

Abstract
Background
It has been found that microRNAs (miRNAs) can function as a regulatory factor across species. For example, food-derived plant miRNAs may pass through the gastrointestinal (GI) tract, enter into the plasma and serum of mammals, and interact with endogenous RNAs to regulate their expression. Although this new type of regulatory mechanism is not well understood, it provides a fresh look at the relationship between food consumption and physiology. To investigate this new type of mechanism, we conducted a systematic computational study to analyze the potential functions of these dietary miRNAs in the human body.
Results
In this paper, we predicted human and plant target genes using RNAhybrid and set some criteria to further filter them. Then we built the cross-species regulatory network according to the filtered targets, extracted central nodes by PageRank algorithm and built core modules. We summarized the functions of these modules to three major categories: ion transport, metabolic process and stress response, and especially some target genes are highly related to ion transport, polysaccharides and the lipid metabolic process. Through functional analysis, we found that human and plants have similar functions such as ion transport and stress response, so our study also indicates the existence of a close link between exogenous plant miRNA targets and digestive/urinary organs.
Conclusions
According to our analysis results, we suggest that the ingestion of these plant miRNAs may have a functional impact on consuming organisms in a cross-kingdom way, and the dietary habit may affect the physiological condition at a genetic level. Our findings may be useful for discovering cross-species regulatory mechanism in further study.
Electronic supplementary material
The online version of this article (doi:10.1186/s12918-016-0292-1) contains supplementary material, which is available to authorized users.
Keywords: miRNA, Cross-species regulation, Core module, Functional analysis
Background
As a novel mechanism of coevolution, cross-kingdom interactions have recently been discovered in many studies [1–3]. Cell communication can cross species through transmitting signals such as hormones, cytokines and small RNAs (sRNAs) [1]. For instance, Pseudomonas aeruginosais, a symbiotic gram-negative bacterium, can produce HSL autoinducers to modulate gene expression in humans [2]. Some of these autoinducers activate epithelial cells to induce generation of neutrophil chemotatic factors and then, these migrated factors will be triggered to produce toxin which are detrimental to the bacteria [2]. Meanwhile, some hosts’ miRNAs can also influence these bacteria. miR-451 and let-7i, which are highly enriched in human HbAS and HbSS erythrocytes, can negatively regulate the growth of the malaria parasite Plasmodium falciparum [4]. Certain Botrytis cinerea small RNAs (Bc-sRNA), like Bc-siR3.1, can silence Arabidopsis and tomato genes to inhibit their hosts’ immunity [3]. It is also reported that milk-derived miRNAs can target infants’ specific transcripts that are involved in cytokines and immunity [5]. Such a cross-species communication also exists in virus-host interaction. Studies have shown that viruses can utilize the hosts’ miRNA machinery to produce their own miRNAs, and further manipulate both virus and host gene expression [6]. Similarly, a cluster of cellular miRNAs, including miR-28, miR-125b, miR-150, miR-223 and miR-382, target the 3′ ends of HIV-1 mRNAs in cultivated resting CD4+ T cells to affect HIV-1 latency, and potently inhibit HIV-1 production [7]. Recently, Zhang et al. [8] reported that the exogenous plant miRNA, miR168a, which is enriched in rice, could target the human/mouse low-density lipoprotein receptor adapter protein 1 (LDLRAP1) mRNA, and inhibit LDLRAP1 expression in the liver. Further research [9–11] has also demonstrated that a variety of exogenous plant RNAs can be found in plasma and serum of mammals through ingestion.
Though more and more exogenous plant miRNAs are experimentally detected in the serum and plasma of human and animals, cross-kingdom regulation of human gene by plant miRNAs is not well understood. It is unclear how exogenous miRNAs evade the RNases and phagocytosis, and maintain stable structures and activity in a low-pH environment when passing through the mammalian gastrointestinal tract to reach the target organs. However, all the above evidence suggests that dietary miRNAs might remain active to regulate the ingesters’ specific genes. Although a number of experimental studies have demonstrated the discoveries of cross-species miRNA regulation, the effect of this event and the underlying mechanism are still unclear. Due to the complexity of sRNAs-mRNAs regulations, current experimental studies have some limitations in filling the gaps of those mechanisms. Hence, we carried out a computational study to exploit the functions and effects of plant-mammal cross-kingdom regulations, given the assumption that exogenous miRNAs exist in plasma and serum of mammals. For this purpose, we systematically predicted the targets of the documented cross-species miRNAs, and we conducted function enrichment analysis of the target genes in both humans and plants, by which we could explore the gene sets’ shared functions, such as similar pathways, regulators, or related diseases. In particular, we gathered 25 plant miRNAs, which have been detected in the serum and plasma of human and animals. We collected the entire human mRNAs 3′UTR regions from the UTRdb [12] and predicted the targets of the 25 plant miRNAs from these regions. Interestingly, we found that our predicted cross-species targets might have a close association with the digestive, urinary organs and the daily human metabolic Gene Ontology (GO) process. In order to better understand the functions of the above 25 miRNAs, we also evaluated their targets in plants as a reference. Due to lack of experimental validations on targets and high-quality gene function annotations for common food crops, we used Arabidopsis thaliana for plant target prediction and functional analyses. We found the Arabidopsis targets share some functional similarity to human targets. Our study might provide some useful hypotheses for discovering the cross-species regulatory mechanism in future research.
