

Abstract
Thienoguanosine (thG) is an isomorphic nucleoside analogue acting as a faithful fluorescent substitute of G, with respectable quantum yield in oligonucleotides. Photophysical analysis of thG reveals the existence of two ground-state tautomers with significantly shifted absorption and emission wavelengths, and high quantum yield in buffer. Using (TD)-DFT calculations, the tautomers were identified as the H1 and H3 keto-amino tautomers. When incorporated into the loop of (−)PBS, the (−)DNA copy of the HIV-1 primer binding site, both tautomers are observed and show differential sensitivity to protein binding. The red-shifted H1 tautomer is strongly favored in matched (−)/(+)PBS duplexes, while the relative emission of the H3 tautomer can be used to detect single nucleotide polymorphisms. These tautomers and their distinct environmental sensitivity provide unprecedented information channels for analyzing G residues in oligonucleotides and their complexes.
Keywords: ab initio calculations, fluorescence, molecular modeling, nucleic acids, tautomerism
The structure, acid-base features, and tautomeric equilibria of the canonical and non-canonical nucleobases found in nucleic acids have been the subject of intense investigation for decades.[1] While the role of minor tautomers in mutagenesis has been one of the primary foci,[2] recent observations suggest that such isomeric nucleobases also play key roles in regular nucleic acid structure and function.[3] As the population of distinct tautomeric forms is impacted by their micro-environment, this added level of complexity also provides opportunities to further advance our understanding of nucleic acid structure and dynamics.
In this context, emissive nucleoside analogues, which have become powerful biophysical tools,[4] provide unique prospects. A tautomerizable nucleoside analogue, where the tautomers would have distinct absorption and emission spectra, could be instrumental for investigating the micro-environment of a nucleobase with greater insight compared to tautomerically stable probes. Herein we analyze the photophysics of thienoguanosine, thG, a highly useful G surrogate,[5] and identify two environmentally sensitive ground-state tautomeric forms (Figure 1) which display distinct absorption and emission spectra. The equilibrium between the two tautomers is mainly governed by the hydrogen-bond donor properties of the solvent. Their observed sensitivity to the microenvironment was rationalized by ab initio calculations. By exploring single- and double-stranded thG-containing oligonucleotides, as well as DNA–protein complexes, we illustrate that this probe provides compelling biophysical information and greater insight compared to monochromatic or ratiometric fluorescent nucleosides.
Figure 1.
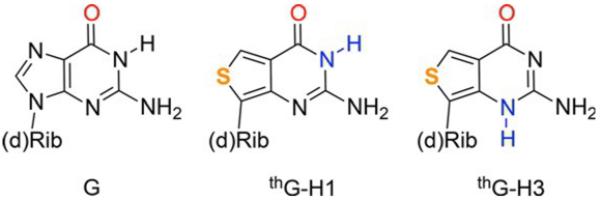
Structures of guanosine (G) and the two emissive tautomers of thienoguanosine (thG–H1 and thG–H3). (d)Rib=d-ribose or 2'-deoxy-d-ribose.
The emission spectra of either thG or dthG in water and methanol are surprisingly complex.[5a,b] Excitation at both λ=360 and 380 nm gives a similar emission spectrum (Figure 2a, orange) centered at λ=468 nm. When the excitation energy is progressively increased, a blue-shifted emission with a maximum at λ=400 nm appears and becomes dominant for excitation below λ=300 nm (Figure 2a, magenta and blue). The simplest interpretation is that thG exhibits two ground-state species with shifted emission spectra. This hypothesis is supported by recording excitation spectra for various emission wavelengths (see Figure S1 in the Supporting Information). Since sample purity was rigorously maintained, the two ground-state species likely correspond to two tautomers, differing by their excitation and emission spectra. This conclusion is highly likely, since tautomers have also been observed for guanosine itself.[6]
Figure 2.
