

Abstract
Hypertension is a leading risk factor for the development and progression of diabetic retinopathy, and contributes to a variety of other retinal diseases in the absence of diabetes. Inhibition of the renin-angiotensin system has been shown to provide beneficial effects against diabetic retinopathy, both in the absence and presence of hypertension, suggesting that angiotensin II and the AT1 receptor may contribute to retinal vascular dysfunction. We investigated the effects of the AT1 receptor antagonist candesartan on retinal vascular permeability (RVP) in normotensive rats with streptozotocin-induced diabetes and in rats with angiotensin II-induced hypertension. We show that candesartan-treatment decreased diabetes- and angiotensin II-stimulated RVP by 58% (P<0.05) and 79% (P<0.05) respectively, compared with untreated controls, suggesting that activation of the AT1 receptor contributes to blood-retinal barrier dysfunction. We found that plasma kallikrein levels are increased in the retina of rats with angiotensin II-stimulated hypertension and that intravitreal injection of either plasma kallikrein or bradykinin is sufficient to increase RVP. We show that a novel small molecule inhibitor of plasma kallikrein, 1-benzyl-1H-pyrazole-4-carboxylic acid 4-carbamimidoyl-benzylamide (ASP-440), delivered systemically via a subcutaneous pump, decreases angiotensin II-stimulated RVP by 70% (P<0.05) and ameliorates angiotensin II-induced hypertension, measured from the carotid artery by telemetry, but did not reduce angiotensin II-induced retinal leukostasis. These findings demonstrate that activation of the AT1 receptor increases RVP and suggest that systemic plasma kallikrein inhibition may provide a new therapeutic approach for ameliorating blood-retinal barrier dysfunction induced by hypertension.
Keywords: Kallikrein, retina, angiotensin II, diabetes, AT1-receptor, hypertension
INTRODUCTION
Diabetic macular edema (DME), which can occur at any stage of diabetic retinopathy, is the leading cause of visual impairment associated with both Type I and Type II diabetes1. The development of DME is thought to be initiated by impaired retinal endothelial cell tight junction integrity and breakdown of the blood-retinal-barrier, leading to increased retinal vascular permeability (RVP) and the accumulation of plasma proteins, lipids, and fluid in the neuroretina2. Increased RVP is among the earliest retinal changes induced by diabetes, and further increases in RVP occur in concordance with the severity of diabetic retinopathy3. Risk factors for DME include hyperglycemia, dyslipidaemia, renal dysfunction, and hypertension4. Patients with hypertension are more likely to progress to DME5, and increasing diastolic blood pressure (BP) is associated with an increase in its incidence6. Moreover, it has been shown that tight BP control reduced the incidence of macular edema by 42% in people with Type II diabetes7. Although the management of clinical risk factors can reduce the incidence of DME, effective treatments for this condition remain a major unmet clinical need.
The UK Prospective Diabetes Study demonstrated that BP reduction using either a beta-blocker or angiotensin-converting enzyme (ACE) inhibitor in patients with both hypertension and diabetes can reduce the occurrence of advanced diabetic retinopathy7. While this study and others suggest that high BP exerts adverse effects on the retina, the molecular mechanisms that mediate these effects on retinal vascular function remain poorly understood. A growing body of evidence suggests that renin-angiotensin system (RAS) inhibition may provide beneficial effects on the retina even in the absence of hypertension. The EUCLID study group has shown that treatment of normotensive Type I diabetics with an ACE inhibitor reduced the progression of diabetic retinopathy, however lower BP and HbA1c were also observed in the treated group8. The Appropriate Blood Pressure Control in Diabetes trial examined the effects of intensive versus standard BP control in patients with Type II diabetes, and found that, even in normotensive patients, intensive BP control decreased the progression of retinopathy9. Furthermore, the DIabetic REtinopathy Candesartan Trials (DIRECT) study reported that the angiotensin AT1 receptor (AT1R) antagonist candesartan reduced the incidence of diabetic retinopathy in people with Type I diabetes without hypertension. However, this study did not observe a beneficial effect on retinopathy progression10. Thus, further understanding of AT1R-mediated actions on the retina could have relevance to diabetic retinopathy both in the presence or absence of hypertension.
We have investigated the effects of candesartan on RVP in normotensive rats with diabetes and in rats with angiotensin II (AngII)-induced hypertension. In this report we also examine the role of the kallikrein-kinin system (KKS) in mediating AngII’s effect on RVP using a novel and highly selective small molecule inhibitor of plasma kallikrein. These studies have revealed a role for plasma kallikrein in mediating AngII-induced RVP and suggest a new approach to treat retinal vascular dysfunction in hypertension.
METHODS
Animals
Diabetes was induced in 8-week old male Sprague-Dawley rats via intraperitoneal injection of 55 mg/kg of streptozotocin (Sigma-Aldrich, St Louis, MO) in 10 mM sodium citrate (pH 4.5), after overnight fast. Following confirmation of hyperglycemia in streptozotocin-injected animals or immediately following the subcutaneous implantation of AngII-loaded osmotic pumps, candesartan-cilexetil in pure powder form (Astrazeneca) was administered ab libitum in drinking water at a concentration of 10 μg/ml. Based upon water consumption, this was equivalent to a dosage of 1.1, 2.0, and 4.0 mg/kg/day for saline-treated rats, rats infused with AngII and diabetic rats respectively. Unless specified as being measured by telemetry, all BP measurements were obtained by tail cuff plethysmography using a non-invasive BP/heart rate monitoring system (UR-5000, Ueda Electronic, Tokyo, Japan) in conscious animals. Blood pressure measurements by telemetry were performed using PA-C40 transmitters (Data Sciences International, St. Paul, MN). Under anesthesia, a telemetric transmitter was fixed to the interscapular area and the pressure sensing catheter was inserted via the external carotid into the common carotid with the tip approximately 3 mm distal to the aortic junction. Rats were housed individually on a receiver pad and systolic and diastolic pressures were monitored continuously and averaged over 15-second intervals every 15 minutes over a 4 hr period from 9:00 AM to 1:00 PM each day. Baseline readings (Day 0) were obtained at 48 hours after catheter implantation. Before RVP measurements, all animals underwent catheterization with a polyvinyl catheter inserted into the left jugular vein as described previously11. All experiments were performed in accordance with the guidelines of the National Institutes of Health Guide for the Care and Use of Laboratory animals and with approval from the Animal Care and Use Committee of the Joslin Diabetes Center.
