

Abstract
Apoptosis plays important roles in the pathophysiology of Type 2 diabetes mellitus (T2DM). The etiology of T2DM is multifactorial, including obesity-associated insulin resistance, defective insulin secretion, and loss of β-cell mass through β-cell apoptosis. β-cell apoptosis is mediated through a milliard of caspase family cascade machinery in T2DM. The glucose-induced insulin secretion is the principle pathophysiology of diabetes and insufficient insulin secretion results in chronic hyperglycemia, diabetes. Recently, hyperglycemia-induced β-cell apoptosis has been extensively studied on the balance of pro-apoptotic Bcl-2 proteins (Bad, Bid, Bik, and Bax) and anti-apoptotic Bcl family (Bcl-2 and Bcl-xL) toward apoptosis in vitro isolated islets and insulinoma cell culture. Apoptosis can only occur when the concentration of pro-apoptotic Bcl-2 exceeds that of anti-apoptotic proteins at the mitochondrial membrane of the intrinsic pathway. A bulk of recent research on hyperglycemia-induced apoptosis on β-cells unveiled complex details on glucose toxicity on β-cells in molecular levels coupled with cell membrane potential by adenosine triphosphate generation through K+ channel closure, opening Ca2+ channel and plasma membrane depolarization. Furthermore, animal models using knockout mice will shed light on the basic understanding of the pathophysiology of diabetes as a glucose metabolic disease complex, on the balance of anti-apoptotic Bcl family and pro-apoptotic genes. The cumulative knowledge will provide a better understanding of glucose metabolism at a molecular level and will lead to eventual prevention and therapeutic application for T2DM with improving medications.
Keywords: Amyloid, apoptosis, β-cells, caspase, hyperglycemia, insulin, islets of langerhans, knockout mouse, pro-apoptotic genes, Type 2 diabetes
INTRODUCTION
The increasing evidence supports that Type 2 diabetes mellitus (T2DM) is modulated through β-cell apoptosis. Apoptosis is a complex biological phenomenon characterized by cell shrinkage, chromatic condensation, internucleosomal DNA fragmentation, and disassembly into membrane-encircled vesicles (apoptotic bodies) [1]. This programmed cell death is implicated in the remodeling of the normal endocrine pancreas after birth, and plays an important role in the development of final β-cell mass [2]. The role of apoptosis in the physiology of normal pancreatic development has been demonstrated in neonatal pancreas, which has a three-fold higher frequency of apoptotic cells than do adult animals [3]. Pancreatic β-cell apoptosis is also a pathological feature that is common to both Type 1 diabetes mellitus (T1DM) and T2DM. In T2DM, insulin resistance with visceral obesity leads to a glucose toxicity effect, which accelerates β-cell death by apoptosis [4].
Defects in apoptotic regulatory machinery are implicated in a variety of pathological status: Excess apoptosis is the underlying cause for β-cell loss for both T1DM and T2DM and inadequate apoptosis may contribute to oncogenesis of pancreatic endocrine tumors (PETs) [5]. Among two apoptosis pathways, including extrinsic (receptor-mediated) and intrinsic (mitochondria-driven) pathway, the extrinsic pathway is activated upon ligation of the cells surface death receptor(s), which in turn activates downstream effector mechanism orchestrated by the caspase family of cysteine proteases (Figure 1) [5]. The prototype example of death signaling via the extrinsic pathway is the Fas death receptor, which instigates assembly of the death-inducing signaling complex (DISC), a multi-protein complex comprising of the cytoplasmic aspects of the Fas receptor, the adaptor protein Fas-associated death domain-containing protein (FADD) and procaspase-8 (Figure 2) [6]. Caspase-3 is a converging point of the apoptotic pathway (Figure 2) [6], and its peptide inhibitors have been shown to prevent islet apoptosis and improve islet graft function [7-9]. Caspases are cysteine-containing aspartic acid-specific proteases which exist as zymogens in the soluble cytoplasm, endoplasmic reticulum (ER), mitochondrial intermembrane space, and nuclear matrix [10]. Apoptosis induced by ligation of cell surface receptors like Fas (CD 95) or tumor necrosis factor (TNF) receptors, “death receptors” represents a pathway controlled by caspases [11]. Ligand binding of the receptor causes assembly of series of proteins of the death-inducing signaling complex (DISC), which then activates an apical caspase, procaspase-8 [12]. The resulting events pursue in cascades that caspase-8 induces activation of caspase-3 [11]. One of these proteins is a caspase-dependent endonuclease (caspase-activated DNase [CAD]), which is freed from its inhibitor (ICAD) by caspase-3 and subsequently cuts DNA into oligonucleosomal (180-bp) fragments [12,13].
FIGURE 1.
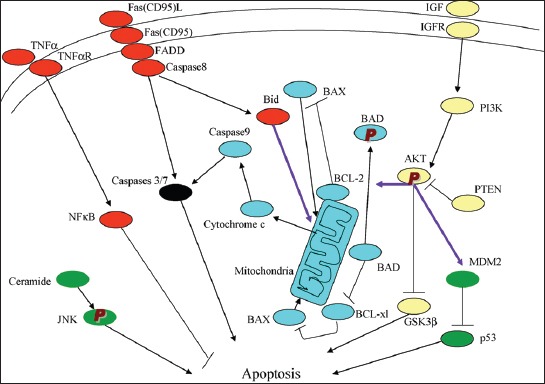
There are the extrinsic (receptor-mediated, red) and intrinsic (mitochondria-driven, blue) apoptosis pathways as opposed to the survival proteins such as the P13/Akt signaling circuity (yellow). From Lee SC, et al., Int J Biochem 2007;39:497-504.
FIGURE 2.
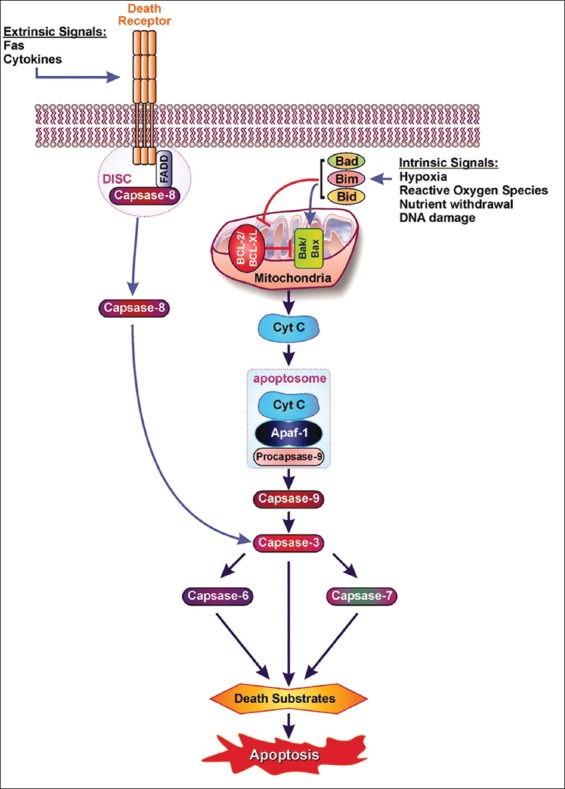
Extrinsic and intrinsic pathways leads to apoptosis via cytochrome c and “apoptosome.” Extrinsic pathway: Fas-Fas L binding leads to the death-inducing signaling complex (DISC) where DISC-caspase-8 complex is activated, leading to caspase-3 activation. Intrinsic pathway: Pro-apoptotic Bcl proteins (Bad, Bid, Bik, Bim) become activated and translocate to the mitochondria, where they bind or inactivate Bcl proteins or form pores in the mitochondrial membrane, which facilitates the release of cytochrome c into cytosol. Once cytochrome c accumulates in the cytosol, it complexes with apocaspase-9 and Apaf-1 to form the “apoptosome,” which, in turn, activates caspase-3. From Emamaulee and Shapiro, Diabetes 2006;55:1907-14.
Apoptosis manifests in two major execution programs, downstream of the death signal: The caspase pathway [14] and upstream of irreversible cellular damage reside in the Bcl family members, which are proteins with both proapoptotic and antiapoptotic properties, playing a pivotal role in life and death of β-cells (Figures 1 and 2) [6,12]. Antiapoptotic member of the Bcl family, including Bcl-2 and Bcl-xL, blunts intrinsic death signaling by blocking the recruitment of proapoptotic members to the mitochondria [14,15]. The cumulative data support that high glucose modulates the balance of proapoptotic caspase family and antiapoptotic Bcl proteins toward apoptosis, thus leading to β-cell death [6,16].
Apoptosis in pancreatic β-cells by the immunologic and immunocytochemical study
Apoptosis is a cause of absolute β-cell deficiency in T1DM and relative β-cell deficiency in T2DM. Fas (CD 95) is a member of the TNF receptor superfamily characterized by the presence of a death domain motif in the cytoplasmic end (Figure 1) [5,17]. Fas L (CD 178) is a membrane bound protein upregulated on the activated T-cells [18]. Expression of dominant-negative Fas or neutralizing antibody to Fas significantly blocks apoptosis, manifests adequate β-cell function, blocks adoptive transfer of diabetes by primed T-cells and retards the course of T1DM development [19].
T2DM is characterized by insulin resistance, defective insulin secretion, loss of β-cell mass with increased β-cell apoptosis, and islet amyloid deposits [20]. In T2DM, insulin resistance with obesity precedes insulin deficiency and plays a considerable role [21], followed by a failure of β-cell insulin production against the progressive insensitivity to insulin. β-cell mass fluctuates according to the body need of insulin [22]: (1) β-cell mass can increase during insulin resistance, (2) progressive β-cell loss is present in T2DM, (3) β-cell deficiency correlates with the glucose intolerance, (4) β-cell death may directly lead to insulin deficiency when loss of 60% or more is accompanied by the presence of insulin resistance with visceral obesity [22].
β-cell mass is regulated by a balance of β-cell replication and apoptosis, and islet hyperplasia and new islets formation from exocrine pancreatic ducts [23]. Elevated caspase-3 and -8 activates in β-cells from T2DM subjects, which can be inhibited by the antidiabetic agent metformin [24]. The β-cells are continuously remodeling through the delicate balance of β-cell replication and apoptosis, which is mediated through a balance between matrix metalloproteinase (MMP)-1 and -2 and tissue inhibitor of MMP (TIMP)-1 and -2 and is dispensable for islet formation and function in vivo [25,26]. Hyperglycemia-induced β-cell apoptosis has been implicated and has been studied mainly in T2DM [22]. Butler et al., extensively studied 124 cases of pancreata from autopsy, including 91 obese cases: 91 obese cases (body mass index (BMI) > 27 kg/m2: 41 cases - T2DM, 15 cases - impaired fasting glucose and 35 cases - non-DM cases) and 33 lean cases (BMI <25 kg/m2: 16 cases - T2DM, 17 cases - non-diabetic cases). The authors measured relative β-cell mass volume using Image-Pro Plus software (Media Cybermetric, Silber Springs, MD), the frequency of β-cell apoptosis by terminal deoxynucleotydyl transferase (TdT)-mediated dUTP nick-end labeling (TUNEL) and replication index using Ki-67 immunocytochemical staining [22]. By TUNEL staining, only discernible cells with TUNEL-positive nuclei were included as positive cells [22]. Obese humans with impaired fasting glucose and T2DM subjects showed 40% and 63% less β-cell volume compared with non-diabetic obese and lean controls, respectively [22]. The frequency of β-cell replication was very low at 0.04-0.06% of β-cell mass, but frequency of β-cell apoptosis by TUNEL was increased 10-fold in lean DM (0.47% β-cell area) and threefold in obese DM (0.31% β-cell area) compared with respective non-diabetic control subjects [22]. It appears that β-cell replication by Ki-67 is underestimated, and β-cell apoptosis by TUNEL is overestimated since replication and apoptosis rate should be about the same to maintain the β-cell mass at a delicate balance. The authors conclude that β-cell mass in T2DM is decreased and that the mechanism underlying the β-cell loss is increased β-cell apoptosis [22].