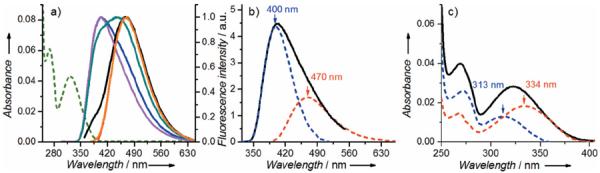
Absorption and emission spectra of thG in TRIS-HCl buffer 25 mm, pH 7.5, 30 mm NaCl, 0.2 mm MgCl2. a) Absorption (green dashed line) and emission spectra of thG at different excitation wavelengths: λ=283 nm (magenta line); λ=298 nm (blue); λ=320 nm (green); λ=345 nm (black); and λ=360 nm (orange). The emission spectra were normalized at their maxima. The normalized emission spectrum at λexc=380 nm fully overlaps that at λexc=360 nm and is not shown. b) Deconvoluted emission spectrum of thG, obtained at λex=283 nm. c) Deconvolution of the absorption spectrum of thG (black line) in its two ground-state forms (colors as in b).
Spectral deconvolution yields well separated emission and absorption spectra of the individual tautomers in buffer (Figure 2b,c). Thus, by judiciously selecting the excitation and emission wavelengths, each tautomer can be individually excited and observed. The fluorescence quantum yield (QY) is found to be constant (0.49±0.03) over a large range of excitation wavelengths (λ=290–375 nm) and close to earlier reported values,[5a,c] thus indicating that the two forms possess similar QY values. The individual absorption spectrum of the red-shifted form (Figure 2c) is very similar to the spectra of thG and dthG in 1,4-dioxane.[5a,b] By using the molar extinction coefficient of thG in 1,4-dioxane (ε333=4530m−1 cm−1),[5a] it is possible to calculate the concentration of the red-shifted tautomer in buffer and deduce the proportion (44%) and the molar extinction coefficient (ε313≈4600m−1 cm−1) of the blue-shifted tautomer. Importantly, the red-shifted tautomer, excited at λ=350–380 nm, is highly photostable (see Figure S2). In contrast, extended illumination at higher energies (e.g., λ=325 nm), where both tautomers absorb, show continuously diminished emission, thus suggesting that the blue-shifted tautomer is less photostable and that the two tautomers are equilibrating.
The spectroscopic properties of thG were comparatively characterized in various solvents (see Table S1). In methanol, ethanol, and n-butanol (see Figures S3 and S4) spectra comparable to those in buffer are obtained, thus indicating that thG also exists in the two tautomeric forms in these solvents. In all other tested solvents, the emission spectra are independent on the excitation wavelength and indicate that only the red-shifted tautomer is present (see Figure S3c). Its emission maximum correlates well with the empirical polarity index ET(30) of the solvent (see Figure S5). Interestingly, although N,N-dimethylformamide, acetonitrile, and methanol are all rather polar [ET(30) >43 kcalmol−1, ε>32], the blue-shifted tautomer is seen only in methanol, thus strongly suggesting hydrogen-bonding stabilization. This proposal is further substantiated by the deconvoluted absorption spectra in polar protic solvents (see Figure S6), as they show that the relative concentration of the blue-shifted isomer linearly increases with solvent proticity (see Figure S6d and Table S1). Thus, the equilibrium between the two thG tautomers is dependent on the hydrogen-bond donor ability of the solvent.
To assist in identifying the two emissive isomers, the ground-state energy minima of five hypothetical thG tautomers were optimized (Figure 3) in the gas phase, 1,4-dioxane, and water at the DFT level, by using PBE0 and M052X functionals and including solvent effects with the Polarizable Continuum Model (PCM; see Tables S2 and S3). The keto-amino thG–H1 tautomer (Figure 3a) appears largely dominant over the other tautomers (Figures 3b–e), with the exception of water where it is only 0.11 eV more stable than thG–H3, when including only bulk solvent effects. Therefore, the two thG keto-amino tautomers, analogous to the most stable tautomer of guanine in solution,[7] are likely populated in water.
Figure 3.
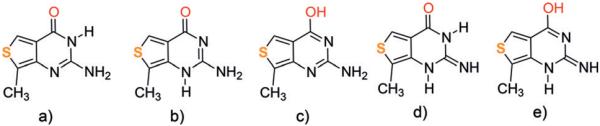
Schematic drawing of the thG tautomers which have been calculated: a) keto-amino thG–H1, b) keto-amino thG–H3, c) enol-amino, d) keto-imino, and e) enol-imino.
