

Supplemental Digital Content is available in the text.
Keywords: adipose tissue, aorta, losartan, mineralocorticoid receptor
Abstract
Sustained stimulation of β-adrenoceptors (β-ARs) and activation of renin–angiotensin–aldosterone system are common features of cardiovascular diseases with rising sympathetic activation, including essential hypertension, myocardial infarction, and heart failure. In this study, we investigated the role of AT1 receptor and mineralocorticoid receptor (MR) in the vascular alterations caused by β-AR overstimulation. β-AR overstimulation with associated cardiac hypertrophy and increased vasoconstrictor response to phenylephrine in aorta were modeled in rats by 7-day isoproterenol treatment. The increased vasoconstrictor response to phenylephrine in this model was blunted by the MR antagonist spironolactone, but not by the AT1 receptor antagonist losartan, despite the blunting of cardiac hypertrophy with both drugs. Spironolactone, but not losartan, restored NO bioavailability in association with lower endothelial nitric oxide synthase–derived superoxide production, increased endothelial nitric oxide synthase dimerization, and aortic HSP90 upregulation. MR genomic and nongenomic functions were activated in aortas from isoproterenol-treated rats. Isoproterenol did not modify plasma levels of MR ligands aldosterone and corticosterone but rather increased perivascular adipose tissue–derived corticosterone in association with increased expression of 11β-hydroxysteroid dehydrogenase type 1. The anticontractile effect of aortic perivascular adipose tissue was impaired by β-AR overstimulation and restored by MR blockade. These results suggest that activation of vascular MR signaling contributes to the vascular dysfunction induced by β-AR overstimulation associated with endothelial nitric oxide synthase uncoupling. These findings reveal an additional explanation for the protective effects of MR antagonists in cardiovascular disorders with sympathetic activation.
Sympathetic overactivity with rising catecholamines levels and adrenergic receptors stimulation is a common feature of many cardiovascular disorders, including hypertension, myocardial infarction (MI), congestive heart failure, and acute cerebrovascular events. In these conditions, the hyperadrenergic state has a major and independent prognostic impact.1 Although the importance of β-adrenoceptor (β-AR) overstimulation in the pathogenesis of left ventricular dysfunction has been widely studied, less is known about its effects on vascular function. Following in vivo β-AR overstimulation, we and others have demonstrated abnormal vasoconstrictor response to agonists in aorta,2 coronary artery,3 and cerebral artery.4 In thoracic aorta, increased vasoconstrictor response induced by isoproterenol treatment was associated with increased reactive oxygen species generation and uncoupling of endothelial nitric oxide synthase (eNOS).2,5 These studies demonstrate that β-AR overstimulation induces vascular dysfunction but the molecular mechanisms remain to be elucidated.
Activation of the renin–angiotensin–aldosterone system is also involved in the pathogenesis of cardiovascular and metabolic diseases, including hypertension, MI, heart failure, and obesity. It is known that β-AR signaling in juxtaglomerular cells stimulates renin release, thereby stimulating renin–angiotensin–aldosterone system. In addition, β-AR agonist isoproterenol increases cardiac expression of angiotensin-converting enzyme6 and antagonism or deficiency of AT1 receptor (AT1R) attenuate isoproterenol-induced cardiac remodeling in mice.7 Elevated circulating aldosterone is also associated with isoproterenol-induced heart failure,8 and blockade of aldosterone-binding mineralocorticoid receptor (MR) is protective from cardiac hypertrophy and diastolic dysfunction induced by chronic isoproterenol treatment in rats.9 Together, these studies have suggested beneficial cardiac effects of AT1R and MR blockade in preventing isoproterenol-induced cardiac remodeling and dysfunction. In the vasculature, either AT1R or MR activation induces proinflammatory, profibrotic, and pro-oxidative vascular signaling pathways.10,11 However, whether AT1R and MR contribute to the vascular abnormalities caused by β-AR overstimulation has not been explored.
Vascular function is also known to be modified by perivascular adipose tissue (PVAT). Angiotensin II, via AT1R promotes aldosterone secretion from adipocytes, acting in a paracrine manner to regulate vascular function and contributing to endothelial dysfunction in obesity.12 PVAT of the thoracic aorta also releases adipocyte-derived relaxing factors that exhibit anticontractile effects.13,14 Acute β-AR activation stimulates the release of adipocyte-derived relaxing factors from PVAT of mesenteric artery.15 However, it is not known whether β-AR overstimulation could regulate the anticontractile effects of PVAT.
Therefore, in this study, we investigated a possible role of AT1R, MR, and PVAT in the vascular dysfunction induced by in vivo administration of isoproterenol as measured by enhanced vasoconstriction to phenylephrine. We hypothesized that MR activation induces uncoupling of eNOS, oxidative stress, and reduces anticontractile role of PVAT after β-AR overstimulation.
Methods
Animals
This study was approved by the Ethics Committee on Animal Use of the University of Campinas (protocol no. 2609-1) and carried out in accordance with the ethical principles for animal experimentation adopted by the Brazilian Society of Laboratory Animal Science (SBCAL/COBEA).
