

Abstract
Background and Purpose
α‐ and β‐melanocyte‐stimulating hormones (MSH) are derived from pro‐opiomelanocortin (POMC) and are the natural agonist ligands of the melanocortin 4 receptor, a key regulator of energy homeostasis. Recent rodent and human data have implicated the MAGEL2 gene, which may regulate activation of POMC neurons, as a significant contributor to the metabolic symptoms observed in Prader–Willi Syndrome (PWS). Firstly, patients with protein truncating mutations in MAGEL2 exhibit numerous clinical characteristics of PWS. Secondly, Magel2‐null mice may not normally activate MC4 receptors, as they are defective in the activation of their POMC neurons and hence may fail to normally release the POMC‐derived MC4 receptor agonist ligands α‐ and β‐MSH. Magel2‐null mice represent a tractable animal model for the metabolic and appetitive imbalance seen in patients with PWS.
Experimental Approach
We tested a dose titration of the MC4 receptor agonist setmelanotide, in development for rare monogenic forms of obesity, in Magel2‐null mice.
Key Results
We show that Magel2‐null mice are hypersensitive to the appetite suppressing and metabolic effects of setmelanotide.
Conclusion and Implications
Setmelanotide may be a useful investigational hormone/neuropeptide replacement therapy for PWS and rare monogenic forms of obesity exhibiting impaired function of POMC neurons.
Abbreviations
- VCO2
carbon dioxide production
- DIO
diet‐induced obesity
- PWS
Prader–Willi Syndrome
- POMC
pro‐opiomelanocortin
- Magel2
melanoma antigen family L2
- Magel2
Magel2‐null
- VO2
oxygen consumption
- RER
respiratory exchange ratio
Tables of Links
| TARGETS |
|---|
| GPCRs |
| MC1 receptor |
| MC2 receptor |
| MC3 receptor |
| MC4 receptor |
| MC5 receptor |
| LIGANDS |
|---|
| Leptin |
| Melanotan II, MT‐II |
| Setmelanotide |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Introduction
Aberrations in the leptin–melanocortin pathway are involved in distinct genetic causes of obesity (Yeo and Heisler, 2012). Rhythm's compound setmelanotide (RM‐493) is a melanocortin MC4 receptor agonist in development for treatment of rare monogenic forms of obesity and Prader–Willi syndrome (PWS) (ClinicalTrials.gov: NCT02041195 and NCT02311673). Administration of setmelanotide to diet‐induced obese (DIO) Rhesus monkeys over an 8 week period, resulted in significant (13.5%) weight loss, increased energy expenditure (14%) and reduced caloric intake (Kievit et al., 2013). In a randomized, double‐blind, placebo‐controlled crossover study, short‐term administration of setmelanotide in obese subjects increased resting energy expenditure and increased the utilization of fat as fuel (Chen et al., 2015). Setmelanotide is devoid of the adverse cardiovascular adverse effects observed with other MC4 receptor agonists as determined in monkeys and in human phase I/II trials (Kievit et al., 2013; Gottesdiener et al., 2015). Five‐day administration of setmelanotide in DIO mice increased resting energy expenditure and increased the utilization of fat as fuel (Kumar et al., 2009; Clemmensen et al., 2015). DIO rodents that are insensitive to the anorexigenic effects of leptin may be sensitive, or even hypersensitive to MC4 receptor agonists (Hansen et al., 2001; Scarpace et al., 2003; Li et al., 2004). Evidence from a mouse model carrying a deletion of the Magel2 (melanoma antigen family L2) gene, one of the genes inactivated in PWS (Lee et al., 2000; Schaaf et al., 2013), suggests that hypothalamic deficiencies in the leptin–melanocortin pathway contribute to obesity in PWS (Bischof et al., 2007; Mercer et al., 2013; Pravdivyi et al., 2015). We previously demonstrated that mice lacking Magel2 are insensitive to the anorexigenic effects of peripherally administered leptin because of an intrinsic defect in pro‐opiomelanocortin (POMC) neurons, but respond robustly to the synthetic melanocortin receptor agonist melanotan II with a reduction in food intake. In this study, we tested a dose titration of setmelanotide on metabolic outcomes in Magel2‐null mice and show that Magel2‐null mice are hypersensitive to metabolic control by setmelanotide when compared with wild‐type (WT) mice. We conclude that setmelanotide warrants evaluation as a possible hormone replacement therapy in PWS or obesity caused by POMC deficiency.