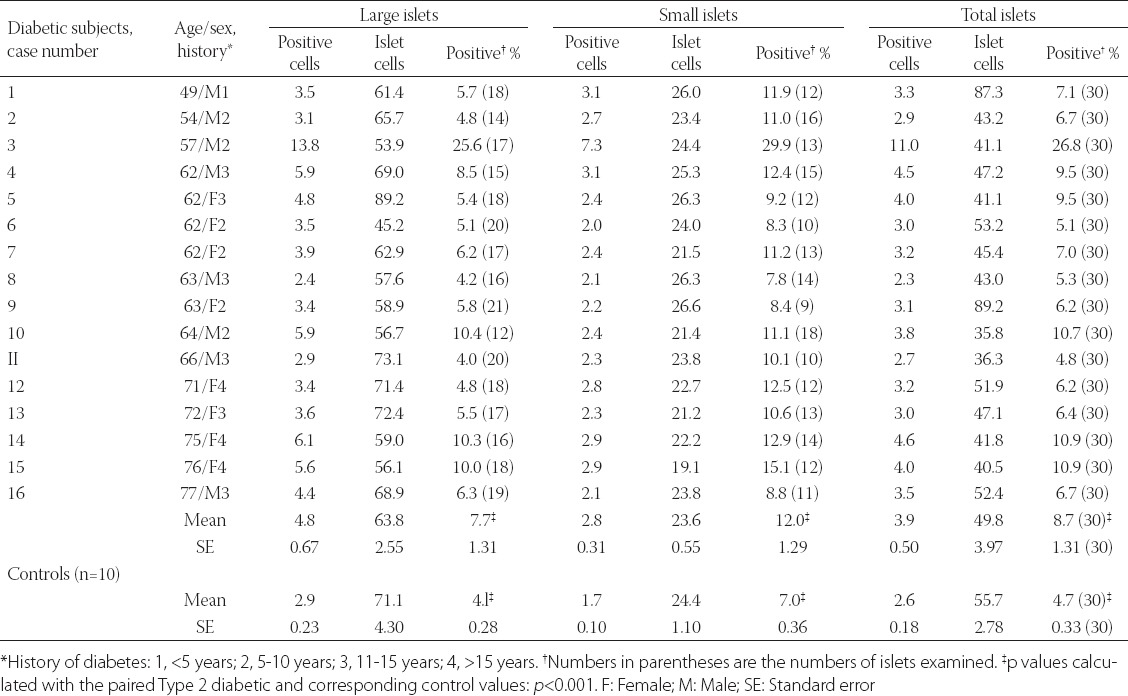
Another immunocytochemical marker for apoptosis is cleaved caspase-3: Each caspase family protease becomes active when the precursor is cleaved into a large subunit with a molecular mass of ~20 kDa and a small subunit with a molecular mass of ~10 kDa, which then forms a tetramer consisting of two large and two small units [27,28]. One of these cleaved caspases is present on the activated caspase-3, a ubiquitously distributed caspase which is the main effector caspase of the apoptotic cascade within cells [24,27]. The commercially available polyclonal anti-cleaved caspase-3 detects endogenous levels of the large (17/19 kDa) cleaved caspase-3 resulting from cleavage adjacent to Asp 175 and does not recognize the full length or other cleaved caspases (Cell Signaling Technology Publication, Beverly, MA, USA, 2006) [29]. Recently, an involvement of caspase-3 in both T1DM and T2DM was implicated: In T1DM, Fas (CD 95)-Fas L (CD 178) may be critical for β-cell destruction as apoptosis in β-cell clone expressing the human Fas β-cell line is mediated by elevated caspase-3 like activity in tissue culture [30] and the frequency of β-cell apoptosis in T2DM pancreatic tissues from autopsy is increased using TUNEL as described before [22]. Our group studied 16 cases of T2DM pancreata compared with 10 control pancreata using rabbit anti-human cleaved caspase-3 (Cell Signaling Technology Publication, Beverly, MA, USA, 2006) for immunocytochemical staining: The control islets revealed 4.7% cleaved caspase-3 positive islet cells in the total islet cells with large and small islets being positive at 4.1% and 7.0%, respectively (Figure 3 and Table 1) [31], whereas T2DM islets showed a higher positive cells at 8.7% in the total islet cells with large and small islets positive at 7.7% and 12%, respectively, at about twice that of the control values (Figure 3 and Table 1) [31]. A double immunochemical staining for insulin and cleaved caspase-3 supported that β-cell nuclei in the degranulated cytoplasm were positive for cleaved caspase-3 (Figure 3) [31]. Cleaved caspase-3 positive islet cells were more in the less amyloid deposited islets than in the islet cells containing more amyloid deposits, the latter corresponded to the end-stage T2DM islets, which have completed apoptotic process (Figure 3) [31]. Thus, the more cleaved caspase-3 positive islets from T2DM subjects may implicate an accelerated apoptotic cascade, accompanied by increasing amyloid deposits, before proceeding to ultimate β-cell death by overwhelming interstitial amyloid deposits [31]. In adult islets, β-cells have an estimated life span of about 60 days [23,32]. Under normal conditions, 0.5% of control adult β-cells were reportedly to undergo apoptosis [22,32-34]. By cleaved caspase-3 immunostaining, about 5% of control β-cells are positive for this apoptosis marker [35]. Thus, there is a wide range of variation between 0.5 and 5% of apoptotic islet cells in the control islets by TUNEL and cleaved caspase immunochemical staining [22,31,35]. Yet both TUNEL and cleaved caspase-3 immunostaining showed much more apoptotic cells in both T2DM and control islets than the β-cells with typical apoptotic bodies [22,31]. Furthermore, a presence of larger islets in T2DM than control islets reflects a more robust balance for regenerating hyperplasic islets in T2DM to compensate for more insulin secretion against an ongoing β-cell apoptosis [31].
FIGURE 3.
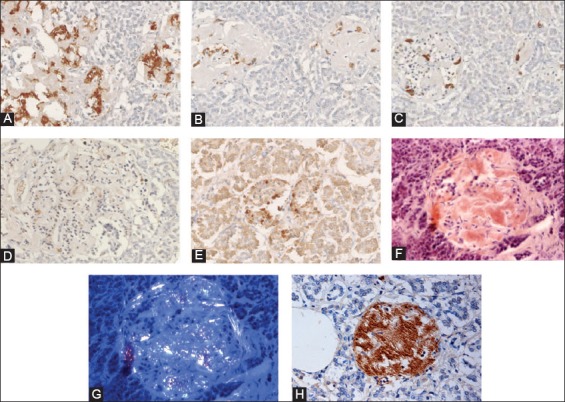
Amyloid deposited Type 2 diabetes mellitus islets (A-H). These amyloid deposited islets have fewer islet cells, consisting of major β-cells (A, 80%), α-cells (B, 10%) and δ-cells (C, 10%) and less densely stained cleaved caspase-3 positive cells (D,* at about 2%) as compared with about 2% cleaved caspase-3 positive cells in the control islets (Table 1). The nuclear positive staining for cleaved caspase-3 (E) is smaller than that of cytoplasmic positive staining of insulin, glucagon, and somatostatin (A-C). The stroma of amyloid deposited islets is Congo red positive (F) and is birefringent under polarized light (G). The recently amyloid deposited islet is also positive for islet amyloid polypeptide (IAPP) (H). The less amyloid deposited islets from the same subject reveal more cleaved caspase-3 positive islet cells at about 15% (E) than trabecular more amyloid deposited islets at about 2% (D). Immunostained for insulin (A), glucagon (B), somatostatin (C), cleaved caspase-3 (D, E), Congo red (F, G), and IAPP (H). A: Insulin, B: Glucagon, C: SRIF, D and E: Cleaved caspase -3 and H: IAPP immunostained. F and G: Congo red. From Tomita T. Pathology 2010;42:432-7, with permission.
TABLE 1.
Cleaved caspase-3 immunocytochemical staining in type 2 diabetic islets. From Tomita, T, pathology, 2010, 42:432-7, with permission
Regarding the two immunocytochemical staining for apoptosis, each method has advantages and disadvantages: The TUNEL assay is very sensitive and widely used but it is prone to some pitfalls. The TUNEL technique can label non-apoptotic nuclei showing signs of active gene transcription [34]. Tumor necrosis and autolysis generate a significantly higher numbers of DNA ends which can be positively labeled under certain conditions [36,37]. The technical problem of TUNEL is mostly related to DNA strand-breaks associated with excessive levels of protease digestion, fixation, and processing procedures [37]. Therefore, techniques that detect DNA fragmentation are not specific for apoptosis and frequently generate erroneously higher results [22,37].
Duan et al., carefully studied apoptosis in histological sections of prostatic cancer cell line PC-3 using both TUNEL and cleaved caspase-3 immunostaining [37]. TUNEL staining depends on the optimal concentration of terminal deoxynucleotydyl transferease (TdT), with which less diluted (1:7) solution positively stained the majority (>90%) of the transplanted cancer cells as compared to an optimally diluted solution (1:16) stained the adequate numbers (<2%) of TUNEL positive cancer cells [37]. By comparing the TUNEL and cleaved caspase-3 immunostaining, the authors concluded that cleaved caspase-3 immunostaining was an easy, sensitive, and reliable method for detecting and quantifying apoptosis and that a good correlation of apoptotic indices existed between the caspase-3 immunostaining and TUNEL assay [37]. We agree with Duan et al. that cleaved caspase-3 immunostaining is an easier and more reliable immunostaining than TUNEL, although the former is not as commonly used as the latter. Cleaved caspase-3 immunostaining has also its own pitfalls as for the immunostaining staining in general: Good fixation with an optimal tissue preservation and proper tissue processing are essential using an adequate concentration of antibody for optimal cleaved caspase-3 immunostaining and the stained sections have to be critically evaluated for discernible nuclear positive staining by excluding false-positive staining in tissue debris and autolytic tissues [22,36-38].
Our group studied cleaved caspase-3 immunostaining in 37 cases of pancreatic endocrine tumors (PETs) [38]: Among 15 cases of insulinomas, five cases were positive, and 10 cases were negative for cleaved caspase-3 (67%). Among non-β-cell PETs, 2 of 2 glucagonomas (100%), 6 of 9 pancreatic polypeptidomas (67%), 10 of 12 gastrinomas (83%), and 3 of 3 non-functioning PETs (100%) were negative for cleaved caspase-3 immunostaining at a total of 21 of 24 non-β-cell tumors (88%) being negative. These results support that 88% of non-β-cell PETs are potentially malignant by the absence of cleaved caspase-3 immunostaining [38] and that negative cleaved caspase-3 immunostaining may be a candidate of malignant marker for PETs [38]. So far, positive activated caspase-3 immunostaining appears to be a good marker for β-cell apoptosis.
Amyloid theory on β-cell apoptosis in T2DM
Ample amyloid deposit in pancreatic islets is a characteristic finding for T2DM [39-41]. The chief constituent of amyloid deposits is islet amyloid polypeptide (IAPP) [39,42-44]. IAPP is concomitantly co-secreted with insulin from β-cells into the blood in response to glucose-induced insulin secretion [43,45]. IAPP hyposecretion is well-established in T1DM and insulin-requiring T2DM [43,45]: A synthetic IAPP, pramlintide [28-30] (pro-hIAPP) has been used for treating both T1DM and insulin-requiring T2DM together with insulin replacement for a better glycemic control [46-49].
We had studied a transformation of water-soluble IAPP in β-cell granules to water-insoluble amyloid deposits in islet stroma by immunocytochemical staining for IAPP [50]: A ratio of IAPP-positive cells against insulin-positive cells was 43% in control islets compared to 25% in T2DM islets (Figure 4) [50], which supports the decreased IAPP serum levels in T2DM since the source of serum IAPP is the β-cell granules [50]. Pancreatic extracts from normal humans contained less IAPP at 10% that of insulin and the fasting serum IAPP level in non-obese controls was 2.0 µmol/l at 5% that of the insulin level of 48 µmol/l [51]. As IAPP-positive β-cell cytoplasm decreased, stromal islet amyloid deposits increased in T2DM islets [50]. In advanced stages of amyloid deposited islets, weakening IAPP-immunostaining in the residual β-cells co-existed with the adjacent fine amyloid fibrils, which were lesser positively immunostained for IAPP (Figure 4) [50]. This finding supports that disappearing water-soluble IAPP in β-granules are transformed to water-insoluble amyloid fibrils within the islets. Freshly prepared IAPP oligomers can form nonselective iron permeable membrane pores, leading to increased Ca2+ concentration, endoplasmic reticulum (ER) stress and apoptosis [20,51]. In the early stage of islet amyloidosis, round to sickle-shaped β-cell cytoplasm without a nucleus was strongly immunostained for IAPP (Figure 5) [50]. This cytoplasm probably represents an early form of amyloidogenic β-cell cytoplasmic proteins consisting of smaller IAPP polymers, which may subsequently form extracellular amyloid β-sheets. IAPP has the propensity to form membrane permeable toxic oligomers, but it remains unclear why amyloidogenic proteins form oligomers in vivo, what their exact structure is and what extent these oligomers play a role in the cytotoxicity and this is now often called unfolded protein disease [51-55].
