

Abstract
Nitric oxide (NO) increases glutamate release to the second-order neurons in the nucleus tractus solitarius (NTS). N-type Ca2+ channel is essential for triggering glutamate release at synaptic terminals. In this study, we determined the role of Cav2.2 subunit in NO-induced increase in glutamate synaptic inputs to NTS neurons. The second-order NTS neurons and nodose ganglionic (NG) neurons were identified by applying DiA, a fluorescent lipophilic tracer, on aortic depressor nerve in rats. NO donor DEA/NO significantly increased tractus solitarius (TS)-evoked excitatory postsynaptic currents (EPSCs) in second-order NTS neurons, an effect was abolished by pretreatment of slice with ODQ, an inhibitor for soluble isoform of guanylyl cyclase. DEA/NO decreased the paired-pulse ratio of TS-evoked EPSCs, while increased the frequency, but not the amplitude, of miniature EPSCs in second-order NTS neurons. Furthermore, DEA/NO significantly increased Ba2+ currents in identified baroreceptor NG neurons. However, DEA/NO had little effect on the Ba2+ currents in the presence of specific N-type Ca2+ blocker ω-conotoxin GVIA. In addition, immunecytochemistry staining revealed that Cav2.2 subunit immunoreactivates were colocalized with DiA-labeled baroreceptor nerve terminals in the NTS. Collectively, these findings suggest that NO stimulates glutamatergic synaptic inputs to second-order NTS neurons through augmentation of Cav2.2-mediated N-type Ca2+ currents.
Keywords: nitric oxide, glutamate, baroreceptor, Ca2+ channels, brainstem
Introduction
The nucleus tractus solitarius (NTS) is a critical brain region involved in the integration and assimilation of visceral sensory processes related to cardiovascular function [5]. The baroreceptor afferent fibers terminate in the intermediate portion of the NTS and form asymmetric synaptic contacts with second-order NTS neurons [1]. The cell bodies of the aortic depressor nerve afferent fibers are located in the nodose ganglion (NG) and project centrally to the NTS and release glutamate to second-order NTS neurons [13]. Nitric oxide (NO) importantly regulates cardiovascular autonomic function in the NTS. For example, overexpression of eNOS in the NTS decreases blood pressure in spontaneously hypertensive rats (SHR) [7]. In contrast, chronic blockade of eNOS activity by transfer of a dominant-negative form of eNOS into the NTS decreases blood pressure in SHR [17]. NO produces its biological actions through distinct signal transduction pathways such as soluble isoform of guanylyl cyclase (sGC), 3′,5′-cyclic guanosine monophosphate (cGMP), and protein kinase G (PKG) [16]. In this regard, blockade of the NO-sGC-cGMP-PKG cascade with the sGC inhibitor eliminates the depressor and bradycardiac response elicited by injection of L-arginine into the NTS.
Previous studies have shown that NO donor diethylammonium (Z)-1-(N,N-diethylamino) diazen-1-ium-1,2-diolate (DEA/NO) increases tractus solitarius (TS)-evoked glutamatergic excitatory postsynaptic potentials in the NTS [18]. Furthermore, NO donor increases the extracellular glutamate level, and NOS inhibitor reduces the basal level of glutamate in the NTS [9]. On the other hand, NO-induced depressor and bradycardic effects in the NTS were largely attenuated by blocking ionotropic glutamate receptors [10]. Because the Ca2+ channel subtypes in NG neuron soma are closely correlated with the Ca2+ channels responsible for neurotransmitter release at their central synaptic terminals in the NTS [6], in this study, we determined the effect of NO on Ca2+ channels in identified baroreceptor NG neurons.
Voltage-gated calcium channels (VGCCs) is critical for triggering neurotransmitter release at synaptic terminals. VGCCs are classified into multiple types based on their amino acid sequences, biophysics, and pharmacological properties. N (Cav2.2)-type Ca2+ channels play a predominant role in neurotransmitter release in the brain [14]. NO increases N-type Ca2+ currents in dorsal root ganglion neurons and sympathetic cervical ganglion neurons [3]. In this study, we determined the role of Cav2.2-mediated N-type Ca2+ in NO-induced increase in glutamate synaptic inputs to second-order NTS neurons.
