

Significance
All photosynthetic organisms face the challenge of absorbing solar energy and regulating its flow through their light-harvesting antennas across widely varying photic conditions. For anoxygenic phototrophs, this process is complicated by the need to downregulate photosynthetic output when oxygen is encountered. The Fenna–Matthews–Olson protein from green sulfur bacteria is able to quench excitations in aerobic conditions effectively despite its apparent lack of photoprotective accessory molecules, indicating a previously unidentified type of energy transfer regulation. In this study, we provide evidence for a novel energy-quenching mechanism involving cysteine–bacteriochlorophyll photochemistry. This interaction should be able to be programed into other natural or bio-inspired antennas, opening new possibilities for regulating these systems in response to excess light.
Keywords: photosynthesis, Fenna–Matthews–Olson protein, excitation quenching, thiyl radical, light-harvesting
Abstract
Light-harvesting antenna complexes not only aid in the capture of solar energy for photosynthesis, but regulate the quantity of transferred energy as well. Light-harvesting regulation is important for protecting reaction center complexes from overexcitation, generation of reactive oxygen species, and metabolic overload. Usually, this regulation is controlled by the association of light-harvesting antennas with accessory quenchers such as carotenoids. One antenna complex, the Fenna–Matthews–Olson (FMO) antenna protein from green sulfur bacteria, completely lacks carotenoids and other known accessory quenchers. Nonetheless, the FMO protein is able to quench energy transfer in aerobic conditions effectively, indicating a previously unidentified type of regulatory mechanism. Through de novo sequencing MS, chemical modification, and mutagenesis, we have pinpointed the source of the quenching action to cysteine residues (Cys49 and Cys353) situated near two low-energy bacteriochlorophylls in the FMO protein from Chlorobaculum tepidum. Removal of these cysteines (particularly removal of the completely conserved Cys353) through N-ethylmaleimide modification or mutagenesis to alanine abolishes the aerobic quenching effect. Electrochemical analysis and electron paramagnetic resonance spectra suggest that in aerobic conditions the cysteine thiols are converted to thiyl radicals which then are capable of quenching bacteriochlorophyll excited states through electron transfer photochemistry. This simple mechanism has implications for the design of bio-inspired light-harvesting antennas and the redesign of natural photosynthetic systems.
Photosynthesis can be performed in the presence of oxygen (oxygenic photosynthesis, in which water is the electron source) or in its absence (anoxygenic photosynthesis, in which other reduced species such as sulfide are the electron sources) (1–3). Many bacteria performing anoxygenic photosynthesis contain type I reaction center (RC) protein complexes that use Fe-S clusters to transfer electrons to ferredoxin or related molecules after photoinduced charge separation (4–6). These Fe-S clusters are easily damaged by molecular oxygen. Therefore, anoxygenic phototrophs must use a pathway either to remove oxygen or to reduce the rate of photosynthesis whenever oxygen is encountered (1, 5).
Phototrophic members of the bacterial phylum Chlorobi (green sulfur bacteria, GSBs) are anoxygenic phototrophs that contain such a type I RC. They also contain two peripheral antenna complexes that aid in regulating light absorption and energy transfer: the chlorosome and the homotrimeric Fenna–Matthews–Olson (FMO) protein complex. When oxygen is encountered, the bacteria are able to decrease the output of photosynthesis. This process involves creating trapping sites in the antenna complexes where de-excitation processes outcompete the rate of energy transfer to the RC, preventing RC damage. This process is well understood in the chlorosome, involving redox-sensitive quinones dispersed among the self-assembled bacteriochlorophyll (BChl) pigments (7–10). The photoprotective mechanism in the FMO protein, in contrast, has thus far been enigmatic.
Studies in the 1980s and 1990s first described a “dithionite effect,” in which adding the strong reductant sodium dithionite [midpoint potential vs. natural hydrogen electrode (NHE) approximately −400 mV] to an oxygenated solution of FMO increases the fluorescence emission response and lengthens the dominant fluorescence lifetime of the complex’s BChl a pigments (11, 12). In oxidizing conditions, the fast lifetime of the quenching pathway (60 ps) is thought to compete with the rate of energy transfer to the RC. This pathway effectively disappears in reducing conditions, allowing the pigments to fluoresce with lifetimes similar to that of monomeric BChl a (2 ns) (13). It was suggested that the quenching effect might be controlled by a modified aromatic amino acid (e.g., tyrosylquinone or trytophylquinone) that plays a role similar to that of the excitation-quenching quinones found in the chlorosome (12). However, to this point, no such amino acid has been found via crystallography or other methods.
The goals of this study were to describe the molecular source of this redox-sensing effect, determine the midpoint potential of the redox-sensing species, and establish a viable excitation-quenching mechanism based on the results. We discovered that the two cysteine residues (C49 and C353) in each FMO protein from the model organism Chlorobaculum (Cba.) tepidum TLS were highly sensitive to redox conditions. These residues are situated near BChls 2 and 3, the lowest-energy BChls in the complex, but are spatially separated from each other by >20 Å, precluding disulfide bond formation. Chemical modification of these cysteines in vitro substantially changed the energy-transfer profile and the redox modulation of the complex. Producing cysteine-deficient mutants of the protein (14) allowed further evaluation of the quenching effect. A double mutation (C49A/C353A) produced a protein lacking the redox effect altogether. We characterized these changes by using absorption spectroscopy, steady-state and time-resolved fluorescence (TRF) spectroscopy, direct electrochemistry, and electron paramagnetic resonance (EPR) spectroscopy. We propose a mechanism in which the charge state of the cysteines (thiyl radical vs. thiol, dictated by redox conditions) affects the photophysical behavior of the BChl a pigments.
Results
Identification of Cysteines as Residues of Interest.
In the paper first describing the FMO protein’s redox-dependent quenching effect in detail (12), it was suggested that an aromatic amino acid residing near a BChl a in the interior of the protein may be modified into a photoactive quinone-like species capable of quenching a BChl a excited state. Crystallographic evidence of air-oxidized FMO protein collected since that time does not support this hypothesis (15, 16). Modern MS techniques have the ability to identify modifications at the residue level (17), allowing us to reevaluate the presence or absence of modified amino acids in the FMO protein. Software such as PEAKS (18) allows de novo peptide sequencing, which searches for peptide modifications that may be missed by referencing online peptide databases.
We prepared the FMO protein for LC-MS/MS in two environments. In the aerobic sample, protein precipitation, proteolytic digestion, and peptide purification were performed in open air. In the anaerobic sample, these steps were repeated in an anaerobic chamber with deoxygenated solutions and with the addition of the reductant sodium dithionite to 10 mM. Subsequent peptide mapping indicated that nearly every amino acid in the protein existed in the same, unmodified form equally across both aerobically and anaerobically prepared samples. The only amino acids that showed significant changes were the two cysteine residues Cys49 and Cys353. These cysteines were found to contain more oxidative modifications (i.e., additions of oxygen atoms to the thiol, resulting in moieties such as cysteic acid) when prepared in aerobic conditions than in anaerobic conditions (Fig. S1). Crystallographic evidence (Fig. 1) shows that Cys49 and Cys353 are within 5 Å of the C-3 acetyl groups of BChls 2 and 3, respectively, and are too separated spatially to form a disulfide bridge (15). BChls 2 and 3 are considered to have two of the lowest pigment-site energies in the complex (19).
Fig. S1.

Peptide coverage and modification identification from PEAKS for FMO protein in reducing conditions (A) and in oxidizing conditions (B). The protein preparation scheme for each condition is shown above each map, and the PTM-identified legend is shown at the right of each map. Modifications that appear on these maps represent greater than 5% of the ion intensity of that particular tryptic peptide.
Fig. 1.

Crystal structure of the FMO protein from Cba. tepidum (PDB code 3ENI, visualized in PyMOL), highlighting the cysteine residues of interest and their proximity to BChl a 2 and 3 in a single protein monomer. BChl a phytyl tails are omitted for clarity. Black dotted lines are labeled with interatomic distances of interest. Brown dotted lines represent protein-to-pigment coordination bonds.
Derivatization of Cysteine Residues with N-Ethylmaleimide.
Although the effect of electrospray ionization in the mass spectrometer on the appearance of these oxidative changes cannot be quantified easily, we decided to assay the role of the cysteines on the protein’s redox effect. We covalently maleylated the cysteines using N-ethylmaleimide (NEM), which permanently prevents thiol groups from undergoing further redox chemistry (20–22). MS (Figs. S2 and S3) confirmed successful modification of 18.6% of Cys49-containing peptides and 29.3% of Cys353-containing peptides. The low modification efficiency likely results from the limited accessibility of exogenous large molecules to the interior of the protein.
Fig. S2.