Results
Target prediction on human and Arabidopsis for 25 miRNAs
Due to the limitation of existing studies, it is difficult to exactly define the matching model between plant miRNAs and human targets. Given the notion that most mature miRNAs act as the RNA interference (RNAi) mechanism by binding to certain sites on target mRNAs in both plants and animals [13], we took a basic target prediction approach to retain the maximum potential targets on humans by using RNAHybrid [14] (see Methods). Meanwhile, we used the same protocol to predict the miRNAs’ targets in Arabidopsis thaliana for comparative analysis and then, we achieved an initial dataset with nearly 380,000 possible human targets and 5700 Arabidopsis targets. After a screening process conducted by selecting filtering parameters regarding targeting attributes, such as the minimum free energy, p-value, the length of bulge and loop and so on, finally about 3000 human and 1800 plant targets were selected for further study.
Predicted target validation on Arabidopsis
In order to verify the reliability of our target prediction method, we collected 170 validated Arabidopsis targets of these 25 miRNAs from TAIR [15] and PMRD [16]. The results of the Arabidopsis target prediction not only showed a high consistency but also shared the exact same binding regions (coding DNA sequence (CDS) or 3′UTR) with validated targets. Out of 170 validate targets, 135 (81.8 %) ranked within the top 50 of each miRNA. More strikingly, 123 out of 170 validated targets ranked in the top ten among the predicted targets. For example, miR156 and miR157 are from the same family, which mainly target SQUAMOSA-promoter binding protein-like (SPL) genes’ coding sequence in Arabidopsis thaliana, except for SPL3, 4, 5 located in the 3′UTR [17, 18]. It is reported that members of a plant miRNA gene family often share high sequence similarity and the target site [19]. As shown in Table 1, not only the filtered target genes and the target regions are consistent with this report, but also all validated targets rank on top of predicted targets.
Table 1.
miR156 target validation
| Validated target genes | Accession number | mRNA | Primary targets | Refined CDS targets | Rank | Refined 3′UTR targets | Rank | ||
|---|---|---|---|---|---|---|---|---|---|
| CDS | 3′UTR | CDS | 3′UTR | ||||||
| SPL2 | AT5G43270 | Y | Y | Y | N | Y | 6 | N | |
| SPL3 | AT2G33810 | Y | Y | N | Y | N | - | Y | 1 |
| SPL4 | AT1G53160 | Y | Y | N | Y | N | - | Y | 2 |
| SPL5 | AT3G15270 | Y | Y | N | Y | N | - | Y | 3 |
| SPL6 | AT1G69170 | Y | Y | Y | N | Y | 5 | N | |
| SPL9 | AT2G42200 | Y | Y | Y | N | Y | 2 | N | |
| SPL10 | AT1G27370 | Y | Y | Y | N | Y | 4 | N | |
| SPL11 | AT1G27360 | Y | Y | Y | N | Y | 3 | N | |
| SPL13-1 | AT5G50570 | Y | Y | Y | N | Y | 7 | N | |
| SPL13 | AT5G50670 | Y | Y | Y | N | Y | 8 | N | |
| SPL15 | AT3G57920 | Y | Y | Y | N | Y | 1 | N | |
We examined whether the validated genes exist in the original mRNA dataset, the primary predicted target set by RNAHybrid, and the refined targets after screening. We also listed the rank of each validated target in our refined target set, which is sorted by minimum free energy (MFE) and p-value.
Reconstruction of cross-species regulatory network
We extracted experimentally verified interactions and signaling networks/pathways for our 531 filtered targets (see Additional file 1) using GeneMania [20], and then created a primary regulatory/interaction network containing 782 genes and 2444 interactions in total (Fig. 1).
Fig. 1.
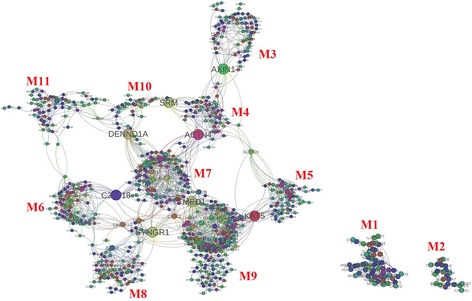
Integrated network of human target genes. The nodes with bigger size represent the bridge genes (AXIN1, SRM, DENND1A, ACTN4, C3orf18, TMED1, KAT5, and SYNGR1) in the reconstructed network
The PageRank algorithm was adopted to assign weights to the 531 nodes and thereby measure their importance. The results were convergent after 25 iterations (Additional file 2: Figure S1), when the perturbation of the weights could be controlled within 0.005. Then, we selected the top 15 nodes as the bridge genes to reconstruct the initial network by extracting modules. We extracted 11 modules by taking the bridge genes as the central nodes to decompose the whole network, which can be regarded as a critical link in the biological network.