Materials and Methods
In vivo anterograde labeling of second-order NTS neurons
Male Sprague-Dawley rats (8–12 weeks old) were anesthetized with isoflurane (2% in oxygen). The aortic depressor nerve (ADN) was identified, isolated, and placed on a section of parafilm. Crystals of the fluorescent dye DiA were carefully placed in contact with the ADN. To insulate the ADN and the tracer, the area was embedded with polyvinylsiloxane gel. The surgical area was closed, and the animal was returned to its home cage to recover for 3–6 weeks.
NTS slice preparation and Electrophysiology recordings
Rats were anesthetized with isoflurane (2% in oxygen) and decapitated. The brainstem was quickly removed and placed in ice-cold artificial cerebrospinal fluid (aCSF) containing 125 mM NaCl, 3 mM KCl, 1.25 mM NaH2PO4, 2.4 mM CaCl2, 1 mM MgCl2, 10 mM glucose, and 25 mM NaHCO3 saturated with 95% O2 and 5% CO2. Horizontal slices (250–300 μm thick) containing the NTS was cut and then preincubated in aCSF continuously gassed with 95% O2 and 5% CO2 and maintained at 34°C.
The DiA-labeled baroreceptor terminals (green) in the NTS slices were briefly identified by an upright microscope (BX50 WI; Olympus) with epifluorescence illumination. The recording pipettes were pulled using borosilicate capillaries (OD: 1.2 mm, ID: 0.86 mm) with a resistance of 3–6 MΩ when it was filled with solution containing 130 mM potassium gluconate, 1 mM MgCl2, 10 mM HEPES, 10 mM EGTA, 1 mM CaCl2, and 4 mM ATP-Mg (pH 7.25, osmolarity 290–300 mOsm). After formed a tight GΩ seal and ruptured the cell membrane, recordings began 5 min after whole-cell access was established. Electronic signals were processed by a multiclamp 700B amplifier, and a Digidata 1440. The evoked excitatory postsynaptic currents (EPSCs) miniature EPSCs (in the presence of tetrodotoxin, 1 μM) were recorded in the presence of 20 μM bicuculline at a holding potential of −70 mV. The evoked EPSCs were elicited by electrical stimulation of the TS ipsilateral to the recording site at a frequency of 0.1 Hz. The distance between the stimulating electrode and the recorded neuron was about 200 μm. Monosynaptic postsynaptic currents were identified on the basis of short onset latencies (≤ 4.5 ms), variation of the latencies (< 0.5 ms) of eEPSCs, and absence of conduction failure of eEPSCs in response to a 20-Hz electrical stimulation. A sodium channel blocker QX-314(5 mM) was included in the recording pipette solution.
Isolation of NG neurons and electrophysiological recordings in NG neurons
Rats were anesthetized by isoflurane (2% in oxygen) and decapitated. The NG with connective tissue capsule was removed and transferred into ice-cold Eagle’s medium containing trypsin (type I, 0.5 mg/ml; Sigma), collagenase (type IA, 1 mg/ml; Sigma), and DNase (type I, 0.1 mg/ml; Sigma). Soybean trypsin inhibitor (type II-s, 1.25 mg/ml) was added into the medium to stop trypsin digestion. Cells were plated onto a 35-mm culture dish for electrophysiological recordings.
A recording pipette with a resistance of 1.5 – 2 M Ω was pulled from glass capillaries (OD: 1.5 mm; ID 1.17 mm) and filled with solution containing 120 mM CsCl, 1 mM MgCl2, 10 mM HEPES, 10 mM EGTA, 4 mM Mg-ATP, and 0.3 mM Na-GTP (pH 7.2 adjusted with CsOH, osmolarity 300 mOsm). DiA-labeled NG neurons were identified by epifluorescence illumination on an inverted IX70 microscope (Olympus) and were recorded in the whole-cell configuration using an EPC-10 amplifier (HEKA Instruments) at room temperature (~25°C). Signals were filtered and acquired by Pulse program (HEKA). Barium was used as the charge carrier and was elicited by a series of potentials from −70 to +50 mV for 150 ms in 10-mV steps (5-s intervals) from a holding potential of −90 mV. The extracellular solution contained 140 mM TEA, 2 mM MgCl2, 3 mM BaCl2, 10 mM glucose, and 10 mM HEPES (pH 7.4, osmolarity 320 mOsm).