Product-ion spectra for the fragmentation of a tryptic peptide containing Cys49 from a sample of NEM-modified FMO. (Upper) An identified peptide that contains a free thiol cysteine. (Lower) An identified peptide that contains NEM-modified cysteine.
Fig. S3.

Extracted ion chromatograms for the peptides VNAPPASPLLPADCDVK (containing Cys49) and WVEHECK (containing Cys353) with various cysteine modifications identified from the MS1 spectra. Note that each panel has a different y-axis scale.
Upon reaction of FMO protein with NEM, changes in the steady-state optical spectra were observed (Fig. 2). Absorption spectra at 298 K (Fig. 2A, Inset) showed no change, but at 77 K (Fig. 2A) slight shifts are seen in the Qy region. Because we believe from MS evidence that 30%, at most, of the cysteines were successfully treated in the NEM-modified sample, then 70% of the sample must be unmodified. We can decompose the NEM-modified protein absorption spectrum into a 100% NEM-modified spectrum by normalizing modified and unmodified spectra and subtracting an NEM-unmodified spectrum scaled at 70% (Fig. 2A, green line). This spectrum shows that the NEM-induced changes primarily affect the spectral region of 795–805 nm. The presence of the maleimide group has likely perturbed the orientation or polarity of the pigment environment corresponding to this region, shifting the site energies to lower energy. The 825-nm band, representing the lowest-energy pigments in the complex and those pigments responsible for steady-state fluorescence, is largely unchanged by NEM modification, indicating that the orientation or polarity of the pigment environment in that region is largely unchanged. Importantly, the increase of fluorescence emission usually seen upon the addition of dithionite is significantly reduced (Fig. 2B) upon NEM modification. Finally, the fluorescence intensity in oxidizing conditions is slightly higher in the NEM-modified protein than in the unmodified protein.
Fig. 2.

Optical spectroscopy of NEM-modified and unmodified FMO. (A) 298 K (Inset) and 77 K absorption in the Qy region. Because the success of NEM modification was calculated by MS to be ∼30%, we subtracted the unmodified FMO absorption spectrum scaled at 70% from that of the NEM-modified FMO spectrum. The resultant plot (green line) estimates the spectrum of a 100% NEM-modified FMO sample. (B) Fluorescence emission (298 K) of NEM-modified and unmodified FMO in air-oxidized and dithionite-reduced conditions.
Steady-State Fluorescence of FMO Cysteine Mutants.
To probe further the role of cysteine residues in the fluorescence response of the FMO protein caused by the redox condition, we constructed three site-directed mutants: C49A, C353A, and the double mutant C49A/C353A. We have reported elsewhere (14) a method for genetic engineering of the FMO protein and an in-depth analysis of the absorption changes in the protein upon cysteine mutation. At cryogenic temperature, the absorption spectra of each mutant are subtly different from that of the WT, allowing an in-depth reanalysis of the FMO energetic landscape. An X-ray crystal structure solved for the C49A/C353A double mutant [Protein Data Bank (PDB) ID code 5H8Z] showed that the overall protein structure and pigment orientations are unchanged upon mutation (14). Here we focus on the fluorescence response of the mutants to the addition of dithionite (Fig. 3).
Fig. 3.

Fluorescence emission response of WT FMO and the C49A, C353A, and C49A/C353A mutants to excitation at 602 nm. All samples had identical absorbance levels (OD602 nm = 0.06) and were treated with the same amount of sodium dithionite (final concentration: 10 mM) so that all samples could be plotted on identically scaled axes for comparison. Dotted lines, air-oxidized solution; solid lines, dithionite-reduced solution.
Compared with WT, the C49A mutation alone has no discernible effect on the steady-state fluorescence response upon the addition of dithionite (Fig. 3, Upper Right). The C353A mutation (Fig. 3, Lower Left) has an intermediate effect: The fluorescence intensity of the reduced form resembles that of the WT and the C49A mutant, but the intensity of the oxidized form is higher than in the WT and C49A mutant. This mutant also shows a distinctive peak splitting not present in the WT. Last, the C49A/C353A double mutant (Fig. 3, Lower Right) does not show a fluorescence response that can be attributed to the simple addition of the individual mutations; it shows both intermediate fluorescence intensity and an abolished response to dithionite addition.
TRF.
To understand why the steady-state fluorescence spectrum changes so drastically upon NEM modification or cysteine mutation, we performed TRF studies on all protein samples. Because the intensity of steady-state fluorescence is directly linked with the fluorescence decay lifetime (the shorter the lifetime, the less intense the fluorescence), these experiments should clearly demonstrate how the abovementioned mutations/modifications have affected the excited-state properties of the BChls. Representative fluorescence decay traces taken at 815 nm are shown in Fig. 4. Fitting of the traces with a sum of exponential decays, convoluted by the instrument response function (IRF), revealed multiple kinetic components. Their lifetimes and contributions are listed in Table 1.
Fig. 4.

TRF decay kinetics at 815 nm for WT, NEM-modified WT, and mutant FMOs after excitation at 602 nm. (A) Air-oxidized and dithionite-reduced WT protein. (B) Air-oxidized and dithionite-reduced NEM-modified WT protein. (C) Air-oxidized mutant protein. (D) Dithionite-reduced mutant protein. Note that the differences in the x-axis scales are caused by the use of different temporal windows for better resolution of data; the Gaussian approximation of each IRF is included for clarity.
Table 1.
TRF spectral-kinetic component lifetimes (τn) and contribution to the overall fit (An)
| Parameter | WT | WT-NEM | C49A | C353A | C49A/C353A | |||||
| Ox. | Red. | Ox. | Red. | Ox. | Red. | Ox. | Red. | Ox. | Red. | |
| A1 | 1 | – | 0.446 | – | 0.893 | – | 0.478 | – | 0.478 | – |
| τ1, ps | 65 | – | 81.5 | – | 79.2 | – | 114 | – | 114 | – |
| A2 | – | – | 0.285 | 0.306 | 0.107 | 0.337 | 0.523 | 0.668 | – | 0.668 |
| τ2, ps | – | – | 555 | 554 | 691 | 926 | 1,780 | 1,640 | – | 1,640 |
| A3 | – | 1 | 0.269 | 0.694 | – | 0.663 | – | 0.332 | 0.523 | 0.332 |
| τ3, ps | – | 2,200 | 2,220 | 2,270 | – | 2,310 | – | 3,020 | 2,650 | 3,560 |
Ox., oxidized; Red., reduced.
The oxidized FMO from the C49A mutant and WT demonstrate similar kinetic characteristics of BChl a fluorescence decay: a dominating fast kinetic component with a lifetime in the 65–79 ps range (Fig. 4 A and C). The fluorescence decay traces of the FMO from the C353A and C49A/C353A mutants additionally show the substantial contribution of a kinetic component with a significantly longer lifetime. Upon dithionite reduction, the FMOs from all the mutants and from WT show essentially the same fluorescence dynamics (Fig. 4 A and D). Fitting of the decay traces has shown that all mutants gained some new intermediate kinetic component not present in the WT FMO (700–900 ps in C49A and 1.6–1.8 ns in C353A and C49A/C353A). Additionally, in the C353A and C49/C353A mutants the archetypical long lifetime component [2.0–2.2 ns in the WT (12, 23)] lengthens to 2.6–3.6 ns (Table 1).
Most interestingly, an intermediate, ∼550 ps kinetic component is present in the NEM-modified FMO (Fig. 4B) in both oxidizing and reducing conditions. Moreover, its contribution of ∼30% in the overall signal is not sensitive to environmental change (reduction or oxidation), strongly suggesting that this is the intrinsic fluorescence dynamics of the pool of FMOs that have cysteines successfully labeled by NEM. Again, MS has shown that, at most, ∼30% of the all FMOs are successfully NEM labeled (see above and SI Materials and Methods). The ∼500-ps kinetic component likely derives from one or more BChl a molecules perturbed by the presence of the NEM moiety near their binding region within the protein.
Protein Film Voltammetry Analysis.
To measure the redox potential of the redox-active groups in the WT and mutant FMO proteins, we applied protein onto a pyrolytic graphite edge (PGE) working electrode and performed protein film voltammetry using the three-electrode apparatus (reference electrode: Ag/AgCl; counter electrode: platinum wire) described previously (24–27). Fig. 5A shows the faradaic current of the unmodified FMO protein at pH 7.4 when the potential is linearly scanned from −100 mV to −400 mV (potential converted to NHE) and back again. After baseline subtraction, reductive and oxidative peaks are observed. The average midpoint potential is −272 mV vs. NHE at pH 7.4. In addition, the stoichiometric number of electrons in the process, n, was calculated by analyzing the FWHM of the reductive and oxidative peaks, respectively (28). At pH 7.4, the FWHM is 71 mV, representing an n value of 1.28. The average FWHM for WT FMO across all pH values measured is 69 mV, representing an n value of 1.32.