FIGURE 4.
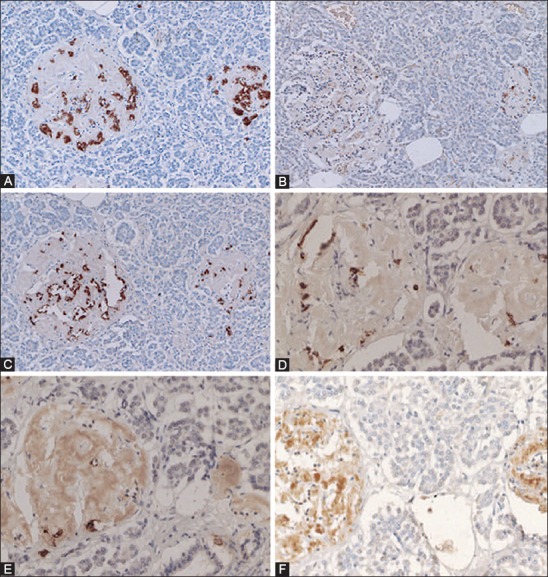
Amyloid deposits in Type 2 diabetes mellitus diabetic islets: Case 10 (A-C) with amyloid deposits in 95% of the islets and Case 14 (D-F) with amyloid deposits >99% of the islets. In the Case 10 islets, the residual β-cells with plump cytoplasm (A) are minor cells and σ-cells with small and compact cytoplasm (C) are major islet cells. Islet amyloid polypeptide (IAPP) immunostaining with 1:400 diluted IAPP antibody reveals weak staining in the islets occupied by amyloid at 95%. (B) In the islets occupied by amyloid >99% reveal a few β-cells (D) of which large cytoplasm is immunostained strongly for insulin whereas amyloid deposits are moderately immunostained for IAPP. (E) In the Case 14, lesser amyloid deposited islets reveal strong immunostaining for the β-cell cytoplasm without nucleus with continuous moderate positive immunostaining to the fibrous amyloid stroma using 1:400 dilution of anti-IAPP antibody. (F) A and D: Insulin, C: Glucagon, B, E, and F: IAPP antibody at 1:400 immunostained for IAPP. From Tomita T. Islets, 2012;4:223-32, with permission.
FIGURE 5.
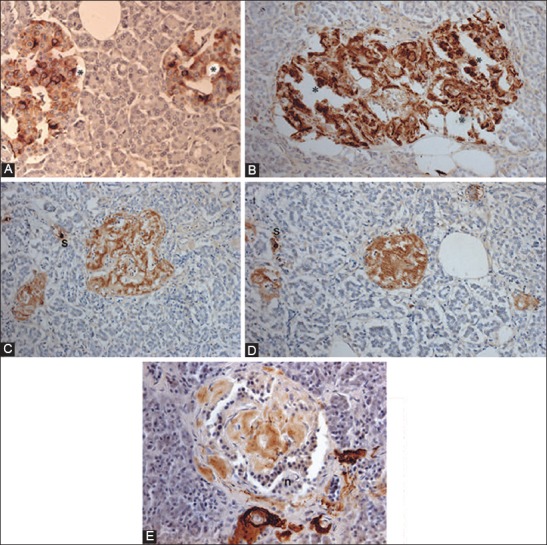
Islet amyloid polypeptide (IAPP) immunocytochemical staining for control (A) and Type 2 diabetes mellitus (T2DM) islets, Case 10 (B-D) and Case 14 (E) IAPP immunostaining was performed using 1:400 for control islets (A) and using 1:200 dilution for T2DM islets (B-D). Control islets were strongly immunostained for the majority of islet cell cytoplasm (A). Both a few control and T2DM islet cytoplasm without nucleus was strongly immunopostive for IAPP (*) (A and B). T2DM islets occupying 95% amyloid deposits were immunostained moderately for IAPP in trabecular amyloid deposits whereas viable islet cells surrounded by amyloid deposits were negative for IAPP (B). In the islets occupied by >99% massive amyloid deposits, which were strongly immunostained for IAPP containing no IAPP-positive β-cells, instead strongly IAPP-positive, scattered single islet cells (s) were noted (C and D). By amyloid p immunostaining, stromal amyloid deposits were moderately positive and perivascular amyloid deposits were strongly positive for amyloid p (E). A-D: IAPP, E: Amyloid P immunostained. From Tomita T. Islets 2012;4:423-232, with permission.
According to the toxic oligomer theory, β-cells in T2DM are killed through IAPP-induced damage of the β-cell membrane [56-58]. These toxic oligomers (not monomer or mature amyloid fibrils) eventually form the end product of β-sheets containing IAPP, Aβ, synuclein, transthyrenin, and prion protein, and share a common epitope (Figure 5) [50,59,60]. Amyloid p is a universal immunologic and immunocytochemical marker for all amyloidosis (Figure 5) [50,59]. Freshly, prepared intermediate IAPP polymers (25-6000 IAPP molecules) have a toxic effect on β-cells but do not exhibit a toxic effect on σ, δ, and PP islet cells since non-β-cells do not contain enough endogenous IAPP [20,50-52,61]. Water-soluble IAPP with low molecular weight in β-cell granules is readily and densely immunostained for IAPP, whereas water-insoluble amyloid fibrils containing IAPP polymers are only weakly immunostained for IAPP (Figure 4) [45,50]. This lack of strong IAPP immunostaining for amyloid fibrils is not clear, but one likely reason is that the unexposed epitope of IAPP polymers within the water-insoluble amyloid fibrils forms β-sheet conformation, to which IAPP antibody cannot penetrate to bind [50,56]. Antibodies generated to this epitope using toxic Aβ1-40 bind toxic oligomers generated from the other amyloidogenic protein in cell culture and block the cytotoxic effects of each of these diverse oligomers [59,60].
Butler and his associates tested the hypothesis that β-cells are preferably vulnerable to hIAPP-induced apoptosis with isolated human islets in tissue culture: Apoptotic cells by TUNEL were increased fivefold after incubation with 40 µmol/l hIAPP compared with control islets [60]. Further, in T2DM islets, the apoptotic cells in islets were adjacent each other containing two separate nuclei, suggesting that cells underwent apoptosis shortly after mitosis [60]. The question that whether amyloid deposit is the cause or the result of T2DM still remains but islet amyloid is clearly linked to the pathophysiology of T2DM [44]. As in an early stage of T2DM, a combined depletion of insulin and IAPP in β-granules causes an early stage of hyperglycemia, and IAPP in β-granules is somehow transformed to cytotoxic IAPP oligomer, which kills β-cells, and subsequent formation of amyloid sheets in islets is the end stage of T2DM islets (Figure 5) [50]. The amyloid sheets are no longer cytotoxic to the remaining β-cells. Thus, a loss of IAPP in T2DM islets is a cause of an early stage of hyperglycemia and displacement of T2DM islets occupied by amyloid sheets is the end stage of β-cell apoptosis and contributes to chronic sustaining hyperglycemia by physically displacing the remaining β-islet cells and obstructing insulin secretion and transport.
Hyperglycemia-induced apoptosis in β-cells
Glucose is the main fuel, which stimulates insulin secretion and mechanism of glucose-induced insulin secretion is the fundamental principle for the pathophysiology of insulin secretion [62,63], and chronic hyperglycemia causes β-cell glucose toxicity and eventually leads to β-cell apoptosis [16]. Obese T2DM subjects are often associated with elevated serum fatty acids and chronic exposure to elevated levels of glucose and free fatty acids causes β-cell dysfunction and may induce β-cell apoptosis [64]. Obese T2DM is often a part of the metabolic syndrome and is accompanied by dyslipidemia, and increased circulating leptin and cytokine levels [65]. Glucose-induced insulin secretion with isolated perifused islets from control rats typically presents both an initial insulin secretion within several minutes and a larger second sustained secretion in about 20 minutes after exposed to a high glucose medium, in which the initial small secretion occurs before glucose is metabolized [62,63]. This early peak of insulin secretion was suggestive of the glucose sensor theory by Matshinsky et al., who proposed glucokinase (GCK, hexokinase IV) as a glucose sensor [65-68]. GCK constitutes a key component of mammalian glucose-sensing machinery [67-69]. In the liver, GCK controls glycogen synthesis and glucose output, whereas in pancreatic islets it regulates insulin secretion [67]. Subsequent studies showed that glucose-sensing mechanisms in the β-cells are divided into the two components [70]: (1) Proximal events of glucose entry and metabolism, (2) the distal mechanisms of insulin secretion, spanning from mitochondrial signal generation and initiation of electrical activity to the ultimate effectors of β-granule exocytosis.
The proximal sensing and metabolic signal generation includes [69]: (1) Glucose equilibrates rapidly across the β-cell membrane due to expression of the high capacity, low-affinity glucose-transporter-2 (GLUT-2), (2) after glucose has entered the β-cells, it is phosphorylated to glucose-6-phosphate by the high KM GCK, which constitutes the flux determining step of glycolysis and is considered as glucose sensor, (3) once, glucose is phosphorylated, glucose is metabolized by glycolysis to pyruvate, nicotinamide adenine dinucleotide (NADH) and adenosine triphosphate (ATP) [70-72]. Pyruvate is the main end product of glycolysis in β-cells, essential for mitochondrial ATP synthesis and is suggested to be an important modulator of insulin secretion [70]. In the mitochondrial matrix, pyruvate is oxidized by pyruvate dehydrogenase to form acetyl-CoA. Acetyl-CoA enters the tricarboxylic acid (TCA) cycle to undergo additional oxidation steps generating CO2 and reducing equivalents, flavin adenine dinucleotide (FADH2) and NADH [71-73]. Oxidation of reducing equivalents by the respiratory chain is coupled to the extrusion of protons from the matrix to the outside of mitochondria, thereby establishing the electrochemical gradient across the inner mitochondrial membrane [74]. The final electron acceptor of these reactions is molecular oxygen [74].
The distal sensing of metabolic signals includes: (1) In the absence of stimulatory glucose (<5 mmol/l) rodent β-cells are electrically silent, with a resting membrane potential of ~70 mV, (2) reduction of the resting K+ conductance by stimulated glucose leads to membrane depolarization and initiation of electrical activity characterized by slow wave depolarization, (3) ATP-sensitive K+ channels set the β-cell membrane potential and closure of these leads to depolarization, (4) membrane depolarization triggers action potential firing and opening voltage-dependent Ca2+ channels, leading to Ca2+ influx, which triggers β-granule exocytosis, (5) action potential is terminated by the opening of voltage-dependent K+ channels, which limit Ca2+ entry and thus insulin release [74]. A comprehensive mathematical model of β-cell sensitivity to glucose predicts a special role for mitochondrial control mechanism in insulin secretion and reactive oxygen species (ROS) generation in the β-cells [71,74]. A failure of insulin secretory machine results in insulin deficiency and subsequent hyperglycemia.
