantly higher in neutrophils from cPLA2α−/− compared to cPLA2α+/+mice, although dectin-2 (Clec4n) expression was 10-fold lower than dectin-1 (Clec7a) in neutrophils. We also evaluated the number of GFP-C. albicans that were engulfed by alveolar macrophages in vivo, which were isolated by lavage from cPLA2α+/+ and cPLA2α−/− mice 6 h after challenge with GFP-C. albicans. Alveolar macrophages from cPLA2α−/− mice had significantly lower numbers of internalized GFP-C. albicans than macrophages from cPLA2α+/+ mice (Fig. 7b). The results suggest that the higher levels of C. albicans in cPLA2α−/− mice may in part be due to a reduced capacity of alveolar macrophages and neutrophils to kill C. albicans.
Fig. 7.
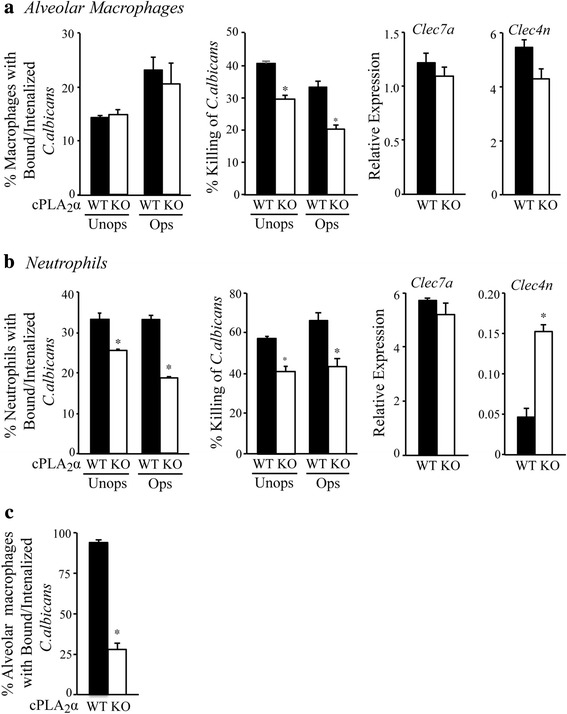
Recognition and killing of C. albicans by alveolar macrophages and neutrophils from cPLA2α−/− and cPLA2α+/+ mice. The ability of (a) alveolar macrophages and (b) neutrophils from untreated cPLA2α+/+ (WT) and cPLA2α−/− (KO) mice to recognize and kill GFP-C. albicans in vitro was compared. Levels of clec7a (dectin-1) and clec4n (dectin-2) expression were determined by real-time PCR. (c) Alveolar macrophages were isolated by lavage from WT and KO mice 6 h after challenge with GFP-C. albicans and the % macrophages containing internalized GFP-C. albicans determined by microscopy. Results are mean ± SEM (n = 3–4). *p < 0.05 compared to cells from WT mice
Discussion
cPLA2α is a highly conserved enzyme that is widely expressed throughout all tissues in mice and humans, and is rapidly activated by diverse agonists through common signaling pathways [1]. It is the only mammalian PLA2 that preferentially releases sn-2 arachidonic acid from phospholipids and its role in initiating the production of eicosanoids is well documented [40, 41]. Identification of humans with cPLA2α deficiency has confirmed that it mediates eicosanoid production and functions in homeostatic processes important for human health [42–45]. cPLA2α has been implicated in regulating both normal physiological processes and disease pathogenesis in many organ systems from studies using cPLA2α−/− mice, however, the specific mechanisms involved in many cases have not been elucidated [1, 46, 47]. In models of lung disease, cPLA2α−/− mice are protected from pulmonary fibrosis, acute lung injury and allergic responses [48–50]. Since lung fibrosis and allergic lung responses are exacerbated in COX-1−/− and COX-2−/− mice but reduced in 5-LO−/− mice, the results suggest that in certain pro-inflammatory disease states cPLA2α contributes to disease through a dominant role for pro-inflammatory leukotrienes [51–54]. By comparing cPLA2α+/+ and cPLA2α−/− mice in this study, we are probing the primary mechanism for eicosanoid production in vivo in response to exposure of the lung to the opportunistic pathogen C. albicans. This model reflects the collective influence of lipid mediators resulting from cPLA2α activation in regulating innate immune responses. Immune competent mice are resistant to infection from intratracheal instillation of C. albicans, which is rapidly cleared from the lungs with minimal health effects due to contributions from both alveolar macrophages and neutrophils in host defense [35]. Our results suggest that cPLA2α contributes to innate immune defense mechanisms in the lung to control C. albicans infection and dampen inflammation.