Fig. 5.

Protein film voltammetry (cyclic voltammetry) of WT FMO protein adsorbed onto a PGE electrode. (A) Voltammogram showing the raw faradaic (dotted line) and baseline-corrected (solid line) current at pH 7.4. (B) Dependence of midpoint potential on pH.
Additionally, the midpoint potential of the redox species in the WT FMO protein is dependent on solution pH (Fig. 5B). This plot is linear across four to five pH units, with nonlinear tailing occurring only at pH values below 3.0 and above 9.0. The slope of this pH dependence in the linear region is −52 mV per pH unit, which, coupled with the n ∼1 value described above, indicates a 1:1 proton-coupled electron transfer (PCET) mechanism for the redox-active species (29).
Similar electrochemical profiles could not be observed in any of the three cysteine mutants, likely indicating that the electrochemical activity of the protein is abolished upon cysteine mutation. It is unlikely the cysteine mutations would alter proper protein adsorption to the electrode surface because the solved crystal structure of the C49A/C353A double mutant (PDB ID code 5H8Z) shows no obvious structural changes to the exterior of the protein (14).
EPR.
A 1:1 PCET-type mechanism of redox transformation is a curious finding for cysteines acting alone with no other cofactors. A possible explanation for this type of mechanism would be the formation of a thiyl radical. We propose that in the transition from reducing to oxidizing conditions a cysteine thiol in the FMO protein may lose a proton and an electron to an oxidizing agent to become a thiyl radical, which should be detectable via EPR. In the 1994 paper first describing the FMO redox effect (12), EPR indicated an organic radical singlet centered at g = 2.001. This signature is present in oxidizing conditions but disappears in reducing conditions. We have repeated these measurements in oxidizing conditions for the WT FMO protein and the three mutants. The resulting spectra (Fig. 6) indicate that the organic radical seen in the WT protein effectively disappears upon mutation of either or both cysteine residue(s). This EPR signature is consistent with previously published spectra of thiyl radicals (30–33).
Fig. 6.

Blank-corrected EPR spectra collected in oxidizing conditions at 77 K (10 30-s scans, signal-averaged) of the WT FMO protein and the three mutants. Protein was concentrated to OD808 nm = 150 in 20 mM CAPS, pH 10.5 + 50% (vol/vol) glycerol.
SI Materials and Methods
Protein Preparation.
The GSB Chlorobaculum (Cba.) tepidum was grown and its FMO protein were isolated as previously described (14, 16, 48, 49), except that 20 mM CAPS buffer, pH 10.5 was substituted for Tris⋅HCl buffer in all chromatography steps. We have found that this higher-pH buffer improves the FMO protein’s stability and solubility and the resolution of chromatographic purification. All chemicals used in this study were of the highest grade and were purchased from Sigma-Aldrich. FMO protein considered oxidized was used as-purified, in equilibrium with laboratory atmosphere. FMO considered reduced was first placed into an anaerobic chamber (95% N2 + 5% H2 gases) and allowed to equilibrate open to the chamber for 0.5 h. Sodium dithionite then was added to a final concentration of 10 mM, and the solution was incubated in the chamber in the dark for an additional 0.5 h. Three mutants of the FMO complex—C49A, C353A, and the double mutant C49A/C353A—were constructed and purified in the same manner as the WT.
Mutagenesis of the FMO Protein.
Our method for mutagenesis of the FMO protein has been described previously (14). In brief, chloramphenicol (25 μg/mL) in LB medium (Sigma-Aldrich) was used for selection and maintenance of pSB1C3 in Escherichia coli strain DH5α. For gentamicin selection of E. coli, 10 µg/mL gentamicin (Sigma-Aldrich) was used. The cloning and assembly of the pSB_FMO-EX plasmid for the generation of mutant FMO expression vectors are described in Saer et al. (14). All oligonucleotides (Integrated DNA Technologies) used for the construction of the expression vectors are provided in Table S2.
Table S2.
Oligonucleotides used in this study
| Oligonucleotide | Target | Sequence (5′ → 3′) |
| FMO-F | fmoA | ATGGCTCTTTTCGGCTCAAACGACG |
| FMO-R | fmoA | TTACTGGGCGTAGAGAATTTCAAACTGC |
| FMO_PSB1 | pSB1C3 | ATTCTCTACGCCCAGTAATACTAGTAGCGGCCG |
| FMO_PSB2 | pSB1C3 | TGAGCCGAAAAGAGCCATCTCTAGAAGCGGCCG |
| C49A1 | fmoA (C49) | CTGCTTCCTGCAGACGCCGATGTTAAGCTGAATG |
| C49A2 | fmoA (C49) | CATTCAGCTTAACATCGGCGTCTGCAGGAAGCAG |
| C353A1 | fmoA (C353) | CTGGGTCGAGCACGTCGCCAAAGGTGGCGTCGGG |
| C353A2 | fmoA (C353) | CCCGACGCCACCTTTGGCGACGTGCTCGACCCAG |
| FMOp-F | fmoA upstream | CCAACAAAGGTTGCGAGGCATACCC |
| Gent_FMOp-R | fmoA upstream | GTTTACGAACCGAACAGGCTTATGTCAATTCAATGATTCTCCTTTTTCTGGTG |
| Gent-F | aacC1 | GAATTGACATAAGCCTGTTCGGTTCGTAAAC |
| FMOd_Gent-R | aacC1 | CGAAATTGTTGTTTCTCGCTTAGGTGGCGGTACTTGGGT |
| FMOd-F | fmoA downstream | GCGAGAAACAACAATTTCG |
| PSB_FMOd-R | fmoA downstream | CAGCGGCCGCTACTAGTAGCTCCGCTACTGGGAGGAC |
| FMOp-PSB1 | pSB1C3 | CCTCGCAACCTTTGTTGGCTCTAGAAGCGGCCG |
| PSB2 | pSB1C3 | TACTAGTAGCGGCCG |
| FMO_FMOp-R | fmoA upstream | GTTTGAGCCGAAAAGAGCCATAATGATTCTCCTTTTTCTGGTG |
| FMO_Gent-F | aacC1 | GCAGTTTGAAATTCTCTACGCCCAGTAAGAATTGACATAAGCCTGTTCGGTTCGTAAAC |
| FMO-seq | fmoA upstream | GGAAGCAACCCAGTGTTTTTCCTGAAAATAC |
For the cloning and assembly of the pSB_FMO-EX plasmid for generation of mutant FMO expression vectors, see Saer et al. (14).
To transform Cba. tepidum, the natural transformation protocol of Saer et al. (14), which is a modification of the Frigaard et al. (50) protocol, was used. All transformations were performed on Cba. tepidum strain WT2321, a strain tolerating growth on solid medium, which was a kind gift from Donald Bryant at The Pennsylvania State University, State College, PA. Briefly, a 20-µL SpeI restriction digest containing 1 μg of pSB_FMO-EX plasmid was combined with 80 µL of Cba. tepidum cells (a concentrated suspension from a 500-µL culture at OD660 = 1.2–1.5). The mixture was transferred to an anaerobic chamber (95% N2 + 5% H2 gases) and spotted on a Chlorobium plating (CP) agar plate (50). The plates were placed in a GasPak anaerobic jar (BD) with a GasPak EZ anaerobe pouch (BD) and thioacetamide (Sigma-Aldrich) to provide CO2 and H2S, respectively. The jar was placed overnight in a cabinet illuminated by incandescent bulbs at 33–35 °C with a photosynthetically active radiation (PAR) photon flux of 2.3 μE⋅m−2⋅s−1. The following day, the cells were resuspended and spread on CP plates containing 100 μg/mL gentamicin. The plates were incubated in the illuminated cabinet as described above for 1 wk. The resulting cell lawns were streaked on fresh CP plates containing gentamicin and were incubated for ∼3 wk until individual colonies appeared. Colonies of Cba. tepidum cells with FMO mutations were screened by PCR using GoTaq Green Master Mix (Promega).
Initial Peptide Modification Analysis with LC-MS/MS and PEAKS Software.
Oxidized FMO samples and reduced FMO samples from the WT bacteria (each ∼200 μg total protein) were precipitated using acetone, resuspended in urea, and digested in solution using trypsin as described previously (51), except that iodoacetamide was not added to any digestion mixture. For the oxidized sample the procedure was performed in open air using air-equilibrated acetone. For the reduced sample the procedure was performed in the anaerobic chamber using acetone that had been degassed using at least six freeze–pump–thaw cycles. Reduced samples were introduced to the air only briefly at the time of injection into an MS instrument.