Independent of the inclusion of one solvent molecule in the computational model (see Figures S7 and S8, and Table S2), the thG–H1 tautomer appears as the main contributor to the observed spectroscopic properties of thG in 1,4-dioxane and is therefore assigned to the red-shifted isomer.[8] The thG–H1 and thG–H3 tautomers are found to be almost isoenergetic in water when solute–solvent hydrogen bonds are considered (see Figure S7 and Table S2). These data suggest that both tautomers likely contribute to the spectroscopic properties of thG in water and that the blue-shifted isomer corresponds to the thG–H3 tautomer. The computed energy difference between the two tautomers (<0.05 eV, that is, <400 cm−1) is beyond the expected accuracy of our method, thus explaining why the molar fraction of the thG–H3 tautomer (see Table S2) does not perfectly match with the experimental value (0.44).[9] The computed vertical absorption and emission energies (see Table S4)[10] indicate that, independent of the functional, the lowest-energy transition in water for both tautomers corresponds to a bright ππ* S0→S1 transition attributed to a HOMO→LUMO excitation.[11]
Interestingly, small differences in the shape of the frontier orbitals involved in the electronic transition (see Figure S9) result in fairly large differences in the computed vertical excitation energy, so that the absorption maximum of the thG–H1 tautomer in water is red-shifted by 30–40 nm, with respect to that of thG–H3. The absorption maxima predicted for the two forms, namely λ=330 ~ 350 nm (depending on the solvation model) for thG–H1 and λ=300 ~ 310 nm for thG–H3, are very close to the experimental ones (see Figures S10a, 2c and Table S1).[12] TD-DFT excited-state geometry optimizations (employing either PBE0 or M052X) predict a stable S1 minimum for both tautomers in all examined solvents.[13] This S1 minimum is characterized by a fairly large oscillator strength, of about 80% of the value computed for absorption. This minimum contrasts guanosine, for which the same functionals predict a barrierless decay to S0, through an effective conical intersection.[14] Both thG tautomers therefore show promising electronic features with potentially robust emissive states. The computed emission wavelengths of λ=448 and 383 nm for the thG–H1 and thG–H3 tautomers, respectively,[15] (see Figure S10b), are in good agreement with the spectroscopic data in buffer (Figure 2b). Moreover, by weighting the contribution of the different tautomers with a simple Boltzmann equation, the computed fluorescence spectra (see Figure S10b) are consistent with the experimental ones. Taken together, PCM/(TD)DFT calculations indicate that the thG–H1 and thG–H3 tautomers are responsible of the observed photophysical features of thG.
To examine the ground-state equilibrium between the tautomers of thG in oligonucleotides, the DNA equivalent of the 18-mer primer binding site of HIV-1 was selected as a biologically relevant model (see Figure S11). It forms a stem-loop of known three-dimensional structure[16] and is involved in the second strand transfer of HIV-1 reverse transcription.[17] Deoxy-thG (dthG), which exhibits spectroscopic properties very similar to thG,[5b,c] substitutes the dG7 residue in the loop [labeled dthG7(−)PBS; Figure 4a inset]. Comparing the emission spectra at various excitation wavelengths clearly shows that both dthG tautomers are present in the (−)PBS loop (Figure 4a). In contrast, when dthG7(−)PBS is annealed to its complementary (+)PBS strand (see Figure S11), forming the dthG7(−)/(+)PBS duplex (Figure 4c, inset), the normalized emission spectra obtained at different excitation wavelengths all overlap, thus indicating that the dthG–H1 tautomer is predominant in the double-stranded form (Figure 4c). Although not attributed to the two tautomers disclosed here, a similar switch from a two-band to a single-band emission was previously observed upon transition from single- to double-stranded structures in model thG- and dthG-labeled sequences,[5a,b] thus indicating that the tautomeric shift reported here is not unique for (−)PBS.
Figure 4.

Emission spectra of dthG7(−)PBS (a, b) and dthG7(−)/(+)PBS duplexes (c, d). a) Emission spectra of dthG7(−)PBS recorded at different excitation wavelengths: λ=298 nm (blue), λ=320 nm (green), λ=345 nm (black), λ=360 nm (orange), and 380 nm (red). Inset: structure of dthG7(−)PBS, the G7 residue (red) is replaced by dthG. b) Emission spectra of dthG7(−)PBS in the absence (black) and in the presence of 1 to 6 equivalents of NC(11-55) protein (red to violet) at λ=320 nm excitation wavelength. c) Emission spectra of the matched dthG7(−)/(+)PBS duplex at the same excitation wavelengths as in (a). Inset: structure of dthG7(−)/(+)PBS duplex. In mismatched duplexes, the C residue in green is replaced by a A, G or T. (d) Emission of the matched and mismatched dthG7-labeled (−)/(+)PBS duplexes at λ=310 nm excitation wavelength. The base opposite to dthG is C (black, native duplex), T (blue), G (red), or A (green). Inset: zoom of the blue part of the spectra. The buffer was as in Figure 2.