Male Wistar rats (12-week old) were obtained from the Multidisciplinary Center for Biological Research of the University of Campinas (Campinas, Brazil). Animals were housed at a constant room temperature (22°C), 12:12 hour light:dark cycle, and with normal chow and water provided ad libitum. Isoproterenol (0.3 mg/kg per day, sc) or the vehicle were administrated once daily for 7 days, concomitantly with the treatment or not via oral gavage with the AT1R antagonist losartan (40 mg/kg per day), or with the MR antagonist spironolactone (200 mg/kg per day).
Vascular Reactivity, Blood Pressure, and Biochemical and Molecular parameters
Detailed methods are available in the only-online Data Supplement.
Statistical Analysis
Data are presented as mean±SEM. Data were analyzed by the Student t test or two-way ANOVA followed by the Bonferroni post-test by using GraphPad Prism 5.0 software (GraphPad Software Corp, San Diego, CA). Values of P<0.05 were considered significantly different.
Results
Losartan and Spironolactone Treatments Similarly Reduce Isoproterenol-Induced Cardiac Hypertrophy
To confirm the efficacy of isoproterenol treatment in inducing long-term β-AR stimulation, the ventricular weight:body weight ratio was measured as an index of myocardial hypertrophy. Isoproterenol treatment increased ventricular weight:body weight ratio without affecting body weight that was similarly reduced by losartan and spironolactone (Table S1). No effect of either isoproterenol or spironolactone on blood pressure or heart rate was observed, but losartan decreased diastolic blood pressure in both control and isoproterenol-treated rats (Table S1).
Spironolactone, but Not Losartan, Prevented the Increased Aortic Reactivity to Phenylephrine in Isoproterenol-Treated Rats
Aortic rings from isoproterenol-treated rats showed an increased contractile response to phenylephrine compared with the control group (Figure 1A). This high contractility was not altered by cotreatment with losartan (Figure 1B), whereas it was fully prevented by spironolactone cotreatment (Figure 1C). These data support a role for MR, but not the AT1R, in the increased aortic contractile response induced by β-AR overstimulation. The relaxation to either acetylcholine or sodium nitroprusside was not modified by the treatments (Figure S1).
Figure 1.
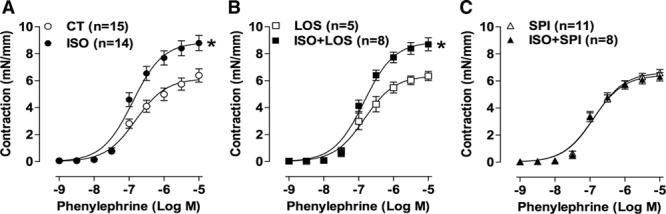
Spironolactone, but not losartan, prevented the enhanced contraction to phenylephrine induced by β-adrenergic overstimulation. Concentration–response curves to phenylephrine obtained in aortic rings from rats treated with vehicle (CT) or isoproterenol (ISO; A) combined with losartan (LOS; B) or with spironolactone (SPI; C). Data are expressed as mean±SEM; number of animals is indicated in parenthesis. Two-way ANOVA: *P<0.001 vs CT or LOS.
MR Antagonist Restored NO Bioavailability, NOS Dimerization, and HSP90 Protein Levels in Aortas of Isoproterenol-Treated Rats
Incubation with a nonselective NOS inhibitor, Nω-nitro-L-arginine methyl ester, enhanced the contractile response to phenylephrine in the aorta of all the groups (Figure 2A). The NOS-dependent anticontractile component of phenylephrine response (as measured by difference of the area under the curve before and after Nω-nitro-L-arginine methyl ester) was impaired in the isoproterenol group, whereas spironolactone reversed this effect (Figure 2B). Pretreatment of aortas with superoxide dismutase (SOD, superoxide scavenger) or tetrahydrobiopterin (BH4, eNOS cofactor) reduced vascular contraction in the isoproterenol group but not in the control and spironolactone-treated groups (Figure 2C and 2E). There was an increased difference of the area under the curve to phenylephrine in the presence of SOD and BH4 in isoproterenol-treated rats, which was normalized by spironolactone (Figure 2D and 2F). These data support a role for MR in increasing superoxide and reducing NO production and bioavailability after β-AR overstimulation. Indeed, aortic NO levels (evaluated by the fluorescence to diaminofluorescein) were decreased in rats exposed to long-term isoproterenol and spironolactone treatments enhanced NO to levels similar to the control group (Figure 3A). By contrast, losartan treatment did not prevent the impairment in NO bioavailability induced by isoproterenol (Figure S2).
Figure 2.