cPLA2α−/− mice do not clear C. albicans from the lung as efficiently as cPLA2α+/+ mice and exhibit greater signs of inflammation including excessive weight loss, increased production of pro-inflammatory cytokines and increased neutrophil recruitment to the lung. Pro-inflammatory cytokines (TNFα, IL1α, IL1β) are higher in cPLA2α−/− than cPLA2α+/+ mice 6–24 h after C. albicans infection. In mouse models of bacterial pneumonia these cytokines are produced by alveolar macrophages from initial interaction with pathogens and signal to epithelial cells and neutrophils to mount responses to infection [55–57]. Alveolar macrophages, isolated 6 h after intratracheal instillation, contain engulfed GFP-C. albicans indicating that the fungi reach the alveoli shortly after instillation. Pro-inflammatory cytokines have been shown to induce the production of neutrophilic chemokines such CXCL1, which is higher in cPLA2α−/− mice and correlates with the elevated neutrophil influx [56, 58]. C. albicans infection in cPLA2α+/+ mice leads to a small but significant increase in production of TNFα, IL1α and IL1β, and induces neutrophil influx, although at lower levels than in cPLA2α−/− mice. It is likely that these innate immune responses in cPLA2α+/+ mice are important for host defense resulting in clearance of C. albicans from the lung. It has been shown that TNFα, IL1α and IL1β are important for host defense against invasive C. albicans infection in mice [59, 60]. However, the exaggerated responses to C. albicans infection in cPLA2α−/− mice point to an important role for cPLA2α in regulating the balance of cytokines produced for effective microbial clearance without excess inflammation that may cause tissue injury and dissemination of C. albicans from the lung. This may in part be due to higher levels of PGE2 in cPLA2α+/+ mice since prostaglandins suppress the production of TNFα, IL1α and IL1β [15, 61–63]. PGE2 is also important in maintaining endothelial barrier function, promoting wound healing and inhibiting neutrophil migration [64]. PGI2 also has anti-inflammatory properties [3]. Our results show relatively high levels of endogenous PGI2 in BALF suggesting constitutive production perhaps by vascular endothelial cells and smooth muscle cells reflecting its important role in maintenance of the vasculature [65]. PGI2 levels were similar in BALF from cPLA2α+/+ and cPLA2α−/− mice, and not increased by C. albicans infection, suggesting another PLA2 is involved in its production and that it is not involved in the phenotypic differences observed during C. albicans infection.
Of the cytokines measured in BALF, IL6 showed the greatest increase in cPLA2α−/− mice early after C. albicans instillation reaching levels 10-fold higher than in cPLA2α+/+ mice. IL6 is an indicator of disease severity, reflecting the more pronounced effect of C. albicans on the health of cPLA2α−/− compared to cPLA2α+/+ mice, which show only a small increase in IL6 production [66]. IL6 is considered a pleiotropic cytokine made by immune and stromal cells in response to diverse agonists that has a homeostatic function and regulates immunity [67]. IL6 regulates the recruitment of leukocytes during infection and may contribute to the higher neutrophil influx in cPLA2α−/− mice [67, 68]. Although IL6 can be induced by prostaglandins, its higher level in cPLA2α−/− mice suggests that it is directly made by cells in response to C. albicans perhaps through the early production of TNFα, IL1α, and IL1β [66, 69, 70]. In contrast to the results of this study, cPLA2α−/− mice are protected during Pseudomonas aeruginosa lung infection that correlates with decreased IL6 production [71]. Therefore, cPLA2α can exacerbate infection or have a protective role in the lung depending on the type of pathogen.
Leukotrienes also regulate immunity in the lung during infection by promoting trafficking of neutrophils, T lymphocytes, dendritic cells and vascular permeability [2, 7]. Mice deficient in leukotriene production are more susceptible to bacterial (Klebsiella pneumonia, Mycobacterium tuberculosis) and fungal (Histoplasmosis) lung infection showing impaired microbial clearance and survival [72–74]. However there are differences in the responses of leukotriene-deficient mice to bacterial and fungal infection. Following bacterial challenge, 5-LO−/− mice have reduced neutrophil influx in the lung [72]. However, Histoplama capsulatum lung infection in 5-LO−/− mice results in increased neutrophil recruitment and greater production of pro-inflammatory cytokines than in wild type mice, as we observed in C. albicans-infected cPLA2α−/− mice. Leukotrienes regulate innate immune responses in part by enhancing alveolar macrophage phagocytosis and microbial killing [72, 74].
Our results demonstrate that alveolar macrophages and neutrophils from uninfected cPLA2α−/− mice have a reduced capacity to kill C. albicans than cells from cPLA2α+/+ mice. We previously reported that C. albicans poorly activates cPLA2α in alveolar macrophages from cPLA2α+/+ mice and induces very little eicosanoid production, although it is enhanced by priming with GM-CSF due to increased expression of dectin-1 [12]. Therefore it is not likely that this inherent difference in the killing capacity of alveolar macrophages from uninfected cPLA2α+/+ and cPLA2α−/− mice is due to production of endogenous eicosanoids during the killing assay in vitro. The basis for this inherent difference in C. albicans killing is not known but the lack of eicosanoids during development of cPLA2α−/− mice may affect gene expression that influences killing of C. albicans. The results also showed that alveolar macrophages isolated from cPLA2α−/− mice 6 h after instillation of GFP-C. albicans have fewer engulfed GFP-C. albicans than macrophages from cPLA2α+/+ mice. It is likely that cells are primed by cytokines in vivo to enhance production of eicosanoids and regulate killing of C. albicans.