Three functional categories of modules
In our study, functional enrichment analysis was applied to the above modules individually using Mosaic and NOA Cytoscape apps [21, 22] and DAVID [23], and we found seven modules highly enriched in certain functions. These modules could be classified into three functional categories: 1) Transport: ion transport and homeostasis process, 2) Other metabolic process: macromolecule biosynthetic and metabolic process and 3) Stress response: immune and stress response (Table 2). Major commonly enriched biological processes functions of the main modules are shown in Fig. 2 (M9, M10 and M11 are excluded, since none of the listed functions are enriched in these modules). The ion transportation and homeostasis process includes chemical, di- and tri-valent inorganic compounds (such as boron and sulfur), which are often enriched in fruits, leafy vegetables, and cereal. Metal cations such as iron, calcium, and sodium are also present. All of these compounds are closely linked with dietary habits. Meanwhile, we found a tight connection between the predicted human tissue targets as they relate to fatty acids, amino acids, alcohol, glycerolipids, cellular polysaccharide, and oxoacid metabolic processes. For the response process, one is the metal ion, inorganic ion and the nutrient substance related response, and the other is a type of defense response to the external stimulus, such as virus, bacterium and wounding. For the immune process, the identified modules show positive regulation of leukocyte and lymphocyte activation with response to the immune effector process as well as inflammation. M6 is related to vesicle targeting and transportation as well, which has been regarded as a carrier of the uptake miRNAs in the human body.
Table 2.
Parts of the functions of selected modules
| Function | Specific process | BP accession number | Module |
|---|---|---|---|
| Transport | iron ion homeostasis | GO:0055072 | M1, M3 |
| calcium ion transport | GO:0006816 | M6 | |
| vesicle-mediated transport | GO:0016192 | M8 | |
| vesicle targeting, to, from or within Golgi | GO:0048199 | M8 | |
| vesicle targeting | GO:0006903 | M8 | |
| regulation of sodium ion transport | GO:0002028 | M8 | |
| Other metabolic process | glycerolipid biosynthetic process | GO:0045017 | M4 |
| biogenic amine metabolic process | GO:0006576 | M4 | |
| alcohol metabolic process | GO:0006066 | M4 | |
| cellular polysaccharide biosynthetic process | GO:0033692 | M4 | |
| polyamine metabolic process | GO:0006595 | M5 | |
| fatty acid biosynthetic process | GO:0006633 | M7 | |
| fatty acid metabolic process | GO:0006631 | M7 | |
| Stress response | innate immune response | GO:0045087 | M1, M2 |
| inflammatory response | GO:0006954 | M1, M2 | |
| response to molecule of bacterial origin | GO:0002237 | M2 | |
| response to wounding | GO:0009611 | M2 | |
| response to chemical stimulus | GO:0042221 | M2,M7 | |
| leukocyte chemotaxis involved in inflammatory response | GO:0002232 | M2 | |
| response to ethanol | GO:0045471 | M4 | |
| response to heat | GO:0009408 | M7 | |
| response to temperature stimulus | GO:0009266 | M7, M8 |
Fig. 2.
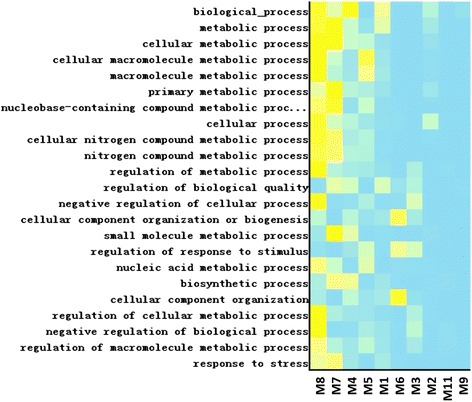
Heatmap of common biological process functions in different modules
Functional analysis of tissue-specific predicted human targets
The range of the protein expression level varies greatly in different tissues; thus, we took tissue specificity as a basis to measure our targets, which could largely reduce the false positive rate of the predicted results. To investigate the relationship between our cross-species targets and the eight tissues (brain, heart, kidney, liver, lung, spleen, stomach, and small intestine), we categorized our target genes into three levels, the ubiquitous level (house keeping genes, HKG), for which genes would express in most of the tissues; highly expressed level, meaning these genes would have a high expression level in several but not all tissues; and the tissue-specific level, which can be specifically expressed within one certain tissue. To characterize the set of three-level expressed genes that we had identified, we conducted functional enrichment analysis, and for the third level we collected verified specific gene sets of the eight tissues from TisGed [24] as the background. The protein products of the first level expressed genes were more likely to be involved in the actin filament-based process, ion transport and signaling (calcium, sulfate, and organic anion), metabolism process such as those found in macromolecule (polysaccharide, carbohydrate, alcohol, and ATP) biosynthetic and metabolic processes, general cell morphogenesis and apoptosis processes, and vesicles localization and targeting.
As shown in Fig. 3, genes with a specific tissue expression level in the brain and heart were more likely to be connected with general cell development as well as the neurological system related process, and cardiac muscle tissue morphogenesis. While the second and third level targets in kidney, liver, and spleen were often involved in specialized biological processes, like metal ion transportation, homeostasis and response in kidney, immune system process and wounding response in spleen, alcohol and macromolecule metabolic processes, and insulin signaling pathway, as well as the catabolic process in the liver, which suggests a high enrichment both in the digestive and urinary system, and also indicates a close link between our targets and these systems.
Fig. 3.