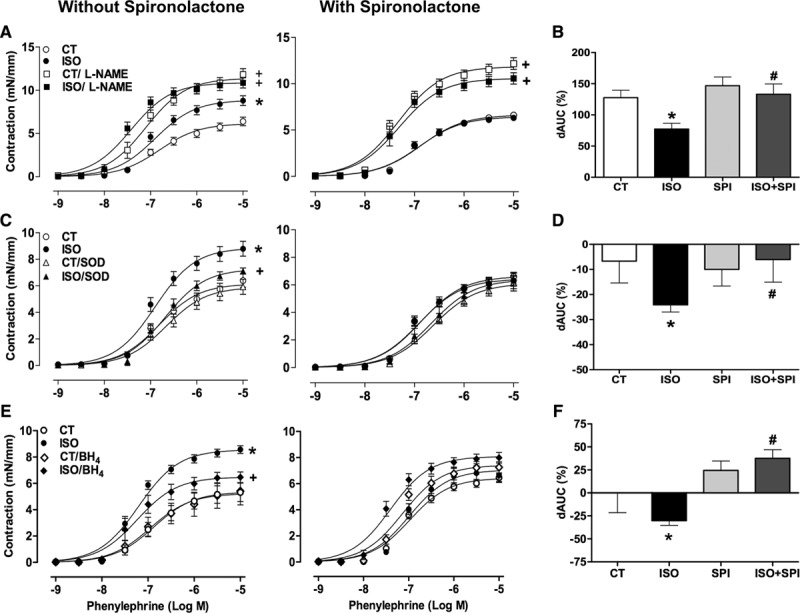
Effect of Nω-nitro-L-arginine methyl ester (L-NAME) (A, square symbols), superoxide dismutase (superoxide dismutase (SOD, superoxide scavenger); C, triangle symbols), and tetrahydrobiopterin (BH4, eNOS cofactor) (E, diamond symbol) on the concentration–response curves to phenylephrine of aortic rings from control (CT) and isoproterenol (ISO) groups without or with spironolactone (SPI) treatment. B, D, and F, The difference of the area under the curve (dAUC) to phenylephrine in the presence and absence of L-NAME, SOD, or BH4, respectively. Data are expressed as mean±SEM (n=4–15 in each group). Two-way ANOVA: P<0.05 +vs respective group without incubation; *vs CT; #vs ISO.
Figure 3.
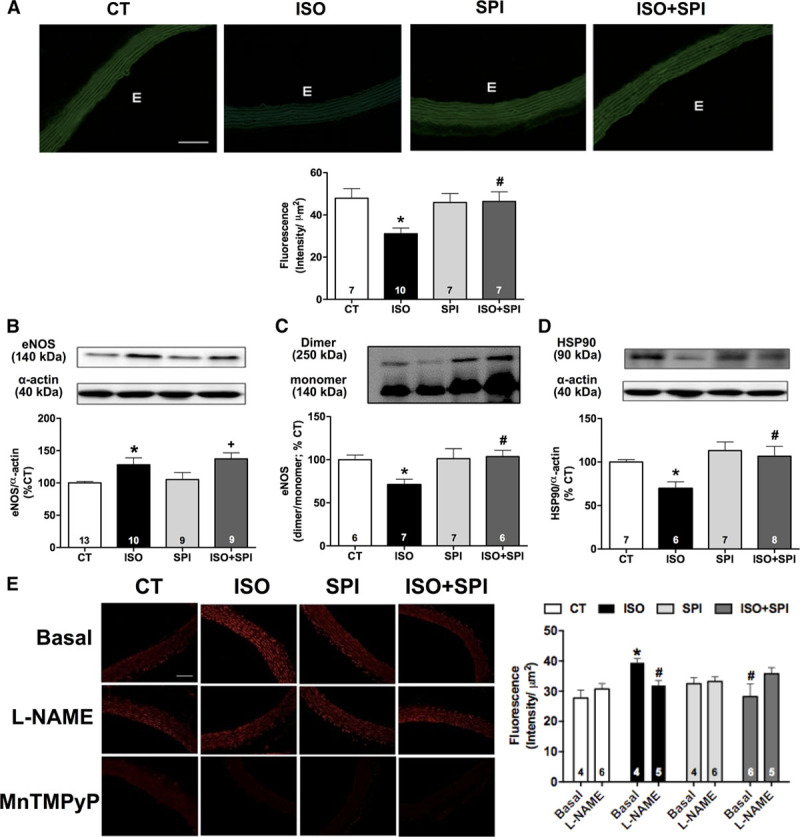
Reduced HSP90 expression and endothelial nitric oxide synthase (eNOS) uncoupling after β-AR overstimulation: prevention by mineralocorticoid receptor blockade. Diaminofluorescein (DAF-2) fluorescence (A), protein expression of total eNOS (B), dimer:monomer eNOS ratio (C), HSP90 (D), and hidroethidine (DHE) fluorescence (E) obtained in aorta from control (CT) and isoproterenol (ISO) groups without or with spironolactone (SPI) treatment. Protein expression was calculated as percentage of CT group. Representative images (20×, bar=100 µm) of DAF-2 and DHE fluorescence are shown in left side of A and E, respectively. In A, E=endothelium. The DHE fluorescence signal was evaluated under basal condition or after incubation with Nω-nitro-L-arginine methyl ester (L-NAME) (1 mmol/L) or MnTMPyP (25 µmol/L). Data represent mean±SEM; number of animals used for each group is indicated in the bars. Two-way ANOVA: P<0.05 *vs CT; +vs SPI; #vs ISO.