Angiotensin II, ASP-440 and HOE-140 treatment
Treatments were achieved by the use of subcutaneous implantation of Alzet mini-osmotic pumps (DURECT corporation, Cupertino CA). AngII (EMD Chemicals Inc, La Jolla, CA) was delivered at 300 ng/kg/min and control rats received saline vehicle. ASP-440 was delivered at 16 μg/kg/hr, with control pumps filled with vehicle (10% polyethylene glycol, 90% PBS). HOE-140 (Sigma-Aldrich, St Louis, MO) was infused at 1 μg/kg/hr, with control pumps filled with saline.
Retinal Vascular Permeability (RVP)
Video fluorescein angiography (VFA) was performed using a scanning laser ophthalmoscope (Rodenstock Instruments) as described previously11. Retinal angiograms and first phase RVP were visualized by VFA immediately following an 80 μl bolus injection of fluorescein in anaesthetized animals via left jugular vein catheter. RVP was quantified using Vitreous fluorescein photometry (VFP) as detailed previously12. RVP was examined in rats at two-weeks of diabetes with or without candesartan treatment, at 6-days post saline or AngII infusion with or without candesartan treatment, at three days post-treatment with HOE-140 or ASP-440, or 40 minutes following intravitreal injections of either plasma kallikrein (EMD Chemicals Inc, La Jolla, CA), or bradykinin 1–9 (Sigma-Aldrich, St Louis, MO), with control eyes receiving a 10 μl balanced salt solution (BSS).
Statistical analysis
Statistical analysis was performed using a one-way analysis of variance or paired Student’s t-test (SigmaStat, Systat Software). Values of p<0.05 were considered statistically significant.
Details on the synthesis, purification and characteristics of ASP-440, measurement of retinal leukostasis, and western blot protocol are available in an online, please see http://hyper.aha.journals.org.
RESULTS
Increased retinal vascular permeability in diabetes is attenuated by AT1R blockade
We characterized RVP in rats with two-weeks of diabetes in the absence or presence of treatment with candesartan. RVP to fluorescein, measured by VFP, was increased 97% from 4.6 arbitrary units (au) in nondiabetic (NDM) rats to 9.1 au in diabetic (DM) rats (P<0.001) (Figure 1A). We show that RVP in candesartan-treated NDM and DM rats were 5.76 au and 6.46 au, respectively. This study revealed that candesartan reduced RVP in DM rats by 58% (relative to NDM) and 79% (relative to candesartan-treated NDM), both with P<0.05. Candesartan treatment also reduced systolic BP (SBP) measured by tail cuff plethysmography in both NDM and DM groups at 2 weeks post treatment (P<0.05, Table 1), and both SBP and diastolic BP (DBP) in DM rats measured from the carotid artery using telemetry (P<0.05, Figure 1B). Similar decreases in SBP (110.9±6.8 to 96.5±10.2mmHg) and DBP (91.4±7.6 to 67.2±9.9mmHg, mean±SEM) measured by telemetry were observed in NDM rats pre- and post-treatment with candesartan for 3 days.
Figure 1. Effect of AT1-receptor blockade on RVP and blood pressure in diabetic rats.
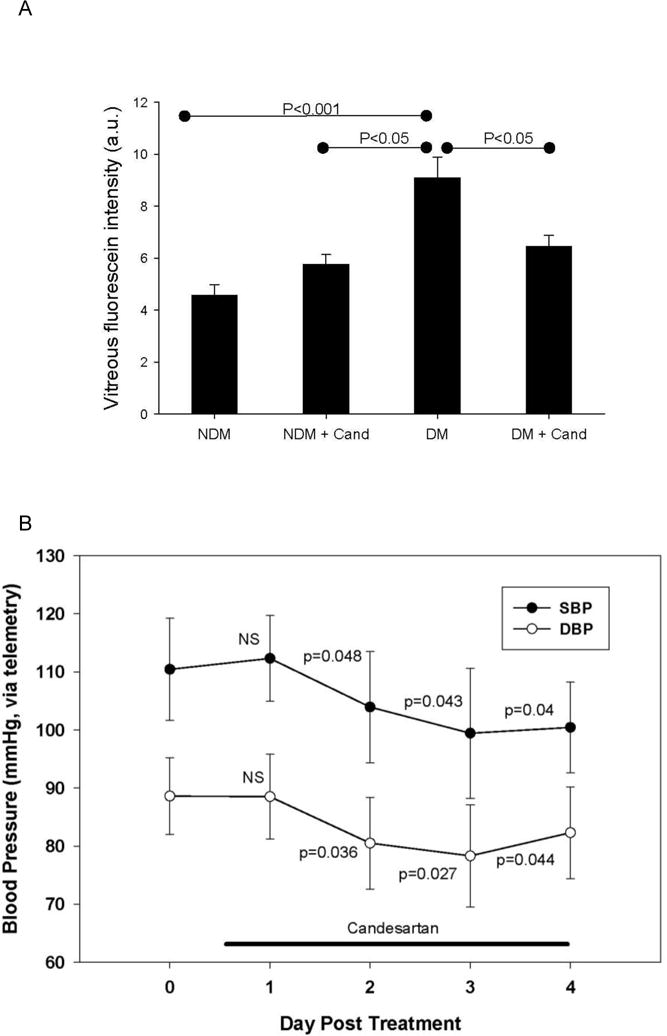
(A) NDM (non-diabetic, n=8), NDM+Cand (non-diabetic + candesartan, n=5), DM (2-week diabetes, n=12), DM+Cand (2-week diabetes + candesartan, n=11). Bars represent mean ±SEM, a.u. (arbitrary units). (B) Systolic (SBP) and diastolic (DBP) blood pressure measurements (mean ± SEM) using telemetry in diabetic rats before and after treatment with candesartan. P values indicate comparisons with Day 0.
Table 1.
Systolic BP (SBP) measured by tail cuff plethysmography, weight, and blood glucose (BG) data for non-diabetic (NDM, n=8), non-diabetic + candesartan (NDM+Cand, n=5), diabetic (DM, n=12), and diabetic + candesartan (DM+Cand, n=11) animals. Values represent mean ± SEM.
| Characteristic | NDM | NDM + Cand | DM | DM + Cand |
|---|---|---|---|---|
| SBP (mmHg) | 136.3±3.0 | 124.7±2.9 | 141.7±3.3*† | 122.0±3.2‡ |
| Weight (g) | 365.6±4.7 | 344±10.6 | 249.6±9.0§† | 248.8±6.0‡‖ |
| BG (mg/dL) | 95.4±2.8 | 115.3±2.4 | 444.3±32.1§ | 459.3±25.2‡ |
p<0.05 DM vs DM+Cand,
p<0.05 DM vs NDM+Cand,
p<0.05 NDM vs DM+Cand,
p<0.05 NDM vs DM,
p<0.05 NDM+Cand vs DM+Cand.