A role for the epithelium during C. albicans lung infection is suggested by results showing that cPLA2α−/− mice have higher levels of CCL20 and CSF2 than cPLA2α+/+ mice. During lung infection CCL20 and CSF2 (GM-CSF) are derived from lung epithelium and contribute to recruitment of dendritic cells and neutrophils [55, 58, 75]. The lung epithelium may also contribute to production of pro-inflammatory cytokines since C. albicans stimulates oral and vaginal epithelial cells to produce chemokines and cytokines including IL1α, IL1β and TNFα [76, 77]. Although this has not been investigated in lung epithelial cells, there may be a local immune response at the lung mucosa for combating C. albicans in cPLA2α+/+ mice. It is interesting that C. albicans disseminates to the kidney in cPLA2α−/− mice suggesting there is damage to the epithelial/endothelial barrier possibly due to the increased inflammation. Since alveolar epithelium damage can be sensed by alveolar macrophages this may lead to heightened pro-inflammatory responses as we observed in cPLA2α−/− mice [78]. The results suggest that cPLA2α activation is an important mechanism for regulating the function of immune and stromal cells in the lung to protect from C. albicans infection.
Conclusions
This study demonstrates that cPLA2α plays a role in protecting the lung from C. albicans infection. Since production of lipid mediators occurs rapidly in response microbial infection we focused on how this pathway regulates the early innate immune responses to C. albicans in the lung in an attempt to assess the more immediate effects of this pathway. The results suggest that cPLA2α contributes to lung homeostasis and the immunosuppressive environment in the lung. There may be tonic pattern receptor signaling resulting in cPLA2α activation and lipid mediator production in the lung by low-level colonization or exposure to commensal organisms such as C. albicans from the oral cavity. This promotes clearance of the relatively avirulent commensal fungus that limits infection and inflammation preventing more pathogenic effects. It is likely that the balance of products from both cyclooxygenase and lipoxygenase pathways is important in immune surveillance in the lung contributing to mucosal integrity and the function of phagocytes for efficient clearance of infectious agents and regulating the extent of inflammation.
Abbreviations
5-LO, 5-lipoxygenase; BALF, bronchoalveolar lavage fluid; CFU, colony forming units; COX, cyclooxygenase; cPLA2α, Group IVA cytosolic phospholipase A2; DAPI, 4’,6-diamidino-2-phenylindole; FC, flow cytometry; GFP, green fluorescent protein; GM-CSF, granulocyte macrophage colony-stimulating factor; HPLC, high performance liquid chromatography; XTT, 2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-5-[(phenylamino)carbonyl]-2H-tetrazolium hydroxide
Acknowledgements
We acknowledge and thank Dr. Joseph V. Bonventre for originally providing the cPLA2α−/− mouse breeders.
Funding
This work was supported by a grant from the National Institutes of Health Grant (HL34303 to CCL and RCM).
Availability of data and materials
The data supporting the conclusions are included within the article.
Authors’ contributions
SJ, AD, and CCL conceived, designed and coordinated the study; SJ, AD, BY, and HL performed and analyzed experiments; CLU and RCM performed and analyzed experiments using mass spectrometry; MG, and EFR provided analytical expertise; SJ, AD, and CCL interpreted the data, wrote the manuscript and provided intellectual input. All authors read and approved the final manuscript.
Competing interests
The authors declare they have no conflicts of interest.
Consent for publication
Not applicable.
Ethics approval and consent to participate
The work with mice was approved by the Institutional Animal Care and Use Committee (IACUC) at National Jewish Health and conducted in accordance with their guidelines. The study does not involve the use of human data or tissue.
Additional files
Flow cytometry gating strategy for cell identification in lung digests from cPLA2α+/+ (WT) and cPLA2α−/− (KO) mice challenged with C. albicans for 24 h. Cells were isolated from enzymatically digested mouse lungs, and after exclusion of doublets and debris, immune cells were identified by CD45 staining. A sequential gating strategy was used to identify populations expressing specific markers: a alveolar macrophages (AM) (CD45+ CD24− CD11b− SiglecF+), (b) tissue macrophages (TM) (CD45+ CD24− CD11b+), (c) neutrophils (PMN) (CD45+ CD11b+ Ly6G+) and (d) CD11b+ dendritic cells (CD11b+ DCs) (CD45+ MHCII+ CD11c+ CD11b+). (TIF 954 kb)
Expression of cytokines and chemokines in lung tissue from cPLA2α+/+ and cPLA2α−/− mice during C. albicans infection. Real-time PCR was carried out using the Mouse Cytokines & Chemokines RT2 Profiler PCR Array to compare expression in lungs of cPLA2α−/− (KO) and cPLA2α+/+ (WT) mice challenged with saline or 106 C. albicans (CA) for 12 and 24 h (n = 6-10 mice/group in 3–5 experiments). *P < 0.05 compared to WT saline control; ϕ P < 0.05 compared to WT saline control, # P < 0.05 compared to KO saline control; **P < 0.05 compared to WT with CA. (TIF 859 kb)
Contributor Information
Sabarirajan Jayaraja, Email: l2sabari@gmail.com.