The individuals with autosomal dominant mutations in GCK gene on chromosome 7p originally displayed an abnormal glucose sensing with stable mild hyperglycemia in maturity onset diabetes of the young T2DM (MODY) [75,76], which occurs at an age onset of 25 years or younger [77,78]. Patients with MODY are non-obese, and hyperglycemia is due to impaired glucose-induced insulin secretion [77,78]. More than six types of monogenic MODY are recognized [77,78]. MODY3 and MODY2 are the most common forms and MODY2 does not predispose to the complication of microvascular diabetic complications [77,78]. This fact further supports a role for GCK as a glucose sensor in T2DM [75,76]. A bulk of detailed information on hyperglycemia-induced apoptosis has been derived from the controlled cultured human and rodent islets in vitro [16,65,79]. In order to study glucose toxicity as a deleterious effect of chronic hyperglycemia on β-cells in vitro, Federici et al., cultured 400 isolated human islets per batch for 5 days in a low glucose (5.5 mmol/L) and a high glucose medium (16.6 mmol/L) for studying possible high glucose effects on Bcl-2 (β-cell lymphoma 2) family gene expression by reverse transcription polymerase chain reaction (RT-PCR) [16]. (1) Bcl-2 was highly expressed in both low and high glucose media, and expression did not change between a high and low glucose condition (Figure 6A) [16], (2) however, Bcl-xL was reduced by 45% in the high glucose cultured islets compared with that of the low glucose cultured islets supporting that a reduction in Bcl-xL protein expression was due to high glucose exposure (Figure 6B) [16], and (3) Bad, Bid, and Bik were expressed in low glucose medium at low levels. The Bad gene expression was greatly increased with a high glucose medium and Bad protein level increased 80% as well, compared with that of low-level glucose (Figure 6B-D) [16]. Bid gene expression was markedly increased with the high glucose medium and so were Bik protein (Figure 6E) [16]. Thus, antiapoptotic Bcl-2 was unaffected by high glucose but proapoptotic genes, Bad, Bid, and Bik markedly increased in high glucose cultured islets (Figure 6) [16]. These data support that chronic high glucose incubation in vitro modulates the balance of pro-apoptotic and antiapoptotic Bcl proteins toward apoptosis, thus leading to eventual β-cell death [16]. Using MIN6N8 cells, which are SV40 transformed insulinoma cells derived from NDO mice, Kim et al., extensively studied the effects of chronic hyperglycemia in the tissue culture: when MIN6N8 cells were cultured in different concentrations of glucose for varying time periods: The high glucose (33.3 mmol/l) induced marked genomic DNA fragmentation in a time- and dose-dependent manner and caused a significant increase of TUNEL-positive cells compared to the low glucose (5.5 mmol/l) cultured cells, concomitant cleavage of poly (adenosine diphosphate [ADP]-ribose) polymerase (RARP) similar to caspase-3 cleavage (Figure 7A) [80]. Pre-treatment with a specific caspase-3 inhibitor (z-DEVD-CHO) completely reduced a high glucose-induced RARP cleavage (Figure 7C) [80] and apoptosis. Culturing with a high glucose significantly increased Fas and P53 expression whereas decreased Bcl-2 and Bcl-xL expression (Figure 7B) [81]. A significant mitochondrial release of cytochrome C into cytosol was observed after 2 days (Figure 7C) [80]. Translocation of Bax was further confirmed by immunostaining using fluorescein isothocyanate (FITC) Bax and Mito-Tracker CMXRos: FITC Bax was located primarily in the cytosol of 5.5 mmol/l glucose treated cells whereas treatment with 33.3 mmol/l glucose increased Bax translocation to mitochondria by 4.3 folds (Figure 7D). Bax interacted with voltage-dependent anion channels (VDAC) (Figure 7E) [80] and Bax oligomerization substantially increased 2 days in a high glucose medium (Figure 7F) [80]. Thus, chronic exposure to a high glucose increased through Bax oligomerization, cytochrome C release, and caspase-3 activation, leading to β-cell apoptosis [80]. Glucose toxicity on β-cell apoptosis was studied with chronic exposure to a high glucose, which markedly reduced GCK expression in a time- and dose-dependent manner but hexokinase expression was unaffected [81]. Immunocytochemistry on tissue culture cells showed that glucose reduces GCK expression dose dependently decreasing by 40% in a high glucose medium. Chronic exposure to high glucose for 4 days abolished to stimulate insulin content and inhibited ATP production [80]. Main studies of apoptotic Bcl proteins had been performed in artificially forced overexpression experiments in vitro [80-84]. Caspases are activated in a hierarchy order, in which initiator caspases (caspase-8 and -10) function to cleave effector caspases (caspase-3 and -7), the latter in turn degrade a number of intercellular protein substrates and lead to the classical morphological changes of apoptosis (Figures 1 and 2) [5,6]. Extracellular events present during the inflammatory response through the release of cytokines, including interferon (INF)-σ, Il-1β, and INF-¡ by infiltrating leukocytes or direct cytotoxic T-cell engagement, can initiate apoptosis [6]. These intrinsic cues function via surface molecule in the death receptor pathway, where specific ligand-receptor binding such as TNF-TNF receptor binding, Fas (CD 95)-Fas L (CD 178) binding lead to receptor clustering, adaptor molecule recruitment and formation of DISC (Figure 2) [6]. Caspase-8 associates with DISC complex, where it is activated, released, and leads to effector activation of caspase-3 [85,86]. Intracellular cues such as DNA damage, hypoxia, nutrient deprivation, or reactive oxygen species (ROS) function via the mitochondrial pathway, which is tightly modulated by the Bcl-2 proteins (Figure 2) [6]. In healthy cells, proapoptotic Bcl-2 proteins (Bim, Bid, Bad, Bax, and Bak) are present in the mature form, while antiapoptotic Bcl-2 proteins (Bcl-2 and Bcl-xL) are constitutively active and reside in the outer membrane of mitochondria (Figure 2) [6,86,87]. Following an intrinsic cue, proapoptotic Bcl proteins become activated and translocate to the mitochondria, where they bind to inactivate antiapoptotic Bcl-2 proteins or form pores in the mitochondrial membrane, which facilitates the release of cytochrome c into the cytosol (Figure 2) [6]. When cytochrome c accumulates in the cytosol, it complexes with procaspase-9 and Apaf-1 to form the “apoptosome”, which in turn activates caspase-3 (Figure 2) [6]. Both intrinsic and extrinsic signaling cascades converge at the point of caspase-3 activation, which is often considered as the “point of no return” in apoptosis [6]. Apoptosis can only occur when the concentration of pro-apoptotic Bcl proteins exceeds that of anti-apoptotic proteins at the mitochondrial membrane of the intrinsic pathway [6].
FIGURE 6.
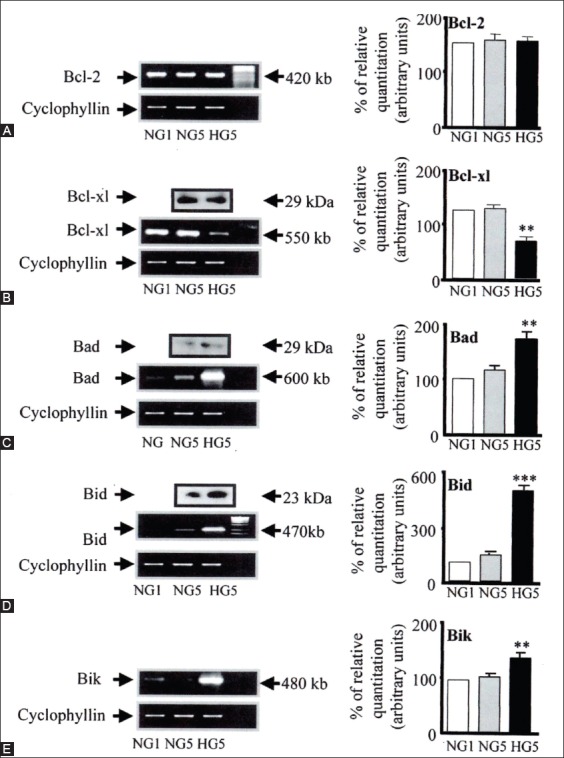
A-E: Bcl family gene regulation in human islets cultured in high versus normal glucose. Expression of Bcl-2, Bcl-xl, Bad, Bid, and Bik mRNA was detected by RT-PCR and quantified by FluorImager analysis of ethidium signal. In each experiment, band densities were normalized against cyclophyllin, and the result are expressed as mRNA level to NGI control islets (NGI = 100%). A: Bcl-2. Bcl-x (HG5 vs. NG5, **P <0.01). C: Bad (HG5 vs. NG5, **P <0.01). D: Bid (HG5 vs. NG5, ***P <0.01) E: Bik (HG5 vs. NG5, **P <0.01). One representative gel is also shown. Islets from six donors were analysed. Means ± SD of relative expression of the genes are shown in bar graph. Statistical analysis was performed by ANOVA
FIGURE 7.
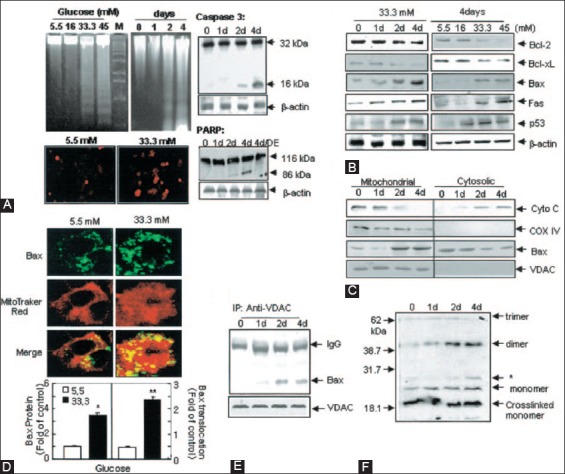
Effects of high glucose on apoptosis in MIN6NB cells. The MIN6N8 cells were treated with different glucose concentrations (5.5-45 mM) for the indicated times. (A) DNA fragmentation (upper left) and TUNEL assay (lower left). The cleavage of caspase-3 (upper right) and poly(ADP-ribose) polymerase (PARP) (lower left) was analyzed. (B) Expression on apoptotic proteins. (C) Release of cytochrome C and Bax translocation. The blots were reprobed with antibodies to cytochrome C oxidase and voltage-dependent anion channels (VDAC). (D) Bax immunocytochemistry. Fluorescent microscopic images for Bax (green), Mito Tracker CMXRos (red) and final merged images (localization of Bax at mitochondria) are shown (upper). Fold of cells exhibiting punctuate ABax and percentage of Bax colocalization with mitochondria was determined by counting ~20-100 cells for each condition (lower). Results represent the average ± SE from three independent experiments (*p < 0.05, **p < 0.01) > (E): Interaction of Bax with VDAC, (F) Bax oligomerization. From Kim et al., Diabetes 2005;54:2601-11.
Recent studies on β-cell apoptosis with knockout mouse models
Recent studies unveiled new roles to certain Bcl-2 family members in other physiological pathway, including glucose metabolism [82], Ca2+ homeostasis [87-89] and mitochondrial morphology [89]. BAD nucleates a core complex at the mitochondria containing GCK, the product of the gene associated with MODY2 [77,78]. BAD resides in a GCK-containing complex which regulates glucose-driven mitochondrial respiration [88]. Danial et al. studied new insight into the role of BAD in glucose-stimulated insulin secretion by β-cells from Bad−/− compared with Bad+/+ islets (Figure 8) [88]: Perifused islets from Bad−/− mice secreted significantly less insulin in response to 25 mM glucose compared to that of Bad+/+ mouse islets at about 40% for the first phase secretion (minutes 0-15) and at 60% for the second phase secretion (minutes 15-40) than those of Bad+/+ islets (Figure 8A and B) [88], however, the total insulin secretion by 30 mM KCl (Figure 8A) [88], glucose-induced (25 mM) changes of ATP/ADP ratio (Figure 8C) [85,88] and insulin secretion in response to 10 mM KIC and 25 mM tolbutamide were compatible in both Bad+/+ and Bad−/− islets (Figure 8D) [88]. GCK activity in homogenates of Bad−/− islets was about 25% that of Bad+/+ islets (Figure 8E) [88]. Insulin secretion by Bad−/− islets was considerably lower than that of Bad+/+ islets in response to 15-25 mM glucose concentration (Figure 8F) [88]. A signature of β-cell dysfunction associated with impaired GCK activity was a loss on glucose sensing (Figure 8E and F) [88] and Bad−/− islets required more glucose to secrete insulin than wild-type islets (Figure 8E and F) [88]. Glucose-induced changes in mitochondrial membrane potential were significantly reduced in Bad−/− β-cells [88]. The reduction was not reduced to a global impairment of mitochondrial respiratory chains, as both genotypes (Bad+/+ and Bad−/−) showed comparable changes in membrane potential to KIC [88]. Basal (Ca2+)i at 3 mM glucose was compatible for both genotypes, indicating that basic control mechanisms for Ca2+ handling were presented in Bad−/− cells [89]. Bad−/− islets did not present a stepwise increase in insulin secretion when exposed to incrementally increased glucose concentration (Figure 8F) [88]. The efficiency of glucose and other fuel secretagogues to stimulate insulin secretion correlated with their capacity to hyperpolarize the mitochondrial potential [88,89]. In β-cells, the characteristic features of glucose-driven mitochondrial respiration corresponded to that of glucose phosphorylation by GCK [89,90]. Glucose-induced changes in the mitochondrial membrane potential were significantly reduced in Bad−/− β-cells and the average (Ca2+)i response to 11 mM glucose was significantly lower in Bad−/−β-cells [86]. The BH3 domain of BAD is an amphipatic σ-helix, which binds to Bcl-2 and Bcl-xL and neutralizes the apoptotic activity of BAD [88]. An intact BH3 domain is required for glucose-stimulated insulin secretion by its binding to Bcl-2 and Bcl-xL. Treatment with BAD SAHBA (stabilized σ-helix of Bcl-2 domain) restored the secretion defect in Bad−/− islets [88], underscoring the sequence specificity of the BAD SAHB effect. Mutating the conserved leucine and aspartic acid residues of BAD BH3 sequence (BAD SAHBA(L,D-->A) abrogated its effect on insulin secretion [88]. By their extensive elegant study, Danial et al., identified GCK as a novel and direct physiologic target of the BAD BH3 domain in β-cells and that phosphorylation within the BH3 domain drives the metabolic functionality of BAD and serves as a physiologic switch its apoptotic and metabolic effects [86]. They demonstrated that Bad has a physiological role in β-cells, aside from its role in β-cell apoptosis and specifically that Bad phosphorylated at serine 155 promotes glucose-stimulated insulin secretion via interactions with GCK. The therapeutic application of BAD SAHBs and other BAD BH3 may be applied in restoring insulin secretion. A phosphorylated BAD SAHB that activates glucose-stimulated insulin secretion but does not affect the survival function of Bcl-xL, which may serve as a prototype therapeutic in diabetes and islet transplantation [88-90].