Methods
Animals
All animal care and experimental procedures complied with the Canadian Council on Animal Care Guidelines and Policies and were approved by the Animal Care and Use Committee: Health Sciences for the University of Alberta. All efforts were made to minimize animal suffering. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath & Lilley, 2015). We chose this model as PWS is a disorder of brain development and function, making it impractical to study pharmacologically outside the live animal. To date, mice are the only species in which there are gene‐targeted strains lacking the Magel2 gene, which is one of several candidate genes for PWS (Kozlov et al., 2007; Resnick et al., 2013).
Experimental procedures
C57BL/6‐Magel2tm1Stw/J mice (The Jackson Laboratory stock #009062) were weaned between 3 and 4 weeks of age, then group‐housed by sex (2–3 per cage) and fed standard rodent chow ad libitum (PicoLab Laboratory Rodent Diet 5L0D). Mice were genotyped by PCR of ear notch biopsies at weaning (Bischof et al., 2007). At 7–9 weeks of age, an experimentally naive cohort of six healthy male mice of each genotype (Magel2‐null ‘Magel2’ and wild‐type ‘WT’) were singly housed in individual ventilated cages in conventional animal housing and fed the same chow in ground form and weighed several times during this first experimental week to ensure that they had adapted to the ground chow (mean weight ± SD 25.2 ± 2.1 g, no difference between genotypes). At 8–10 weeks of age, the mice were transferred into Oxymax metabolic cages (Columbus Instruments) that recorded food intake, activity (laser beam breaks), oxygen consumption (VO2) and carbon dioxide production (VCO2) every 14 min. VO2 and VCO2 readings were unstable for 2–3 intervals after the cages were opened for treatment, so these values were not used for the respiratory exchange ratio (RER) and energy expenditure analyses. VO2 and VCO2 used to calculate heat (CV × VO2, where CV = 3.815 × 1.232 × RER) and RER (VCO2/ VO2). Percent fat utilization was derived from RER using the equation (Jequier et al., 1987) % fat utilization = 1 − (RER − 0.707)/(1 − 0.707). Mice were singly housed and had access to powdered standard chow and water ad libitum and were maintained on a 12 h light/dark cycle (on at 6:00h/off at 18:00h), at 18–22°C.
Setmelanotide treatment
After 30 h acclimatization to the metabolic chambers, mice were injected i.p. with 120 μL SSD (0.9% saline, 2% heat‐inactivated normal mouse serum, 0.5% DMSO ‘vehicle’) between 16:30h and 17:00h. After another 24 h, mice were injected i.p. with setmelanotide (Rhythm Pharmaceuticals, Boston, MA, USA) in 120 μL SSD. Metabolic cage measurements before the SSD injection were used for baseline data, and measurements after setmelanotide were compared with measurements after vehicle for effect of treatment analyses. Four groups of mice were tested with descending doses of setmelanotide (1, 0.2, 0.1 and 0.04 mg·kg−1).
Data and statistical analysis
The data and statistical analysis in this study comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Data from the post‐compound treatment were compared with data from the post‐vehicle injection in the same group of mice. Group differences using two factors (genotype and lighting phase or genotype and treatment) were evaluated by two‐way ANOVA, followed by Bonferroni post testing to determine whether there was an effect of treatment or lighting phase within each genotype when the main effects of factors were positive by ANOVA using an F‐statistic and a significance value of P < 0.05. For food intake, an additional analysis was performed that compared food intake after compound with food intake after vehicle, for each mouse. The sample size was set using significance level α = 0.05, power 1‐β = 0.8, two‐tailed distribution, an attrition rate of 15% and an expected difference between outcomes 2*(STDEV) based on preliminary data. For most groups, n = 6 mice were included for the analyses except for the WT group treated with 0.2 mg·kg−1 setmelanotide (n = 5 analysed out of 6 experimental animals), the Magel2 group treated with 0.1 mg·kg−1 setmelanotide (n = 5 analysed out of 6 experimental animals) and both the WT and Magel2 groups treated with 0.04 mg·kg−1 setmelanotide (both n = 5 analysed out of 6 experimental animals). These four animals were excluded because excessive food was found at the bottom of their cages at the end of the experiment, making their food intake measurements unreliable. Experimenters were blinded with respect to the genotypes of the mice but not with respect to treatment.