Azzeddine Dakhama, Email: adakhama@centurylink.net.
Bogeon Yun, Email: yunb@njhealth.org.
Moumita Ghosh, Email: ghoshm@njhealth.org.
HeeJung Lee, Email: leeh@njhealth.org.
Elizabeth F. Redente, Email: redentee@njhealth.org
Charis L. Uhlson, Email: charis.uhlson@ucdenver.edu
Robert C. Murphy, Email: robert.murphy@ucdenver.edu
Christina C. Leslie, Email: lesliec@njhealth.org
References
- 1.Leslie CC. Cytosolic phospholipase A2: physiological function and role in disease. J Lipid Res. 2015;56(8):1386–1402. doi: 10.1194/jlr.R057588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kanaoka Y, Boyce JA. Cysteinyl leukotrienes and their receptors: Cellular distribution and function in immune and inflammatory responses. J Immunol. 2004;173(3):1503–10. doi: 10.4049/jimmunol.173.3.1503. [DOI] [PubMed] [Google Scholar]
- 3.Woodward DF, Jones RL, Narumiya S. International union of basic and clinical pharmacology. LXXXIII: classification of prostanoid receptors, updating 15 years of progress. Pharmacol Rev. 2011;63(3):471–538. doi: 10.1124/pr.110.003517. [DOI] [PubMed] [Google Scholar]
- 4.Back M, Powell WS, Dahlen SE, Drazen JM, Evans JF, Serhan CN, Shimizu T, Yokomizo T, Rovati GE. Update on leukotriene, lipoxin and oxoeicosanoid receptors: IUPHAR Review 7. British J Pharmacol. 2014;171(15):3551–74. doi: 10.1111/bph.12665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haeggstrom JZ, Funk CD. Lipoxygenase and leukotriene pathways: biochemistry, biology, and roles in disease. Chemical Rev. 2011;111(10):5866–98. doi: 10.1021/cr200246d. [DOI] [PubMed] [Google Scholar]
- 6.Smith WL, Urade Y, Jakobsson PJ. Enzymes of the cyclooxygenase pathways of prostanoid biosynthesis. Chemical Rev. 2011;111(10):5821–5865. doi: 10.1021/cr2002992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peters-Golden M, Canetti C, Mancuso P, Coffey MJ. Leukotrienes: Underappreciated mediators of innate immune responses. J Immunol. 2005;174(2):589–94. doi: 10.4049/jimmunol.174.2.589. [DOI] [PubMed] [Google Scholar]
- 8.Ricciotti E, Fitzgerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. 2011;31(5):986–1000. doi: 10.1161/ATVBAHA.110.207449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tilley SL, Coffman TM, Koller BH. Mixed messages: modulation of inflammation and immune responses by prostaglandins and thromboxanes. J Clin Invest. 2001;108(1):15–23. doi: 10.1172/JCI200113416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kalinski P. Regulation of immune responses by prostaglandin E2. J Immunol. 2012;188(1):21–8. doi: 10.4049/jimmunol.1101029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alvarez Y, Valera I, Municio C, Hugo E, Padron F, Blanco L, Rodriguez M, Fernandez N, Crespo MS. Eicosanoids in the innate immune response: TLR and non-TLR routes. Mediators Inflamm. 10.1155/2010/201929. [DOI] [PMC free article] [PubMed]
- 12.Parti RP, Loper R, Brown GD, Gordon S, Taylor PR, Bonventre JV, Murphy RC, Williams DL, Leslie CC. Cytosolic phospholipase A2 activation by Candida albicans in alveolar macrophages: Role of dectin-1. Am J Respir Cell Mol Biol. 2009;42(4):415–23. doi: 10.1165/rcmb.2009-0110OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Suram S, Brown GD, Ghosh M, Gordon S, Loper R, Taylor PR, Akira S, Uematsu S, Williams DL, Leslie CC. Regulation of cytosolic phospholipase A2 activation and cyclooxygeanse 2 expression in macrophages by the β-glucan receptor. J Biol Chem. 2006;281(9):5506–14. doi: 10.1074/jbc.M509824200. [DOI] [PubMed] [Google Scholar]