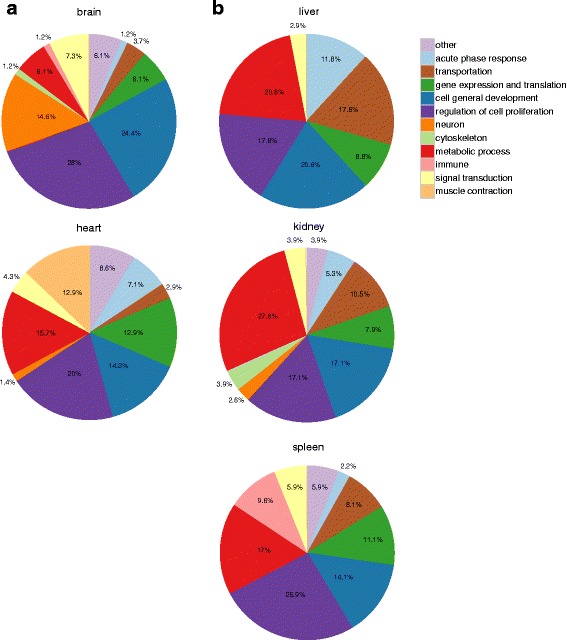
Function distribution of the high level targets among five tissues. Pie graphs show fractions of cellular Biological Processes (BP) derived from genes belonging to the second expression level in six human tissues. Names of BP categories are shown at the right. a The BP terms of brain and heart are more likely to be related with regulation of cell proliferation. b For liver, kidney and spleen, functions related to transportation, the metabolic processes and immune-related processes are highly enriched
Functional similarity: ion transport and stress response between Arabidopsis and human
As Arabidopsis and human are two very different species, there is no established method to compare gene functions of them and no single cross-species platform or algorithm can perform the pathway analysis on both Arabidopsis and human. The “functions” mentioned in this paper refers protein functions annotated in Gene Ontology. According to our enrichment analysis results (Additional files 3, 4 and 5), ion transport and stress response exist in most modules from both Arabidopsis and human. In plants, there are two ways to acquire iron ions: based on iron reduction and iron chelation. Arabidopsis activates the reduction-based strategy of Fe uptake upon Fe deficiency, and this process is partly induced by an acidification of the root hair zone through the extrusion of protons to solubilize Fe chelates; hence, this iron ion transport pathway can regulate the growth of root. Also, some transporters are expressed in shoots and seedlings. Fe distributes in three main organelles in cell: vacuole, chloroplast and mitochondria, and the iron ion transport process can influence mitochondrial respiration and chloroplast photosynthesis [25]. In Arabidopsis, the Fe uptake pathway appears to be regulated at the transcriptional and post-transcriptional level. At the transcriptional level, the transcription of IRT1genes is strongly up-regulated, IRT1 is the founding member of the large ZIP family [26], and our predicted target genes involved in metal ion transport include ZIP family gene. Two genes AtNRAMP3 and AtNRAMP4 are also involved in this pathway [27]. In human, extracellular Fe3+ is reduced to the more soluble Fe2+ by reductases embedded in the cell’s plasma membrane. The Fe2+ generated then becomes the substrate for two different uptake systems, a high-affinity system expressed in iron-limited cells and a low-affinity system active in iron-replete cells. The high-affinity system is just the same as in Arabidopsis. In the human iron transport pathway, some genes involved may influence organs such as livers and so on [25]. One mainly gene is NRAMP2 [28], and AtNRAMPs show homology to the NRAMP family [27]. We find a predicted target gene FER involved in human iron transport is also involved in the plant iron transport pathway according to our prediction result.
Plants and animals both suffer from external stimuli and pressure all the time, which helps them to constantly adapt to environmental stressors by improving the ability to respond to threats during evolution [29]. By applying the same module extraction and functional analysis method to our Arabidopsis targets (see Additional file 3), we found two aspects of stress response: 1) response to stimulus, (e.g., probable histone H2A variant 3), and 2) the more specific defense response as exhibited by the protein MOS2 resisting bacterium. The putative DNA repair protein RAD23-1 responds to DNA damage with stimulus reactions. The other defense response could be seen in the pyruvate dehydrogenase E1 component subunit alpha-2 and mitochondrial protein, which helps to repel salt stress and osmotic stress. Meanwhile, we found that predicted human targets could play a prominent role in both the stress response and immune process related to 106 relevant GO processes, of which 72 pieces are external response processes, which shows a high similarity to that of plants. Similarities can be seen in plant responses to metal ions, inorganic salt and unhealthy nutrient levels as well as starvation. All of these processes can be compared to biotic and abiotic stresses whose main factors are water and nutrients. We also found a high rate of external stimulus response in plants including cellular responses to molecules of bacterial origin, responses to lipopolysaccharides (LPSs), which are the main component of the gram-negative bacteria that can protect them from chemical attack. Gram-negative bacteria also respond positively to wounding and inflammation. Module M3/AXIN1 (Axis inhibition protein 1), which is associated with 26 genes, which involve myeloid leukocyte mediated immunity, activation of blood coagulation via a clotting cascade, glucagon secretion, and immune response-inhibiting signal transduction, which forms the immune system process.