Materials
Setmelanotide (l‐cysteinamide, N2‐acetyl‐l‐arginyl‐l‐cysteinyl‐d‐alanyl‐l‐histidyl‐d‐phenylalanyl‐l‐arginyl‐l‐tryptophyl‐, cyclic (2‐ > 8)‐disulfide, UNII: N7T15V1FUY) is a synthetic 8 amino‐acid, cyclic peptide MC4 receptor agonist with a molecular formula of C49H68N18O9S2 (anhydrous free base) and molecular weight of 1117.3 Da (anhydrous free base); this compound was supplied by Rhythm Pharmaceuticals (Boston MA). Setmelanotide binds with high affinity to the human MC4 receptor (inhibitory constant = 2.1 nmol·L−1) and is efficient in activating MC4 receptors (EC50 = 0.27 nmol·L−1). Activation of other melanocortin receptors (MC1, MC3 and MC5) requires concentrations that are more than 20‐fold or higher than for MC4 receptors. Setmelanotide displayed no activity for the MC2 receptor, also known as the adrenocorticotropic hormone receptor.
Results
Baseline measurements
Mice [Magel2‐null (referred to as Magel2) and WT littermate controls, 8–10 weeks old] were placed in metabolic cages and allowed to acclimatize for 6 h before the start of baseline measurements. There were no significant differences in initial body weight between genotypes. As expected, food intake, activity, energy expenditure and RER were lower in the light (inactive) period as detected by two‐factor ANOVA (Figure 1A–C). Magel2 mice consumed 17% less food in the dark phase than WT mice (Figure 1A). Horizontal activity did not differ between genotypes (Figure 1B). However, Magel2 mice made only 58% (dark) and 44% (light) of the number of vertical movements compared with WT mice (Figure 1C). VO2 and VCO2 were used to calculate energy expenditure (kcal·h−1). Total energy expenditure was reduced equally in both genotypes in the light phase (Figure 1D). The RER (VCO2/VO2) provides an indirect measurement of the relative contribution of carbohydrate versus fat to whole‐body energy expenditure and is lower in the light phase reflecting increased usage of fat for fuel (Figure 1E). RER was lower in Magel2 mice compared with WT mice during both light and dark phases (Figure 1E), indicating that a larger percentage of the total energy expenditure of Magel2 mice is being produced by fat oxidation (Figure 1F).
Figure 1.
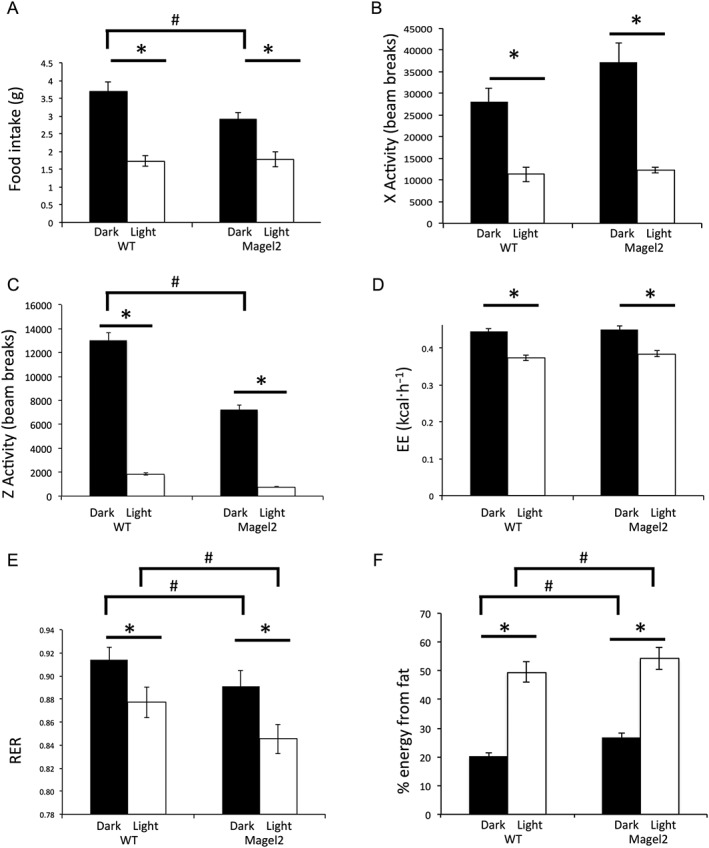