- 14.Suram S, Gangelhoff TA, Taylor PR, Rosas M, Brown GD, Bonventre JC, Akira S, Uematsu S, Williams DL, Murphy RC, et al. Pathways regulating cytosolic phospholipase A2 activation and eicosanoid production in macrophages by Candida albicans. J Biol Chem. 2010;285(40):30676–85. doi: 10.1074/jbc.M110.143800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suram S, Silveira LJ, Mahaffey S, Brown GD, Bonventre JV, Williams DL, Gow NA, Bratton DL, Murphy RC, Leslie CC. Cytosolic phospholipase A2alpha and eicosanoids regulate expression of genes in macrophages involved in host defense and inflammation. PLoS One. 2013;8(7):e69002. doi: 10.1371/journal.pone.0069002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yun B, Lee H, Jayaraja S, Suram S, Murphy RC, Leslie CC. Prostaglandins from cytosolic phospholipase A2alpha/cyclooxygenase-1 pathway and mitogen-activated protein kinases regulate gene expression in Candida albicans-infected macrophages. J Biol Chem. 2016;291(13):7070–86. doi: 10.1074/jbc.M116.714873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davies LC, Jenkins SJ, Allen JE, Taylor PR. Tissue-resident macrophages. Nat Immunol. 2013;14(10):986–95. doi: 10.1038/ni.2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Whitsett JA, Alenghat T. Respiratory epithelial cells orchestrate pulmonary innate immunity. Nat Immunol. 2015;16(1):27–35. doi: 10.1038/ni.3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ghannoum MA, Jurevic RJ, Mukherjee PK, Cui F, Sikaroodi M, Naqvi A, Gillevet PM. Characterization of the oral fungal microbiome (mycobiome) in healthy individuals. PLoS Pathog. 2010;6(1):e1000713. doi: 10.1371/journal.ppat.1000713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huffnagle GB, Noverr MC. The emerging world of the fungal microbiome. Trends Microbiol. 2013;21(7):334–41. doi: 10.1016/j.tim.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Charlson ES, Diamond JM, Bittinger K, Fitzgerald AS, Yadav A, Haas AR, Bushman FD, Collman RG. Lung-enriched organisms and aberrant bacterial and fungal respiratory microbiota after lung transplant. Am J Respir Crit Care Med. 2012;186(6):536–45. doi: 10.1164/rccm.201204-0693OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nguyen LD, Viscogliosi E, Delhaes L. The lung mycobiome: an emerging field of the human respiratory microbiome. Frontiers Microbiol. 2015;6:89. doi: 10.3389/fmicb.2015.00089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hube B. From commensal to pathogen: stage- and tissue-specific gene expression of Candida albicans. Curr Opin Microbiol. 2004;7(4):336–41. doi: 10.1016/j.mib.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 24.Poulain D. Candida albicans, plasticity and pathogenesis. Crit Rev Microbiol. 2013;41(2):208–17. doi: 10.3109/1040841X.2013.813904. [DOI] [PubMed] [Google Scholar]
- 25.Yao Z, Liao W. Fungal respiratory disease. Curr Opin Pulm Med. 2006;12(13):222–7. doi: 10.1097/01.mcp.0000219272.57933.01. [DOI] [PubMed] [Google Scholar]
- 26.Muthig M, Hebestreit A, Ziegler U, Seidler M, Muller FM. Persistence of Candida species in the respiratory tract of cystic fibrosis patients. Med Mycol. 2010;48(1):56–63. doi: 10.3109/13693780802716532. [DOI] [PubMed] [Google Scholar]
- 27.Bonventre JV, Huang Z, Taheri MR, O’Leary E, Li E, Moskowitz MA, Sapirstein A. Reduced fertility and postischaemic brain injury in mice deficient in cytosolic phospholipase A2. Nature. 1997;390(6660):622–5. doi: 10.1038/37635. [DOI] [PubMed] [Google Scholar]
- 28.Wheeler RT, Kombe D, Agarwala SD, Fink GR. Dynamic, morphotype-specific Candida albicans beta-glucan exposure during infection and drug treatment. PLoS Pathog. 2008;4(12):e1000227. doi: 10.1371/journal.ppat.1000227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real time quantitative PCR and the 2-ΔΔCT method. Methods. 2001;25(4):402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 30.Misharin AV, Morales-Nebreda L, Mutlu GM, Budinger GR, Perlman H. Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am J Respir Cell Molec Biol. 2013;49(4):503–10. doi: 10.1165/rcmb.2013-0086MA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hall LM, Murphy RC. Electrospray mass spectrometric analysis of 5-hydroperoxy and 5-hydroxyeicosatetraenoic acids generated by lipid peroxidation of red blood cell ghost phospholipids. J Am Soc Mass Spectrom. 1998;9(5):527–32. doi: 10.1016/S1044-0305(98)00013-0. [DOI] [PubMed] [Google Scholar]
- 32.Ermert D, Niemiec MJ, Rohm M, Glenthoj A, Borregaard N, Urban CF. Candida albicans escapes from mouse neutrophils. J Leuk Biol. 2013;94(2):223–36. doi: 10.1189/jlb.0213063. [DOI] [PubMed] [Google Scholar]