Tryptic fragments were injected into an Ultimate 3000 Nano LC system (Thermo Scientific Dionex) equipped with a home-packed Michrom Magic C-18 UPLC column. Fragments were analyzed with a Q-Exactive Plus Orbitrap mass spectrometer (Thermo Scientific).
Peptides were identified from the accurate masses of precursor ion (MS1) spectra and product ion (MS2) sequencing by either searching against the NCBI database using Mascot (Matrix Science) or sequencing de novo using PEAKS 7.0 (Bioinformatics Solutions Inc.). Using PEAKS, quantitative information about the level of chemical modification in each set of tryptic peptides could obtained (18) and compared among all samples.
For analysis using PEAKS, the MS2 data were directly loaded into the software for peptide reconstruction (de novo sequencing) by using an error tolerance of 10 ppm for the precursor (parent) ion and 0.01 Da for the fragment (daughter) ions. All common amino acid modifications available within the software were considered during sequencing. Peptide identification was accepted if the peptide-spectrum match’s −10logP confidence score was higher than 30 (i.e., the chance of misidentification was unacceptably high at −10logP <30), the matching product-ion spectrum had more than 25% of b or y ion peaks identified, and the minimum fragment-ion relative intensity for a peptide with modification was larger than 2%.
Cysteine Modification with NEM and MS Confirmation of Modification.
After identification of the cysteine residues in the FMO protein as biochemical targets for analysis, samples of WT FMO protein were modified with NEM, which permanently derivatizes any free, accessible protein thiols. The modification procedure was performed as follows: A concentrated 200-μL solution (containing ∼15 μM protein) of sample was dialyzed to 150 mM potassium phosphate buffer, pH 7.0. This sample was reduced using sodium dithionite in the anaerobic chamber as described previously (presumably, any accessible cysteines in the protein then would exist in the free thiol form). Then, 20 µL of 125-mM NEM was added to the sample. The derivatization proceeded in the dark at room temperature for 2 h. The derivatized protein samples then were thoroughly buffer-exchanged into fresh air-equilibrated phosphate buffer.
After modification of cysteines, ∼5 μg of modified protein in 100 μL of buffer was prepared for LC-MS/MS. The protein was precipitated using a 2D Clean-Up Kit (GE Healthcare Biosciences). The protein pellets were gently dried in open air and then were resuspended in 100 mM Tris⋅HCl (pH 8.5) with 0.1% RapiGest detergent (Waters Corp.) and were digested in solution with trypsin using a previous protocol (51). Tryptic peptides were purified using C-18 zip-tip pipettes, diluted 10 times with water + 0.1% formic acid, and injected into the aforementioned LC system and Q-Exactive Plus Orbitrap mass spectrometer system for LC-MS/MS. Peptide sequencing and reconstruction were performed by loading the MS2 data into the PEAKS software as described above. Quantitative information about the level of NEM modification in each set of tryptic peptides was obtained by comparing the MS1 intensities (parent MS1 ions were identified by the chemical modifications found in the MS2 daughter ions) among all samples. All common cysteine modifications available within the software were considered during sequencing.
Steady-State Absorption and Fluorescence Spectroscopy.
Steady-state absorption spectroscopy was performed in a UV-1800 UV/Vis/NIR spectrophotometer (Shimadzu Corp. USA). Cryogenic measurements were performed using a liquid nitrogen Optistat DN-2 optical cryostat (Oxford Instruments). Before cryogenic experiments, pure glycerol was added to each sample to a final concentration of 50% (vol/vol) to act as a glassing agent.
Steady-state fluorescence experiments were performed using a NanoLog Spectrofluorometer (Horiba Scientific) equipped with a Vis-NIR grating and CCD detector. All FMO samples were excited into the Qx absorption band at 602 nm. In addition to sodium dithionite, various other reductants were tested for their ability to affect the fluorescence of the FMO protein, including reduced glutathione, β-mercaptoethanol, sodium sulfide, DTT, Tris(2-carboxyethyl)phosphine (TCEP), cysteine, sodium thiosulfate, and sodium sulfate. When used, these reductants were added to a 10-mM final concentration.
Picosecond Time-Resolved Fluorescence, Data Processing, and Global Fitting.
TRF measurements were carried out using the laser system and universal streak camera setup described previously (52) with minor modifications. Excitation pulses were generated at 602 nm with a frequency of 8 MHz (125 ns between subsequent excitations). The energy of the excitation beam was less than 0.002 μJ, corresponding to a photon flux of less than 2.0 × 1011 photons per square centimeter per pulse. In the streak camera (Hamamatsu Corp.), either a slow M5677 single-sweep unit (time window 1 ns to milliseconds) or a fast M5676 single-sweep unit (time window 100 ps to 1 ns) was used, depending on the temporal resolution required.
The TRF spectra were cleared from random noise by recomposing the data from the dominant principal components by using the singular value decomposition (SVD). Then, the spectra were globally fit with a combination of monoexponentially decaying spectral-kinetic components convoluted by Gaussian approximation of the IRF using the program ASUfit (www.public.asu.edu/∼laserweb/asufit/asufit.html) (13). The IRF of the streak camera tube was confirmed by recording a profile of the scattered excitation laser beam in an appropriate streak camera time window.
Protein Film Voltammetry.
Before use, a PGE working electrode was cleaned using 1-μm alumina slurry on cotton wool, followed by thorough sonication in water. The electrode then was passed into the aforementioned anaerobic chamber, where 10 μL of a highly concentrated FMO solution (OD808 nm >50) was pipetted onto the graphite tip. After a 2-min equilibration on the electrode, the remaining protein solution was pipetted off, and the PGE electrode was placed into an open glass cell containing a 75-mM mixed buffer [15 mM each of Hepes, 2-(cyclohexylamino)ethanesulfonic acid (CHES), 3-[[1,3-dihydroxy-2-(hydroxymethyl)propan-2-yl]amino]propane-1-sulfonic acid (TAPS), Mes, and sodium acetate] titrated to the desired pH with concentrated NaOH or HCl, with 100 mM NaCl added as a supporting electrolyte. An Ag/AgCl reference electrode (+197 mV vs. the NHE) and a platinum wire counter electrode were inserted into the open glass cell as well. A custom plastic cap was 3D printed to hold the electrodes in place so that they were at the same vertical level and did not touch. All electrodes were connected to a Model 600 Series Electrochemical Analyzer workstation (CH Instruments, Inc.). At a given pH, the potential between the working and reference electrode was cycled twice from +197 to −603 mV vs. Ag/AgCl and back at a constant scan rate of 100 mV/s while measuring the change in current between the working and counter electrodes (i.e., cyclic voltammetry using a triangular potential waveform). The second scan was retained for analysis. All data were baseline corrected, converted to potentials vs. NHE, and analyzed with the program SOAS (bip.cnrs-mrs.fr/bip06/software.html) (53). A baseline was obtained by scanning voltage with no protein adsorbed onto the electrode. Scanning to higher oxidative potential did not reveal any other protein-derived redox couples.
Searching for Cysteines in Other Antenna Complexes.
In a cursory search, 127 antenna proteins from anoxygenic phototrophs were surveyed for the presence of cysteine residues using the NCBI database. Of the 127 proteins, 20 were from the green bacterial CsmA antenna, 13 were from the filamentous anoxygenic phototrophic B-808/866 antenna (seven α and six β chains), 45 were from the purple bacterial LH1 complex (19 α and 26 β chains), and 49 were from the purple bacterial LH2 complex (24 α and 25 β chains). Within each antenna protein group, the proteins came from a representative selection of organisms; e.g., within the purple bacterial LH2 protein group, various groups of purple bacteria, such as purple nonsulfur bacteria, purple sulfur bacteria, and aerobic anoxygenic marine purple bacteria, were represented. Table S1 shows that only a small group of LH2 and LH1 proteins, primarily from aerobic purple bacteria, contain any cysteines among the anoxygenic photosynthetic antenna complexes (Note: the subject of cysteine utilization among FMO antenna proteins is described in the main text).
Table S1.
Cysteine utilization among a representative group of anoxygenic phototroph photosynthetic antenna complexes
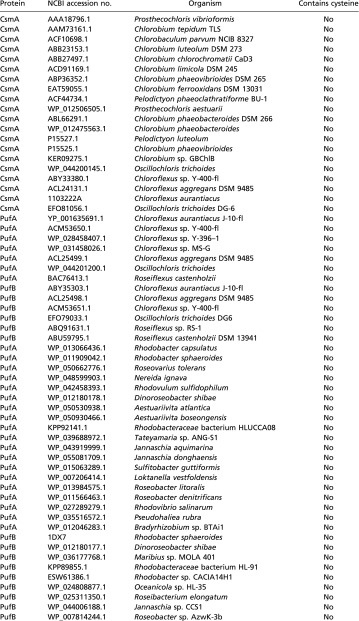 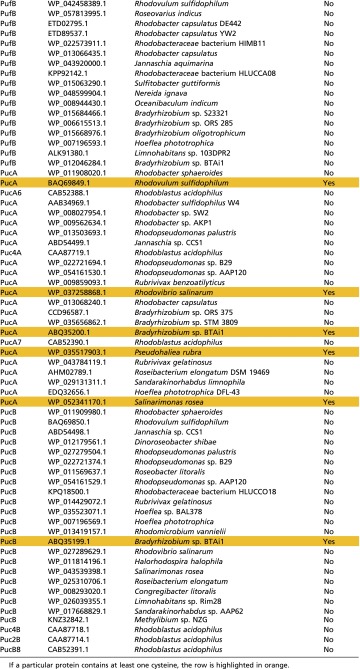
|
Discussion
Cysteines Play a More Important Role in Energy Transfer Through the FMO Protein than Previously Thought.