Baseline measurements in metabolic cages during the dark (active) and light phases. (A) 12 h food intake, (B) X‐activity, (C) Z‐activity, (D) energy expenditure (EE), (E) RER and (F) % fat utilization, derived from RER. The two‐way ANOVA showed a significant effect of lighting on food intake [F(1,18) = 302, P < 0.0001], X‐activity [F(1,18) = 257, P < 0.001], Z‐activity [F(1,18) = 102, P < 0.0001], energy expenditure [F(1,18) = 408, P < 0.001] and RER [F(1,18) = 40, P < 0.0001]. The two‐factor ANOVA also showed a significant effect of genotype on food intake [F(1,18) = 17, P < 0.001], Z‐activity [F(1,18) = 54, P < 0.01] and RER [F(1,18) = 20, P < 0.0005]. Bars represent mean ± SEM; *P < 0.05, significant difference between light and dark phases; # P < 0.05, significant difference between genotypes; two‐way ANOVA with Bonferroni post hoc test.
Effect of the MC4 receptor agonist setmelanotide
Experimental design
A single crossover design was used. Mice were tested at 8–10 weeks in order to minimize the possible confounding factor of excess fat accumulation that occurs in adult Magel2 mice (Mercer et al., 2009). After acclimatization to the metabolic cages, mice were injected i.p. with vehicle between 16:30h and 17:00h; 24 h later, mice were injected i.p. with setmelanotide (0.04, 0.1, 0.2 or 1 mg·kg−1). Measurements taken over a 22 h period after setmelanotide administration, and were compared with measurements taken over a 22 h period after vehicle administration.
Cumulative food intake
Cumulative food intake after setmelanotide treatment was compared with food intake over the same time interval on the previous day after vehicle. The results are represented as a ratio of cumulative food intake within each mouse or as cumulative food intake over a 3 h time frame (Figure 2E). The 0.1 mg·kg−1 setmelanotide dose showed the greatest difference between genotypes showing a profound effect on food intake over 3 h in Magel2 mice, while having no effect on WT mice (Figure 2A and E). Although all four doses of setmelanotide reduced 3 h food intake in Magel2 mice, only the setmelanotide treatment with the two higher dosages (1 and 0.2 mg·kg−1) effectively reduced 3 h food intake in WT mice (illustrated in Figure 3).
Figure 2.