Immunocytochemistry staining
Under deep anesthesia with sodium pentobarbital (60 mg/kg, ip), rats were intracardially perfused and fixed with 4% paraformaldehyde. Coronal slices of brainstem were sectioned to 25 μm in thickness. Sections containing NTS were incubated with the primary antibody (rabbit anti-CaV2.2, 1:200 dilution; Alomone labs) for 2 h at room temperature and overnight at 4°C. Subsequently, the sections were incubated with goat anti-rabbit IgG conjugated with Alexa fluor 594 (1:100 dilution; Jackson ImmunoResearch) for 1.5 h at room temperature. Finally, the sections were incubated with Cyanine3-conjugated tyramide (1:100 dilution; PerkinElmer Life Sciences) for 10 min at room temperature. Then sections were mounted on slides, and viewed using a confocal microscope.
Data analysis
Data was presented as means ± S.E. The effects of drugs on the amplitude of TS-evoked EPSCs was analyzed by Clampfit software. The frequency and amplitude of mEPSCs were determined by using Minianalysis. The IBa currents for NG neurons were analyzed by PulseFit software. Wilcoxon signed-rank test or nonparametric ANOVA (Kruskal – Wallis) with Dunn’s post hoc test. P < 0.05 will be considered to be statistically significant.
Results
NO donor increases glutamatergic synaptic inputs to second-order NTS neurons
We first determined the effect of NO donor on glutamatergic synaptic inputs to the second-order NTS neurons. DEA/NO (100 μM) significantly increased the amplitude of TS-evoked EPSCs in 8 labeled NTS neurons (Figure 1). Bath application of CNQX (20 μM) abolished evoked EPSCs (Figure 1A and B). The DEA/NO-induced increase in TS-evoked EPSCs was reproducible (data not show). After testing the initial effect of DEA/NO (100 μM) on TS-evoked EPSCs, 10 μM ODQ (sGC inhibitor) was added to the recording chamber. Subsequent application of DEA/NO failed to increase the amplitude of TS-evoked EPSCs in the presence of ODQ in these 8 labeled NTS neurons (Figure 1).
Figure 1. DEA/NO increased evoked EPSCs in second-order NTS neuron.
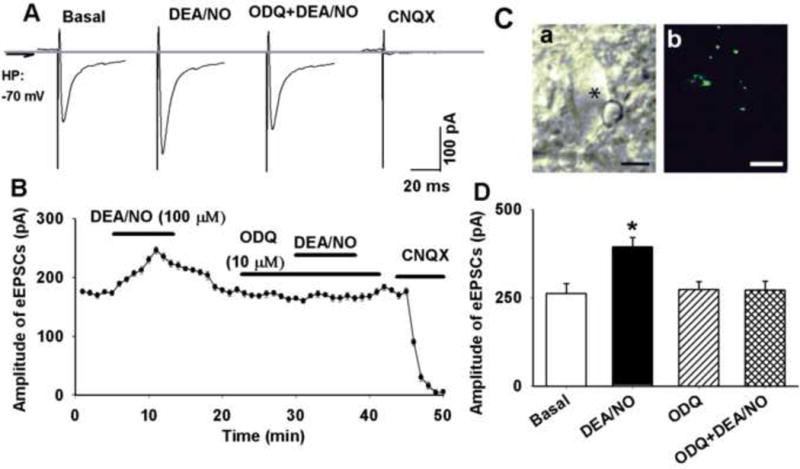
A: Raw tracings showing TS-eEPSCs in a labeled NTS neuron during control and application of 100 μM DEA/NO, DEA/NO plus ODQ (10 μM), and CNQX (20 μM). The traces are averages of 10 consecutive responses. B: ODQ abolished DEA/NO-induced increase in eEPSCs in a labeled NTS neuron. C: an NTS neuron (indicated by *) viewed with infrared light and differential interference contrast optics (a) and the attached DiA-labeled baroreceptor afferents terminals (b, green color). D: summary data showing that DEA/NO increased amplitude of TS-evoked EPSCs in second-order NTS neuron, an effect was abolished by ODQ. Data were presented as means ± SEM. * P < 0.05 compared with control value. Scale bars in C a and b: 10 μm.