In this study we have revisited the molecular source of the FMO protein’s redox-dependent modulation of energy transfer. An initial MS analysis of the protein nominated the cysteine residues for further study. Cysteine modification with NEM resulted in a protein sample with altered absorption and fluorescence properties. Additionally, a 1:1 PCET transformation was electrochemically detected at a potential of −272 mV vs. the NHE at the physiological pH of 7.4. Individual and tandem mutation of the cysteine residues provided further evidence for their involvement with redox cycling in the protein. The fluorescence-quenching effect is attenuated in the C353A single mutant and is abolished in the C49A/C353A double mutant. Electrochemical signatures similar to the WT could not be observed in any of the three mutants. Finally, EPR shows that a thiyl radical signature present in the oxidized protein disappears upon cysteine mutation. Interestingly, the C49A mutant behaves similarly to the WT in terms of its fluorescence activity but lacks electrochemical and EPR response; although C49 and C353 are situated distantly from each other, these two residues may exert some cooperative effect on one another. Our recent determination of the C49A/C353A mutant’s 3D structure via X-ray crystallography (PDB code 5H8Z) shows no evidence for secondary effects of the mutations on the structure of the protein or orientation of the pigments within their binding pockets (14). Therefore, the fluorescence, electrochemical, and EPR responses of the mutants seem to be genuinely tied to the presence of protein thiol groups.
Because excitations are delocalized within the FMO protein among its various pigments, there are many possible paths of energy transfer through the complex, with some paths being more favorable than others (19, 34–37). A recent report modeled several favorable paths for energy flow through the complex in silico, indicating that BChls 2 and 4 contribute most strongly to the final steps of energy transfer to the lowest-energy BChl a, BChl 3 (38). From a regulatory standpoint, it is logical for regulatory species to be positioned near two low-energy pigments (i.e., BChls 2 and 3). The mutagenesis of the cysteine residues near BChls likely changes the electrostatic environments of the pigment-binding pockets, in turn altering the observed fluorescence lifetimes of the final energy acceptors. Compelling evidence for electrostatic effects taking a role comes from the TRF experiments, which show the appearance of a new intermediate decay component in the 1-ns regime although there are no large-scale changes in protein structure upon cysteine mutation (14).
Conservation of Cysteines Across FMO and Other Antenna Complexes.
The regions surrounding the two cysteines are highly conserved across GSBs (which all contain FMO) and Chloracidobacterium (Cab.) thermophilum (the only outgroup known to contain the FMO protein) (Fig. 7). Cys353 is retained among all members, but Cys49 is conserved only among the closely related members of the family Chlorobiaceae (containing the genera Chlorobium, Chlorobaculum, and Prosthecochloris) and genus Chloroherpeton. “Candidatus (Ca.) Thermochlorobacter aerophilum” (a GSB outgroup to the family Chlorobiaceae) and Cab. thermophilum (an outgroup to the whole phylum Chlorobi) contain a shifted cysteine in this region or no cysteine at all, respectively. The organisms lacking the N-terminal cysteine have the capability to grow in microaerobic conditions, conditions in which other GSBs do not thrive (39, 40). If this cysteine contributes to the repression of FMO-to-RC energy transfer in oxidizing conditions, it is logical for a microaerobe to lose this regulation to photosynthesize better in the presence of oxygen. Indeed, a past study on Cab. thermophilum indicated that its FMO protein is less responsive to the addition of sodium dithionite (41). This observation provides precedence for the aforementioned cooperativity between C49 and C353 in the redox effect; with Cab. thermophilum FMO missing one of these cysteines, its redox effect is merely attenuated, not abolished.
Fig. 7.

Alignments of the FMO protein N-terminal and C-terminal regions from various members of the GSB phylum Chlorobi and the chlorophototrophic Acidobacterium Chloracidobacterium thermophilum. The alignment was constructed using Clustal Omega (www.ebi.ac.uk/Tools/msa/clustalo/). The sequence from “Ca. Thermochlorobacter aerophilum” was provided by Donald Bryant of The Pennsylvania State University, State College, PA (40). The arrows indicate conserved positions 49 and 353; cysteines are colored red.
Surprisingly, cysteine residues are severely underrepresented among anoxygenic photosynthetic light-harvesting complexes. Using the National Center for Biotechnology Information (NCBI) protein database, we examined the LH2 proteins from 32 unique photosynthetic purple bacteria, LH1 proteins from 38 unique purple bacteria, the B806–866 complex from 10 Chloroflexi, and the CsmA baseplate protein from 20 Chlorobi and Chloroflexi (for a list of proteins and organisms, see Table S1). The total number of proteins surveyed was 127 (some organisms contained multiple isoforms of the same antenna protein). None of the CsmA or B806–866 proteins surveyed contained any cysteine residues. Of the Proteobacteria, only five species (Rhodovulum sulfidophilum, Rhodovibrio salinarum, Bradyrhizobium sp. BTAi1, Salinarimonas rosea, and Pseudohaliea rubra) contained one or two cysteines in their LH2 α-polypeptide, and only one (Bradyrhizobium sp BTAi1) contained a cysteine in its LH2 β-polypeptide. None of the Proteobacteria contained any cysteines their LH1 α- or β-polypeptides. In the case of the photosynthetic Proteobacteria, the cysteine-containing LH2 proteins come from marine aerobes (Rhodovulum, Rhodovibrio, and Pseudohaliea) or green plant symbionts (Salinarimonas and Bradyrhizobium). These organisms are capable of heterotrophy and may be inclined to decrease, but not stop, photosynthetic output in aerobic conditions. Most photosynthetic Proteobacteria in general have a well-described genetic repression system to halt photosynthesis in aerobic conditions at the transcription level (42, 43); therefore a future direction would be to determine the presence or absence of such a system in the above-mentioned Proteobacteria.
A Cysteine–BChl Photochemistry Mechanism.
The use of sodium dithionite in our experiments likely represents ideal reduction of the protein complex. Although sodium dithionite is an excellent industrial low-potential chemical reductant, it is not a reductant found in biology, in part because of its instability in trace amounts of water (44, 45). Therefore it is not the naturally acting reductant within the cell in vivo. A previous report from our group maintained that other reductants such as sodium borohydride and various sulfur-based reductants did not have an appreciable effect on FMO fluorescence (12). We revisited this statement, retesting the ability of other reductants (including more sulfur-based chemicals than before) to elicit optical changes in the protein, possibly illuminating its natural, endogenous reductant. The results (Fig. S4) indicate that many reductants do indeed have the ability to increase the fluorescence emission of the FMO protein, although sodium dithionite is the most effective. The best physiologically relevant reductants are reduced glutathione and sulfide, each nearly doubling the fluorescence intensity of the FMO protein. Glutathione is a compelling candidate for endogenous reductant because it has known radical activity, is present in micromolar concentrations in GSBs, and is sensitive to dissolved oxygen concentration (46, 47). Even small changes in the quenching activity of the protein are likely to result in large-scale metabolic changes. Therefore, we see this quenching mechanism as a photosynthetic “volume knob” rather than an “on/off switch.”
Fig. S4.

An assay of the relative increase in fluorescence (represented by the ratio of the fluorescence intensity of the reduced protein versus the fluorescence intensity of the oxidized protein, Fred/Fox) conferred to the FMO protein after the addition of various chemical reductants to a 20-mM final concentration. TCEP is the only reductant used that is not sulfur-based. β-ME, β-mercaptoethanol.
Our current mechanistic interpretation of the data is that the redox-modulation process is an electron transfer/recombination event between a thiyl radical and excited BChl a that de-excites the BChl a (Fig. 8). In this mechanism, a thiyl radical in close proximity to an excited BChl a becomes a good oxidizing agent, with an excited-state electron-transfer reaction resulting in a neutral thiol (or thiolate) and a BChl a radical cation. The BChl a radical cation, now a good oxidizing agent, abstracts one electron from the thiol (or thiolate), regenerating the thiyl radical and BChl a, now in the ground state. The excitation energy the BChl a originally possessed is therefore released as heat. Dithionite may be an unusually good reducing agent for the cysteine thiyl radical because it itself is in equilibrium with the [SO2]− radical anion in solution (44), affording a substrate for radical termination.