Distinct behavior was seen for mismatched duplexes between dthG7(−)PBS and complementary (+)PBS oligonucleotides, where dthG was placed opposite A, T, or G (Figure 4d). In contrast to the fully complementary duplex, where the dthG–H3 tautomer is nearly absent, its relative contribution as estimated by the ratio of the fluorescence intensities at λ=375 and 550 nm, I375/I550, increases by a factor of three and five in the mismatched duplexes with opposite dG and dA, respectively (see Table S5). For the mismatched duplex with opposite dT, the difference with the matched duplex is marginal, but the two duplexes can be easily discriminated by the twofold difference in their quantum yield (see Table S5). This difference likely results both from a change in polarity (as suggested by the changes in the positions of the dthG–H1 emission maximum; see Table S5) and in the quenching by the flanking nucleobases, as a result of the different geometries adopted by dthG and its neighbors in the two duplexes. The relative emission of the two dthG tautomers and the dthG quantum yield are therefore highly sensitive to the nature of the opposite base and can thus be used in combination to detect single nucleotide polymorphisms.
To further illustrate the potential applications of the two spectrally distinct dthG tautomers when in oligonucleotides, we investigated their response to binding of the HIV-1 nucleocapsid NC(11-55) peptide to the (−)PBS loop.[16b] Titration with NC(11-55) protein resulted in a strong increase of the dthG–H1 peak of dthG7(−)PBS, but only a marginal increase in the dthG–H3 peak (shift in the H3/H1 emission ratio from 1.1 to 0.8), thus indicating that the relative emission of the two tautomers is sensitive to protein binding (Figure 4b). As NC(11-55) was reported to direct the G7 base toward the exterior of the loop[16b] and restrict its collisions with the neighboring bases,[18] it appears that the dthG–H1 tautomer is more sensitive than dthG–H3 to these changes.
To shed light on the biophysical observations, molecular dynamics (MD) simulations using the ff12SB AMBER force field were performed on the NMR structure of ΔP(−)PBS DNA,[16b] a truncated form of (−)PBS lacking the 3′ overhang (see Figure S11). Analysis of MD trajectories (0.2 μs of unbiased MD trajectory production) and thermodynamic parameters unequivocally shows that there are no differences in the behavior of either dG or the two dthG tautomers in two representative ΔP(−)PBS structures (see Figures S12, S13a,b, and S14). In contrast, analysis of local motion within the Watson–Crick base pair established by either dG or dthG at position 7 in the (−)/(+)PBS DNA duplex clearly shows that dthG–H1 has the same behavior as dG, whereas dthG–H3 pairs with the counterpart dC with lower stability (see Figure S13d). A local structural analysis of MD trajectories further confirms that dthG–H1 forms the three canonical hydrogen-bonds with dC as observed for guanine (Figure S15a,b), while dthG–H3 contacts dC in multiple non-canonical complexes (see Figures S15c and S16). Overall, and consistently with experimental observations, the replacement of dG with dthG–H3 in the (−)/(+)PBS DNA duplex is noticeably less favorable than the replacement with dthG–H1, from a thermodynamic and conformational viewpoint.[19] Finally, MD simulations reveal that the two tautomers are mainly in the anti-conformation in both the stem-loop and the duplex (see Table S6).
In summary, through a careful analysis of its spectroscopic properties as a free nucleoside and when incorporated into oligonucleotides, thienoguanosine thG was observed to exhibit two ground-state tautomers with significantly shifted absorption and emission spectra. Quantum mechanical calculations unambiguously identified the two tautomers as being the keto-amino tautomers, thG–H1 and thG–H3. MD studies further suggested thG–H1 behaves similarly to its native counterpart in both the single- and double-stranded structures studied here, whereas the thG–H3 tautomer behaves comparably to G only in the loop of a stem-loop DNA. When incorporated into double-stranded sequences, thG–H3 tautomerizes to the favorable and benign thG–H1 tautomer, which forms a stable Watson–Crick base pair. The ratio of the two tautomers and their relative emission were found to be highly sensitive to the nucleic acid strandedness, to the nature of the opposite base in DNA duplexes, as well as to protein binding. The tautomerism of the isomorphic thG, which is associated with distinct and highly emissive states, thus constitutes a highly useful additional channel of information that provides an unprecedented window into features of substituted G residues in oligonucleotides.