Although aortic expression of total eNOS protein was increased in isoproterenol-treated rats (Figure 3B), the abundance of its dimeric form was reduced (Figure 3C), as well as its phosphorylation in Ser1177 (Figure S3). Spironolactone did not affect the isoproterenol-induced increase in total eNOS protein levels, but it normalized eNOS dimerization (Figure 3B and 3C). eNOS phosphorylation were not affected by spironolactone or losartan treatment (Figure S3). Because HSP90 is an eNOS chaperone that augments NO production and inhibits superoxide formation,16 we investigated HSP90 expression. Isoproterenol treatment significantly reduced HSP90 protein expression, which was restored by spironolactone (Figure 3D).
eNOS-Derived Superoxide Anion Production After β-AR Overstimulation Is Prevented by Spironolactone
We measured reactive oxygen species production by quantification of the fluorescence formed after exposing aortic slices to hidroethidine. The fluorescent signal was almost undetectable after incubation with the SOD mimetic MnTMPyP, indicating superoxide as the main reactive oxygen species detectable by hidroethidine in aortas (Figure 3E). Isoproterenol induced an increase in vascular superoxide production that was inhibited by Nω-nitro-L-arginine methyl ester incubation, indicating enhanced eNOS-derived superoxide production (Figure 3E). Importantly, spironolactone, but not losartan, blocked this increase (Figure 3E; Figure S4). The data indicate that β-AR overstimulation induces vascular oxidative stress by eNOS, dependent on MR activation.
β-Adrenergic Overstimulation Activates Aortic MR Genomic and Nongenomic Activity
MR functions by translocating to the nucleus to regulate gene transcription (genomic mechanisms) and also by activating cytoplasmic signaling pathways (nongenomic mechanisms). Isoproterenol treatment increased the nuclear:cytoplasmic MR ratio in the aorta (Figure 4A) and increased gene expression of the smooth muscle cell MR target gene, osteopontin (Figure 4B) and the endothelial MR target protein, the γ-subunit of the epithelial sodium channel (ENaC; Figure 4C) when compared with the control group. Spironolactone prevented isoproterenol-induced MR nuclear translocation, osteopontin mRNA, and ENaC protein expression (Figure 4A through 4C) consistent with genomic MR activity. In addition, aorta from isoproterenol-treated rats demonstrated higher Src and ERK1/2 phosphorylation, which was prevented by spironolactone (Figure 4D and 4E). Thus, chronic isoproterenol treatment seems to activate both MR genomic and nongenomic activities in the aorta.
Figure 4.
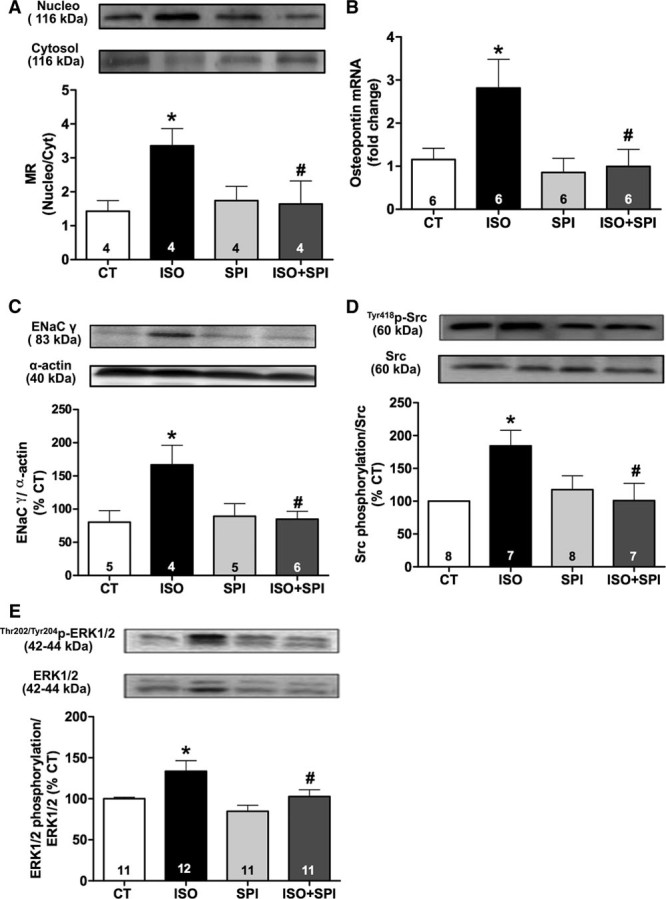
Genomic and nongenomic pathways of vascular mineralocorticoid receptor (MR) activity after isoproterenol treatment. Nuclear translocation of MR (A), mRNA expression of osteopontin (B), and protein expression of γ-epithelial sodium channel (γENaC; C), Src (D), and ERK1/2 (E) phosphorylation in thoracic aorta of control (CT) and isoproterenol (ISO) groups without or with spironolactone (SPI) treatment. Data represent mean±SEM; number of animals used in each group is indicated into the bars. Two-way ANOVA: P<0.05 *vs CT; #vs ISO.