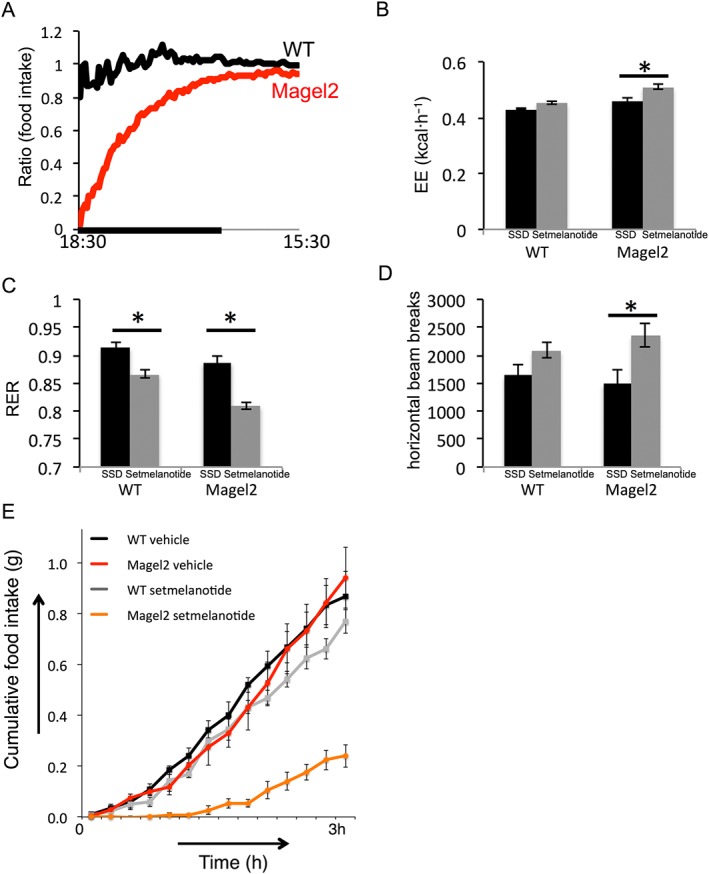
Effects on cumulative food intake suppression, energy expenditure, activity and RER following setmelanotide versus vehicle in Magel2 and WT mice. Vehicle or setmelanotide (0.1 mg·kg−1) was administered i.p. to mice within 1 h before the start of the dark cycle. (A) Ratio of cumulative food intake after setmelanotide to cumulative food intake after vehicle, plotted versus time of day. A value of ‘1’ indicates no difference between cumulative food intake after setmelanotide and after vehicle. The black bar indicates the dark phase. (B) Energy expenditure over 3 h post‐injection, (C) RER over 3 h post‐injection and (D) horizontal activity over 1 h post‐injection, after treatment with 0.1 mg·kg−1 setmelanotide compared with treatment with vehicle (SSD). Bars represent mean ± SEM; * P < 0.05, significant difference between setmelanotide and vehicle; two‐way ANOVA with Bonferroni post hoc test. (E) 3 h cumulative food intake at 0.1 mg·kg−1 in Magel2 and WT mice (mean ± SEM). The two‐factor ANOVA showed a significant effect of genotype on energy expenditure [F(1,18) = 5.9, P < 0.05], RER [F(1,18) = 12, P < 0.005], 1 h horizontal activity [F(1,18) = 7.2, P < 0.05] and 3 h food intake [F(1,18) = 7.5, P < 0.02].
Figure 3.
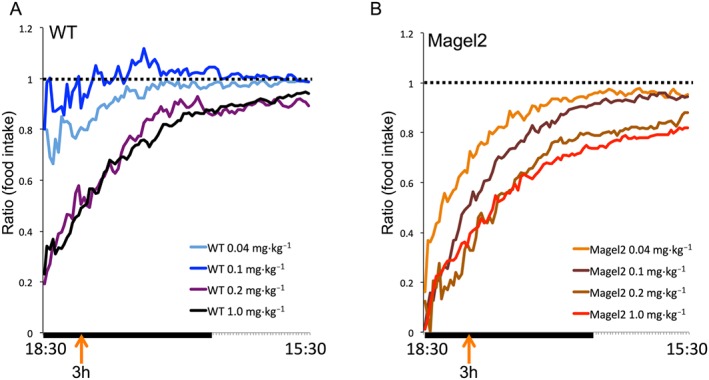
Graphical representation of the effects on cumulative food intake suppression following setmelanotide (0.04–1 mg·kg−1) or vehicle in Magel2 and WT mice. Ratio of cumulative food intake after setmelanotide to cumulative food intake after vehicle, plotted against the time of day for each of four doses, within each genotype. Ratios were calculated for each mouse and then averaged within genotypes. A value of ‘1’ (dotted line) indicates equal cumulative food intake after treatment with compound, compared with values after vehicle treatment.
Energy expenditure
Average energy expenditure over 3 h after setmelanotide (i.e. 17:30h–20:30h) was compared with energy expenditure after vehicle. MC4 receptor activation with setmelanotide over 5 days had been shown to increase energy expenditure in DIO mice (Clemmensen et al., 2015). In our experiments, setmelanotide increased energy expenditure in WT and Magel2 mice at 1 and 0.2 mg·kg−1. Setmelanotide also increased energy expenditure in Magel2 mice at the lower dose of 0.1 mg·kg−1 (Figure 2B), where an effect on WT mice was not observed.