FIGURE 8.
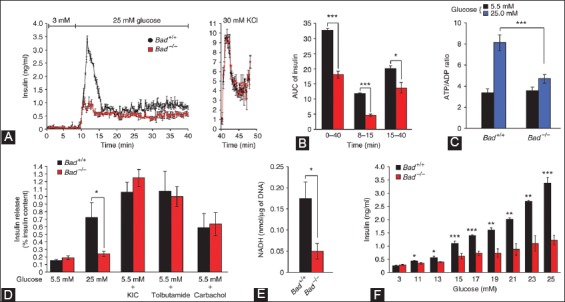
Characterization of the insulin secretion defect in Bad−/− islets. (A) Perifused islets from Bad−/− mice (red) with 25 mM glucose secreted significantly less insulin compared with that of Bad+/+ islets (black). (B) Insulin secretion throughout the perifusion (min 0-40), first phase (min 8-15) and second phase (min 15-40). (C) Glucose-induced changes in ATP/ADP ratio in Bad+/+ and Bad−/− islets - 5.5 mM (black), 25 mM (blue). (D) Insulin secretion in response to glucose 5.5 mM and 25 mM, 10 mM σ-ketoisocaproate (KIC), 0.25 mM tolbutamide and carbachol. (E) GCK activity in homogenates of primary islets isolated from Bad+/+ (black) and Bad−/− mice (red). (F) Insulin secretion by Bad+/+ (black) and Bad−/− (red) islets perifused with incrementally increasing concentration of glucose. From Daniel NN, et al. Nature 2003;424:952-6.
More recently, Luciani et al. have demonstrated that chemical and genetic loss-of-function of antiapoptotic Bcl-2 and Bcl-xL significantly augments glucose-dependent metabolic and Ca2+ signals by extending the role of Bcl-2 and Bcl-xL through suppressing glucose signaling in pancreatic β-cells [91]. Prolonged Bcl antagonism induced dose- and time-dependent cell death in human and mouse islet cells [92]. The enhancement of β-cell glucose response by Bcl-2 was studied using genetic ablated mice as genetic loss-of-function approach. Real-time PCR confirmed the loss of Bcl-2−/− β-cells and Bcl-2+/− islets compared to Bcl+/+ controls islets with no compensatory increase in Bcl-xL (Figure 9A) [92]. Bcl-2−/− and Bcl+/− β-cells showed enhanced sensibility to glucose (Figure 9B and C) [92]. Intact islets from Bcl-2−/− mice also showed increased Ca2+ and metabolic NAD (P)H response to glucose (Figure 9E and F) [92]. Perifused Bcl-2−/− islets to glucose revealed significantly increased insulin secretion compared with Bcl-2+/+ islets (Figure 9G) [92]. Loss of Bcl-2 had no effect on responses to depolarization with KCl (Figure 9D and H) [92]. The immediate augmentation of glucose-induced Ca2+ responses in Bcl-2 heterozygous β-cells indicates that effects were dependent on gene dosage (Figure 9B and C) [92]. Inducible deletion of Bcl-xL mouse β-cells also increased glucose-stimulated NAD(H)P and Ca2+ responses and resulted in an improved in vivo glucose tolerance in the Bcl-xL knockout mice [92]. These results suggest that prosurvival Bcl proteins normally dampen the β-cell response to glucose and physiology of β-cells [92]. The above two studies using knockout mice are somewhat conflicting in some places: Bad−/− islets secrete less insulin than Bad+/+ islets [88], whereas another study reported that Bad−/− and Bad+/− islets secreted more insulin than Bad+/+ islets [92] and this conflict should be resolved by future studies.
FIGURE 9.
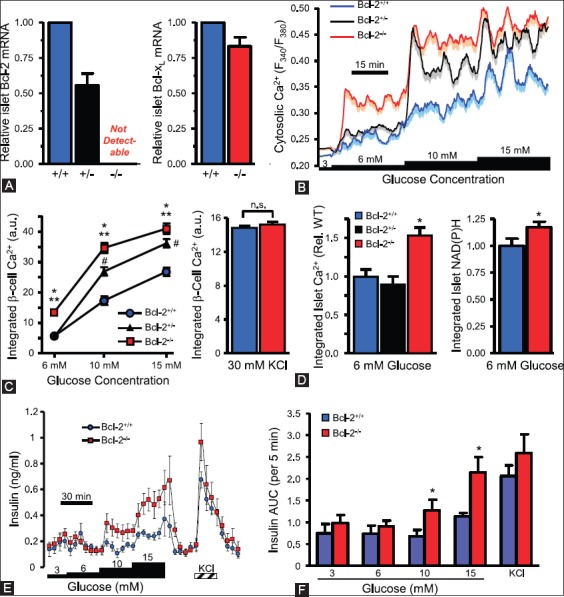
Loss of Bcl-2 enhances β-cell glucose responses. (A) Quantitative PCR quantification of Bcl-2 and Bcl-xL mRNA levels in islets from Bcl-2+/- and Bcl-2−/− islets compared to Bcl-2+/+ islets. (B) Average cytosolic Ca2+ levels of dispersed islet cells from Bcl-2+/+, Bcl-2+/−, and Bcl-2−/− islets. (C) Incremental area under the curve of Ca2+ responses by Bcl-2+/+, Bcl-2+/−, and Bcl-2−/− islets. (D) Integrated cytosolic Ca2+ responses of Bcl-2−/− and Bcl-2+/+ β-cells depolarized with 30 mM KCl, (E and F) integrated Ca2+ and NAD(P)H autofluorescence increases of intact islet cells, normalized Bcl-2+/+ control islet cells. (G) Insulin secretion profiles of perifused islets from Bcl-2+/+ and Bcl-2−/− islets. (H) Quantified area under the curve of insulin secretion profiles by Bcl-2+/+ and Bcl-2−/− islets. From Luciani DS, et al. Diabetes 2013;62:170-82.
Bcl proteins directly affect mitochondrial proteins in the β-cells. The study of antiapoptotic activates of Bcl-2 and Bcl-xL has shown that they interact with mitochondria via their BH4 domain [92,93]. A cell-permeable Bcl-xL BH4 domain peptide triggers cytosolic and mitochondrial Ca2+ fluctuations in β-cells, which may result from direct mitochondrial actions of the BH4 domain and ER Ca2+ release [93]. Bcl-xL can lower acetyl-CoA levels independently of Bax and Bak [93]. Antiapoptotic Bcl-2 family proteins can modulate β-cell function, and thus has implications for the pathophysiology of DM. The reduction in Bcl-2 and Bcl-xL under prediabetic conditions can affect β-cell function and insulin hypersecretion and is an early marker of human DM [94-97]. These results suggest that endogenous Bcl-2 and Bcl-xL suppress the β- cell response to glucose. The emerging evidence places Bcl family proteins at the intersection of β-cell function and survival. The involvement of apoptosis-regulating proteins provides fertile ground for future insights into the pathophysiology of diabetes and other diseases.
GCK is a novel and direct physiologic target of the Bad BH3 domain in β-cells and genetic evidence combined with the pharmacological activity of novel stapled Bad BH3 peptides indicates that phosphorylation within the BH3 domain drives the metabolic function of Bad and serves as a physiologic switch between its apoptotic and metabolic effects [93]. The molecular targeting of GCK activation holds therapeutic promise and leads to the development of several GCK activator compounds and further there is a therapeutic application of Bad, SAHBs, and other Bad BH3 mimics in restoring insulin secretion [92]. The recent reports on β-cell apoptosis have come from well-funded laboratories with many researchers of different disciplines to cooperate in this competitive area of research.
The current and ongoing research focuses on insulin secretion coupled with glucose metabolism using isolated islets from genetically ablated mice. These studies explore the antiapoptotic effects of Bcl-2 and Bcl-xL proteins versus apoptotic proteins, Bim, Bid, Bad, and Bik [87]. Saturated fatty acids including palmitate induce apoptosis and activation of the ER stress in β-cells [98]. Free fatty acids together with a high glucose induce β-cell apoptosis so called glucolipotoxicity [99]. β-cells synthesize and secrete large amounts of insulin, thus β-cells have highly developed ER which is susceptible to ER stress [98]. ER stress-induced apoptosis is controlled by the apoptotic proteins, Bim, Bid, Bad, and Bik and is protected by overexpressed Bcl-xL in β-cells [100].
Glucose induces insulin secretion through its glucose sensor and metabolism, and insulin secretion, in turn, modulates glucose metabolism through its myriad of glycolytic reactions in mitochondria, leading to ATP generation, closure of ATP-regulated K+ channels, plasma membrane depolarization, an opening of voltage-dependent Ca2+ channels, and an increase in free cytosolic Ca2+ concentration, resulting in insulin secretion [80,100,101]. Chronic hyperglycemia also induces myriad of biological reactions causing glucose toxicity, which in turn causes β-cell apoptosis [99-101]. How to prevent and revert β-cell apoptosis by regulating the balance of Bcl family and apoptotic genes against apoptosis may lead to prevention and therapeutic application for T2DM [92].
Improving medications for T2DM
In T2DM, visceral obesity is still the most important factor causing insulin resistance, and dieting and exercising are the initial step for controlling the diabetes. There are several kinds of oral medications for treating T2DM and their mechanisms to lower blood glucose levels are modulated through a myriad of complicated biological processes: (1) Metformin (Biaguanides): The first line of medication of choice, particularly in those overweight and obese individuals with normal kidney function. It also helps to lower blood glucose levels by making liver and muscle tissue more sensitive to insulin through increasing insulin receptor. Metformin decreases hyperglycemia by suppressing glucose production by the liver, where it rapidly increases insulin receptor activation in the human liver and signals preferentially through insulin-receptor substrate-2 and increased glucose uptake via increased GLUT-2 translocation [101-103]. The molecular mechanisms are inhibiting the mitochondrial respiratory chain, activation AMP-activated protein kinase, inhibition of glycogen-induced elevation of cAMP and consequent activation of protein kinase A and inhibition of mitochondrial glycerophosphate dehydrogenase [101-103]. (2) Sulfonylureas: Stimulate insulin secretion by binding to the sulfonylurea receptor (SUR) subunit of an ATP-sensitive K+ channel (KATP) on the β-cell membrane and inhibit a hyperpolarizing efflux of K+, thus causing the electric potential over the β-cell membrane to become more positive. This depolarization opens voltage-gate Ca++ channels. The rise in intracellular Ca++ leads to increase exocytosis of β-granules with the cell membrane, resulting in increased insulin secretion. The first generation of sulfonylureas are tolbutamide, tolazamide, and actohexamide and the second generation of sulfonylureas are glyburide, glipizide, gliclazide, glimepiride, and others. Tolbutamide and gliclazide block channels containing SUR1 (β-cell type) but not SUR2 (cardiac, smooth muscle types), whereas glibenclamide, glimepiride, repaglinide, and meglitinide block both types of the channels [104].
Combination medications of metformin and sulfonylureas are also available:
Metformin and glyburide (Glucovance)
Metformin and glipizide (Metaglip)
Metformin and rapaglinide (Prandi-Met)
Metformin XR and saxagliptin (Kombiglyze XR).