Discussion
Arabidopsis and human are very different species, and it is hard to define “functional similarity” between them according to current computational tools and databases. We used three most comprehensive function/pathway databases, GO, KEGG and BioCyc to perform function enrichment analysis. We understand that most GO terms present very generic functions, but so far GO is the most comprehensive functional annotation database for both Arabidopsis and human. In KEGG database, the number of pathways for Arabidopsis and human are 133 and 299, and those pathways only covered 4818 and 6997 genes, respectively. Due to the unbalance of annotations between Arabidopsis and human, we cannot compare involved pathways between them. But according to our enrichment analysis results and literature search, we still find some interesting results, which infer that the pathways between Arabidopsis and human are not only common at the generic level, but also have a similar mechanism in the specific process.
According to our enrichment analysis results, one of the enriched functions is ion transport (GO: 0006811) in Arabidopsis and some GO terms about metal ion transport also exist in human, such as iron ion transport (GO: 0006826). In plants, iron is potentially highly toxic to cells, and hence, iron homeostasis needs to be tightly regulated. Similarly, dietary Fe deficiency affects many human beings on earth, leading to asthenia, increased sensitivity to diseases, and even death. We predict human targets using Arabidopsis miRNAs in our research, and then we find some predicted targets involved in iron transport process both in human and plant, such as FER. As we use Arabidopsis miRNAs to predict human targets, we find a predicted target gene FER involved in human iron transport, and this gene is also involved in the plant iron transport pathway.
Plants have to endure various stresses such as drought, salt, low temperature, etc. These adverse abiotic and biotic environmental factors force plants to develop their stress response mechanisms through cell signaling, genetic regulatory adaptation, and other defense responses. The human body can also respond to stresses and external stimuli, a state of perceived threat to homeostasis, by activating the immune system. In our research, we found that potential cross-species targeted by the exogenous miRNAs are highly related to the immune system and stress response process in the human body, such as the response to chemical stimulus and the defense response to bacterium. These responses are in the same category of the genes targeted by miRNAs in native plants. In other words, the exogenous miRNAs in humans may mimic the indigenous miRNAs in plants in terms of biological functions. So far, there exists no connection between these two response mechanisms. It is reported the stress response is generally transient because its accompanying effects can be harmful in the long term. However, if these exogenous miRNAs exist in the human chronically from the daily food consumption, they may have lasting evolutionary effects on a human population with similar food sources.
The cross-species regulatory mechanism was not proposed until 2012, and significant discussion (both pro and con) continues on the validity of this proposal. To the best of our knowledge—up to now—no validated method has been developed to predict miRNA targets across species for the exact binding site. Given the common mechanism of mature miRNAs:RNAi binding, we only applied basic target binding principles in our primary prediction step. Meanwhile, we used the same method to predict the Arabidopsis targets as a test benchmark. The significant consistency between our predicted plant targets and the validated ones for this benchmark strongly supports the effectiveness of our method and parameters. Our method tuned using Arabidopsis target validation gives a remarkable reduction of the noisy points in our data (Additional file 6: Figure S2) and, hence, provides a valid guidance to explore the new mechanism of cross-species miRNA targets.
Conclusions
In conclusion, we present a novel computational method to study the cross-species regulation between human genes and plant miRNAs. According to the target genes in both human and Arabidopsis we predicted for the same 25 miRNAs and the cross-species regulatory network we built, we summarized the functions to several major categories. From these functions, we found that there are some similar functions between human and plant target genes, such as ion transport and stress response. These findings may provide a hint of transcriptional regulatory interactions between human and plants through miRNAs. And it might point a new direction to understand the biological processes in human body through the cross-species regulatory mechanism in the future.
Methods
Plant species selection and original datasets
Related studies have detected miRNAs of Oryza sativa and Glycine max in animal serum, but we chose miRNAs in Arabidopsis thaliana for our analyses for the following reasons. A miRNA family often shares similar nucleotide sequences among different plant species. Arabidopsis is a model organism [30–32], and compared with other plants, it has more data and annotations. A total of 25 Arabidopsis miRNA datasets and 35,173 mRNAs were retrieved from the Arabidopsis Information Resource (TAIR) [15] and PMRD [16], and 34,619 human 3′UTR were from UTRdb [12]. We observed that most of these 25 miRNAs widely exist in many plants with high expression levels (Table 3), which means that human can easily intake significant quantity from common plant foods.
Table 3.