Enhanced Corticosterone Levels and Increased Expression of 11β-HSD1 in PVAT After Isoproterenol Treatment
MR can be activated by the mineralocorticoid aldosterone and also by some corticosteroids. To investigate if isoproterenol activates the MR by modulating levels of endogenous ligands, we measured plasma and PVAT levels of aldosterone and corticosterone (the main glucocorticoid in rodents). Plasma and PVAT levels of aldosterone were increased by spironolactone treatment, consistent with a feedback mechanism associated with effective MR blockade (Figure 5A and 5B). Neither isoproterenol nor spironolactone treatment affected plasma corticosterone levels (Figure 5C). However, corticosterone content was enhanced in PVAT after isoproterenol treatment, whereas spironolactone did not alter this effect (Figure 5D). Treatments did not affect aortic PVAT weight (data not shown). These data suggest that enhanced PVAT-derived corticosterone could be a mechanism leading to paracrine activation of vascular MR after β-adrenergic overstimulation.
Figure 5.
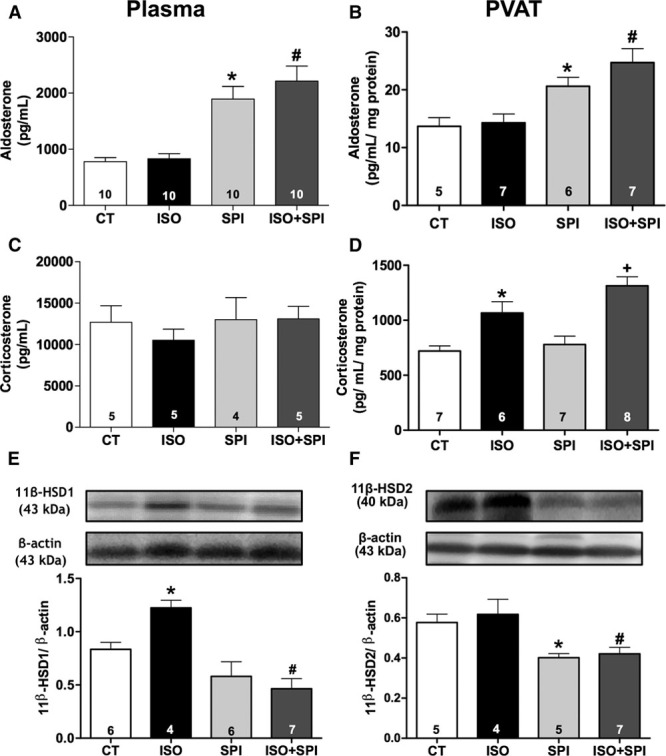
β-AR overstimulation enhanced corticosterone content and protein expression of 11β-HSD1 in perivascular adipose tissue (PVAT) of aorta. Aldosterone (A and B) and corticosterone (C and D) content measured in plasma and aortic PVAT from control (CT) and isoproterenol (ISO) groups without or with spironolactone (SPI) treatment. Protein expression of 11β-HSD1 (E) and 11β-HSD2 (F) were evaluated in PVAT from CT, ISO, SPI, and ISO+SPI groups. Data represent mean±SEM; number of animals is indicated in the bars. Two-way ANOVA: P<0.05 *vs CT; +vs SPI; #vs ISO.
PVAT protein expression of aldosterone synthase (CYP11B1), a final enzyme required for glucocorticoid synthesis, was not affected by isoproterenol treatment (Figure S5A); whereas, isoproterenol increased the PVAT expression of 11β-hydroxysteroid dehydrogenase type-1 (11β-HSD1), the enzyme that generates active glucocorticoids from their inactive 11-keto derivatives (Figure 5E), with no effect in 11β-hydroxysteroid dehydrogenase type-2 (11β-HSD2) expression (Figure 5F). Spironolactone decreased the expression of 11β-HSD1 and 11β-HSD2 (Figure 5E and 5F). Glucocorticoid synthase (CYP11B2) protein expression in PVAT did not differ from the control and isoproterenol groups (Figure S5B).
Spironolactone Restores the Anticontractile Effect of Perivascular Tissue, Which Is Impaired by Isoproterenol
Next, we investigated the role of PVAT on vascular contraction to phenylephrine. As expected, the presence of PVAT reduced the contraction to phenylephrine in aortas from control rats (Figure 6A). However, this anticontractile effect of PVAT was impaired by isoproterenol treatment (Figure 6B and 6E). Spironolactone did not affect the basal role of PVAT on the contractile response to phenylephrine (Figure 6C and 6E) but rather, in the presence of MR antagonist, the anticontractile effect of PVAT was restored in rats cotreated with isoproterenol and spironolactone (Figure 6D and 6E).