Chronic angiotensin II infusion causes increased RVP
In order to further investigate the role of the AT1R on RVP, we examined the effect of chronic AngII infusion in control and candesartan-treated rats. We infused Sprague-Dawley rats with 300 ng/kg/min AngII via subcutaneous pump for 1, 3, or 6 days. This study showed a 2.9 fold-increase in RVP was present after one day of AngII infusion, which was sustained at 3 and 6 days (Figure 2A). AngII’s effects on SBP continued to increase during this time (Saline vs AngII: Day 1, 130.4±2.3 vs 157.0±1.8; Day 3, 130.6±4.9 vs 190.8±11.1 (P<0.001), Day 6, 139±2.4 vs 209±11.4 mmHg (P<0.001 Saline vs AngII, p<0.001 AngII day 1 vs AngII day 6)). First phase venous RVP was increased in rats infused with AngII for 6 days compared with saline-treated controls (Figure 2B). Treatment of AngII-infused rats with candesartan for 6 days prevented the increases in SBP (Saline 134.7±1.7, AngII 188.3±6.2, AngII+Cand 126.2±1.7 mmHg; mean±SEM, P<0.05 Saline vs AngII and AngII vs Cand) and decreased RVP by 81% compared with the untreated AngII group (Figure 2C). Vitreous fluorescein levels were 4.5±0.3 au, 10.4±1.2 au, and 5.6±0.4 au for saline, AngII alone, and AngII+Cand groups, respectively (Figure 2C). Candesartan treatment of rats receiving saline vehicle infusion was 5.1±0.8 au (n=3, data not shown), which was similar to untreated rats with a saline pump.
Figure 2. Effects of chronic AngII-infusion and AT1-receptor blockade on RVP.
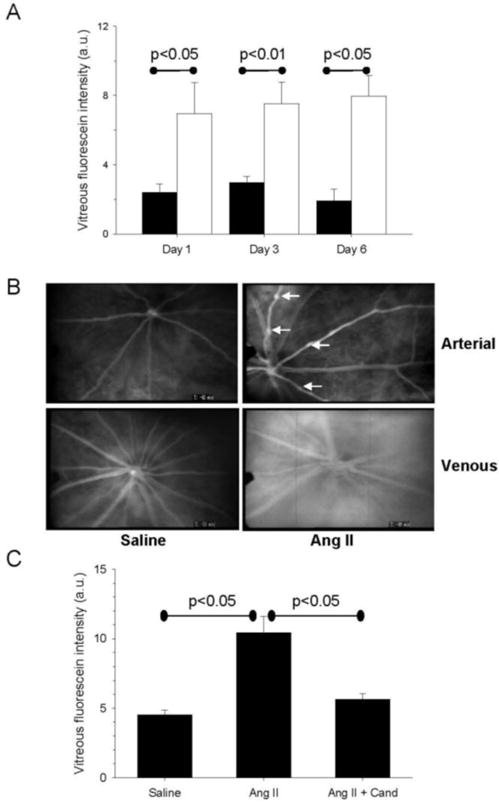
(A) The effect of systemic AngII infusion on RVP measured by vitreous fluorescein intensity at 1, 3 and 6 days post-treatment in AngII (unfilled bars, n=4) and saline control animals (filled bars, n=5). (B) Representative fluorescein angiograms from 6-day saline- and AngII-treated rats. Upper panels show the arterial phase, with arrows indicating arterial caliber abnormalities. Lower panels show diffuse vascular leakage of the AngII animal in the venous phase compared with the saline control. (C) Effect on RVP following 6-days of saline (n=19), AngII (n=16), or AngII + candesartan (n=10) treatment. Bars represent mean ± SEM, a.u. (arbitrary units).
Protein quantification of components of the KKS
Since the RAS and KKS interact at multiple levels13 and both plasma kallikrein and the bradykinin B2 receptor (B2-R) have been implicated in vasogenic edema14;15, we investigated the effects of AngII infusion on components of the KKS in the retina. This study showed that B2-R and B1-receptor (B1-R) levels in the retina were similar in rats receiving AngII and saline infusion (Figure 3), consistent with studies that have examined B1-R and B2-R mRNA levels in the retina of control and diabetic mice16. In contrast, we detected an increase in a 25 kDa heavy chain fragment of plasma kallikrein (P<0.05), and a trend for increased cleaved high-molecular weight kininogen heavy chain (cleaved HK, P=0.064) in AngII-infused animals (Figure 3), suggesting that active plasma kallikrein and its product, cleaved HK, are present in the retina with their levels increased in AngII-infused animals.
Figure 3. Western blot analysis of KKS components in retinal lysates.
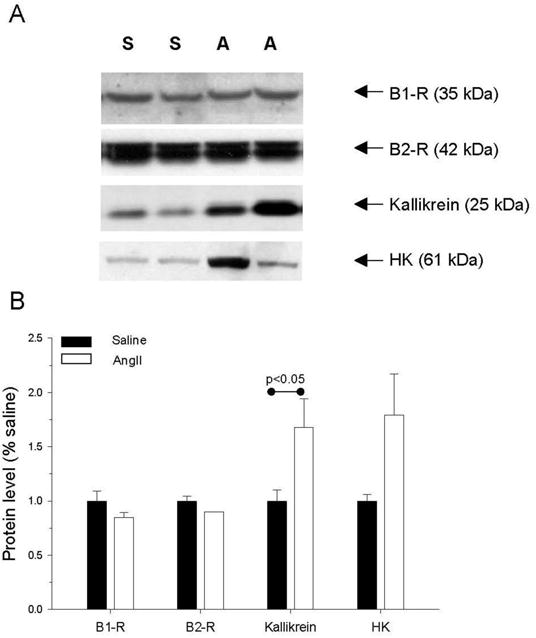
(A) Representative western blots showing immunoreactivity of proteins in saline (S)- and AngII (A)-treated animals. B1-R (bradykinin B1-receptor), B2-R (bradykinin B2-receptor), HK (cleaved high molecular weight kininogen). (B) Quantification of western blot results in saline (n=4 B-1R, n=3 B-2 R, n=11 kallikrein, n=12 HK) and AngII (n=4 B-1R, n=4 B-2 R, n=12 kallikrein, n=11 HK) animals infused for 6 days. Bars represent mean ± SEM.