Recently, flood gate of new medications have been introduced and are now in the market: (3) Thiazolidinediones: Target insulin resistance through improving insulin action in muscle, adipose, and hepatic tissue by acting as agonists of peroxisome proliferator-activated receptor-¡ (PPAR-¡) nuclear receptors. Activation of PPAR-¡ results in a myriad of both metabolic and vascular effects by upregulating and downregulating expression of numerous genes, including genes known to regulate lipid and glucose metabolism and increase non-oxidative glucose disposal, increase triglyceride synthesis, and improve free fatty acid metabolism. These drugs also lower blood pressure, improve lipid metabolism by raising high-density lipoprotein (HDL) cholesterol levels and reducing triglyceride levels [105]. Rosiglitazone (Avandia) and pioglitazone (ACTOS) are approved by the Federal Drug Administration (FDA). (4) DPP (Dipeptidyl peptide)-4 inhibitor: Increases incretin, GLP-1 (glucagon-like peptide-1) levels as well as β-cell neogenesis, inhibits β-cell apoptosis, inhibits glucagon secretion, delays gastric emptying and induces satiety [106]. Sitagliptin (Januvia), saxagiptin (Onglyza) and linaglipin (Tradgenta) were approved by the FDA. (5) SGLT (sodium/glucose contrasporter)-2: Leads to reduction on blood glucose levels by enhancing glycemic control through glycosuria and lowered blood glucose levels, reducing body weight and lowering systemic and diastolic blood pressure [107]. Canagliflozin (Invokana) and dapaglifrozin (Farxiga) were approved by the FDA. More recently, combinations of metformin and new glycemic medications including thiazolidnediones and SGLT were approved by the FDA and were introduced into the market. The further studies on the molecular levels will help better understanding the mechanisms of new medications and combinations of new medications and the established ones, and eventually lead to better glycemic controls and prevention of T2DM in the near future.
DECLARATION OF INTERESTS
The author declares no conflict of interests.
REFERENCES
- 1.Kerr JF, Wyllie AH, Currie AR. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26(4):239–57. doi: 10.1038/bjc.1972.33. http://dx.doi.org/10.1038/bjc.1972.33 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Finegood DT, Scaglia L, Bonner-Weir S. Dynamics of beta-cell mass in the growing rat pancreas: Estimation with a simple mathematical model. Diabetes. 1995;44(3):249–56. doi: 10.2337/diab.44.3.249. http://dx.doi.org/10.2337/diab.44.3.249 . [DOI] [PubMed] [Google Scholar]
- 3.Scaglia L, Cahill CJ, Finegood DT, Bonner-Weir S. Apoptosis participates in the remodeling of the endocrine pancreas in the neonatal rat. Endocrinology. 1997;138(4):1736–41. doi: 10.1210/endo.138.4.5069. http://dx.doi.org/10.1210/endo.138.4.5069 . [DOI] [PubMed] [Google Scholar]
- 4.Mandrup-Poulsen T. beta-cell apoptosis: Stimuli and signaling. Diabetes. 2001;50(Suppl 1):S58–63. doi: 10.2337/diabetes.50.2007.s58. http://dx.doi.org/10.2337/diabetes.50.2007.S58 . [DOI] [PubMed] [Google Scholar]
- 5.Lee SC, Pervaiz S. Apoptosis in the pathophysiology of diabetes mellitus. Int J Biochem Cell Biol. 2007;39(3):497–504. doi: 10.1016/j.biocel.2006.09.007. http://dx.doi.org/10.1016/j.biocel.2006.09.007 . [DOI] [PubMed] [Google Scholar]
- 6.Emamaullee JA, Shapiro AM. Interventional strategies to prevent beta-cell apoptosis in islet transplantation. Diabetes. 2006;55(7):1907–14. doi: 10.2337/db05-1254. http://dx.doi.org/10.2337/db05-1254 . [DOI] [PubMed] [Google Scholar]
- 7.Nakano M, Matsumoto I, Sawada T, Ansite J, Oberbroeckling J, Zhang HJ, et al. Caspase-3 inhibitor prevents apoptosis of human islets immediately after isolation and improves islet graft function. Pancreas. 2004;29(2):104–9. doi: 10.1097/00006676-200408000-00004. http://dx.doi.org/10.1097/00006676-200408000-00004 . [DOI] [PubMed] [Google Scholar]
- 8.Brandhorst D, Kumarasamy V, Maatoui A, Alt A, Bretzel RG, Brandhorst H. Porcine islet graft function is affected by pretreatment with a caspase-3 inhibitor. Cell Transplant. 2006;15(4):311–7. doi: 10.3727/000000006783981936. http://dx.doi.org/10.3727/000000006783981936 . [DOI] [PubMed] [Google Scholar]
- 9.Cheng G, Zhu L, Mahato RI. Caspase-3 gene silencing for inhibiting apoptosis in insulinoma cells and human islets. Mol Pharm. 2008;5(6):1093–102. doi: 10.1021/mp800093f. http://dx.doi.org/10.1021/mp800093f . [DOI] [PubMed] [Google Scholar]
- 10.Nicholson DW, Thornberry NA. Caspases: Killer proteases. Trends Biochem Sci. 1997;22(8):299–306. doi: 10.1016/s0968-0004(97)01085-2. http://dx.doi.org/10.1016/S0968-0004(97)01085-2 . [DOI] [PubMed] [Google Scholar]
- 11.Chandra J, Zhivotovsky B, Zaitsev S, Juntti-Berggren L, Berggren PO, Orrenius S. Role of apoptosis in pancreatic beta-cell death in diabetes. Diabetes. 2001;50(Suppl 1):S44–7. doi: 10.2337/diabetes.50.2007.s44. http://dx.doi.org/10.2337/diabetes.50.2007aS44 . [DOI] [PubMed] [Google Scholar]
- 12.Peter ME, Krammer PH. Mechanisms of CD95 (APO-1/Fas)-mediated apoptosis. Curr Opin Immunol. 1998;10(5):545–51. doi: 10.1016/s0952-7915(98)80222-7. http://dx.doi.org/10.1016/S0952-7915(98)80222-7 . [DOI] [PubMed] [Google Scholar]
- 13.Sakahira H, Enari M, Nagata S. Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature. 1998;391(6662):96–9. doi: 10.1038/34214. http://dx.doi.org/10.1038/34214 . [DOI] [PubMed] [Google Scholar]
- 14.Gross A, McDonnell JM, Korsmeyer SJ. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 1999;13(15):1899–911. doi: 10.1101/gad.13.15.1899. http://dx.doi.org/10.1101/gad.13.15.1899 . [DOI] [PubMed] [Google Scholar]
- 15.Green DR. Apoptotic pathways: Ten minutes to dead. Cell. 2005;121(5):671–4. doi: 10.1016/j.cell.2005.05.019. http://dx.doi.org/10.1016/j.cell.2005.05.019 . [DOI] [PubMed] [Google Scholar]
- 16.Federici M, Hribal M, Perego L, Ranalli M, Caradonna Z, Perego C, et al. High glucose causes apoptosis in cultured human pancreatic islets of Langerhans: A potential role for regulation of specific Bcl family genes toward an apoptotic cell death program. Diabetes. 2001;50(6):1290–301. doi: 10.2337/diabetes.50.6.1290. http://dx.doi.org/10.2337/diabetes.50.6.1290 . [DOI] [PubMed] [Google Scholar]
- 17.Wajant H. The Fas signaling pathway: More than a paradigm. Science. 2002;296(5573):1635–6. doi: 10.1126/science.1071553. http://dx.doi.org/10.1126/science.1071553 . [DOI] [PubMed] [Google Scholar]
- 18.Suda T, Takahashi T, Golstein P, Nagata S. Molecular cloning and expression of the Fas ligand, a novel member of the tumor necrosis factor family. Cell. 1993;75(6):1169–78. doi: 10.1016/0092-8674(93)90326-l. http://dx.doi.org/10.1016/0092-8674(93)90326-L . [DOI] [PubMed] [Google Scholar]
- 19.Allison J, Thomas HE, Catterall T, Kay TW, Strasser A. Transgenic expression of dominant-negative Fas-associated death domain protein in beta cells protects against Fas ligand- induced apoptosis and reduces spontaneous diabetes in nonobese diabetic mice. J Immunol. 2005;175(1):293–301. doi: 10.4049/jimmunol.175.1.293. http://dx.doi.org/10.4049/jimmunol.175.1.293 . [DOI] [PubMed] [Google Scholar]
- 20.Haataja L, Gurlo T, Huang CJ, Butler PC. Islet amyloid in type 2 diabetes, and the toxic oligomer hypothesis. Endocr Rev. 2008;29(3):303–16. doi: 10.1210/er.2007-0037. http://dx.doi.org/10.1210/er.2007-0037 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes. 1988;37(12):1595–607. doi: 10.2337/diab.37.12.1595. http://dx.doi.org/10.2337/diab.37.12.1595 . [DOI] [PubMed] [Google Scholar]
- 22.Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52(1):102–10. doi: 10.2337/diabetes.52.1.102. http://dx.doi.org/10.2337/diabetes.52.1.102 . [DOI] [PubMed] [Google Scholar]
- 23.Bonner-Weir S. Islet growth and development in the adult. J Mol Endocrinol. 2000;24(3):297–302. doi: 10.1677/jme.0.0240297. http://dx.doi.org/10.1677/jme.0.0240297 . [DOI] [PubMed] [Google Scholar]
- 24.Marchetti P, Del Guerra S, Marselli L, Lupi R, Masini M, Pollera M, et al. Pancreatic islets from type 2 diabetic patients have functional defects and increased apoptosis that are ameliorated by metformin. J Clin Endocrinol Metab. 2004;89(11):5535–41. doi: 10.1210/jc.2004-0150. http://dx.doi.org/10.1210/jc.2004-0150 . [DOI] [PubMed] [Google Scholar]
- 25.Tomita T, Iwata K. Gelatinases and inhibitors of gelatinases in pancreatic islets and islet cell tumors. Modern Pathol. 1997;10(1):47–54. [PubMed] [Google Scholar]
- 26.Perez SE, Cano DA, Dao-Pick T, Rougier JP, Werb Z, Hebrok M. Matrix metalloproteinases 2 and 9 are dispensable for pancreatic islet formation and function in vivo. Diabetes. 2005;54(3):694–701. doi: 10.2337/diabetes.54.3.694. http://dx.doi.org/10.2337/diabetes.54.3.694 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hirata H, Takahashi A, Kobayashi S, Yonehara S, Sawai H, Okazaki T, et al. Caspases are activated in a branched protease cascade and control distinct downstream processes in Fas-induced apoptosis. J Exp Med. 1998;187(4):587–600. doi: 10.1084/jem.187.4.587. http://dx.doi.org/10.1084/jem.187.4.587 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tewari M, Quan LT, O’Rourke K, Desnoyers S, Zeng Z, Beidler DR, et al. Yama/CPP32 beta, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell. 1995;81(5):801–9. doi: 10.1016/0092-8674(95)90541-3. http://dx.doi.org/10.1016/0092-8674(95)90541-3 . [DOI] [PubMed] [Google Scholar]
- 29.Gown AM, Willingham MC. Improved detection of apoptotic cells in archival paraffin sections: Immunohistochemistry using antibodies to cleaved caspase 3. J Histochem Cytochem. 2002;50(4):449–54. doi: 10.1177/002215540205000401. http://dx.doi.org/10.1177/002215540205000401 . [DOI] [PubMed] [Google Scholar]
- 30.Martin SJ, Green DR. Protease activation during apoptosis: Death by a thousand cuts? Cell. 1995;82(3):349–52. doi: 10.1016/0092-8674(95)90422-0. http://dx.doi.org/10.1016/0092-8674(95)90422-0 . [DOI] [PubMed] [Google Scholar]
- 31.Tomita T. Immunocytochemical localisation of caspase-3 in pancreatic islets from type 2 diabetic subjects. Pathology. 2010;42(5):432–7. doi: 10.3109/00313025.2010.493863. http://dx.doi.org/10.3109/00313025.2010.493863 . [DOI] [PubMed] [Google Scholar]
- 32.Bonner-Weir S. Life and death of the pancreatic beta cells. Trends Endocrinol Metab. 2000;11(9):375–8. doi: 10.1016/s1043-2760(00)00305-2. http://dx.doi.org/10.1016/S1043-2760(00)00305-2 . [DOI] [PubMed] [Google Scholar]