- 33.Suratt BT, Young SK, Lieber J, Nick JA, Henson PM, Worthen GS. Neutrophil maturation and activation determine anatomic site of clearance from circulation. Am J Physiol Lung Cell Mol Physiol. 2001;281(4):L913–21. doi: 10.1152/ajplung.2001.281.4.L913. [DOI] [PubMed] [Google Scholar]
- 34.Sawyer RT. Experimental pulmonary candidiasis. Mycopathol. 1990;109(2):99–109. doi: 10.1007/BF00436790. [DOI] [PubMed] [Google Scholar]
- 35.Sawyer RT, Harmsen AG. The relative contribution of resident pulmonary alveolar macrophages and inflammatory polymorphonuclear neutrophils in host resistance to pulmonary infection by Candida albicans. Mycopathol. 1989;108(2):95–105. doi: 10.1007/BF00436059. [DOI] [PubMed] [Google Scholar]
- 36.Spellberg B, Ibrahim AS, Edwards JE, Jr, Filler SG. Mice with disseminated candidiasis die of progressive sepsis. J Infect Dis. 2005;192(2):336–43. doi: 10.1086/430952. [DOI] [PubMed] [Google Scholar]
- 37.Sabesin SM. Renal failure and disseminated candidiasis. Arch Intern Med. 1962;110:526–34. doi: 10.1001/archinte.1962.03620220118020. [DOI] [PubMed] [Google Scholar]
- 38.Barth MW, Hendrzak JA, Melnicoff MJ, Morahan PS. Review of the macrophage disappearance reaction. J Leuk Biol. 1995;57(3):361–67. doi: 10.1002/jlb.57.3.361. [DOI] [PubMed] [Google Scholar]
- 39.Jose RJ, Williams AE, Mercer PF, Sulikowski MG, Brown JS, Chambers RC. Regulation of neutrophilic inflammation by proteinase-activated receptor 1 during bacterial pulmonary infection. J Immunol. 2015;194(12):6024–34. doi: 10.4049/jimmunol.1500124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hanel AM, Schüttel S, Gelb MH. Processive interfacial catalysis by mammalian 85-kilodalton phospholipase A2 enzymes on product-containing vesicles: application to the determination of substrate preferences. Biochemistry. 1993;32(23):5949–58. doi: 10.1021/bi00074a005. [DOI] [PubMed] [Google Scholar]
- 41.Clark JD, Lin L-L, Kriz RW, Ramesha CS, Sultzman LA, Lin AY, Milona N, Knopf JL. A novel arachidonic acid-selective cytosolic PLA2 contains a Ca2+-dependent translocation domain with homology to PKC and GAP. Cell. 1991;65(6):1043–51. doi: 10.1016/0092-8674(91)90556-E. [DOI] [PubMed] [Google Scholar]
- 42.Kirkby NS, Reed DM, Edin ML, Rauzi F, Mataragka S, Vojnovic I, Bishop-Bailey D, Milne GL, Longhurst H, Zeldin DC, et al. Inherited human group IVA cytosolic phospholipase A2 deficiency abolishes platelet, endothelial, and leucocyte eicosanoid generation. FASEB J. 2015;29(11):4568–78. doi: 10.1096/fj.15-275065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Faioni EM, Razzari C, Zulueta A, Femia EA, Fenu L, Trinchera M, Podda GM, Pugliano M, Marongiu F, Cattaneo M. Bleeding diathesis and gastro-duodenal ulcers in inherited cytosolic phospholipase-A2 alpha deficiency. Thromb Haemost. 2014;112(6):1182–89. doi: 10.1160/TH14-04-0352. [DOI] [PubMed] [Google Scholar]
- 44.Adler DH, Cogan JD, Phillips JA, 3rd, Schnetz-Boutaud N, Milne GL, Iverson T, Stein JA, Brenner DA, Morrow JD, Boutaud O, et al. Inherited human cPLA(2alpha) deficiency is associated with impaired eicosanoid biosynthesis, small intestinal ulceration, and platelet dysfunction. J Clin Invest. 2008;118(6):2121–31. doi: 10.1172/JCI30473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brooke MA, Longhurst HJ, Plagnol V, Kirkby NS, Mitchell JA, Ruschendorf F, Warner TD, Kelsell DP, MacDonald TT. Cryptogenic multifocal ulcerating stenosing enteritis associated with homozygous deletion mutations in cytosolic phospholipase A2-alpha. Gut. 2014;63(1):96–104. doi: 10.1136/gutjnl-2012-303581. [DOI] [PubMed] [Google Scholar]
- 46.Uozumi N, Shimizu T. Roles for cytosolic phospholipase A2a as revealed by gene-targeted mice. Prostaglandin Other Lipid Mediat. 2002;68–69:59–69. doi: 10.1016/S0090-6980(02)00021-7. [DOI] [PubMed] [Google Scholar]
- 47.Bonventre JV, Sapirstein A. Group IV cytosolic phospholipase A2 (PLA2) function: insights from the knockout mouse. Adv Exp Med Biol. 2002;507:25–31. doi: 10.1007/978-1-4615-0193-0_5. [DOI] [PubMed] [Google Scholar]