Expression levels of exogenous plant miRNAs in common plant foods
| Exogenous miRNAs | Brassica napus | Glycine max | Oryza sativa | Sorghum bicolor | Solanum lycopersicum | Vitis vinifera | Zea mays |
|---|---|---|---|---|---|---|---|
| miR156a | √ | √ | 2.72E+05 | 3.09E+02 | 2.98E+03 | 2.08E+03 | 1.24E+03 |
| miR156g | √ | √ | 2.72E+05 | 1.34E+03 | NA | 1.78E+03 | 572 |
| miR157a | NA | NA | NA | NA | NA | NA | NA |
| miR157d | NA | NA | NA | NA | NA | NA | NA |
| miR159a | NA | √ | 5.91E+03 | 966 | NA | 8.02 | 6.41E+03 |
| miR160a | √ | √ | 1.32E+03 | 3.1 | 75.6 | 32.7 | 12.7 |
| miR162a | √ | √ | NA | NA | NA | 1.04E+04 | NA |
| miR164a | √ | √ | 3.99E+03 | 81.3 | 1.81E+04 | 301 | 88 |
| miR164c | √ | √ | 3.41E+03 | 22.9 | NA | 1.24E+04 | 51.1 |
| miR165a | NA | NA | NA | NA | NA | NA | NA |
| miR166a | √ | √ | 4.03E+04 | 643 | 2.92E+05 | 2.25E+04 | 8.00E+03 |
| miR167a | √ | √ | 1.51E+04 | 1.66E+03 | 1.42E+04 | 1.34E+03 | 1.29E+03 |
| miR167d | √ | √ | 1.57E+04 | 3.02E+03 | NA | 548 | 180 |
| miR168a | √ | √ | 3.58E+05 | NA | √ | NA | 1.47E+05 |
| miR169a | √ | √ | 8.17E+03 | 387 | 47.4 | 23 | 318 |
| miR169b | √ | √ | 9.83E+03 | 120 | NA | 4.59 | 58.8 |
| miR169h | √ | NA | NA | NA | NA | 104 | NA |
| miR171a | √ | √ | 3.24E+03 | 12.3 | 206 | 212 | √ |
| miR171c | √ | √ | 2.71E+03 | NA | 85.3 | 536 | 17 |
| miR172a | √ | √ | 1.72E+04 | 6.44E+03 | 1.27E+04 | 8.6 | 73.3 |
| miR172c | √ | √ | 1.48E+04 | 91.3 | NA | 3.21E+03 | 5.28E+03 |
| miR390a | √ | √ | NA | NA | 2.83E+03 | NA | 236 |
| miR394a | √ | √ | NA | 155 | NA | 145 | 56.9 |
| miR397a | √ | √ | 827 | NA | NA | 1.25E+03 | NA |
| miR408 | NA | NA | 790 | 8.50E+03 | NA | 1.69E+03 | NA |
Here the numbers represent the reads per million; “√” means that the miRNA is found in the species but its expression level is unavailable; ‘NA’ means that there is no evident to support this miRNA
Target prediction by RNAhybrid
RNAhybrid [14] was chosen as the target prediction tool with criteria of “seed region rules” (perfect nucleotide match at the core sequence that encompassed the first two to eight bases of the mature miRNA); the free energy of the hybrid was expected to be within the range of the authentic miRNA-target pairs, typically lower than −25 kcal/mol. For Arabidopsis target prediction, we also applied an additional restriction on the length of loop and bulge within 5 and 9 nt, respectively.
Plant target validation
To test the reliability of our targets, we designed a three-level validation method. First, we checked whether this validated target existed in our input mRNA dataset; second, we checked the primarily predicted targets to ensure the effectiveness of our predicting method; third, we checked whether it was excluded after the screening process to evaluate the performance of our screening parameters. After the three-level test, we ranked the target by its p-value first, then the MFE, and set a threshold to include most of the validated targets.
Node weight assignment
According to the connectivity among all the N nodes in the network, we used the PageRank [33] algorithm to evaluate each node by constructing an N*N matrix A. As a famous algorithm used by Google, PageRank has proved to be a good measurement of the importance of website pages. If there is a connection between two nodes, we set a value of one to the corresponding position of matrix with all the rest set at zero. Through this process, we recorded all edges in A. We also used a 1*N-dimensional matrix B0 to store each node’s original weight, which is designed equally as one. Then, we multiplied the matrix A and B0 to obtain B iteratively. After about ten iterations, we obtained a converged matrix B as the final weight of each node.
Core module selection
According to the relationship between any two nodes, we took the entire network as an equivalent of an undirected, connected graph G. The core module selection process is based on the bridge gene v, to extract sub-graph G’ from G. The bridge genes rank as nodes with the highest connectivity; thus, we derived the modules by simply deleting these bridges from G, so the G graph was changed into 11 separate highly connected components.
Functional analysis on network and modules
Two Cytoscape apps, Mosaic [21] and NOA [22], were used to analyze the functional similarity among the different human target sub-networks. The GO biological process terms were assigned to each gene in the module using Mosaic, and then all sub-networks were analyzed together under the “Batch mode” of NOA with the complete network as the background. In order to control the type I error rate of multiple hypothesis testing, the Benjanmini & Hochberg method was employed to adjust P-values, so that GO terms were considered as statistically significant in overrepresented functions with adjusted-P <0.1. The functional heatmap (Fig. 2) was generated by NOA.
Since NOA app does not have the annotation for Arabidopsis, in order to compare the functional similarities of modules between Arabidopsis and human, we performed the functional enrichment analysis on each detected modules/sub-networks for both Arabidopsis (Additional file 3) and human (Additional file 4) with DAVID [23]. The GO biological process terms were considered to be statistically significant with DAVID’s default threshold p-value <0.1. Then a web-based tool CateGOrizer [34] was used for simplifying enriched functions and grouping them into more generic GO categories for (Additional file 5). The functional similarity between Arabidopsis and human was measured by calculating the overlaps of two generic GO categories obtained from the CateGOrizer.