Intravitreal injections of plasma kallikrein and bradykinin increase RVP
To directly investigate the effect of intraocular plasma kallikrein on RVP, we injected 20 ng of activated purified plasma kallikrein into the vitreous chamber of rats and measured vitreous fluorescein levels 40 minutes after injection, utilizing the contralateral eye as a control (10 μl injection of BSS). We show that activated plasma kallikrein increased RVP by 60% compared with BSS injected eyes (Figure 4A). Similarly, we demonstrated that intravitreal injection of 10 μM bradykinin (2 μM final vitreous concentration) increased RVP by 86% (P<0.01) compared with BSS injected control eyes (Fig 4B), and that this bradykinin-induced increase in RVP was decreased in rats infused with HOE-140.
Figure 4. Role of the kallikrein-kinin pathway in RVP and SBP.
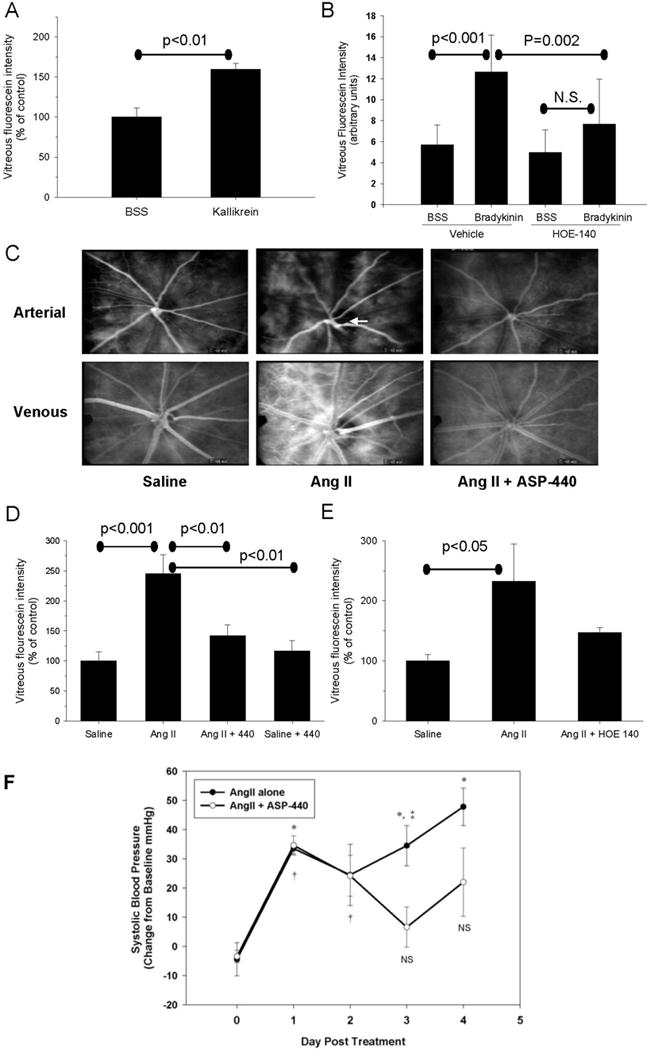
(A) Effect of intravitreal injection of BSS (balanced salt solution, n=4) or purified activated plasma kallikrein (n=5) on RVP measured by VFP. (B) Effect of intravitreal injection of BSS (n=5) or bradykinin (n=8) on RVP in the absence or presence of systemic infusion with HOE-140. (C) Fluorescein angiograms from representative animals treated for 3 days with saline, AngII and AngII + ASP-440 in the arterial (top panels) and venous (bottom panels) phases. Arrow indicates area of arterial caliber abnormalities in the AngII treated animal. (D) Effect of 3-day treatment with ASP-440 on AngII-mediated RVP increases. Saline (n=5), AngII (n=7), AngII + ASP-440 (n=7). (E) Effect of 3-day treatment with HOE-140 on AngII mediated RVP increases. Saline (n=8), AngII (n=7), AngII + HOE-140 (n=8). Bars represent mean ± SEM. (F) Time course of SBP measured using telemetry in rats infused with AngII in the absence or presence of ASP-440. * and † indicate P<0.05 for Day 0 vs AngII alone (n=4) and AngII+ASP-440 (n=7), respectively. ‡ indicates P<0.05 for AngII alone vs AngII+ASP-440.
Plasma kallikrein inhibitor (ASP-440) attenuates retinal vascular leakage in rats with AngII-induced hypertension
We investigated the effects of a novel plasma kallikrein catalytic activity inhibitor, ASP-440 (Figure S1, Table S1, please see http://hyper.aha.journals.org), on AngII-induced RVP by the infusion of rats with 300 ng/kg/min AngII with or without co-infusion with 16 μg/kg/hr ASP-440, via separate subcutaneous pumps for three days. Analysis of RVP by VFA showed that 3-day infusion of AngII alone increased retinal vascular fluorescein leakage and vessel caliber abnormalities, and that co-administration with ASP-440 attenuated these changes (Figure 4C). Quantification by VFP showed that treatment with ASP-440 decreased RVP in AngII-infused rats by 70% (P<0.001) compared with saline-treated control animals (Figure 4D). Treatment of control animals with ASP-440 did not alter RVP (Figure 4D). In order to investigate the possibility that the B2-R may contribute to the increased RVP caused by AngII, we treated animals with both 300 ng/kg/min AngII and 1 μg/kg/hr HOE-140. We show that AngII-stimulated RVP was reduced in HOE-140 treated rats by 64% compared with animals receiving AngII alone (Figure 4E). Neither ASP-440 (Saline+Veh 130.25.1, AngII+Veh 184.4±8.5, AngII+ASP-440 195.8±9.0 mmHg P<0.05 Saline vs AngII, P<0.05 Saline vs AngII+ASP-440) nor HOE-140 treatment (Saline 137.3±3.3, AngII+Veh 183.2±8.5, AngII+HOE-140 175.9±9.2 mmHg p<0.05 Saline vs AngII, P<0.05 Saline vs AngII+HOE-140) reduced SBP in AngII animals when BP was measured via tail cuff at Day 3, although animals treated with ASP-440 and AngII showed a trend for higher SBP compared with AngII alone using this method. Comparable effects of AngII on RVP and SBP were observed with Long-Evans rats, and the co-infusion of AngII+ASP-440 reduced RVP but did not reduce SBP (measured via tail cuff plethysmography) or retinal leukostasis (Figure S2, please see http://hyper.aha.journals.org).