Fig. 8.

Proposed electron transfer/recombination mechanism for de-excitation of BChl pigments near thiyl radicals. The y-axis scale relates to the potential of the BChl a species. (A) Oxidizing conditions. (B) Reducing conditions. A, absorption; F, fluorescence; R, redox-active group.
Programming a Similar Mechanism into Other Light-Harvesting Antennas.
Our proposed mechanism can be envisioned as a tool for the construction of bio-inspired light-harvesting antennas or for the redesign of natural light-harvesting antennas. As an example, we examined the X-ray crystal structure (PDB code 1KZU) for the LH2 antenna complex from Rhodoblastus acidophilus (formerly Rhodopseudomonas acidophila) strain 10050. In this structure, W40 of the β-polypeptide (Puc3B) coordinates within 6 Å of the C-3 acetyl group of a B850 ring BChl a. In silico W40C mutagenesis predicts that the resultant cysteine will coordinate to the BChl a in a geometry similar to that of the cysteines from FMO (Fig. S5). In situations where energy-transfer attenuation is desired in response to changing oxygen levels, the simplicity of the cysteine–BChl interaction is an attractive option for protein engineering. An example situation in which this mechanism may be warranted would be modification of potential photosynthetic nitrogen-fixing crop symbionts (i.e., anoxygenic purple bacteria such as Salinarimonas and Bradyrhizobium) to allow them to grow more efficaciously in aerobic conditions, providing essential nitrogen to the host plant. Another example would be using the cysteines to modulate the photosynthetic rate in cyanobacterial or algal biofuel cultures to control their growth rate better and thus maximize the output of the desired fuel (e.g., oil, hydrogen, and others).
Fig. S5.

In silico W40C single-point mutagenesis in the Puc3B β-protein of LH2 from Rbs. acidophilus (PDB code 1KZU) using the mutagenesis function in PyMOL.
Conclusion
We conclude that the cysteine-dependent excitation-quenching mechanism in the FMO protein is previously unreported in the field of photosynthesis. The FMO protein itself is unique among photosynthetic antennas in that it does not contain photoprotective carotenoids, which normally quench BChl a triplet states and prevent singlet oxygen sensitization. With evidence emerging that the energy level of the triplet-excited state of the FMO protein is too low to sensitize singlet oxygen (23), it is appropriate to ask why the FMO protein needs a quenching mechanism in the first place. The answer likely lies in the susceptibility of the RC to oxidative damage in the presence of oxygen. The FMO protein’s redox-dependent quenching mechanism serves not to protect itself from damage but to protect the RC from receiving excitations in oxidizing conditions.
Materials and Methods
A detailed description of the materials and methods is given in SI Materials and Methods. The GSB Cba. tepidum was grown and its FMO protein was isolated as previously described (16, 48, 49), except that 20 mM CAPS (N-cyclohexyl-3-aminopropanesulfonic acid) buffer, pH 10.5 was substituted for Tris⋅HCl buffer in all chromatography steps. Oxidized FMO was used as purified in laboratory atmosphere, whereas reduced FMO was treated with 10 mM sodium dithionite in an anaerobic chamber. Three mutants of the FMO protein, C49A, C353A, and the double mutant C49A/C353A, were constructed and purified using a previously described method (14, 50). All oligonucleotides used in the construction of the mutants are provided in Table S2. For modification with NEM, 15 µg of WT FMO protein was reduced with 10 mM sodium dithionite and treated with 10 mM final concentration of NEM in the dark for 2 h.
All protein samples for MS were precipitated with acetone, trypsin digested, peptide purified, and injected into an Ultimate 3000 Nano LC system (Thermo Scientific Dionex) equipped with a Michrom Magic C-18 UPLC column (51). Fragments were analyzed in a Q-Exactive Plus Orbitrap mass spectrometer (Thermo Scientific). Modified peptides were identified from their accurate masses of precursor ion (MS1) spectra and product ion (MS2) spectra by either searching the NCBI database using Mascot (Matrix Science) or sequencing de novo using PEAKS 7.0 (Bioinformatics Solutions, Inc.) (18). Quantitative information about the level of chemical modification in each set of tryptic peptides was obtained from the precursor ion intensities of the peptides of interest and was compared among all samples.
Steady-state absorption spectroscopy was performed in a UV-1800 UV/Vis/NIR spectrophotometer (Shimadzu Corp. USA). Steady-state fluorescence experiments were performed using a NanoLog Spectrofluorometer (Horiba Scientific) equipped with a Vis-NIR grating and CCD detector, exciting at 602 nm. When required, samples [in 2:3 (vol:vol) buffer:glycerol] were cooled to 77 K in an Optistat DN liquid nitrogen cryostat (Oxford Instruments). TRF measurements were carried out using the laser system and universal streak camera setup described in detail previously (52), exciting at 602 nm. The spectra were cleared from noise and globally fit using the program ASUfit (13).
Protein film voltammetry was performed in an anaerobic chamber using a PGE working electrode, Ag/AgCl reference electrode, and platinum counter electrode. FMO protein was adsorbed to the working electrode surface, all electrodes were submerged in a mixed buffer electrolyte system, and cyclic voltammetry was performed with a Model 600 Series Electrochemical Analyzer workstation (CH Instruments, Inc.). All data were baseline corrected and analyzed with the program SOAS (53).
EPR was performed using a JEOL JES-FA X-BAND (9.2 GHz) or Bruker EMX-PLUS (9.2 GHz) EPR spectrometer at 77 K with quartz sample tubes. Protein was concentrated to OD808 nm = 150 in 200 µL in 50:50 (vol/vol) 20 mM CAPS buffer, pH 10.5, and glycerol. Ten 30-s scans were accumulated and averaged, and the following parameters were used during the collection of the EPR spectra: amplitude, 2,000; time constant, 0.3 s; modulation width, 0.5 mT; sweep width, 7.5 mT; frequency, 9094.105 MHz; power, 1 mW.
SI Results
MS Determination of Cysteine Residues as Residues of Interest.
A peptide map of the initial PEAKS searches for posttranslational modifications (PTMs) in oxidized or reduced WT Cba. tepidum FMO protein is shown in Fig. S1. Sequence coverage of the protein in each condition was >98%, with only a small peptide from the N-terminal domain, VKARA, missing. The N-terminal methionine residue also was not found, as is consistent with common bacterial protein processing in vivo.
Because separating out real in vivo PTMs from those caused by instrumental conditions is difficult, we focused our search not on the identity of the PTMs but rather on the general extent of modification on certain amino acids in the two protein samples. For example, various oxidations of tryptophan residues and methionine residues were found throughout the sequence for each sample, but their relative abundance did not change appreciably between the samples. The only peptides that showed considerable change in the extent of modification between the two samples were one N-terminal peptide, VNAPPASPLLPADCDVK (residues 36–52), and one C-terminal peptide, WVEHVCK (residues 348–354). These peptides contained the only cysteine residues (Cys49 and Cys353) in the protein, with the extent of the cysteine modification varying between protein conditions: Cys49 was 20% modified in reducing conditions and 50% modified in oxidizing conditions; Cys353 was 10% modified in reducing conditions and 60% modified in oxidizing conditions. The modifications found were oxidative modifications such as sulfenic acid and cysteinylation transformations. As shown in Fig. 1 in the main text, the two cysteine residues are too spatially separated to form intra- or intermonomer disulfide bonds with one another.
MS Confirmation of NEM Modification of FMO Protein Cysteine Residues.
The MS2 (product ion) data from digested NEM-modified FMO identified two tryptic peptides containing the cysteines of interest: VNAPPASPLLPADCDVK (Cys49) and WVEHECK (Cys353). NEM-derivatized cysteine was identified from product-ion spectra generated from the fragmentation of each peptide as an addition of 125.05 Da to the b and y ions attributed to the cysteine. Representative product-ion spectra for one of the peptides of interest, VNAPPASPLLPADCDVK, one showing Cys49 in the free thiol form and one showing Cys49 in NEM-derivatized form, are shown in Fig. S2.
Derivatized and underivatized peptides could be found in the NEM-modified FMO sample, indicating that the NEM modification was not 100% effective. De novo sequencing surveyed all types of common cysteine modifications in the sample, not only the free thiol or NEM-derivatized forms. In both the unmodified and NEM-derivatized samples, product-ion spectra were found that are consistent with Cys49 and Cys353 in the free thiol form, dehydroalanine form (elimination of sulfur atom during fragmentation), persulfide form, cysteic acid form, β-methylthiolated form, NEM-derivatized form, and NEM-derivatized form bound to one extra water molecule.