Abbreviations
AXIN1, Axis inhibition protein 1; Bc-sRNA, Botrytis cinerea small RNA; CDS, coding DNA sequence; GI, gastrointestinal; GO, gene ontology; HKG, house keeping genes; LDLRAP1, low-density lipoprotein receptor adapter protein 1; LPSs, lipopolysaccharides; MFE, minimum free energy; miRNAs, microRNAs; RNAi, RNA interference; SPL, SQUAMOSA-promoter binding protein-like; sRNAs, small RNAs; TAIR, the Arabidopsis Information Resource
Acknowledgements
This work was supported by Natural Science Foundation of Jilin Province (20150101056JC and 20140101194JC), China Postdoctoral Science Foundation (2015M570273), National Natural Science Foundation of China (61471181), and the Postdoctoral Research Station of College of Computer Science and Technology, Jilin University.
Availability of data and material
The datasets supporting the results of this article are included within the article and its additional files.
Authors’ contributions
HZ, YPL, CZ and DX conceived the initial study and prepared relevant data and their pre-processing. HZ, YPL and YNL designed the prediction model. YPL, HML, YMZ, WJ and HYW implemented the prediction algorithm. HZ, YPL, CZ and DX performed the data analyses. All authors wrote and reviewed the manuscript. All authors read and approved the final manuscript.
Authors’ information
HZ, YNL and DX are Professors at Symbol Computation and Knowledge Engineering of Ministry of Education, College of Computer Science and Technology, Jilin University of China and Department of Computer Science and Christopher S. Bond Life Sciences Center, University of Missouri, USA. YPL, HML, HYW, WJ and YMZ are masters at Symbol Computation and Knowledge Engineering of Ministry of Education, College of Computer Science and Technology, Jilin University of China. CZ is a researcher at Institute of Computational Biomedicine, Weill Cornell Medical College, New York, USA.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Additional files
Supplementary Information. It contains the input of network construction (Data S1) and verified tissue-specific gene selection (Data S2). (PDF 132 kb)
Node weight assignment. (a) Node weight distribution of all 531 nodes. (b) Weight changes during iterations. The change was reduced to within 0.003 after 15 iterations. (PDF 8264 kb)
The technical details and p-values to describe the enrichment of GO terms for Arabidopsis. (XLS 109 kb)
The technical details and p-values to describe the enrichment of GO terms for human. (XLS 96 kb)
The result of grouping generic GO categories for Arabidopsis and human individually by CateGOrizer. (XLS 23 kb)
Comparison between plant and human target distributions after the filtering process. (a) Original Arabidopsis targets. (b) Arabidopsis targets after screening. (c) Original Human targets. (d) Human targets after screening. There is a remarkable reduction of the noisy points between (a) and (b), and between (c) and (d), which strongly supports the effectiveness of our method and parameters, and provides a valid guide that can help explore the mechanism of cross-species miRNA targets. (PDF 8744 kb)
Contributor Information
Hao Zhang, Email: zhangh@jlu.edu.cn.
Yanpu Li, Email: pierce1024@qq.com.
Yuanning Liu, Email: lyn@jlu.edu.cn.
Haiming Liu, Email: 499787141@qq.com.
Hongyu Wang, Email: 459850889@qq.com.
Wen Jin, Email: 1005028631@qq.com.
Yanmei Zhang, Email: 763871836@qq.com.
Chao Zhang, Email: chz2009@med.cornell.edu.
Dong Xu, Email: xudong@missouri.edu.
References
- 1.Jiang M, Sang X, Hong Z. Beyond nutrients: food-derived microRNAs provide cross-kingdom regulation. Bioessays. 2012;34(4):280–4. doi: 10.1002/bies.201100181. [DOI] [PubMed] [Google Scholar]
- 2.Hughes DT, Sperandio V. Inter-kingdom signalling: communication between bacteria and their hosts. Nat Rev Microbiol. 2008;6(2):111–20. doi: 10.1038/nrmicro1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weiberg A, et al. Fungal small RNAs suppress plant immunity by hijacking host RNA interference pathways. Science. 2013;342(6154):118–23. doi: 10.1126/science.1239705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.LaMonte G, et al. Translocation of sickle cell erythrocyte microRNAs into Plasmodium falciparum inhibits parasite translation and contributes to malaria resistance. Cell Host Microbe. 2012;12(2):187–99. doi: 10.1016/j.chom.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gu Y, et al. Lactation-related microRNA expression profiles of porcine breast milk exosomes. PLoS One. 2012;7(8):e43691. doi: 10.1371/journal.pone.0043691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liang H, Zen K, Zhang J, Zhang CY, Chen X. New roles for microRNAs in cross-species communication. RNA Biol. 2013;10(3):367–70. doi: 10.4161/rna.23663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang J, et al. Cellular microRNAs contribute to HIV-1 latency in resting primary CD4+ T lymphocytes. Nat Med. 2007;13(10):1241–7. doi: 10.1038/nm1639. [DOI] [PubMed] [Google Scholar]