RER
Average RER over 3 h after setmelanotide was compared with RER after vehicle (Figure 2C). MC4 receptor activation with setmelanotide (RM‐493) was known to decrease RER in DIO mice (Clemmensen et al., 2015). In the present work, setmelanotide treatment had a significant effect on RER at all four doses and lowered RER compared with vehicle for both genotypes.
Activity
Total activity (horizontal beam breaks) over 1 h after setmelanotide (i.e. 17:30h–18:30h) was compared with activity after vehicle. Setmelanotide treatment increased 1 h activity compared with vehicle treatment in mice of both genotypes at 1 and 0.2 mg·kg−1. Setmelanotide treatment also increased 1 h activity compared with vehicle treatment in Magel2 mice at a lower dose (0.1 mg·kg−1; Figure 2D).
Discussion
Setmelanotide is a novel MC4 receptor agonist in development as hormone/neuropeptide replacement therapy in the treatment of obesity in rare genetic severe obesity disorders involving the MC4 receptor pathway, such as POMC deficiency. Melanocortin receptor agonists have also been shown to be effective in animals that are resistant to the anorexigenic effects of leptin, secondary to diet‐induced obesity. Mice lacking Magel2 are likewise leptin‐resistant because of an intrinsic defect in POMC neurons but retain sensitivity to the melanocortin receptor agonist melanotan II (Mercer et al., 2013). We have now demonstrated that setmelanotide is effective in Magel2‐null mice and in WT mice and indeed is more effective at lower doses in the mutant mice compared with mice with an intact Magel2 gene. Magel2 mice respond to setmelanotide at doses below those that evoke a response in WT mice, when compared with saline injection. Setmelanotide reduced food intake, increased energy expenditure and increased activity in both genotypes at two doses: 1 and 0.2 mg·kg−1. Mice lacking Magel2 responded at lower dosages of setmelanotide (0.1 and 0.04 mg·kg−1), which demonstrates that Magel2 mice are hypersensitive to the effects of setmelanotide. At 0.1 mg·kg−1, setmelanotide treatment reduced 3 h food intake by 75%, reduced 22 h food intake by 7%, increased energy expenditure by 11% and decreased RER by 9% compared with treatment with vehicle, in Magel2 mice, while WT mice failed to respond at this dose of setmelanotide. Although we analysed only male mice in this study, we would expect to find similar results with female mice as no notable sex‐related differential responses to setmelanotide have been observed in rodents, primates or humans to date.
We noted a lower basal RER in Magel2 mice. However, Magel2 mice have intact metabolic flexibility as they maintain higher RER in active compared with inactive phases. Low RER is also seen in underfeeding, muscle wasting and mitochondrial myopathies where the capacity of muscle to use glucose as fuel is impaired (Lecker et al., 2004). It is possible that an atrophic condition could contribute to the reduced RER observed in Magel2 mice, consistent with the reduced lean mass observed in adult Magel2 mice (Mercer et al., 2009). Our previous studies had detected decreased running wheel activity at 8 months of age (Kozlov et al., 2007) and lower food intake in the home cage in Magel2 mice. We did not detect any difference between genotypes in either horizontal activity or energy expenditure, possibly because our mice were young and not yet obese.
Based on these data and earlier findings, an order of sensitivity to setmelanotide treatment can be predicted where defects upstream of the MC4 receptor that lower agonist tone predispose to melanocortin agonist hypersensitivity. These defects are predicted to include leptin and leptin receptor deficiency (setmelanotide has shown efficacy in the Zucker diabetic fatty rats and db/db mice, which lack a functional leptin receptor; Rhythm Pharmaceuticals, unpublished data), POMC deficiency (POMC‐null mice had been shown to be hypersensitive to melanocortin agonists; Yaswen et al., 1999) and now Magel2 gene defects in POMC neurons, which based on the data presented here predict hypersensitivity to setmelanotide‐mediated MC4 receptor activation. As expected in this pathway, the heterozygous Mc4r+/− mice (which have a lower level of MC4 receptors) appear more resistant than WT mice to setmelanotide treatment, and the deletion of MC4 receptors in Mc4r−/− mice abolished responses to setmelanotide (Rhythm Pharmaceuticals, unpublished).