Figure 6.
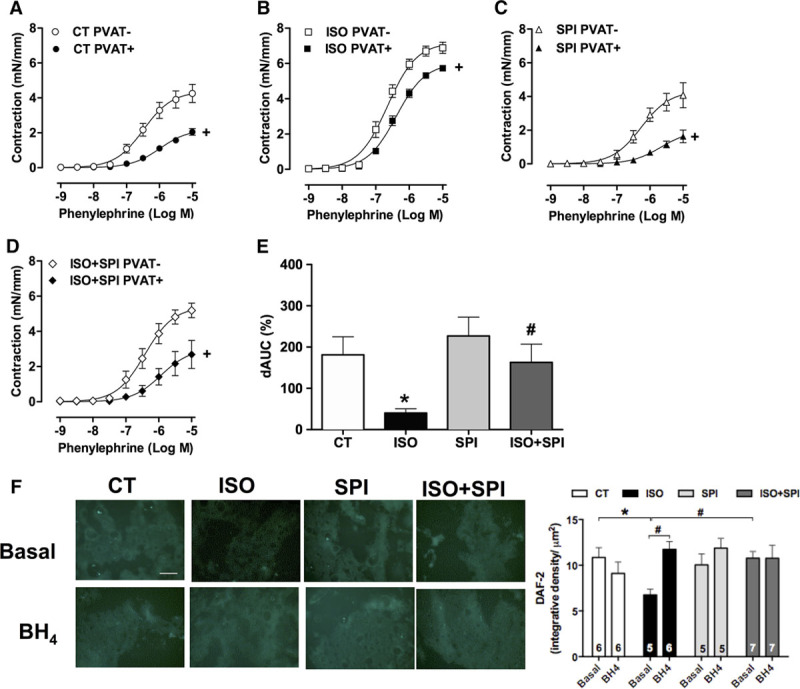
Impaired anticontractile function of perivascular adipose tissue (PVAT) after isoproterenol treatment is rescued by spironolactone. Concentration–response curves to phenylephrine were obtained in aortic rings without (−) or with (+) PVAT from control (CT, A) and isoproterenol (ISO, B) groups without or with spironolactone (SPI) treatment (C and D). E, The difference of the area under the curve (dAUC) to phenylephrine in the absence and presence of PVAT. Fluorescence to NO-sensitive dye diaminofluorescein (DAF-2) was evaluated in thoracic PVAT sections from CT, ISO, SPI, and ISO+SPI groups under basal conditions or after incubation with tetrahydrobiopterin (BH4, eNOS cofactor) (100 µmol/L, F). Representative images (20×, white bar=100 µm) of DAF-2 fluorescence are shown. Data are expressed as mean±SEM (n=5–15 in each group). Two-way ANOVA: P<0.05 +vs PVAT; *vs CT; #vs ISO.
Recently, it was demonstrated that PVAT from thoracic aorta expresses eNOS that produces NO as a PVAT-derived relaxing factor, whereas uncoupled eNOS in this tissue might be a mechanism involved in vascular dysfunction.17 Therefore, we explored eNOS uncoupling in PVAT after β-adrenergic overstimulation. PVAT-derived NO production was impaired by isoproterenol treatment and restored by spironolactone treatment or BH4 incubation (Figure 6F).
Discussion
This study demonstrated that the MR antagonist spironolactone, but not the AT1R blocker losartan, prevented the increased vasoconstrictor response to phenylephrine induced by β-AR overstimulation. The protective vascular effect of spironolactone was associated with (1) increased eNOS dimerization, HSP90 expression and NO production, (2) reduced eNOS-derived superoxide production, (3) inhibition of genomic and nongenomic vascular MR pathways, and (4) restoration of the anticontractile role of aortic PVAT. Furthermore, we found elevated corticosterone content in aortic PVAT after β-adrenergic stimulation. These findings support a model in which chronic β-adrenergic stimulation promotes vascular MR activation, which results in eNOS uncoupling and oxidative stress. This model provides a novel mechanism by which MR antagonists can be protective in patients with cardiovascular disease by preventing vascular dysfunction associated with hyperadrenergic state, such as in heart failure, MI, and essential hypertension.
Renin–angiotensin–aldosterone system inhibitors, including angiotensin-converting enzyme inhibitors and AT1R antagonists, and more recently MR antagonists improve survival in patients with left ventricular dysfunction.18,19 The diuretic effect of the MR antagonist was not sufficient to explain its cardioprotective effects in patients with heart failure.20 Although both losartan and spironolactone reduced ventricular hypertrophy in a similar magnitude, we observed that only MR antagonism was effective in the prevention of increased vasoconstrictor response to phenylephrine, NO bioavailability, and eNOS-dependent oxidative stress. This result demonstrates for the first time a role of MR, but not AT1R, in mediating the major vascular effects of β-adrenergic overstimulation. In addition, losartan, but not spironolactone, reduced diastolic blood pressure measured invasively in anesthetized rats. However, we cannot exclude the possibility of group differences in blood pressure because we did not monitor this parameter 24 hours per day in awake, unrestrained animals.