After identifying all cysteine-bearing parent ion (MS1) m/z ratios from the product-ion spectra, total ion intensity was calculated for each by surveying the LC and mass spectra. By determining how much ion intensity was generated for each peptide containing a different form of cysteine, the percentage of the modification extent could be calculated. The ion-intensity profiles are shown in Fig. S3. For Cys49, the modification extents for unmodified FMO are as follows: free thiol, 74.8%; dehydroalanine, 2.2%; cysteic acid, 9.8%; persulfide, 7.4%; β-methylthiolation, 5.8%. For Cys49, the modification extents for NEM-derivatized FMO are as follows: free thiol, 18.0%; dehydroalanine, 15.7%; cysteic acid, 24.8%; persulfide, 10.2%; β-methylthiolation, 12.7%; NEM-derivatized, 13.7%; NEM-derivatized plus one water molecule, 4.9% (total NEM modification therefore was 18.6%). For Cys353, the modification extents for unmodified FMO are as follows: free thiol, 0%; cysteic acid, 100%. For Cys353, the modification extents for NEM-derivatized FMO are as follows: free thiol, 20.7%; cysteic acid, 50.0%; NEM-derivatized, 29.3%, NEM-derivatized plus one water molecule, 0% (total NEM modification therefore was 29.3%).
Testing of Various Reductants for Their Ability to Interact with the FMO Protein.
A previous report from our group maintained that other reductants such as sodium borohydride and various sulfur-based reductants did not have an appreciable effect on FMO fluorescence (12). We retested the ability of other reductants (including more sulfur-based chemicals than before) to elicit optical changes in the protein, possibly illuminating the natural, endogenous reductant for the protein. The results (Fig. S4) indicate that many reductants do indeed have the ability to increase the fluorescence emission of the FMO protein, although none are as effective as sodium dithionite. Even TCEP, which does not act as a reducing agent based on direct thiol chemistry, showed the ability to increase FMO fluorescence.
Redesigning a Natural Light-Harvesting Antenna with Cysteine–BChl Photochemistry.
The thiyl radical–BChl photochemistry mechanism proposed in the main text can be envisioned as a tool for the construction of bio-inspired light-harvesting antennas or for the redesign of natural light-harvesting antennas. As an example, we examined the X-ray crystal structure (PDB code 1KZU) for the LH2 antenna complex from Rhodoblastus acidophilus (formerly Rhodopseudomonas acidophila) strain 10050. In this structure, W40 of the β-polypeptide (Puc3B) coordinates within 6 Å of the C-3 acetyl group of a B850 ring BChl a. In silico W40C mutagenesis predicts that the resultant cysteine will coordinate to the BChl a in a geometry similar to that of the cysteines from FMO (Fig. S5).
Acknowledgments
We thank Kaitlyn Faries and Prof. Dewey Holten from Washington University for assistance with steady-state fluorescence experiments. All work, except for EPR spectroscopy, was supported as part of the Photosynthetic Antenna Research Center, an Energy Frontier Research Center funded by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under Award DE-SC0001035 (to R.E.B.). The purchase of the Bruker EMX-PLUS EPR spectrometer was supported by National Science Foundation Award MRI, CHE-1429711 (to L.M.M.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
See Commentary on page 8562.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1603330113/-/DCSupplemental.
References
- 1.Blankenship RE. 2014. Molecular Mechanisms of Photosynthesis (John Wiley & Sons, Ltd., Southern Gate, Chichester, West Sussex, UK), 2nd Ed.
- 2.Nicholls DG, Ferguson S. Bioenergetics. 4th Ed Elsevier Ltd.; San Diego: 2013. [Google Scholar]
- 3.Falkowski PG, Raven JA. Aquatic Photosynthesis. 2nd Ed Princeton Univ Press; Prineton, NJ: 2007. [Google Scholar]
- 4.Hohmann-Marriott MF, Blankenship RE. Evolution of photosynthesis. Annu Rev Plant Biol. 2011;62:515–548. doi: 10.1146/annurev-arplant-042110-103811. [DOI] [PubMed] [Google Scholar]
- 5.Hauska G, Schoedl T, Remigy H, Tsiotis G. The reaction center of green sulfur bacteria(1) Biochim Biophys Acta. 2001;1507(1-3):260–277. doi: 10.1016/s0005-2728(01)00200-6. [DOI] [PubMed] [Google Scholar]
- 6.Büttner M, et al. Photosynthetic reaction center genes in green sulfur bacteria and in photosystem 1 are related. Proc Natl Acad Sci USA. 1992;89(17):8135–8139. doi: 10.1073/pnas.89.17.8135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Orf GS, Blankenship RE. Chlorosome antenna complexes from green photosynthetic bacteria. Photosynth Res. 2013;116(2-3):315–331. doi: 10.1007/s11120-013-9869-3. [DOI] [PubMed] [Google Scholar]
- 8.Vassilieva EV, et al. Electron transfer may occur in the chlorosome envelope: The CsmI and CsmJ proteins of chlorosomes are 2Fe-2S ferredoxins. Biochemistry. 2001;40(2):464–473. doi: 10.1021/bi001917d. [DOI] [PubMed] [Google Scholar]
- 9.Oostergetel GT, van Amerongen H, Boekema EJ. The chlorosome: A prototype for efficient light harvesting in photosynthesis. Photosynth Res. 2010;104(2-3):245–255. doi: 10.1007/s11120-010-9533-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li H, Frigaard N-U, Bryant DA. [2Fe-2S] proteins in Chlorosomes: CsmI and CsmJ participate in light-dependent control of energy transfer in Chlorosomes of Chlorobaculum tepidum. Biochemistry. 2013;52(8):1321–1330. doi: 10.1021/bi301454g. [DOI] [PubMed] [Google Scholar]
- 11.Karapetyan NV, Swarthoff T, Rijgersberg CP, Amesz J. Fluorescence emission spectra of cells and subcellular preparations of a green photosynthetic bacterium. Effects of dithionite on the intensity of the emission bands. Biochim Biophys Acta. 1980;593(2):254–260. doi: 10.1016/0005-2728(80)90063-8. [DOI] [PubMed] [Google Scholar]
- 12.Zhou W, LoBrutto R, Lin S, Blankenship RE. Redox effects on the bacteriochlorophyll a-containing Fenna-Matthews-Olson protein from Chlorobium tepidum. Photosynth Res. 1994;41(1):89–96. doi: 10.1007/BF02184148. [DOI] [PubMed] [Google Scholar]
- 13.Niedzwiedzki DM, Blankenship RE. Singlet and triplet excited state properties of natural chlorophylls and bacteriochlorophylls. Photosynth Res. 2010;106(3):227–238. doi: 10.1007/s11120-010-9598-9. [DOI] [PubMed] [Google Scholar]
- 14.Saer RG, et al. Perturbation of bacteriochlorophyll molecules in Fenna-Matthews-Olson protein complexes through mutagenesis of cysteine residues. Biochim Biophys Acta Bioenergetics. 2016;1857(9):1455–1463. doi: 10.1016/j.bbabio.2016.04.007. [DOI] [PubMed] [Google Scholar]
- 15.Tronrud DE, Wen J, Gay L, Blankenship RE. The structural basis for the difference in absorbance spectra for the FMO antenna protein from various green sulfur bacteria. Photosynth Res. 2009;100(2):79–87. doi: 10.1007/s11120-009-9430-6. [DOI] [PubMed] [Google Scholar]
- 16.Li Y-F, Zhou W, Blankenship RE, Allen JP. Crystal structure of the bacteriochlorophyll a protein from Chlorobium tepidum. J Mol Biol. 1997;271(3):456–471. doi: 10.1006/jmbi.1997.1189. [DOI] [PubMed] [Google Scholar]
- 17.Han X, Aslanian A, Yates JR., 3rd Mass spectrometry for proteomics. Curr Opin Chem Biol. 2008;12(5):483–490. doi: 10.1016/j.cbpa.2008.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ma B, et al. PEAKS: Powerful software for peptide de novo sequencing by tandem mass spectrometry. Rapid Commun Mass Spectrom. 2003;17(20):2337–2342. doi: 10.1002/rcm.1196. [DOI] [PubMed] [Google Scholar]