Presynaptic mechanism was responsible for NO donor-induced increase in synaptic inputs
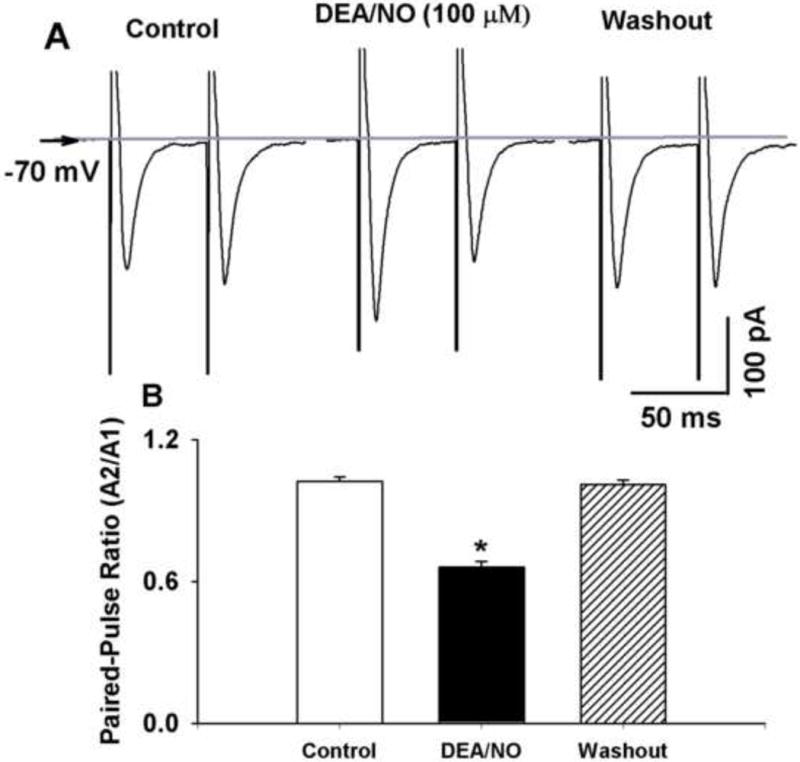
We next examined the effect of 100 μM DEA/NO on the paired-pulse ratio (PPR) of TS-evoked EPSCs. Bath application of 100 μM DEA/NO increased amplitude of the first EPSCs (A1), while produced lesser increase in the amplitude of the second EPSC2 (A2). The PPR (A2/A1) decreased from 1.15 ± 0.05 to 0.71 ± 0.08 in 7 labeled NTS neurons (Figure 2). Furthermore, we determined the effect of 100 μM DEA/NO on miniature EPSCs (mEPSCs) in the presence of 1 μM tetrodotoxin. Bath application of 100 μM DEA/NO increased the frequency of mEPSCs from 4.3 ± 1.2 to 6.2 ± 1.4 Hz without affecting the amplitude of the mEPSCs (20.54 ± 2.3 vs. 22.40 ± 2.65 pA) in 9 labeled NTS neurons (Figure 3).
Figure 2. NO donor decreased paired-pulse ratio (PPR) of eEPSCs in second-order NTS neurons.
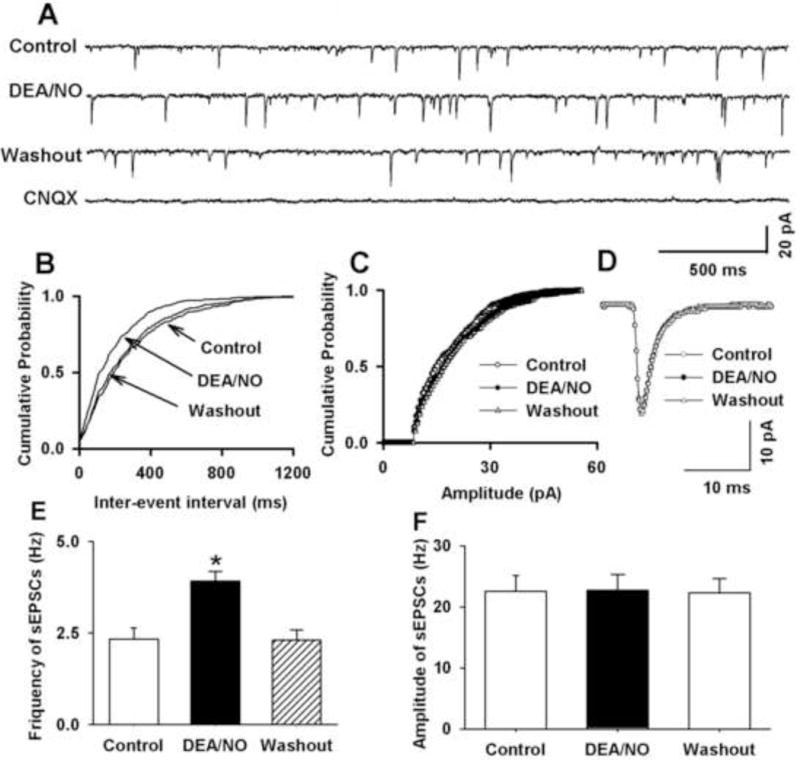
A and B: Raw tracings (A) and summary data (B) showing paired-pulse eEPSCs during control and application of 100 μM DEA/NO in a NTS neuron. Traces are averages of 10 consecutive responses. Data were presented as means ± SEM. * P < 0.05 compared with control value.