- 33.Bonner-Weir S, O’Brien TD. Islets in type 2 diabetes: In honor of Dr. Robert C Turner. Diabetes. 2008;57(11):2899–904. doi: 10.2337/db07-1842. http://dx.doi.org/10.2337/db07-1842 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rhodes CJ. Type 2 diabetes-a matter of beta-cell life and death? Science. 2005;307(5708):380–4. doi: 10.1126/science.1104345. http://dx.doi.org/10.1126/science.1104345 . [DOI] [PubMed] [Google Scholar]
- 35.Tomita T. Immunocytochemical localization of cleaved caspase-3 in pancreatic islets from type 1 diabetic subjects. Islets. 2010;2(1):24–9. doi: 10.4161/isl.2.1.10041. http://dx.doi.org/10.4161/isl.2.1.10041 . [DOI] [PubMed] [Google Scholar]
- 36.Labat-Moleur F, Guillermet C, Lorimier P, Robert C, Lantuejoul S, Brambilla E, et al. TUNEL apoptotic cell detection in tissue sections: Critical evaluation and improvement. J Histochem Cytochem. 1998;46(3):327–34. doi: 10.1177/002215549804600306. http://dx.doi.org/10.1177/002215549804600306 . [DOI] [PubMed] [Google Scholar]
- 37.Duan WR, Garner DS, Williams SD, Funckes-Shippy CL, Spath IS, Blomme EA. Comparison of immunohistochemistry for activated caspase-3 and cleaved cytokeratin 18 with the TUNEL method for quantification of apoptosis in histological sections of PC-3 subcutaneous xenografts. J Pathol. 2003;199(2):221–8. doi: 10.1002/path.1289. http://dx.doi.org/10.1002/path.1289 . [DOI] [PubMed] [Google Scholar]
- 38.Tomita T. Cleaved caspase-3 immunocytochemical staining for pancreatic islets and pancreatic endocrine tumors: A potential marker for biological malignancy. Islets. 2010;2(2):82–8. doi: 10.4161/isl.2.2.10807. http://dx.doi.org/10.4161/isl.2.2.10807 . [DOI] [PubMed] [Google Scholar]
- 39.Ehrlich JC, Ratner IM. Amyloidosis of the islets of Langerhans. A restudy of islet hyalin in diabetic and non-diabetic individuals. Am J Pathol. 1961;38(1):49–59. [PMC free article] [PubMed] [Google Scholar]
- 40.Höppener JW, Ahrén B, Lips CJ. Islet amyloid and type 2 diabetes mellitus. N Engl J Med. 2000;343(9):411–9. doi: 10.1056/NEJM200008103430607. [DOI] [PubMed] [Google Scholar]
- 41.Hull RL, Westermark GT, Westermark P, Kahn SE. Islet amyloid: A critical entity in the pathogenesis of type 2 diabetes. J Clin Endocrinol Metab. 2004;89(8):3629–43. doi: 10.1210/jc.2004-0405. http://dx.doi.org/10.1210/jc.2004-0405 . [DOI] [PubMed] [Google Scholar]
- 42.Butler AE, Janson J, Soeller WC, Butler PC. Increased beta-cell apoptosis prevents adaptive increase in beta-cell mass in mouse model of type 2 diabetes: Evidence for role of islet amyloid formation rather than direct action of amyloid. Diabetes. 2003;52(9):2304–14. doi: 10.2337/diabetes.52.9.2304. http://dx.doi.org/10.2337/diabetes.52.9.2304 . [DOI] [PubMed] [Google Scholar]
- 43.Cooper GJ, Willis AC, Clark A, Turner RC, Sim RB, Reid KB. Purification and characterization of a peptide from amyloid-rich pancreases of type 2 diabetic patients. Proc Natl Acad Sci U S A. 1987;84(23):8628–32. doi: 10.1073/pnas.84.23.8628. http://dx.doi.org/10.1073/pnas.84.23.8628 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Westermark P, Wernstedt C, Wilander E, Sletten K. A novel peptide in the calcitonin gene related peptide family as an amyloid fibril protein in the endocrine pancreas. Biochem Biophys Res Commun. 1986;140(3):827–31. doi: 10.1016/0006-291x(86)90708-4. http://dx.doi.org/10.1016/0006-291X(86)90708-4 . [DOI] [PubMed] [Google Scholar]
- 45.Kahn SE, Andrikopoulos S, Verchere CB. Islet amyloid: A long-recognized but underappreciated pathological feature of type 2 diabetes. Diabetes. 1999;48(2):241–53. doi: 10.2337/diabetes.48.2.241. http://dx.doi.org/10.2337/diabetes.48.2.241 . [DOI] [PubMed] [Google Scholar]
- 46.Buse JB, Weyer C, Maggs DC. Amylin replacement with Pramlintide in type 1 and type 2 diabetes: A physiological approach to overcome a barrier with insulin therapy. Clin Diabetes. 2002;20(3):137–44. http://dx.doi.org/10.2337/diaclin.20.3.137 . [Google Scholar]
- 47.Fineman M, Weyer C, Maggs DG, Strobel S, Kolterman OG. The human amylin analog, pramlintide, reduces postprandial hyperglucagonemia in patients with type 2 diabetes mellitus. Horm Metab Res. 2002;34(9):504–8. doi: 10.1055/s-2002-34790. http://dx.doi.org/10.1055/s-2002-34790 . [DOI] [PubMed] [Google Scholar]
- 48.Kruger DF, Gatcomb PM, Owen SK. Clinical implications of amylin and amylin deficiency. Diabetes Educ. 1999;25(3):389–97. doi: 10.1177/014572179902500310. http://dx.doi.org/10.1177/014572179902500310 . [DOI] [PubMed] [Google Scholar]
- 49.Weyer C, Maggs DG, Young AA, Kolterman OG. Amylin replacement with pramlintide as an adjunct to insulin therapy in type 1 and type 2 diabetes mellitus: A physiological approach toward improved metabolic control. Curr Pharm Des. 2001;7(14):1353–73. doi: 10.2174/1381612013397357. http://dx.doi.org/10.2174/1381612013397357 . [DOI] [PubMed] [Google Scholar]
- 50.Tomita T. Islet amyloid polypeptide in pancreatic islets from type 2 diabetic subjects. Islets. 2012;4(3):223–32. doi: 10.4161/isl.20477. http://dx.doi.org/10.4161/isl.20477 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Clark A, Nilsson MR. Islet amyloid: A complication of islet dysfunction or an aetiological factor in Type 2 diabetes? Diabetologia. 2004;47(2):157–69. doi: 10.1007/s00125-003-1304-4. http://dx.doi.org/10.1007/s00125-003-1304-4 . [DOI] [PubMed] [Google Scholar]
- 52.Clark A, Wells CA, Buley ID, Cruickshank JK, Vanhegan RI, Matthews DR, et al. Islet amyloid, increased A-cells, reduced B-cells and exocrine fibrosis quantitative changes in the pancreas in type 2 diabetes. Diabetes Res. 1988;9(4):151–9. [PubMed] [Google Scholar]
- 53.Jurgens CA, Toukatly MN, Fligner CL, Udayasankar J, Subramanian SL, Zraika S, et al. ß-cell loss and ß-cell apoptosis in human type 2 diabetes are related to islet amyloid deposition. Am J Pathol. 2011;178(6):2632–40. doi: 10.1016/j.ajpath.2011.02.036. http://dx.doi.org/10.1016/j.ajpath.2011.02.036 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mirzabekov TA, Lin MC, Kagan BL. Pore formation by the cytotoxic islet amyloid peptide amylin. J Biol Chem. 1996;271(4):1988–92. doi: 10.1074/jbc.271.4.1988. http://dx.doi.org/10.1074/jbc.271.4.1988 . [DOI] [PubMed] [Google Scholar]
- 55.O’Brien TD, Glabe CG, Butler PC. Evidence for proteotoxicity in beta cells in type 2 diabetes: Toxic islet amyloid oligomers from intracellularly in the secretory pathway. Am J Pathol. 2010;176(2):861–9. doi: 10.2353/ajpath.2010.090532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Anguiano M, Nowak RJ, Lansbury PT., Jr Protofibrillar islet amyloid polypeptide permeabilizes synthetic vesicles by a pore-like mechanism that may be relevant to type II diabetes. Biochemistry. 2002;41(38):11338–43. doi: 10.1021/bi020314u. http://dx.doi.org/10.1021/bi020314u . [DOI] [PubMed] [Google Scholar]
- 57.Engel MF, Khemtémourian L, Kleijer CC, Meeldijk HJ, Jacobs J, Verkleij AJ, et al. Membrane damage by human islet amyloid polypeptide through fibril growth at the membrane. Proc Natl Acad Sci U S A. 2008;105(16):6033–8. doi: 10.1073/pnas.0708354105. http://dx.doi.org/10.1073/pnas.0708354105 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jansen A, van Hagen M, Drexhage HA. Defective maturation and function of antigen-presenting cells in type 1 diabetes. Lancet. 1995;345(8948):491–2. doi: 10.1016/s0140-6736(95)90586-3. http://dx.doi.org/10.1016/S0140-6736(95)90586-3 . [DOI] [PubMed] [Google Scholar]
- 59.Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, et al. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300(5618):486–9. doi: 10.1126/science.1079469. http://dx.doi.org/10.1126/science.1079469 . [DOI] [PubMed] [Google Scholar]
- 60.Ritzel RA, Butler PC. Replication increases beta-cell vulnerability to human islet amyloid polypeptide-induced apoptosis. Diabetes. 2003;52:1701–8. doi: 10.2337/diabetes.52.7.1701. http://dx.doi.org/10.2337/diabetes.52.7.1701 . [DOI] [PubMed] [Google Scholar]
- 61.Tomita T. Amylin in human adult pancreatic islets. Pathology. 2003;35(1):34–6. http://dx.doi.org/10.1097/01268031-200335010-00005 . http://dx.doi.org/10.1080/0031302021000062299 . [PubMed] [Google Scholar]
- 62.Tomita T, Lacy PE, Matschinsky FM, McDaniel ML. Effect of alloxan on insulin secretion in isolated rat islets perifused in vitro. Diabetes. 1974;23(6):517–24. doi: 10.2337/diab.23.6.517. http://dx.doi.org/10.2337/diab.23.6.517 . [DOI] [PubMed] [Google Scholar]
- 63.Tomita T, Scarpelli DG. Interaction of cyclic AMP and alloxan on insulin secretion in isolated rat islets perifused in vitro. Endocrinology. 1977;100(5):1327–33. doi: 10.1210/endo-100-5-1327. http://dx.doi.org/10.1210/endo-100-5-1327 . [DOI] [PubMed] [Google Scholar]
- 64.Cnop M, Welsh N, Jonas JC, Jörns A, Lenzen S, Eizirik DL. Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: Many differences, few similarities. Diabetes. 2005;54(Suppl 2):S97–107. doi: 10.2337/diabetes.54.suppl_2.s97. http://dx.doi.org/10.2337/diabetes.54suppl_2.S97 . [DOI] [PubMed] [Google Scholar]
- 65.Donath MY, Ehses JA, Maedler K, Schumann DM, Ellingsgaard H, Eppler E, et al. Mechanisms of beta-cell death in type 2 diabetes. Diabetes. 2005;54(Suppl 2):S108–13. doi: 10.2337/diabetes.54.suppl_2.s108. http://dx.doi.org/10.2337/diabetes.54.suppl_2.S108 . [DOI] [PubMed] [Google Scholar]
- 66.Bedoya FJ, Matschinsky FM, Shimizu T, O’Neil JJ, Appel MC. Differential regulation of glucokinase activity in pancreatic islets and liver of the rat. J Biol Chem. 1986;261(23):10760–4. [PubMed] [Google Scholar]
- 67.Matschinsky FM. Glucokinase as glucose sensor and metabolic signal generator in pancreatic beta-cells and hepatocytes. Diabetes. 1990;39(6):647–52. doi: 10.2337/diab.39.6.647. http://dx.doi.org/10.2337/diab.39.6.647 . http://dx.doi.org/10.2337/diabetes.39.6.647 . [DOI] [PubMed] [Google Scholar]
- 68.Matschinsky F, Liang Y, Kesavan P, Wang L, Froguel P, Velho G, et al. Glucokinase as pancreatic beta cell glucose sensor and diabetes gene. J Clin Invest. 1993;92(5):2092–8. doi: 10.1172/JCI116809. http://dx.doi.org/10.1172/JCI116809 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shimizu T, Knowles BB, Matschinsky FM. Control of glucose phosphorylation and glucose usage in clonal insulinoma cells. Diabetes. 1988;37(5):563–8. doi: 10.2337/diab.37.5.563. http://dx.doi.org/10.2337/diab.37.5.563 . [DOI] [PubMed] [Google Scholar]