- 48.Nagase T, Uozumi N, Ishii S, Kita Y, Yamamoto H, Ohga E, Ouchi Y, Shimizu T. A pivotal role of cytosolic phospholipase A2 in bleomycin-induced pulmonary fibrosis. Nat Med. 2002;8(5):480–4. doi: 10.1038/nm0502-480. [DOI] [PubMed] [Google Scholar]
- 49.Nagase T, Uozumi N, Ishii S, Kume K, Izumi T, Ouchi Y, Shimizu T. Acute lung injury by sepsis and acid aspiration: a key role for cytosolic phospholipase A2. Nat Immunol. 2000;1(1):42–6. doi: 10.1038/76897. [DOI] [PubMed] [Google Scholar]
- 50.Uozumi N, Kume K, Nagase T, Nakatani N, Ishii S, Tashiro F, Komagata Y, Maki K, Ikuta K, Ouchi Y, et al. Role of cytosolic phospholipase A2 in allergic response and parturition. Nature. 1997;390(6660):618–22. doi: 10.1038/37622. [DOI] [PubMed] [Google Scholar]
- 51.Irvin CG, Tu YP, Sheller JR, Funk CD. 5-Lipoxygenase products are necessary for ovalbumin-induced airway responsiveness in mice. Am J Physiol. 1997;272(6 Pt 1):L1053–58. doi: 10.1152/ajplung.1997.272.6.L1053. [DOI] [PubMed] [Google Scholar]
- 52.Gavett SH, Madison SL, Chulada PC, Scarborough PE, Qu W, Boyle JE, Tiano HF, Lee CA, Langenbach R, Roggli VL, et al. Allergic lung responses are increased in prostaglandin H synthase-deficient mice. J Clin Invest. 1999;104(6):721–32. doi: 10.1172/JCI6890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bonner JC, Rice AB, Ingram JL, Moomaw CR, Nyska A, Bradbury A, Sessoms AR, Chulada PC, Morgan DL, Zeldin DC, et al. Susceptibility of cyclooxygenase-2-deficient mice to pulmonary fibrogenesis. Am J Pathol. 2002;161(2):459–470. doi: 10.1016/S0002-9440(10)64202-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Peters-Golden M, Bailie M, Marshall T, Wilke C, Phan SH, Toews GB, Moore BB. Protection from pulmonary fibrosis in leukotriene-deficient mice. Am J Respir Crit Care Med. 2002;165(2):229–35. doi: 10.1164/ajrccm.165.2.2104050. [DOI] [PubMed] [Google Scholar]
- 55.Quinton LJ, Mizgerd JP. Dynamics of lung defense in pneumonia: resistance, resilience, and remodeling. Annu Rev Physiol. 2015;77:407–30. doi: 10.1146/annurev-physiol-021014-071937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.LeibundGut-Landmann S, Weidner K, Hilbi H, Oxenius A. Nonhematopoietic cells are key players in innate control of bacterial airway infection. J Immunol. 2011;186(5):3130–37. doi: 10.4049/jimmunol.1003565. [DOI] [PubMed] [Google Scholar]
- 57.Barry KC, Fontana MF, Portman JL, Dugan AS, Vance RE. IL-1alpha signaling initiates the inflammatory response to virulent Legionella pneumophila in vivo. J Immunol. 2013;190(12):6329–39. doi: 10.4049/jimmunol.1300100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yamamoto K, Ahyi AN, Pepper-Cunningham ZA, Ferrari JD, Wilson AA, Jones MR, Quinton LJ, Mizgerd JP. Roles of lung epithelium in neutrophil recruitment during pneumococcal pneumonia. Am J Respir Cell Mol Biol. 2014;50(2):253–62. doi: 10.1165/rcmb.2013-0114OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vonk AG, Netea MG, van Krieken JH, Iwakura Y, van der Meer JW, Kullberg BJ. Endogenous interleukin (IL)-1 alpha and IL-1 beta are crucial for host defense against disseminated candidiasis. J Infect Dis. 2006;193(10):1419–26. doi: 10.1086/503363. [DOI] [PubMed] [Google Scholar]
- 60.Marino MW, Dunn AR, Grail D, Inglese M, Noguchi Y, Richards E, Jungbluth A, Wida H, Moore M, Williamson B, et al. Characterization of tumor necrosis factor-deficient mice. Proc Natl Acad Sci U S A. 1997;94:8093–98. doi: 10.1073/pnas.94.15.8093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sokolowska M, Chen LY, Liu Y, Martinez-Anton A, Qi HY, Logun C, Alsaaty S, Park YH, Kastner DL, Chae JJ, et al. Prostaglandin E2 inhibits NLRP3 inflammasome activation through EP4 receptor and intracellular cyclic AMP in human macrophages. J Immunol. 2015;194(11):5472–87. doi: 10.4049/jimmunol.1401343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Brandwein SR. Regulation of interleukin 1 production by mouse peritoneal macrophages. Effects of arachidonic acid metabolites, cyclic nucleotides, and interferons. J Biol Chem. 1986;261(19):8624–32. [PubMed] [Google Scholar]