Figure 3. NO donor increased frequency of mEPSCs in second-order NTS neurons.
A: Raw tracings showing mEPSCs during control, application of 100 μM DEA/NO, washout, and 20 μM CNQX in a labeled NTS neuron. B and C: Cumulative plot analysis of mEPSCs of the same neuron showing the distributions of the inter-event interval (B) and amplitude (C) during control, application of DEA/NO, and washout. D: Superimposed averages of 100 consecutive mEPSCs obtained during control, application of DEA/NO, and washout. E and F: summary data showing that DEA/NO increased the frequency, but not amplitude, of mEPSCs in second order NTS neurons. Data were presented as means ± SEM. * P < 0.05 compared with control value.
Localization of Cav2.2 subunits in the baroreceptor nerve terminals in the NTS
Because Cav2.2-mediated N-type Ca2+ current is the most prominent VGCCs in the baroreceptor NG neurons [13], we determined the spatial relationship between Cav2.2 subunits and DiA-labeled afferent terminals in the NTS. The slices containing DiA-labeled afferent terminals were immunostained with a specific antibody against Cav2.2 subunit of Ca2+ channels. The confocal images revealed numerous puncta in the NTS that were DiA-labeled nerve terminals (green) and immunoreactive for Cav2.2 (red). The yellow color indicated that DiA-labeled nerve terminals and Cav2.2 immunoreactivities were colocalized in the NTS (Figure 4C).
Figure 4. Confocal images depicting the spatial distribution of Cav2.2 immunoreactivities in baroreceptor afferent terminals in NTS slice.
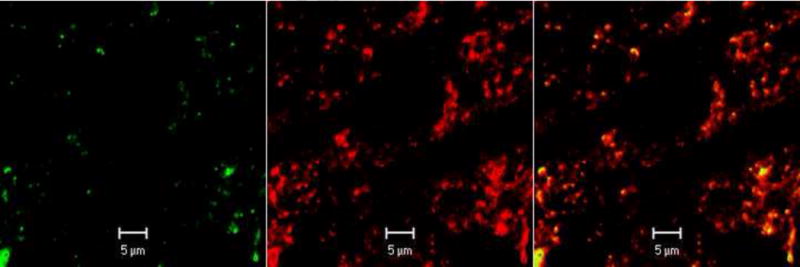
A: Anterograde labeling of baroreceptor afferent terminals (green) in the NTS. B: Immunoreactivities of Cav2.2 subunits (red) under the same view field in NTS slice. C: Merged images of A and B.
NO donor increased N-type Ca2+ currents in identified baroreceptor NG neurons
We next determined the effect of DEA/NO on VGCCs in labeled NG neurons. To avoid the interference of Ca2+ with intracellular signal pathways, we recorded barium current (IBa). DEA/NO (1 mM) increased the current density of IBa from 70.1 ± 7.9 to 97.0 ± 7.0 pA/pF in 9 labeled NG neurons. However, DEA/NO (1 mM) slightly increased the residual IBa after blockade of N-type Ca2+ channels with 1 μM ω-conotoxin GVIA (Figure 5).
Figure 5. NO donor increased IBa in baroreceptor NG neurons.