Abnormal vasoconstrictor responses to agonists including phenylephrine have been demonstrated in several vascular beds2–5 in the isoproterenol-induced left ventricular hypertrophy model. These studies put forward a key role for β-AR overstimulation in inducing vascular dysfunction. We previously demonstrated a role for uncoupled eNOS in the altered vascular contractility induced by β-AR stimulation.5 Because aldosterone-induced MR activation can impair eNOS-derived NO production associated with a reduction in eNOS dimerization in endothelial cells,21 we hypothesized that MR blockade could improve this endothelial pathway. In accordance, improved NO bioavailability, enhanced eNOS dimer expression, and reduced eNOS-derived superoxide were observed in aortas from isoproterenol+spironolactone–treated rats. Although eNOS uncoupling was associated with exacerbated contractile response to phenylephrine, the endothelium-dependent relaxation to acetylcholine was not significantly changed by isoproterenol treatment. This apparently contradictory finding might be explained by the greater sensitivity of basal NO to destruction by superoxide when compared with agonist-stimulated NO production.22
Dimerization of eNOS regulates its catalytic activity and NO production. Association of eNOS with HSP90 has been demonstrated to be an important mechanism regulating eNOS dimerization, rather than eNOS phosphorylation.23 Here, we show that β-AR overstimulation significantly reduced aortic expression of this chaperone, which was prevented by spironolactone. Interestingly, impaired interaction of HSP90 with eNOS results in decreased NO production and superoxide generation.16 Therefore, reduced vascular expression of HSP90 is a potential link between β-AR overstimulation and eNOS dysfunction that can be prevented by MR blockade.
Increased nuclear:cytoplasmic MR ratio was found in aorta from isoproterenol-treated rats. Given that MR dissociation from HSP90 induces MR cytoplasmic-to-nucleus trafficking,24 reduction in HSP90 expression induced by isoproterenol could also contribute to enhance MR transcriptional activity. Spironolactone treatment enhanced HSP90 expression and attenuated nuclear localization of MR, osteopontin mRNA, and ENaC protein levels in isoproterenol-treated rats. Osteopontin is a multifunctional glycophosphoprotein that can be secreted by endothelium and vascular smooth muscle cells; its gene expression is induced by MR and contributes to proinflammatory and profibrotic effects of aldosterone.25,26 In addition, recent data suggest that MR activation induces binding to the ENaC promoter, increasing expression of ENaC mRNA in endothelial cells with associated endothelial dysfunction.27 We observed an increased aortic expression of the regulatory γ-subunit of ENaC in aorta of isoproterenol-treated rats that was prevented by cotreatment with spironolactone, suggesting that β-adrenergic stimulation might upregulate ENaC through MR activation. To our knowledge. this is the first demonstration that osteopontin and ENaC can be upregulated in vascular tissue in response to β-adrenergic stimulation and could be an additional mechanism involved in the vascular pathology associated with sympathetic hyperactivity.
In addition to genomic activity of MR, a nongenomic MR pathway in the vasculature has been demonstrated to contribute to oxidative stress, inflammation, and vascular dysfunction.11 Aldosterone rapidly activates several kinases, including Src and mitogen-activated protein kinases in vascular smooth muscle cells.28 Here, we reported that MR blockade attenuated phosphorylation of Src and ERK1/2 in aortas from isoproterenol-treated rats, which indicates convergence of the β-adrenergic and MR-signaling pathways. However, the observed association between the spironolactone-induced changes in the biochemical and molecular parameters evaluated and the vascular protection in the contractile function may not necessarily reflect cause and effect relationship, representing a limitation of this study.
Both aldosterone and glucocorticoids bind to the MR to activate its genomic and nongenomic functions.29 In this study, β-AR overstimulation did not change either aldosterone or corticosterone plasma levels; however, corticosterone, but not aldosterone, was enhanced in aortic PVAT from isoproterenol-treated rats. Spironolactone did not attenuate the enhanced levels of corticosterone induced by isoproterenol in PVAT, suggesting that the beneficial vascular effects of spironolactone may be downstream and could be because of blockade of glucocorticoid-induced MR activation. Mature adipocytes express CYP11B2 and CYP11B1 and can produce aldosterone and corticosterone.12 The expression of these enzymes was not modified by isoproterenol treatment. Glucocorticoid content is also regulated by 11β-HSD type 1 and 2. 11β-HSD2 converts glucocorticoids into inactive metabolites, which favors aldosterone-MR interaction. However, 11β-HSD1 in the presence of NADPH promotes glucocorticoids regeneration from inert 11-keto metabolites.30 Therefore, an upregulation of 11β-HSD1 in PVAT after chronic β-adrenergic stimulation might be a mechanism associated with high perivascular corticosterone content, thereby leading to paracrine activation of vascular MR. However, as β-AR signal through the small GTPase Rac131 which has been shown to activate MR without ligand,32 we should also consider ligand-independent MR activation as a potential mechanism for isoproterenol-induced MR activation.