Further analysis of blood pressure in Sprague Dawley rats using telemetry showed an increase in SBP and DBP in the carotid artery at Day 1 through Day 4 in rats receiving AngII alone, compared with baseline measurements (Figure 4F). Rats receiving AngII+ASP-440 had an increase in SBP recorded by telemetry that was similar to the AngII alone group at Days 1 and 2 followed by partial normalization at Day 3 (P< 0.05 for Day 3 AngII alone vs AngII+ASP-440, Figure 4F). SBP in the AngII alone and AngII+ASP-440 groups, collected from 9:00 PM to 1:00 AM on the end of Day 3, was increased by 30.6 and 15.5 mmHg, respectively, compared with baseline measurements at this time period, changes similar to that observed during the day. SBP of AngII+ASP-440 infused rats at Day 4 measured using telemetry was 122.8±19.7 mmHg whereas SBP of these rats measured using tail cuff plethysmography was 176.4±6.7 mmHg, which is comparable to 195.8±9.0mmHg SBP via tail cuff in the RVP group (Figure 4D), confirming the discordance between tail cuff plethysmography and telemetry SBP measurements in AngII+440 treated rats.
DISCUSSION
This report shows that systemic AT1R antagonism and plasma kallikrein inhibition ameliorates retinal vascular hyperpermeability. We show that 1) the AT1R antagonist candesartan decreased RVP in diabetic rats, 2) chronic infusion of AngII in rats increased RVP and that this response was blocked by candesartan, and 3) systemic treatment of rats with a new small molecule inhibitor of plasma kallikrein (ASP-440) reduced AngII-induced RVP and SBP measured from the carotid artery using telemetry, but did not reduce SBP measured via tail cuff plethysmography or retinal leukocyte adhesion. These findings demonstrate a novel role for plasma kallikrein in mediating the increase in RVP and the sustained elevation of SBP in rats with AngII-induced hypertension.
AngII signaling is mediated by two receptor subtypes, the AT1R and AT2 receptor (AT2R), both of which are expressed in the retina17. Although AT1R and AT2R interact at multiple levels with vascular permeability factors and their receptors, including the bradykinin system (reviewed in13;18), the role of AT-receptors in vascular permeability, particularly in the retina, have not been elucidated. Since the loss of blood-retinal-barrier function is thought to play a critical role in the etiology of diabetic retinopathy and macula edema, further understanding of the effects of AT1R antagonists on RVP may have important relevance to their use in the treatment of this disease.
AT1R antagonism has been reported to exert dual vascular effects by reducing AT1R signaling while enhancing AT2R-mediated responses19. Indeed, the improvement in endothelium-dependent vasorelaxation by AT1R blockade has been attributed, in part, to increased bradykinin/B2 receptor-mediated eNOS activation and its generation of cGMP20–22. AT1R antagonism has been reported to increase plasma bradykinin levels in people with hypertension23 and increase tissue kininogenase activity in mice20. However, bradykinin and NO also potently increase vascular hyperpermeability and edema24;25, and have been implicated in increasing RVP induced by retinal hemorrhage12. Therefore the augmentation of the AT2R-bradykinin pathway by AT1R blockade could potentially lead to an increase in vascular leakage. We demonstrate that candesartan reduced RVP in normotensive DM rats and in rats with AngII-induced hypertension, suggesting that in these experimental models; the AT1R contributes to the increase in RVP. These results extend previous studies, which have shown that ACE inhibition decreased blood-retinal barrier permeability in hypertensive Type 1 diabetic patients and in normotensive diabetic rats26;27.
While a large body of literature suggests that ACE inhibition potentiates endogenous bradykinin, thought to be beneficial in kidney and heart28;29, the present study and previous reports suggest that bradykinin has deleterious effects on the retina30;31. Indeed, the beneficial effects of ACE inhibition on the retina have also been observed with AT1R antagonism (reviewed in18), suggesting that inhibition of AngII production is the critical aspect of ACE inhibition that is important in diabetic retinopathy. Previous reports have shown that AngII and angiotensinogen are elevated in the vitreous fluid from patients with proliferative diabetic retinopathy compared with NDM subjects12;32, and that serum concentrations of ACE and renin correlate with the severity of diabetic retinopathy (reviewed in18), suggesting the involvement of both local intraocular and systemic RAS in diabetic retinopathy. Studies that examine the effect of AT1R antagonism on retinal abnormalities in diabetes, including the present study, have shown a drug-induced decrease in systemic BP11, with results from the DIRECT-Prevent 1 study showing a decrease in SBP of 2.6 mmHg in Type I diabetic patients with retinopathy when treated with candesartan10. Since the efficacious dose of candesartan used in our studies decreased both RVP and SBP in our diabetic and AngII-infusion rat models, our findings do not characterize the potential individual contributions for the BP-dependent and -independent effects of the AT1R on RVP.
We demonstrate that continuous systemic treatment of rats with the plasma kallikrein inhibitor ASP-440 ameliorated AngII-induced RVP at both 3 days and 7 days (not shown). To our knowledge, this is the first report of a small molecule plasma kallikrein selective inhibitor with in vivo efficacy against vascular hyperpermeability. Since candesartan blocked AngII-induced RVP, our results suggest that plasma kallikrein mediates the increase in RVP induced by AT1R-stimulated hypertension. Moreover, we found that AngII infusion increased inflammatory cell responses (as measured by retinal leukostasis) that were not normalized by treatment with ASP-440, although RVP was attenuated by ASP-440 in these same animals (Figure S2, please see http://hyper.aha.journals.org). This suggests that plasma kallikrein inhibition, while not directly inhibiting leukostasis, does reduce RVP that occurs coincident with inflammatory cell recruitment. Using telemetry, we also observed that ASP-440 decreased SBP in rats exposed to AngII-induced hypertension, however this effect of ASP-440 was not observed until day 3 and 4 of infusion. The 2-day delay prior to the appearance of this BP lowering effect of ASP-440 could suggest that plasma kallikrein contributes to the maintenance of the sustained BP increase in this model. Interestingly, we did not detect a decrease in AngII-induced SBP by ASP-440 using tail cuff plethysmography at 3 days post infusion. Similar discordance between these two measures of BP have been reported in the literature33, attributed to an exaggerated stress response to the conditions of tail-cuff measurement in the presence of Ang-II.