In both obese human subjects and non‐human primates, short‐term administration of setmelanotide increased resting energy expenditure and increased the utilization of fat as fuel (Kievit et al., 2013; Chen et al., 2015). Short‐term (5 day) administration of setmelanotide in DIO mice also increased resting energy expenditure and increased the utilization of fat as fuel (Kumar et al., 2009; Clemmensen et al., 2015). The hypersensitivity to setmelanotide treatment identified in Magel2 mice predicts increased sensitivity to setmelanotide in patients suffering from rare monogenic forms of obesity that exhibit POMC deficiency, potentially including PWS. In these disorders, a lack of endogenous agonist tone at the MC4 receptor by the natural agonist α‐MSH may allow setmelanotide to restore MC4 receptor agonist tone, thus exerting metabolic and behavioural appetite control. Obesity caused by PWS may have several causes, including compulsive food‐seeking behaviour and low resting energy expenditure. In addition, increased fat mass is accompanied by a commensurate increase in circulating leptin, suggesting a leptin‐resistant state. As the MAGEL2 gene is only one of the genes that is inactivated in PWS, further studies are needed to determine whether setmelanotide is an effective treatment for PWS. In conclusion, setmelanotide may be an effective treatment for metabolic and behavioural dysfunction in genetic forms of obesity, including PWS.
Author contributions
J.M.B. performed experiments and reviewed the manuscript. L.H.T.V.d.P. collaborated on experimental design, interpretation of results and writing of the manuscript and participated in the editing of the report and in the decision to submit the article for publication. W.F.C. collaborated on experimental design, interpretation of results and writing of the manuscript. R.W. collaborated on experimental design and interpretation of results and wrote the manuscript.
Conflict of interest
L.H.T.V.d.P. is an employee of Rhythm Pharmaceuticals.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
We acknowledge the expert assistance of Amy Barr and the Cardiovascular Research Centre and Raven Kirschenman for animal care. This work was supported by Rhythm Pharmaceuticals.
Bischof, J. M. , Van Der Ploeg, L. H. T. , Colmers, W. F. , and Wevrick, R. (2016) Magel2‐null mice are hyper‐responsive to setmelanotide, a melanocortin 4 receptor agonist. British Journal of Pharmacology, 173: 2614–2621. doi: 10.1111/bph.13540.
References
- Alexander SP, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015). The concise guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischof JM, Stewart CL, Wevrick R (2007). Inactivation of the mouse Magel2 gene results in growth abnormalities similar to Prader–Willi syndrome. Hum Mol Genet 16: 2713–2719. [DOI] [PubMed] [Google Scholar]
- Chen KY, Muniyappa R, Abel BS, Mullins KP, Staker P, Brychta RJ et al. (2015). RM‐493, a melanocortin‐4 receptor (MC4R) agonist, increases resting energy expenditure in obese individuals. J Clin Endocrinol Metab 100: 1639–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemmensen C, Finan B, Fischer K, Tom RZ, Legutko B, Sehrer L et al. (2015). Dual melanocortin‐4 receptor and GLP‐1 receptor agonism amplifies metabolic benefits in diet‐induced obese mice. EMBO Mol Med 7: 288–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesdiener K, Conners H, Van der Ploeg L, Fiedorek F, Hylan M, Louis W et al. (2015). T‐P‐3134: analysis of the synthetic peptide setmelanotide (RM‐493), a melanocortin‐4 receptor (MC4R) agonist, on cardiovascular parameters in three phase1b/2a studies. Oral abstract presentation at: The Obesity Society Annual Meeting at ObesityWeekSM 2014; November 2-7, 2014; Boston, MA. www.obesityweek.com.