- 19.Milder MTW, Brüggemann B, van Grondelle R, Herek JL. Revisiting the optical properties of the FMO protein. Photosynth Res. 2010;104(2-3):257–274. doi: 10.1007/s11120-010-9540-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hill BG, Reily C, Oh J-Y, Johnson MS, Landar A. Methods for the determination and quantification of the reactive thiol proteome. Free Radic Biol Med. 2009;47(6):675–683. doi: 10.1016/j.freeradbiomed.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rogers LK, Leinweber BL, Smith CV. Detection of reversible protein thiol modifications in tissues. Anal Biochem. 2006;358(2):171–184. doi: 10.1016/j.ab.2006.08.020. [DOI] [PubMed] [Google Scholar]
- 22.Liu Q, Levy EJ, Chirico WJ. N-Ethylmaleimide inactivates a nucleotide-free Hsp70 molecular chaperone. J Biol Chem. 1996;271(47):29937–29944. doi: 10.1074/jbc.271.47.29937. [DOI] [PubMed] [Google Scholar]
- 23.Orf GS, Niedzwiedzki DM, Blankenship RE. Intensity dependence of the excited state lifetimes and triplet conversion yield in the Fenna-Matthews-Olson antenna protein. J Phys Chem B. 2014;118(8):2058–2069. doi: 10.1021/jp411020a. [DOI] [PubMed] [Google Scholar]
- 24.Hwang HJ, Ang M, Lu Y. Determination of reduction potential of an engineered Cu(A) azurin by cyclic voltammetry and spectrochemical titrations. J Biol Inorg Chem. 2004;9(4):489–494. doi: 10.1007/s00775-004-0547-y. [DOI] [PubMed] [Google Scholar]
- 25.Johnson DL, Maxwell CJ, Losic D, Shapter JG, Martin LL. The influence of promoter and of electrode material on the cyclic voltammetry of Pisum sativum plastocyanin. Bioelectrochemistry. 2002;58(2):137–147. doi: 10.1016/s1567-5394(02)00125-1. [DOI] [PubMed] [Google Scholar]
- 26.Johnson D, Norman S, Tuckey RC, Martin LL. Electrochemical behaviour of human adrenodoxin on a pyrolytic graphite electrode. Bioelectrochemistry. 2003;59(1-2):41–47. doi: 10.1016/s1567-5394(02)00188-3. [DOI] [PubMed] [Google Scholar]
- 27.Armstrong FA, Butt JN, Sucheta A. 1993. Voltammetric studies of redox-active centers in metalloproteins adsorbed on electrodes. Methods Enzymol 227:479–500. [DOI] [PubMed]
- 28.Léger C, et al. Enzyme electrokinetics: Using protein film voltammetry to investigate redox enzymes and their mechanisms. Biochemistry. 2003;42(29):8653–8662. doi: 10.1021/bi034789c. [DOI] [PubMed] [Google Scholar]
- 29.Léger C, et al. Enzyme electrokinetics: Energetics of succinate oxidation by fumarate reductase and succinate dehydrogenase. Biochemistry. 2001;40(37):11234–11245. doi: 10.1021/bi010889b. [DOI] [PubMed] [Google Scholar]
- 30.Salgado MT, Ramasamy S, Tsuneshige A, Manoharan PT, Rifkind JM. A new paramagnetic intermediate formed during the reaction of nitrite with deoxyhemoglobin. J Am Chem Soc. 2011;133(33):13010–13022. doi: 10.1021/ja1115088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lassmann G, et al. Protein thiyl radicals in disordered systems: A comparative EPR study at low temperature. Phys Chem Chem Phys. 2003;5(11):2442–2453. [Google Scholar]
- 32.Sevilla MD, Yan MY, Becker D. Thiol peroxyl radical formation from the reaction of cysteine thiyl radical with molecular oxygen: An ESR investigation. Biochem Biophys Res Commun. 1988;155(1):405–410. doi: 10.1016/s0006-291x(88)81100-8. [DOI] [PubMed] [Google Scholar]
- 33.Licht S, Gerfen GJ, Stubbe J. Thiyl radicals in ribonucleotide reductases. Science. 1996;271(5248):477–481. doi: 10.1126/science.271.5248.477. [DOI] [PubMed] [Google Scholar]
- 34.Engel GS, et al. Evidence for wavelike energy transfer through quantum coherence in photosynthetic systems. Nature. 2007;446(7137):782–786. doi: 10.1038/nature05678. [DOI] [PubMed] [Google Scholar]
- 35.Fidler AF, Caram JR, Hayes D, Engel GS. Towards a coherent picture of excitonic coherence in the Fenna–Matthews–Olson complex. J Phys At Mol Opt Phys. 2012;45(15):154013. [Google Scholar]
- 36.Hayes D, et al. Dynamics of electronic dephasing in the Fenna–Matthews–Olson complex. New J Phys. 2010;12(6):065042. [Google Scholar]
- 37.Panitchayangkoon G, et al. Long-lived quantum coherence in photosynthetic complexes at physiological temperature. Proc Natl Acad Sci USA. 2010;107(29):12766–12770. doi: 10.1073/pnas.1005484107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schmidt Am Busch M, Müh F, El-Amine Madjet M, Renger T. The Eighth Bacteriochlorophyll Completes the Excitation Energy Funnel in the FMO Protein. J Phys Chem Lett. 2011;2(2):93–98. doi: 10.1021/jz101541b. [DOI] [PubMed] [Google Scholar]
- 39.Bryant DA, et al. Candidatus Chloracidobacterium thermophilum: An aerobic phototrophic Acidobacterium. Science. 2007;317(5837):523–6. doi: 10.1126/science.1143236. [DOI] [PubMed] [Google Scholar]
- 40.Liu Z, et al. ‘Candidatus Thermochlorobacter aerophilum:’ an aerobic chlorophotoheterotrophic member of the phylum Chlorobi defined by metagenomics and metatranscriptomics. ISME J. 2012;6(10):1869–1882. doi: 10.1038/ismej.2012.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tsukatani Y, Wen J, Blankenship RE, Bryant DA. Characterization of the FMO protein from the aerobic chlorophototroph, Candidatus Chloracidobacterium thermophilum. Photosynth Res. 2010;104(2-3):201–209. doi: 10.1007/s11120-009-9517-0. [DOI] [PubMed] [Google Scholar]
- 42.Masuda S, et al. Repression of photosynthesis gene expression by formation of a disulfide bond in CrtJ. Proc Natl Acad Sci USA. 2002;99(10):7078–7083. doi: 10.1073/pnas.102013099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nickens DG, Bauer CE. Analysis of the puc operon promoter from Rhodobacter capsulatus. J Bacteriol. 1998;180(16):4270–4277. doi: 10.1128/jb.180.16.4270-4277.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rinker RG, Gordon TP, Mason DM, Corcoran WH. The presence of the SO2- radical ion in aqueous solutions of sodium dithionite. J Phys Chem. 1959;63(1):302. [Google Scholar]
- 45.Englander SW, Calhoun DB, Englander JJ. Biochemistry without oxygen. Anal Biochem. 1987;161(2):300–306. doi: 10.1016/0003-2697(87)90454-4. [DOI] [PubMed] [Google Scholar]
- 46.Madej E, Wardman P. The oxidizing power of the glutathione thiyl radical as measured by its electrode potential at physiological pH. Arch Biochem Biophys. 2007;462(1):94–102. doi: 10.1016/j.abb.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 47.Masip L, Veeravalli K, Georgiou G. The many faces of glutathione in bacteria. Antioxid Redox Signal. 2006;8(5-6):753–762. doi: 10.1089/ars.2006.8.753. [DOI] [PubMed] [Google Scholar]
- 48.Gerola PD, Olson JM. A new bacteriochlorophyll a-protein complex associated with chlorosomes of green sulfur bacteria. Biochim Biophys Acta. 1986;848(1):69–76. doi: 10.1016/0005-2728(86)90161-1. [DOI] [PubMed] [Google Scholar]
- 49.Wen J, Zhang H, Gross ML, Blankenship RE. Native electrospray mass spectrometry reveals the nature and stoichiometry of pigments in the FMO photosynthetic antenna protein. Biochemistry. 2011;50(17):3502–3511. doi: 10.1021/bi200239k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Frigaard N-U, Bryant DA. Chromosomal gene inactivation in the green sulfur bacterium Chlorobium tepidum by natural transformation. Appl Environ Microbiol. 2001;67(6):2538–2544. doi: 10.1128/AEM.67.6.2538-2544.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang H, et al. Molecular mechanism of photoactivation and structural location of the cyanobacterial orange carotenoid protein. Biochemistry. 2014;53(1):13–19. doi: 10.1021/bi401539w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Niedzwiedzki DM, et al. Photophysical properties of the excited states of bacteriochlorophyll f in solvents and in chlorosomes. J Phys Chem B. 2014;118(9):2295–2305. doi: 10.1021/jp409495m. [DOI] [PubMed] [Google Scholar]
- 53.Fourmond V, et al. SOAS: A free program to analyze electrochemical data and other one-dimensional signals. Bioelectrochemistry. 2009;76(1-2):141–147. doi: 10.1016/j.bioelechem.2009.02.010. [DOI] [PubMed] [Google Scholar]