Supplementary Material
Acknowledgments
The work was supported by a fellowship from the Ministère de la Recherche (M.S.), the European Project THINPAD “Targeting the HIV-1 Nucleocapsid Protein to fight Antiretroviral Drug Resistance” (FP7-Grant Agreement 601969), Agence Nationale de la Recherche (ANR blanc Fluometadn and FEMTOSTACK), Agence Nationale de Recherche sur le SIDA, French-Ukrainian Dnipro program, the Université de Strasbourg, the Centre National de la Recherche Scientifique (CNRS), the Institut de la Santé et de la Recherche Médicale (INSERM), Progetto Bilaterale CNR/CNRS, and the US National Institutes of Health (GM 069773). Computing time was provided at the French national computing centers by GENCI (Grand Equipement National de Calcul Intensif) and the Meso-center for High Performance Computing at the Université de Strasbourg and supported by the project EQUIP@MESO.
Footnotes
Supporting information for this article can be found under: http://dx.doi.org/10.1002/anie.201601688.
References
- [1].a) Miles HT. Proc. Natl. Acad. Sci. USA. 1961;58:791. doi: 10.1073/pnas.47.6.791. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wong YP, Wong KL, Kearns DR. Biochem. Biophys. Res. Commun. 1972;49:1580. doi: 10.1016/0006-291x(72)90522-0. [DOI] [PubMed] [Google Scholar]; c) Lee GC, Chan SI. J. Am. Chem. Soc. 1972;94:3218. doi: 10.1021/ja00764a054. [DOI] [PubMed] [Google Scholar]; d) Robinson H, Gao YG, Bauer C, Roberts C, Switzer C, Wang AHJ. Biochemistry. 1998;37:10897. doi: 10.1021/bi980818l. [DOI] [PubMed] [Google Scholar]; e) Blas JR, Luque FJ, Orozco M. J. Am. Chem. Soc. 2004;126:154. doi: 10.1021/ja036806r. [DOI] [PubMed] [Google Scholar]
- [2].a) Drake JW, Baltz RH. Annu. Rev. Biochem. 1976;45:11. doi: 10.1146/annurev.bi.45.070176.000303. [DOI] [PubMed] [Google Scholar]; b) Topal MD, Fresco JR. Nature. 1976;263:285. doi: 10.1038/263285a0. [DOI] [PubMed] [Google Scholar]; c) Shugar D, Kierdaszuk B. Proc. Int. Symp. Biomol. Struct. Interactions, Suppl. J. Biosci. 1985;8:657. [Google Scholar]; d) Wang WN, Hellinga HW, Beese LS. Proc. Natl. Acad. Sci. USA. 2011;108:17644. doi: 10.1073/pnas.1114496108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].a) Westhof E. FEBS Lett. 2014;588:2464. doi: 10.1016/j.febslet.2014.06.031. [DOI] [PubMed] [Google Scholar]; b) Singh V, Fedeles BI, Essigmann JM. RNA. 2015;21:1. doi: 10.1261/rna.048371.114. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kimsey I, Al-Hashimi HM. Curr. Opin. Struct. Biol. 2014;24:72. doi: 10.1016/j.sbi.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Sinkeldam RW, Greco NJ, Tor Y. Chem. Rev. 2010;110:2579. doi: 10.1021/cr900301e. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tanpure AA, Pawar MG, Srivatsan SG. Isr. J. Chem. 2013;53:366. [Google Scholar]; c) Phelps K, Morris A, Beal PA. ACS Chem. Biol. 2012;7:100. doi: 10.1021/cb200422t. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Wilhelmsson LM, Rev Q. Biophys. 2010;43:159. doi: 10.1017/S0033583510000090. [DOI] [PubMed] [Google Scholar]; e) Wilson JN, Kool ET. Org. Biomol. Chem. 2006;4:4265. doi: 10.1039/b612284c. [DOI] [PubMed] [Google Scholar]; f) Hawkins ME. Methods Enzymol. 2008;450:201. doi: 10.1016/S0076-6879(08)03410-1. [DOI] [PubMed] [Google Scholar]