- Hansen MJ, Ball MJ, Morris MJ (2001). Enhanced inhibitory feeding response to alpha‐melanocyte stimulating hormone in the diet‐induced obese rat. Brain Res 892: 130–137. [DOI] [PubMed] [Google Scholar]
- Jequier E, Acheson K, Schutz Y (1987). Assessment of energy expenditure and fuel utilization in man. Annu Rev Nutr 7: 187–208. [DOI] [PubMed] [Google Scholar]
- Kievit P, Halem H, Marks DL, Dong JZ, Glavas MM, Sinnayah P et al. (2013). Chronic treatment with a melanocortin‐4 receptor agonist causes weight loss, reduces insulin resistance, and improves cardiovascular function in diet‐induced obese rhesus macaques. Diabetes 62: 490–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG (2010). Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. J Pharm Pharmacol 1: 94–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlov SV, Bogenpohl JW, Howell MP, Wevrick R, Panda S, Hogenesch JB et al. (2007). The imprinted gene Magel2 regulates normal circadian output. Nat Genet 39: 1266–1272. [DOI] [PubMed] [Google Scholar]
- Kumar KG, Sutton GM, Dong JZ, Roubert P, Plas P, Halem HA et al. (2009). Analysis of the therapeutic functions of novel melanocortin receptor agonists in MC3R‐ and MC4R‐deficient C57BL/6J mice. Peptides 30: 1892–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecker SH, Jagoe RT, Gilbert A, Gomes M, Baracos V, Bailey J et al. (2004). Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J 18: 39–51. [DOI] [PubMed] [Google Scholar]
- Lee S, Kozlov S, Hernandez L, Chamberlain SJ, Brannan CI, Stewart CL et al. (2000). Expression and imprinting of MAGEL2 suggest a role in Prader–Willi syndrome and the homologous murine imprinting phenotype. Hum Mol Genet 9: 1813–1819. [DOI] [PubMed] [Google Scholar]
- Li G, Zhang Y, Wilsey JT, Scarpace PJ (2004). Unabated anorexic and enhanced thermogenic responses to melanotan II in diet‐induced obese rats despite reduced melanocortin 3 and 4 receptor expression. J Endocrinol 182: 123–132. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer RE, Kwolek EM, Bischof JM, van Eede M, Henkelman RM, Wevrick R (2009). Regionally reduced brain volume, altered serotonin neurochemistry, and abnormal behavior in mice null for the circadian rhythm output gene Magel2. Am J Med Genet B Part B, Neuropsychiatr Genet 150B: 1085–1099. [DOI] [PubMed] [Google Scholar]
- Mercer RE, Michaelson SD, Chee MJ, Atallah TA, Wevrick R, Colmers WF (2013). Magel2 is required for leptin‐mediated depolarization of POMC neurons in the hypothalamic arcuate nucleus in mice. PLoS Genet 9: e1003207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pravdivyi I, Ballanyi K, Colmers WF, Wevrick R (2015). Progressive postnatal decline in leptin sensitivity of arcuate hypothalamic neurons in the Magel2‐null mouse model of Prader–Willi syndrome. Hum Mol Genet 24: 4276–4283. [DOI] [PubMed] [Google Scholar]
- Resnick JL, Nicholls RD, Wevrick R, Prader‐Willi Syndrome Animal Models Working G (2013). Recommendations for the investigation of animal models of Prader–Willi syndrome. Mamm Genome 24: 165–178. [DOI] [PubMed] [Google Scholar]
- Scarpace PJ, Matheny M, Zolotukhin S, Tumer N, Zhang Y (2003). Leptin‐induced leptin resistant rats exhibit enhanced responses to the melanocortin agonist MT II. Neuropharmacology 45: 211–219. [DOI] [PubMed] [Google Scholar]
- Schaaf CP, Gonzalez‐Garay ML, Xia F, Potocki L, Gripp KW, Zhang B et al. (2013). Truncating mutations of MAGEL2 cause Prader–Willi phenotypes and autism. Nat Genet 45: 1405–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaswen L, Diehl N, Brennan MB, Hochgeschwender U (1999). Obesity in the mouse model of pro‐opiomelanocortin deficiency responds to peripheral melanocortin. Nat Med 5: 1066–1070. [DOI] [PubMed] [Google Scholar]
- Yeo GS, Heisler LK (2012). Unraveling the brain regulation of appetite: lessons from genetics. Nat Neurosci 15: 1343–1349. [DOI] [PubMed] [Google Scholar]