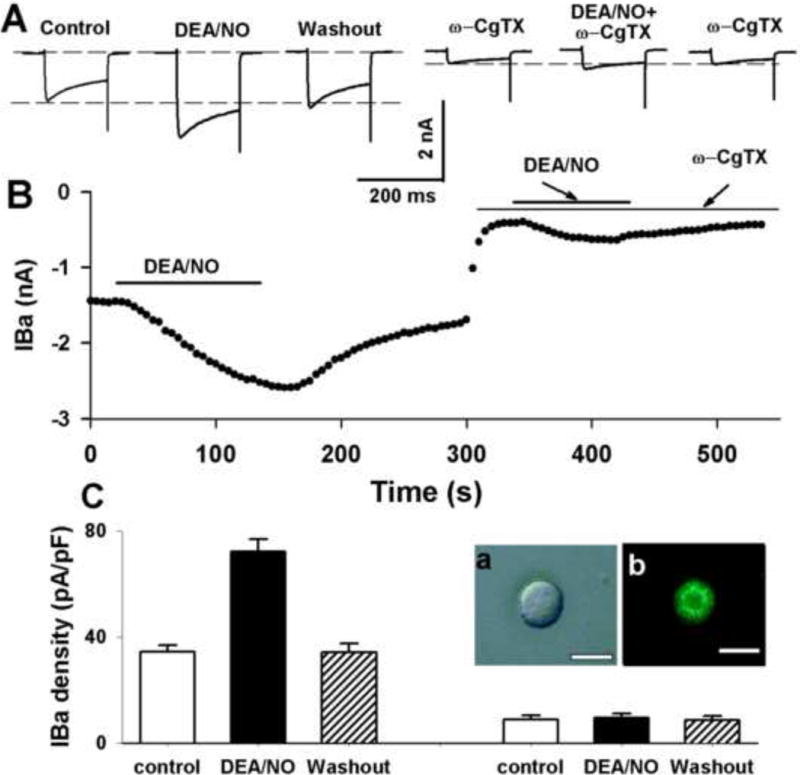
A: Representative raw tracings showing the effect of 1 mM DEA/NO on IBa before and after blockade of N-type Ca2+ channels with ω-conotoxin GVIA in a labeled NG neuron. B: Time course of NO-induced increases in IBa. The IBa currents were elicited by a test pulse from a holding potential of −90 to −10 mV for 150 ms. ω-CgTX: ω-conotoxin GVIA.
Discussion
In this study, we investigated the role of Cav2.2-mediated N-type Ca2+ currents in NO-induced increase in glutamatergic synaptic inputs to second-order NTS neurons. The second-order NTS neurons receiving baroreceptor afferents were identified in brainstem slices by applying a fluorescent lipophilic tracer, DiA, around the surface of AND [4, 13]. It should be noted that some ADN fibers are sensitive to PO2 level but not sensitive to increase in the blood pressure [2]. However, it has been shown that ADN of rats does not contain a significant number of functional chemoreceptor afferent fibers. Furthermore, these ADN chemosensitive afferent fibers cannot generate a chemoreflex-like response [8]. Thus, it is likely that NTS second-order neurons receiving labeled ADN fibers represent inputs from peripheral baroreceptors.
Previous studies have shown that NO-induced depressor response in the NTS is mediated by augmentation of glutamate release. Blocking ionotropic glutamate receptors attenuates NO-induced depressor effect [10]. Consistent with these in vivo studies, we found that NO increases glutamatergic synaptic inputs to second-order NTS neurons, an effect was abolished by sGC inhibitor ODQ, suggesting that sGC-cGMP-PKG pathway is involved in the downstream signal pathways. Because ODQ alone had little effect on evoked EPSCs, it seems that the sGC-cGMP-PKG pathway is not activated in the absence of NO stimulation. We chose DEA/NO from a wide range of NO donors based on the fact that DEA/NO produces stable concentrations of NO and has a consistent effect on the evoked postsynaptic potentials in brainstem slice preparation [18]. To determine if the effect of NO donor on TS-evoked EPSCs was through a presynaptic mechanism, we calculated the PPR for the evoked EPSCs in second–order NTS neurons. If DEA/NO increases the probability of synaptic glutamate release, the ratio of the second EPSC amplitude (A2) to the first EPSC amplitude (A1) should change. In contrast, if DEA/NO act postsynaptically, the ratio of (A2/A1) should remain unchanged since NO increases A1 and A2 to the same degree. We found that DEA/NO decreased the PPR of evoked EPSCs, suggesting that NO increases the probability of presynaptic glutamate release in the synaptic terminal. In support of this notion, we also found that DEA/NO increases the frequency of miniature EPSCs without changing their amplitude. It should be noted that the miniature EPSCs represent glutamate release from all sources of glutamate synaptic inputs to the NTS neurons including baroreceptor terminals, while The TS-evoked EPSCs represent glutamate release from baroreceptor nerve terminals. It is likely that the effect of NO on glutamate release is not limited to the baroreceptor nerve terminals since nNOS-immunoreactive fibers are localized with glutamate-immunoreactive fibers in close apposition in the NTS [11]. However, we cannot completely rule out the potential effect of NO on postsynaptic neurons in the NTS at a circumstance that relatively high concentration of NO of NO is produced and diffused from its site of production to affect distant neurons.