- 70.MacDonald PE, Joseph JW, Rorsman P. Glucose-sensing mechanisms in pancreatic beta-cells. Philos Trans R Soc Lond B Biol Sci. 2005;360(1464):2211–25. doi: 10.1098/rstb.2005.1762. http://dx.doi.org/10.1098/rstb.2005.1762 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.De Vos A, Heimberg H, Quartier E, Huypens P, Bouwens L, Pipeleers D, et al. Human and rat beta cells differ in glucose transporter but not in glucokinase gene expression. J Clin Invest. 1995;96(5):2489–95. doi: 10.1172/JCI118308. http://dx.doi.org/10.1172/JCI118308 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Iynedjian PB. Mammalian glucokinase and its gene. Biochem J. 1993;293:1–13. doi: 10.1042/bj2930001. http://dx.doi.org/10.1042/bj2930001 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Newgard CB, McGarry JD. Metabolic coupling factors in pancreatic beta-cell signal transduction. Annu Rev Biochem. 1995;64:689–719. doi: 10.1146/annurev.bi.64.070195.003353. http://dx.doi.org/10.1146/annurev.bi.64.070195.003353 . [DOI] [PubMed] [Google Scholar]
- 74.Fridlyand L, Philson LH. Glucose sensing in the pancreatic beta cells: A computational systems analysis. Theor Biol Med Model. 2010;7(15):1–44. doi: 10.1186/1742-4682-7-15. http://dx.doi.org/10.1186/1742-4682-7-15 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Froguel P, Vaxillaire M, Sun F, Velho G, Zouali H, Butel MO, et al. Close linkage of glucokinase locus on chromosome 7p to early-onset non-insulin-dependent diabetes mellitus. Nature. 1992;356(6365):162–4. doi: 10.1038/356162a0. http://dx.doi.org/10.1038/356162a0 . [DOI] [PubMed] [Google Scholar]
- 76.Porter JR, Barrett TG. Monogenic syndromes of abnormal glucose homeostasis: Clinical review and relevance to the understanding of the pathology of insulin resistance and beta cell failure. J Med Genet. 2005;42(12):893–902. doi: 10.1136/jmg.2005.030791. http://dx.doi.org/10.1136/jmg.2005.030791 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tattersall R. Maturity-onset diabetes of the young: A clinical history. Diabet Med. 1998;15(1):11–4. doi: 10.1002/(SICI)1096-9136(199801)15:1<11::AID-DIA561>3.0.CO;2-0. http://dx.doi.org/10.1002/(SICI)1096-9136(199801)15:1<11: AID-DIA561>3.0.CO;2-0 . [DOI] [PubMed] [Google Scholar]
- 78.What is Maturity-Onset Diabetes of the Young (MODY)? [Last retrieved on 2008 Jul 29];National Diabetes Information Clearinghouse (NDIC) (National Institute of Diabetes and Digestive and Kidney Diseases) (NIH) [Google Scholar]
- 79.Leahy JL, Bonner-Weir S, Weir GC. Beta-cell dysfunction induced by chronic hyperglycemia. Current ideas on mechanism of impaired glucose-induced insulin secretion. Diabetes Care. 1992;15(3):442–55. doi: 10.2337/diacare.15.3.442. http://dx.doi.org/10.2337/diacare.15.3.442 . [DOI] [PubMed] [Google Scholar]
- 80.Kim WH, Lee JW, Suh YH, Hong SH, Choi JS, Lim JH, et al. Exposure to chronic high glucose induces beta-cell apoptosis through decreased interaction of glucokinase with mitochondria: Downregulation of glucokinase in pancreatic beta-cells. Diabetes. 2005;54(9):2602–11. doi: 10.2337/diabetes.54.9.2602. http://dx.doi.org/10.2337/diabetes.54.9.2602 . [DOI] [PubMed] [Google Scholar]
- 81.Chan SL, Yu VC. Proteins of the bcl-2 family in apoptosis signalling: From mechanistic insights to therapeutic opportunities. Clin Exp Pharmacol Physiol. 2004;31(3):119–28. doi: 10.1111/j.1440-1681.2004.03975.x. http://dx.doi.org/10.1111/j.1440-1681.2004.03975.x . [DOI] [PubMed] [Google Scholar]
- 82.Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407(6805):770–6. doi: 10.1038/35037710. http://dx.doi.org/10.1038/35037710 . [DOI] [PubMed] [Google Scholar]
- 83.Ou D, Wang X, Metzger DL, James RF, Pozzilli P, Plesner A, et al. Synergetic inhibition of tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in human pancreatic beta cells by Bcl-2 and X-linked inhibitor of apoptosis. Hum Immunol. 2005;66(3):274–84. doi: 10.1016/j.humimm.2004.12.002. http://dx.doi.org/10.1016/j.humimm.2004.12.002 . [DOI] [PubMed] [Google Scholar]
- 84.Saldeen J. Cytokines induce both necrosis and apoptosis via a common Bcl-2-inhibitable pathway in rat insulin-producing cells. Endocrinology. 2000;141(6):2003–10. doi: 10.1210/endo.141.6.7523. http://dx.doi.org/10.1210/endo.141.6.7523 . [DOI] [PubMed] [Google Scholar]
- 85.Boatright KM, Salvesen GS. Mechanisms of caspase activation. Curr Opin Cell Biol. 2003;15(6):725–31. doi: 10.1016/j.ceb.2003.10.009. http://dx.doi.org/10.1016/j.ceb.2003.10.009 . [DOI] [PubMed] [Google Scholar]
- 86.Hui H, Dotta F, Di Mario U, Perfetti R. Role of caspases in the regulation of apoptotic pancreatic islet beta-cells death. J Cell Physiol. 2004;200(2):177–200. doi: 10.1002/jcp.20021. http://dx.doi.org/10.1002/jcp.20021 . [DOI] [PubMed] [Google Scholar]
- 87.Danial NN, Gramm CF, Scorrano L, Zhang CY, Krauss S, Ranger AM, et al. BAD and glucokinase reside in a mitochondrial complex that integrates glycolysis and apoptosis. Nature. 2003;424(6951):952–6. doi: 10.1038/nature01825. http://dx.doi.org/10.1038/nature01825 . [DOI] [PubMed] [Google Scholar]
- 88.Danial NN, Walensky LD, Zhang CY, Choi CS, Fisher JK, Molina AJ, et al. Dual role of proapoptotic BAD in insulin secretion and beta cell survival. Nat Med. 2008;14(2):144–53. doi: 10.1038/nm1717. http://dx.doi.org/10.1038/nm1717 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Antinozzi PA, Ishihara H, Newgard CB, Wollheim CB. Mitochondrial metabolism sets the maximal limit of fuel-stimulated insulin secretion in a model pancreatic beta cell: A survey of four fuel secretagogues. J Biol Chem. 2002;277(14):11746–55. doi: 10.1074/jbc.M108462200. http://dx.doi.org/10.1074/jbc.M108462200 . [DOI] [PubMed] [Google Scholar]
- 90.Berggren PO, Larsson O. Ca2+ and pancreatic B-cell function. Biochem Soc Trans. 1994;22(1):12–8. doi: 10.1042/bst0220012. http://dx.doi.org/10.1042/bst0220012 . [DOI] [PubMed] [Google Scholar]
- 91.Liang Y, Bai N, Doliba C, Wang L, Barner DK, Matschinsky FM. Glucose metabolism and insulin release in mouse HC9 cells, as model of wild-type pancreatic beta cells. Am J Physiol. 1996;270:E846–57. doi: 10.1152/ajpendo.1996.270.5.E846. [DOI] [PubMed] [Google Scholar]
- 92.Luciani DS, White SA, Widenmaier SB, Saran VV, Taghizadeh F, Hu X, et al. Bcl-2 and Bcl-xL suppress glucose signaling in pancreatic ß-cells. Diabetes. 2013;62(1):170–82. doi: 10.2337/db11-1464. http://dx.doi.org/10.2337/db11-1464 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Real PJ, Cao Y, Wang R, Nikolovska-Coleska Z, Sanz-Ortiz J, Wang S, et al. Breast cancer cells can evade apoptosis-mediated selective killing by a novel small molecule inhibitor of Bcl-2. Cancer Res. 2004;64(21):7947–53. doi: 10.1158/0008-5472.CAN-04-0945. http://dx.doi.org/10.1158/0008-5472.CAN-04-0945 . [DOI] [PubMed] [Google Scholar]
- 94.Yi CH, Pan H, Seebacher J, Jang IH, Hyberts SG, Heffron GJ, et al. Metabolic regulation of protein N-alpha-acetylation by Bcl-xL promotes cell survival. Cell. 2011;146(4):607–20. doi: 10.1016/j.cell.2011.06.050. http://dx.doi.org/10.1016/j.cell.2011.06.050 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rong YP, Bultynck G, Aromolaran AS, Zhong F, Parys JB, De Smedt H, et al. The BH4 domain of Bcl-2 inhibits ER calcium release and apoptosis by binding the regulatory and coupling domain of the IP3 receptor. Proc Natl Acad Sci U S A. 2009;106(34):14397–402. doi: 10.1073/pnas.0907555106. http://dx.doi.org/10.1073/pnas.0907555106 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Simonson DC. Hyperinsulinemia and its sequelae. Horm Metab Res Suppl. 1990;22:17–25. [PubMed] [Google Scholar]
- 97.Eizirik DL, Cardozo AK, Cnop M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocr Rev. 2008;29(1):42–61. doi: 10.1210/er.2007-0015. http://dx.doi.org/10.1210/er.2007-0015 . [DOI] [PubMed] [Google Scholar]
- 98.Thomas HE, McKenzie MD, Angstetra E, Campbell PD, Kay TW. Beta cell apoptosis in diabetes. Apoptosis. 2009;14(12):1389–404. doi: 10.1007/s10495-009-0339-5. http://dx.doi.org/10.1007/s10495-009-0339-5 . [DOI] [PubMed] [Google Scholar]
- 99.Zhou YP, Pena JC, Roe MW, Mittal A, Levesetti M, Baldwin AC, et al. Overexpression of Bcl-xL in beta cells prevents cell death but impairs mitochondrial signal for insulin secretion. Am J Physiol Endocrinol Metab. 2000;278(2):E340–51. doi: 10.1152/ajpendo.2000.278.2.E340. [DOI] [PubMed] [Google Scholar]
- 100.Lowell BB, Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science. 2005;307(5708):384–7. doi: 10.1126/science.1104343. http://dx.doi.org/10.1126/science.1104343 . [DOI] [PubMed] [Google Scholar]
- 101.Dunn CJ, Peters DH. Metformin. A review of its pharmacological properties and therapeutic use in non-insulin-dependent diabetes mellitus. Drugs. 1995;49(5):721–49. doi: 10.2165/00003495-199549050-00007. [DOI] [PubMed] [Google Scholar]
- 102.Hundal RS, Inzucchi SE. Metformin: New understandings, new uses. Drugs. 2003;63(18):1879–94. doi: 10.2165/00003495-200363180-00001. http://dx.doi.org/10.2165/00003495-200363180-00001 . [DOI] [PubMed] [Google Scholar]
- 103.Gunton JE, Delhanty PJ, Takahashi SI, Baxter RC. Metformin rapidly increases insulin receptor activation in human liver and signals preferentially through insulin-receptor substrate-2 and increase glucose uptake via increased GLUT-2 translocation. J Clin Endocrinol Metab. 2003;88(3):323–32. doi: 10.1210/jc.2002-021394. http://dx.doi.org/10.1210/jc.2002-021394 . [DOI] [PubMed] [Google Scholar]
- 104.Proks P, Reimann F, Green N, Gribble F, Ashcroft F. Sulfonylurea stimulation of insulin secretion. Diabetes. 2002;51(Suppl 3):S368–76. doi: 10.2337/diabetes.51.2007.s368. http://dx.doi.org/10.2337/diabetes.51.2007.S368 . [DOI] [PubMed] [Google Scholar]
- 105.Kendall DM. Thiazolidinediones: The case for early use. Diabetes Care. 2006;29(1):154–7. doi: 10.2337/diacare.29.1.154. http://dx.doi.org/10.2337/diacare.29.01.06.dc05-0711 . [DOI] [PubMed] [Google Scholar]
- 106.Ahrén B, Schmitz O. GLP-1 receptor agonists and DPP-4 inhibitors in the treatment of type 2 diabetes. Horm Metab Res. 2004;36(11-12):867–76. doi: 10.1055/s-2004-826178. http://dx.doi.org/10.1055/s-2004-826178 . [DOI] [PubMed] [Google Scholar]
- 107.Abdul-Ghani MA, Norton L, DeFrenzo RA. Role of sodium-glucose cotransporter 2 (SGLT-2) inhibitors in the treatment of type 2 diabetes. Endocr Rev. 2011;32(4):515–31. doi: 10.1210/er.2010-0029. http://dx.doi.org/10.1210/er.2010-0029 . [DOI] [PubMed] [Google Scholar]