The main effector peptide of the KKS, bradykinin, is generated directly from HK by plasma kallikrein. Bradykinin is also generated from low molecular weight kininogen by the action of an aminopeptidase on the kallidin (Lys-bradykinin) peptide generated by tissue kallikrein. Intravascular delivery of bradykinin in rats has been shown to decrease BP, a response attenuated by HOE-14034. Bradykinin-induced vasorelaxation has been attributed to the activation of bradykinin receptors on the endothelium19–21. In contrast, bradykinin, via it effects on the central nervous system (CNS), has been shown to elevate blood pressure via the B2-R and induce a cardiac sympathetic response34–37. The hypotensive effect of ASP-440 observed here by telemetry suggests that plasma kallikrein-mediated bradykinin formation in the Ang-II model leads to a net increase in BP. The absence of an apparent BP lowering response, using tail cuff plethysmography, to ASP-440 in AngII treated rats shows that plasma kallikrein can exert effects on systemic BP that are not detected by the indirect measurement of BP via tail cuff in heated and restrained animals.
We show that intravitreal injection of either purified activated plasma kallikrein or bradykinin increases RVP. These findings are consistent with the well established effects of the KKS in increasing vascular permeability24;38. We found that both a B2-R antagonist and a plasma kallikrein-selective inhibitor reduced AngII-stimulated RVP. The use of a small molecule plasma kallikrein inhibitor introduces a new strategy to modulate KKS action in vivo. Since this inhibitor is >1000-fold more selective in inhibiting plasma kallikrein compared with tissue kallikrein, this compound provides a new pharmacological opportunity to inhibit bradykinin production mediated by plasma kallikrein. This would suppress pathological effects mediated by increased bradykinin generated by this enzyme, rather than pan-antagonizing all actions of bradykinin at the B2-R level, as done by HOE-140. Although the action of increased bradykinin signaling can be deleterious for the eye31, bradykinin, likely generated by tissue kallikrein, has been shown to have beneficial effects in several other tissues28, particularly the heart29. Although we did observe attenuation of increased RVP by treatment with HOE-140, systemic use of B2-R blockers for the treatment of vascular disease may be limited by their ability to indiscriminately inhibit tissue-specific beneficial effects of bradykinin. Since most of the beneficial effects of the KKS have been attributed to tissue kallikrein, in contrast to plasma kallikrein-mediated bradykinin production associated with angioedema39, treatment of RVP with a specific plasma kallikrein inhibitor would not be expected to have significant adverse effects. Moreover, the rare occurrence of complete deficiencies in plasma kallikrein in humans are usually only detected in later decades by observation of abnormal prolongation of activated partial thromboplastin clotting time, but otherwise with no known clinically significant morbidities40.
While our data shows that plasma kallikrein inhibition by ASP-440 decreases retinal hyper-vasopermeability stimulated by AngII-induced hypertension, and previous work from our lab and others have implicated the KKS in diabetic retinopathy12;30, further studies will be needed to characterize the effects of plasma kallikrein inhibitors on other models of hypertensive- and diabetic retinopathy. Our results showing efficacy of ASP-440 via systemic delivery suggest that plasma kallikrein inhibition may provide a new therapeutic approach to preserve blood-retinal barrier function and decrease BP.
PERSPECTIVES
Recent results from the DIRECT study have found that treatment with an AT1R blocker has beneficial effects on the incidence of diabetic retinopathy in people with Type I diabetes10. Our study has found that plasma kallikrein is involved in the biochemical pathways downstream of AT1R activation and, given that plasma kallikrein, HK and FXII are found in the vitreous of people with diabetic retinopathy12, provides additional support for an alternative pathway by which AT1R blockade may contribute to amelioration of diabetic retinopathy in the absence of ischemia or BP changes. Given the growing prevalence of patients with uncontrolled and resistant hypertension41, particularly in people with diabetes, direct inhibition of plasma kallikrein may provide a new therapeutic approach for the treatment of retinal disorders involving vascular hyperpermeability.
Acknowledgments
Sources of funding: This work was supported in part by the NHMRC Australia CJ Martin Research Fellowship (JAP), the US National Institutes of Health DK 60165, EY019029, and Juvenile Diabetes Research Foundation (EPF), DK 36836 (Joslin’s Diabetes and Endocrinology Research Center), R43HL090132 (TJC), and the Massachusetts Lions Eye Research Fund (SEB).
Footnotes
Conflict of Interest/Disclosure(s): S. Sinha and T.J. Chilcote, ActiveSite Pharmaceuticals, “Inhibitors of plasma kallikrein”, PCT Publication WO/2008/016883, 2008.