In this study, we also determined whether NO-induced increase in glutamatergic synaptic transmission is dependent on VGCCs. Given that the Ca2+ channel subtypes in soma of NG neurons are closely correlated with the Ca2+ channels responsible for neurotransmitter release at their central synaptic terminals in the NTS [12], we determined the effect of NO on Ca2+ channels in identified baroreceptor NG neurons. We found that DEA/NO significantly increased the Ba2+ current in labeled NG neurons. However, the NO-induced increases in Ba2+ current were abolished after blocking N-type Ca2+ channels with ω-conotoxin-GVIA in labeled NG neurons. These data suggest that N-type subtype is the most prominent subtype for VGCCs in the NG neurons mediating the effect of NO. Because N-type Ca2+ current is predominantly mediated by Cav2.2 subunit of VGCCs, we further determined the spatial distribution of nNOS and Cav2.2 subunit of VGCCs in the baroreceptor nerve terminals in the NTS. We found that Cav2.2 subunit immunoreactivity colocalizes with nNOS in the baroreceptor nerve terminals in the NTS. In addition to N-type Ca2+, other Ca2+ channel subtypes such as P/Q (Cav2.1)- and R (Cav2.3)-type Ca2+ channels contributes to neurotransmitter release in the central nervous system [15]. Therefore, we cannot rule out the possibility that subtypes other than N (Cav2.2)-type Ca2+ channels are involved in NO-induced increase in glutamatergic synaptic inputs to NTS neurons.
In summary, findings from this study significantly advance our understanding of the cellular and synaptic mechanisms underlying the regulation of activity of NTS neurons involved in the control of autonomic function. Our data suggests that NO excites second-order NTS neurons through an increase in glutamatergic synaptic inputs. The enhanced glutamate release may lead to an increase in NO production through activation of nNOS by NMDA and AMPA receptors. Thus, glutamate and NO may reciprocally interact as a positive feedback loop in the regulation of excitability of NTS neurons.
Highlights.
Nitric oxide increase presynaptic glutamate release to baroreceptor NTS neurons
Nitric oxide increase N-type Ca2+ currents in baroreceptor NTS neurons in nodose ganglion
Cav2.2 subunit is present on the baroreceptor afferent nerve terminals
Acknowledgments
This work was supported by NIH grant (MH096086).
List of abbreviations
- aCSF
artificial cerebrospinal fluid
- ADN
aortic depressor nerve
- AMPA
α-amino-3-hydroxy-4-isoxazoleproprionic acid
- CNQX
6-cyano-7-nitroquinoxaline-2,3-dione
- DEA/NO
Diethylammonium (Z)-1-(N,N-diethylamino)diazen-1-ium-1,2-diolate
- DiA
4-(4-dihexadecylamino)styryl)-N-methylpyridinium iodide
- EPSCs
excitatory postsynaptic currents
- GABA
γ-aminobutyric acid
- IPSCs
inhibitory postsynaptic currents
- NG
nodose ganglion
- NO
nitric oxide
- NTS
nucleus tractus solitaries
- ODQ
1H-[1,2,4]oxadiazolo[4,3,-a]quinoxalin-1-one
- PPR
paired-pulse ratio
- sGC
soluble guanylyl cyclase
- TS
tractus solitarius
- VACCs
voltage-activated Ca2+ currents
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Aicher SA, Sharma S, Pickel VM. N-methyl-D-aspartate receptors are present in vagal afferents and their dendritic targets in the nucleus tractus solitarius. Neuroscience. 1999;91:119–132. doi: 10.1016/s0306-4522(98)00530-2. [DOI] [PubMed] [Google Scholar]