- 63.Kim SH, Serezani CH, Okunishi K, Zaslona Z, Aronoff DM, Peters-Golden M. Distinct protein kinase A anchoring proteins direct prostaglandin E2 modulation of Toll-like receptor signaling in alveolar macrophages. J Biol Chem. 2011;286(11):8875–83. doi: 10.1074/jbc.M110.187815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Konya V, Ullen A, Kampitsch N, Theiler A, Philipose S, Parzmair GP, Marsche G, Peskar BA, Schuligoi R, Sattler W, et al. Endothelial E-type prostanoid 4 receptors promote barrier function and inhibit neutrophil trafficking. J Allergy Clin Immunol. 2013;131(2):532–40. doi: 10.1016/j.jaci.2012.05.008. [DOI] [PubMed] [Google Scholar]
- 65.Kawabe J, Ushikubi F, Hasebe N. Prostacyclin in vascular diseases. - Recent insights and future perspectives. Circ J. 2010;74(5):836–43. doi: 10.1253/circj.CJ-10-0195. [DOI] [PubMed] [Google Scholar]
- 66.Hunter CA, Jones SA. IL-6 as a keystone cytokine in health and disease. Nat Immunol. 2015;16(5):448–57. doi: 10.1038/ni.3153. [DOI] [PubMed] [Google Scholar]
- 67.Jones SA. Directing transition from innate to acquired immunity: defining a role for IL-6. J Immunol. 2005;175(6):3463–68. doi: 10.4049/jimmunol.175.6.3463. [DOI] [PubMed] [Google Scholar]
- 68.Fielding CA, McLoughlin RM, McLeod L, Colmont CS, Najdovska M, Grail D, Ernst M, Jones SA, Topley N, Jenkins BJ. IL-6 regulates neutrophil trafficking during acute inflammation via STAT3. J Immunol. 2008;181(3):2189–95. doi: 10.4049/jimmunol.181.3.2189. [DOI] [PubMed] [Google Scholar]
- 69.Hinson RM, Williams JA, Shacter E. Elevated interleukin 6 is induced by prostaglandin E2 in a murine model of inflammation: possible role of cyclooxygenase-2. Proc Natl Acad Sci U S A. 1996;93(10):4885–90. doi: 10.1073/pnas.93.10.4885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Quinton LJ, Jones MR, Robson BE, Simms BT, Whitsett JA, Mizgerd JP. Alveolar epithelial STAT3, IL-6 family cytokines, and host defense during Escherichia coli pneumonia. Am J Respir Cell Mol Biol. 2008;38(6):699–706. doi: 10.1165/rcmb.2007-0365OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Guillemot L, Medina M, Pernet E, Leduc D, Chignard M, Touqui L, Wu Y. Cytosolic phospholipase A2alpha enhances mouse mortality induced by Pseudomonas aeruginosa pulmonary infection via interleukin 6. Biochimie. 2014;107 Pt A:95–104. doi: 10.1016/j.biochi.2014.08.018. [DOI] [PubMed] [Google Scholar]
- 72.Bailie MB, Standiford TJ, Laichalk LL, Coffey MJ, Strieter R, Peters-Golden M. Leukotriene-deficient mice manifest enhanced lethality from Klebsiella pneumonia in association with decreased alveolar macrophage phagocytic and bactericidal activities. J Immunol. 1996;157(12):5221–4. [PubMed] [Google Scholar]
- 73.Medeiros AI, Sa-Nunes A, Soares EG, Peres CM, Silva CL, Faccioli LH. Blockade of endogenous leukotrienes exacerbates pulmonary histoplasmosis. Infect Immun. 2004;72:1637–44. doi: 10.1128/IAI.72.3.1637-1644.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Secatto A, Rodrigues LC, Serezani CH, Ramos SG, Dias-Baruffi M, Faccioli LH, Medeiros AI. 5-Lipoxygenase deficiency impairs innate and adaptive immune responses during fungal infection. PLoS One. 2012;7(3):e31701. doi: 10.1371/journal.pone.0031701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kallal LE, Schaller MA, Lindell DM, Lira SA, Lukacs NW. CCL20/CCR6 blockade enhances immunity to RSV by impairing recruitment of DC. Eur J Immunol. 2010;40(4):1042–52. doi: 10.1002/eji.200939778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Whiley RA, Cruchley AT, Gore C, Hagi-Pavli E. Candida albicans strain-dependent modulation of pro-inflammatory cytokine release by in vitro oral and vaginal mucosal models. Cytokine. 2012;57(1):89–97. doi: 10.1016/j.cyto.2011.10.017. [DOI] [PubMed] [Google Scholar]
- 77.Steele C, Leigh J, Swoboda R, Fidel PL., Jr Growth inhibition of Candida by human oral epithelial cells. J Infect Dis. 2000;182(5):1479–85. doi: 10.1086/315872. [DOI] [PubMed] [Google Scholar]
- 78.Hussell T, Bell TJ. Alveolar macrophages: plasticity in a tissue-specific context. Nat Rev Immunol. 2014;14(2):81–93. doi: 10.1038/nri3600. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data supporting the conclusions are included within the article.