- [5].a) Shin D, Sinkeldam RW, Tor Y. J. Am. Chem. Soc. 2011;133:14912. doi: 10.1021/ja206095a. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Park S, Otomo H, Zheng L, Sugiyama H. Chem. Commun. 2014;50:1573. doi: 10.1039/c3cc48297a. [DOI] [PubMed] [Google Scholar]; c) Sholokh M, Sharma R, Shin D, Das R, Zaporozhets OA, Tor Y, Mely Y. J. Am. Chem. Soc. 2015;137:3185. doi: 10.1021/ja513107r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a) Colominas C, Luque FJ, Orozco M. J. Am. Chem. Soc. 1996;118:6811. [Google Scholar]; b) Nir E, Janzen C, Imhof P, Kleinermanns K, de Vries MS. J. Chem. Phys. 2001;115:4604. [Google Scholar]; c) Marian CM. J. Phys. Chem. A. 2007;111:1545. doi: 10.1021/jp068620v. [DOI] [PubMed] [Google Scholar]
- [7].Lee YJ, Jang YH, Kim Y, Hwang S. Bull. Korean Chem. Soc. 2012;33:4255. [Google Scholar]
- [8].Interestingly, the stability of the thG–H3 tautomer increases with polarity and particularly with the hydrogen-bonding ability of the solvent.
- [9].For the examined solvents, PBE0 and M052X provide similar indications, thus suggesting that our conclusions are robust with respect to the choice of the functional. Inclusion of vibrational effects does not substantially affect the conformational equilibrium between thG–H1 and thG–H3 tautomers.
- [10].Computed at the PCM/TD-PBE0 and PCM/TD-M052X level.
- [11].a) Samanta PK, Manna AK, Pati SK. J. Phys. Chem. B. 2012;116:7618. doi: 10.1021/jp301752k. [DOI] [PubMed] [Google Scholar]; b) Samanta PK, Pati SK. New J. Chem. 2013;37:3640. [Google Scholar]
- [12].In addition to the relative position of the lowest energy peak, the general shape of the absorption spectra of the thG-H1 and thG–H3 tautomers was found to be very close to experimental ones, thus supporting their assignment.
- [13].a) Gedik M, Brown A, Photochem J. Photobiol. A. 2013;259:25. [Google Scholar]; b) Samanta PK, Pati SK. Phys. Chem. Chem. Phys. 2015;17:10053. doi: 10.1039/c5cp00134j. [DOI] [PubMed] [Google Scholar]
- [14].a) Karunakaran V, Kleinermanns K, Improta R, Kovalenko SA. J. Am. Chem. Soc. 2009;131:5839. doi: 10.1021/ja810092k. [DOI] [PubMed] [Google Scholar]; b) Improta R. Chem. Eur. J. 2014;20:8106. doi: 10.1002/chem.201400065. [DOI] [PubMed] [Google Scholar]
- [15].According to PCM/6-311+G(2d,2p) calculations on a thG·6H2O model.
- [16].a) Johnson PE, Turner RB, Wu ZR, Hairston L, Guo J, Levin JG, Summers MF. Biochemistry. 2000;39:9084. doi: 10.1021/bi000841i. [DOI] [PubMed] [Google Scholar]; b) Bourbigot S, Ramalanjaona N, Boudier C, Salgado GFJ, Roques BP, Mély Y, Bouaziz S, Morellet N. J. Mol. Biol. 2008;383:1112. doi: 10.1016/j.jmb.2008.08.046. [DOI] [PubMed] [Google Scholar]
- [17].Darlix JL, Godet J, Ivanyi-Nagy R, Fossé P, Mauffret O, Mély Y. J. Mol. Biol. 2011;410:565. doi: 10.1016/j.jmb.2011.03.037. [DOI] [PubMed] [Google Scholar]
- [18].Godet J, Ramalanjaona N, Sharma KK, Richert L, De Rocquigny H, Darlix JL, Duportail G, Mély Y. Nucleic Acids Res. 2011;39:6633. doi: 10.1093/nar/gkr274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].To rule out any possible bias by the selected force field used for the MD simulations (AMBER ff12SB force field) similar computations were performed using the CHARMM all-atom force field for nucleic acids. The results of these calculations are highly comparable and are detailed in the Supporting Information.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.