References
- 1.Klein R, Klein BE, Moss SE, Davis MD, DeMets DL. The Wisconsin epidemiologic study of diabetic retinopathy. IV. Diabetic macular edema. Ophthalmology. 1984;91:1464–1474. doi: 10.1016/s0161-6420(84)34102-1. [DOI] [PubMed] [Google Scholar]
- 2.Antcliff RJ, Marshall J. The pathogenesis of edema in diabetic maculopathy. Semin Ophthalmol. 1999;14:223–232. doi: 10.3109/08820539909069541. [DOI] [PubMed] [Google Scholar]
- 3.Miyamoto K, Khosrof S, Bursell SE, Rohan R, Murata T, Clermont AC, Aiello LP, Ogura Y, Adamis AP. Prevention of leukostasis and vascular leakage in streptozotocin-induced diabetic retinopathy via intercellular adhesion molecule-1 inhibition. Proc Natl Acad Sci U S A. 1999;96:10836–10841. doi: 10.1073/pnas.96.19.10836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Girach A, Lund-Andersen H. Diabetic macular oedema: a clinical overview. Int J Clin Pract. 2007;61:88–97. doi: 10.1111/j.1742-1241.2006.01211.x. [DOI] [PubMed] [Google Scholar]
- 5.Roy MS, Affouf M. Six-year progression of retinopathy and associated risk factors in African American patients with type 1 diabetes mellitus: the New Jersey 725. Arch Ophthalmol. 2006;124:1297–1306. doi: 10.1001/archopht.124.9.1297. [DOI] [PubMed] [Google Scholar]
- 6.Klein R, Klein BE, Moss SE, Cruickshanks KJ. The Wisconsin Epidemiologic Study of Diabetic Retinopathy: XVII. The 14-year incidence and progression of diabetic retinopathy and associated risk factors in type 1 diabetes. Ophthalmology. 1998;105:1801–1815. doi: 10.1016/S0161-6420(98)91020-X. [DOI] [PubMed] [Google Scholar]
- 7.Matthews DR, Stratton IM, Aldington SJ, Holman RR, Kohner EM. Risks of progression of retinopathy and vision loss related to tight blood pressure control in type 2 diabetes mellitus: UKPDS 69. Arch Ophthalmol. 2004;122:1631–1640. doi: 10.1001/archopht.122.11.1631. [DOI] [PubMed] [Google Scholar]
- 8.Chaturvedi N, Sjolie AK, Stephenson JM, Abrahamian H, Keipes M, Castellarin A, Rogulja-Pepeonik Z, Fuller JH. Effect of lisinopril on progression of retinopathy in normotensive people with type 1 diabetes. The EUCLID Study Group. EURODIAB Controlled Trial of Lisinopril in Insulin-Dependent Diabetes Mellitus. Lancet. 1998;351:28–31. doi: 10.1016/s0140-6736(97)06209-0. [DOI] [PubMed] [Google Scholar]
- 9.Schrier RW, Estacio RO, Mehler PS, Hiatt WR. Appropriate blood pressure control in hypertensive and normotensive type 2 diabetes mellitus: a summary of the ABCD trial. Nat Clin Pract Nephrol. 2007;3:428–438. doi: 10.1038/ncpneph0559. [DOI] [PubMed] [Google Scholar]
- 10.Chaturvedi N, Porta M, Klein R, Orchard T, Fuller J, Parving HH, Bilous R, Sjolie AK. Effect of candesartan on prevention (DIRECT-Prevent 1) and progression (DIRECT-Protect 1) of retinopathy in type 1 diabetes: randomised, placebo-controlled trials. Lancet. 2008;372:1394–1402. doi: 10.1016/S0140-6736(08)61412-9. [DOI] [PubMed] [Google Scholar]
- 11.Horio N, Clermont AC, Abiko A, Abiko T, Shoelson BD, Bursell SE, Feener EP. Angiotensin AT(1) receptor antagonism normalizes retinal blood flow and acetylcholine-induced vasodilatation in normotensive diabetic rats. Diabetologia. 2004;47:113–123. doi: 10.1007/s00125-003-1262-x. [DOI] [PubMed] [Google Scholar]
- 12.Gao BB, Clermont A, Rook S, Fonda SJ, Srinivasan VJ, Wojtkowski M, Fujimoto JG, Avery RL, Arrigg PG, Bursell SE, Aiello LP, Feener EP. Extracellular carbonic anhydrase mediates hemorrhagic retinal and cerebral vascular permeability through prekallikrein activation. Nat Med. 2007;13:181–188. doi: 10.1038/nm1534. [DOI] [PubMed] [Google Scholar]
- 13.Schmaier AH. The kallikrein-kinin and the renin-angiotensin systems have a multilayered interaction. Am J Physiol Regul Integr Comp Physiol. 2003;285:R1–13. doi: 10.1152/ajpregu.00535.2002. [DOI] [PubMed] [Google Scholar]
- 14.Schneider L, Lumry W, Vegh A, Williams AH, Schmalbach T. Critical role of kallikrein in hereditary angioedema pathogenesis: a clinical trial of ecallantide, a novel kallikrein inhibitor. J Allergy Clin Immunol. 2007;120:416–422. doi: 10.1016/j.jaci.2007.04.028. [DOI] [PubMed] [Google Scholar]
- 15.Davis AE., III The pathophysiology of hereditary angioedema. Clin Immunol. 2005;114:3–9. doi: 10.1016/j.clim.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 16.Ebrahimian TG, Tamarat R, Clergue M, Duriez M, Levy BI, Silvestre JS. Dual effect of angiotensin-converting enzyme inhibition on angiogenesis in type 1 diabetic mice. Arterioscler Thromb Vasc Biol. 2005;25:65–70. doi: 10.1161/01.ATV.0000149377.90852.d8. [DOI] [PubMed] [Google Scholar]
- 17.Wheeler-Schilling TH, Kohler K, Sautter M, Guenther E. Angiotensin II receptor subtype gene expression and cellular localization in the retina and non-neuronal ocular tissues of the rat. Eur J Neurosci. 1999;11:3387–3394. doi: 10.1046/j.1460-9568.1999.00787.x. [DOI] [PubMed] [Google Scholar]
- 18.Clermont A, Bursell SE, Feener EP. Role of the angiotensin II type 1 receptor in the pathogenesis of diabetic retinopathy: effects of blood pressure control and beyond. J Hypertens Suppl. 2006;24:S73–S80. doi: 10.1097/01.hjh.0000220410.69116.f8. [DOI] [PubMed] [Google Scholar]
- 19.Landmesser U, Drexler H. Effect of angiotensin II type 1 receptor antagonism on endothelial function: role of bradykinin and nitric oxide. J Hypertens Suppl. 2006;24:S39–S43. doi: 10.1097/01.hjh.0000220405.38622.23. [DOI] [PubMed] [Google Scholar]
- 20.Tsutsumi Y, Matsubara H, Masaki H, Kurihara H, Murasawa S, Takai S, Miyazaki M, Nozawa Y, Ozono R, Nakagawa K, Miwa T, Kawada N, Mori Y, Shibasaki Y, Tanaka Y, Fujiyama S, Koyama Y, Fujiyama A, Takahashi H, Iwasaka T. Angiotensin II type 2 receptor overexpression activates the vascular kinin system and causes vasodilation. J Clin Invest. 1999;104:925–935. doi: 10.1172/JCI7886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hornig B, Kohler C, Schlink D, Tatge H, Drexler H. AT1-receptor antagonism improves endothelial function in coronary artery disease by a bradykinin/B2-receptor-dependent mechanism. Hypertension. 2003;41:1092–1095. doi: 10.1161/01.HYP.0000064942.77814.26. [DOI] [PubMed] [Google Scholar]