- 2.Brophy S, Ford TW, Carey M, Jones JF. Activity of aortic chemoreceptors in the anaesthetized rat. J Physiol. 1999;514(Pt 3):821–828. doi: 10.1111/j.1469-7793.1999.821ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen C, Schofield GG. Nitric oxide donors enhanced Ca2+ currents and blocked noradrenaline-induced Ca2+ current inhibition in rat sympathetic neurons. J Physiol. 1995;482(Pt 3):521–531. doi: 10.1113/jphysiol.1995.sp020537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen CY, Bonham AC. Glutamate suppresses GABA release via presynaptic metabotropic glutamate receptors at baroreceptor neurones in rats. J Physiol. 2005;562:535–551. doi: 10.1113/jphysiol.2004.076885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dias AC, Colombari E, Mifflin SW. Effect of nitric oxide on excitatory amino acid-evoked discharge of neurons in NTS. Am J Physiol Heart Circ Physiol. 2003;284:H234–240. doi: 10.1152/ajpheart.00037.2002. [DOI] [PubMed] [Google Scholar]
- 6.Harper AA. Similarities between some properties of the soma and sensory receptors of primary afferent neurones. Exp Physiol. 1991;76:369–377. doi: 10.1113/expphysiol.1991.sp003504. [DOI] [PubMed] [Google Scholar]
- 7.Hirooka Y, Sakai K, Kishi T, Ito K, Shimokawa H, Takeshita A. Enhanced depressor response to endothelial nitric oxide synthase gene transfer into the nucleus tractus solitarii of spontaneously hypertensive rats. Hypertens Res. 2003;26:325–331. doi: 10.1291/hypres.26.325. [DOI] [PubMed] [Google Scholar]
- 8.Kobayashi M, Cheng ZB, Tanaka K, Nosaka S. Is the aortic depressor nerve involved in arterial chemoreflexes in rats? J Auton Nerv Syst. 1999;78:38–48. doi: 10.1016/s0165-1838(99)00054-5. [DOI] [PubMed] [Google Scholar]
- 9.Lin HC, Kang BH, Wan FJ, Huang ST, Tseng CJ. Reciprocal regulation of nitric oxide and glutamate in the nucleus tractus solitarii of rats. Eur J Pharmacol. 2000;407:83–89. doi: 10.1016/s0014-2999(00)00684-1. [DOI] [PubMed] [Google Scholar]
- 10.Lin HC, Wan FJ, Tseng CJ. Modulation of cardiovascular effects produced by nitric oxide and ionotropic glutamate receptor interaction in the nucleus tractus solitarii of rats. Neuropharmacology. 1999;38:935–941. doi: 10.1016/s0028-3908(99)00017-9. [DOI] [PubMed] [Google Scholar]
- 11.Lin LH, Emson PC, Talman WT. Apposition of neuronal elements containing nitric oxide synthase and glutamate in the nucleus tractus solitarii of rat: a confocal microscopic analysis. Neuroscience. 2000;96:341–350. doi: 10.1016/s0306-4522(99)00560-6. [DOI] [PubMed] [Google Scholar]
- 12.Mendelowitz D, Reynolds PJ, Andresen MC. Heterogeneous functional expression of calcium channels at sensory and synaptic regions in nodose neurons. J Neurophysiol. 1995;73:872–875. doi: 10.1152/jn.1995.73.2.872. [DOI] [PubMed] [Google Scholar]
- 13.Mendelowitz D, Yang M, Andresen MC, Kunze DL. Localization and retention in vitro of fluorescently labeled aortic baroreceptor terminals on neurons from the nucleus tractus solitarius. Brain Res. 1992;581:339–343. doi: 10.1016/0006-8993(92)90729-s. [DOI] [PubMed] [Google Scholar]
- 14.Reid CA, Bekkers JM, Clements JD. N- and P/Q-type Ca2+ channels mediate transmitter release with a similar cooperativity at rat hippocampal autapses. J Neurosci. 1998;18:2849–2855. doi: 10.1523/JNEUROSCI.18-08-02849.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reid CA, Bekkers JM, Clements JD. Presynaptic Ca2+ channels: a functional patchwork. Trends Neurosci. 2003;26:683–687. doi: 10.1016/j.tins.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 16.Southam E, Garthwaite J. The nitric oxide-cyclic GMP signalling pathway in rat brain. Neuropharmacology. 1993;32:1267–1277. doi: 10.1016/0028-3908(93)90021-t. [DOI] [PubMed] [Google Scholar]
- 17.Waki H, Murphy D, Yao ST, Kasparov S, Paton JF. Endothelial NO synthase activity in nucleus tractus solitarii contributes to hypertension in spontaneously hypertensive rats. Hypertension. 2006;48:644–650. doi: 10.1161/01.HYP.0000238200.46085.c6. [DOI] [PubMed] [Google Scholar]
- 18.Wang S, Paton JF, Kasparov S. Autonomic Neuroscience: Differential sensitivity of excitatory and inhibitory synaptic transmission to modulation by nitric oxide in rat nucleus tractus solitarii. Exp Physiol. 2007;92:371–382. doi: 10.1113/expphysiol.2006.036103. [DOI] [PubMed] [Google Scholar]