- 8.Zhang L, et al. Exogenous plant MIR168a specifically targets mammalian LDLRAP1: evidence of cross-kingdom regulation by microRNA. Cell Res. 2012;22(1):107–26. doi: 10.1038/cr.2011.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Y, et al. Analysis of plant-derived miRNAs in animal small RNA datasets. BMC Genomics. 2012;13(3):177. doi: 10.1186/1471-2164-13-381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liang H, et al. Regulation of mammalian gene expression by exogenous microRNAs. Wiley Interdiscip Rev RNA. 2012;3(5):733–42. doi: 10.1002/wrna.1127. [DOI] [PubMed] [Google Scholar]
- 11.Vaucheret H, Chupeau Y. Ingested plant miRNAs regulate gene expression in animals. Cell Res. 2012;22(1):3–5. doi: 10.1038/cr.2011.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grillo G, et al. UTRdb and UTRsite (RELEASE 2010): a collection of sequences and regulatory motifs of the untranslated regions of eukaryotic mRNAs. Nucleic Acids Res. 2010;38(38):D75–80. doi: 10.1093/nar/gkp902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bushati N, Cohen S. M. microRNA functions. Annu Rev Cell Dev Biol. 2008;23:175–205. doi: 10.1146/annurev.cellbio.23.090506.123406. [DOI] [PubMed] [Google Scholar]
- 14.Rehmsmeier M, Steffen P, Hochsmann M, Giegerich R. Fast and effective prediction of microRNA/target duplexes. RNA. 2004;10(10):1507–17. doi: 10.1261/rna.5248604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garcia-Hernandez M, et al. TAIR: a resource for integrated Arabidopsis data. Funct Integr Genomics. 2010;2(6):239–53. doi: 10.1007/s10142-002-0077-z. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Z, et al. PMRD: plant microRNA database. Nucleic Acids Res. 2010;38:D806–13. doi: 10.1093/nar/gkp818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rubio-Somoza I, Weigel D. MicroRNA networks and developmental plasticity in plants. Trends Plant Sci. 2011;16(5):258–64. doi: 10.1016/j.tplants.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 18.Zhang B, Pan X, Cobb GP, Anderson TA. Plant microRNA: a small regulatory molecule with big impact. Dev Biol. 2006;289(1):3–16. doi: 10.1016/j.ydbio.2005.10.036. [DOI] [PubMed] [Google Scholar]
- 19.Li A, Mao L. Evolution of plant microRNA gene families. Cell Res. 2007;17(3):212–8. doi: 10.1038/sj.cr.7310113. [DOI] [PubMed] [Google Scholar]
- 20.Montojo J, et al. GeneMANIA Cytoscape plugin: fast gene function predictions on the desktop. Bioinformatics. 2010;26(22):2927–8. doi: 10.1093/bioinformatics/btq562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang C, et al. Mosaic: making biological sense of complex networks. Bioinformatics. 2012;28(14):1943–4. doi: 10.1093/bioinformatics/bts278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang C, et al. NOA: a cytoscape plugin for network ontology analysis. Bioinformatics. 2013;29(16):2066–7. doi: 10.1093/bioinformatics/btt334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 24.Xiao SJ, Zhang C, Zou Q, Ji ZL. TiSGeD: a database for tissue-specific genes. Bioinformatics. 2010;26(9):1273–5. doi: 10.1093/bioinformatics/btq109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thomine S, Lanquar V. Iron transport and signaling in plants. Ann Rev Plant Biol. 2003;54(4):99–131. doi: 10.1146/annurev.arplant.54.031902.135018. [DOI] [PubMed] [Google Scholar]
- 26.Vert G, et al. IRT1, an Arabidopsis transporter essential for iron uptake from the soil and for plant growth. Plant Cell. 2002;14(6):1223–33. doi: 10.1105/tpc.001388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thomine S, Wang R, Ward JM, Crawford NM, Schroeder JI. Cadmium and iron transport by members of a plant metal transporter family in Arabidopsis with homology to Nramp genes. Proc Natl Acad Sci U S A. 2000;97(9):4991–6. doi: 10.1073/pnas.97.9.4991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eide DJ. The molecular biology of metal ion transport in Saccharomyces cerevisiae. Annu Rev Nutr. 1998;18(1):441–69. doi: 10.1146/annurev.nutr.18.1.441. [DOI] [PubMed] [Google Scholar]
- 29.Segerstrom SC, Miller GE. Psychological stress and the human immune system: a meta-analytic study of 30 years of inquiry. Psychol Bull. 2004;130(4):601–30. doi: 10.1037/0033-2909.130.4.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arabidopsis Genome I. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000;408(6814):796–815. doi: 10.1038/35048692. [DOI] [PubMed] [Google Scholar]
- 31.Riechmann JL, et al. Arabidopsis transcription factors: genome-wide comparative analysis among eukaryotes. Science. 2000;290(5499):2105–10. doi: 10.1126/science.290.5499.2105. [DOI] [PubMed] [Google Scholar]
- 32.Schmid M, et al. A gene expression map of Arabidopsis thaliana development. Nat Genet. 2005;37(5):501–6. doi: 10.1038/ng1543. [DOI] [PubMed] [Google Scholar]
- 33.Brin S, Page L. The anatomy of a large-scale hypertextual Web search engine. Computer Networks and ISDN Systems. 1998;30(98):107–17. doi: 10.1016/S0169-7552(98)00110-X. [DOI] [Google Scholar]
- 34.Na D, Son H, Gsponer J. Categorizer: a tool to categorize genes into user-defined biological groups based on semantic similarity. BMC Genomics. 2014;15(1):1091. doi: 10.1186/1471-2164-15-1091. [DOI] [PMC free article] [PubMed] [Google Scholar]