- 22.Yayama K, Hiyoshi H, Imazu D, Okamoto H. Angiotensin II stimulates endothelial NO synthase phosphorylation in thoracic aorta of mice with abdominal aortic banding via type 2 receptor. Hypertension. 2006;48:958–964. doi: 10.1161/01.HYP.0000244108.30909.27. [DOI] [PubMed] [Google Scholar]
- 23.Campbell DJ, Krum H, Esler MD. Losartan increases bradykinin levels in hypertensive humans. Circulation. 2005;111:315–320. doi: 10.1161/01.CIR.0000153269.07762.3B. [DOI] [PubMed] [Google Scholar]
- 24.Groger M, Lebesgue D, Pruneau D, Relton J, Kim SW, Nussberger J, Plesnila N. Release of bradykinin and expression of kinin B2 receptors in the brain: role for cell death and brain edema formation after focal cerebral ischemia in mice. J Cereb Blood Flow Metab. 2005;25:978–989. doi: 10.1038/sj.jcbfm.9600096. [DOI] [PubMed] [Google Scholar]
- 25.Bucci M, Roviezzo F, Posadas I, Yu J, Parente L, Sessa WC, Ignarro LJ, Cirino G. Endothelial nitric oxide synthase activation is critical for vascular leakage during acute inflammation in vivo. Proc Natl Acad Sci U S A. 2005;102:904–908. doi: 10.1073/pnas.0408906102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parving HH, Larsen M, Hommel E, Lund-Andersen H. Effect of antihypertensive treatment on blood-retinal barrier permeability to fluorescein in hypertensive type 1 (insulin-dependent) diabetic patients with background retinopathy. Diabetologia. 1989;32:440–444. doi: 10.1007/BF00271264. [DOI] [PubMed] [Google Scholar]
- 27.Gilbert RE, Kelly DJ, Cox AJ, Wilkinson-Berka JL, Rumble JR, Osicka T, Panagiotopoulos S, Lee V, Hendrich EC, Jerums G, Cooper ME. Angiotensin converting enzyme inhibition reduces retinal overexpression of vascular endothelial growth factor and hyperpermeability in experimental diabetes. Diabetologia. 2000;43:1360–1367. doi: 10.1007/s001250051539. [DOI] [PubMed] [Google Scholar]
- 28.Kakoki M, Takahashi N, Jennette JC, Smithies O. Diabetic nephropathy is markedly enhanced in mice lacking the bradykinin B2 receptor. Proc Natl Acad Sci U S A. 2004;101:13302–13305. doi: 10.1073/pnas.0405449101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Griol-Charhbili V, Messadi-Laribi E, Bascands JL, Heudes D, Meneton P, Giudicelli JF, Alhenc-Gelas F, Richer C. Role of tissue kallikrein in the cardioprotective effects of ischemic and pharmacological preconditioning in myocardial ischemia. FASEB J. 2005;19:1172–1174. doi: 10.1096/fj.04-3508fje. [DOI] [PubMed] [Google Scholar]
- 30.Abdouh M, Talbot S, Couture R, Hassessian HM. Retinal plasma extravasation in streptozotocin-diabetic rats mediated by kinin B(1) and B(2) receptors. Br J Pharmacol. 2008;154:136–143. doi: 10.1038/bjp.2008.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Phipps JA, Feener EP. The kallikrein-kinin system in diabetic retinopathy: Lessons for the kidney. Kidney Int. 2008;73:1114–1119. doi: 10.1038/ki.2008.9. [DOI] [PubMed] [Google Scholar]
- 32.Funatsu H, Yamashita H, Nakanishi Y, Hori S. Angiotensin II and vascular endothelial growth factor in the vitreous fluid of patients with proliferative diabetic retinopathy. Br J Ophthalmol. 2002;86:311–315. doi: 10.1136/bjo.86.3.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pelaez LI, Manriquez MC, Nath KA, Romero JC, Juncos LA. Low-dose angiotensin II enhances pressor responses without causing sustained hypertension. Hypertension. 2003;42:798–801. doi: 10.1161/01.HYP.0000085782.99773.B6. [DOI] [PubMed] [Google Scholar]
- 34.Mukai H, Fitzgibbon WR, Ploth DW, Margolius HS. Effect of chronic bradykinin B2 receptor blockade on blood pressure of conscious Dahl salt-resistant rats. Br J Pharmacol. 1998;124:197–205. doi: 10.1038/sj.bjp.0701797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou W, Fu LW, Tjen ALS, Guo ZL, Longhurst JC. Role of glutamate in a visceral sympathoexcitatory reflex in rostral ventrolateral medulla of cats. Am J Physiol Heart Circ Physiol. 2006;291:H1309–H1318. doi: 10.1152/ajpheart.00202.2006. [DOI] [PubMed] [Google Scholar]
- 36.Zahner MR, Pan HL. Role of paraventricular nucleus in the cardiogenic sympathetic reflex in rats. Am J Physiol Regul Integr Comp Physiol. 2005;288:R420–R426. doi: 10.1152/ajpregu.00563.2004. [DOI] [PubMed] [Google Scholar]
- 37.Privitera PJ, Thibodeaux H, Yates P. Rostral ventrolateral medulla as a site for the central hypertensive action of kinins. Hypertension. 1994;23:52–58. doi: 10.1161/01.hyp.23.1.52. [DOI] [PubMed] [Google Scholar]
- 38.Hulstrom D, Svensjo E. Intravital and electron microscopic study of bradykinin-induced vascular permeability changes using FITC-dextran as a tracer. J Pathol. 1979;129:125–133. doi: 10.1002/path.1711290304. [DOI] [PubMed] [Google Scholar]
- 39.Bork K, Frank J, Grundt B, Schlattmann P, Nussberger J, Kreuz W. Treatment of acute edema attacks in hereditary angioedema with a bradykinin receptor-2 antagonist (Icatibant) J Allergy Clin Immunol. 2007;119:1497–1503. doi: 10.1016/j.jaci.2007.02.012. [DOI] [PubMed] [Google Scholar]
- 40.Katsuda I, Maruyama F, Ezaki K, Sawamura T, Ichihara Y. A new type of plasma prekallikrein deficiency associated with homozygosity for Gly104Arg and Asn124Ser in apple domain 2 of the heavy-chain region. Eur J Haematol. 2007;79:59–68. doi: 10.1111/j.1600-0609.2007.00871.x. [DOI] [PubMed] [Google Scholar]
- 41.Calhoun DA, Jones D, Textor S, Goff DC, Murphy TP, Toto RD, White A, Cushman WC, White W, Sica D, Ferdinand K, Giles TD, Falkner B, Carey RM. Resistant hypertension: diagnosis, evaluation, and treatment: a scientific statement from the American Heart Association Professional Education Committee of the Council for High Blood Pressure Research. Circulation. 2008;117:e510–e526. doi: 10.1161/CIRCULATIONAHA.108.189141. [DOI] [PubMed] [Google Scholar]