Spironolactone treatment prevented the isoproterenol-induced 11β-HSD1 expression in PVAT. Earlier studies demonstrated that suppression of 11β-HSD1 abolishes the inhibitory effect of glucocorticoids on eNOS expression33 and prevents heart failure development after MI.34 Therefore, reduction in 11β-HSD1 expression in PVAT is a potential cardiovascular protective mechanism of spironolactone. Despite this effect, corticosterone levels were still high in PVAT from rats cotreated with isoproterenol and spironolactone, which could be related to the minor expression of 11β-HSD2 induced by spironolactone. However, the cellular origin of corticosterone secretion induced by isoproterenol in PVAT and the signaling pathway involved in this effect remains an open question. In addition, as endothelium, vascular smooth muscle cells, inflammatory cells and adipocytes express functional MR35–38 further investigation using cell-type–specific knockout mice should address which cell-type–specific MR signaling is mediating the vascular effects induced by β-adrenergic stimulation.
In conclusion, our findings revealed a novel mechanism of regulation of vascular dysfunction mediated by β-AR overstimulation inducing PVAT-derived corticosterone production, associated with impaired PVAT anticontractile effect and vascular MR activation. MR blockade with spironolactone protected from increased vasoconstrictor response to phenylephrine, upregulation of ENaC, and downregulation of HSP90 and eNOS uncoupling. The present findings uncover a role for MR blockade in sympathoexcitatory cardiovascular diseases and provide an additional novel vascular mechanism for the protective effects of MR antagonism.
Perspectives
We demonstrated that MR antagonist spironolactone prevents the vascular alterations induced by long-term β-AR stimulation, including enhanced vasoconstriction, uncoupling of eNOS, reduced NO bioavailability, and oxidative stress. The beneficial vascular effects of spironolactone were independent of changes in systemic levels of MR ligand. Instead, we observed increased levels of PVAT-derived corticosterone in response to β-adrenergic stimulation associated with enhanced protein levels of 11β-HSD1 that regenerates glucocorticoids. MR genomic and nongenomic signaling were observed in aortas of isoproterenol-treated rats. Therefore, this study suggests a novel link between β-AR signaling and vascular MR activation in causing vascular dysfunction. Moreover, the results indicate an additional mechanism for the protective vascular effects of MR antagonists in cardiovascular diseases associated with increased sympathetic activity, such as essential hypertension, MI, and heart failure.
Acknowledgments
We thank Dr Gisele K. Couto for help in diaminofluorescein (DAF-2) and hidroethidine (DHE) analysis. We also thank Dr Maria O. de Souza for a generous gift of ENaC antibodies.
Sources of Funding
This work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP Brazil grants 14/07947-6 and 11/15972-2) and Ministerio de Educación Cultura y Deporte (PHBP14/00001). A.P. Davel, D.V. Vassallo, and L.V. Rossoni are research fellows from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, Brazil).
Disclosures
None.
Supplementary Material
Footnotes
The online-only Data Supplement is available with this article at http://hyper.ahajournals.org/lookup/suppl/doi:10.1161/HYPERTENSIONAHA.116.07911/-/DC1.
Novelty and Significance
What Is New?
Spironolactone prevents the increased vasoconstrictor response to phenylephrine and uncoupling of endothelial nitric oxide synthase in aorta of a rat model of chronic β-adrenergic stimulation induced by isoproterenol.
β-AR overstimulation impairs the anticontractile function of perivascular adipose tissue and induces perivascular adipose tissue–derived glucocorticoid production.
What Is Relevant?
Mineralocorticoid receptor blockade with spironolactone prevented the vascular dysfunction induced by β-AR overstimulation, independent of changes in systemic levels of mineralocorticoid receptor ligands, suggesting a novel mechanism for the protective vascular effects of mineralocorticoid receptor antagonists in cardiovascular diseases associated with increased sympathetic activity, such as essential hypertension and heart failure.
Summary
Mineralocorticoid receptor activation is crucial for the vascular alterations induced by long-term β-AR stimulation, including increased vasoconstriction, uncoupling of endothelial nitric oxide synthase, and impaired anticontractile function of PVAT.
References
- 1.Grassi G. Sympathetic neural activity in hypertension and related diseases. Am J Hypertens. 2010;23:1052–1060. doi: 10.1038/ajh.2010.154. doi: 10.1038/ajh.2010.154. [DOI] [PubMed] [Google Scholar]
- 2.Davel AP, Kawamoto EM, Scavone C, Vassallo DV, Rossoni LV. Changes in vascular reactivity following administration of isoproterenol for 1 week: a role for endothelial modulation. Br J Pharmacol. 2006;148:629–639. doi: 10.1038/sj.bjp.0706749. doi: 10.1038/sj.bjp.0706749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim N, Chung J, Kim E, Han J. Changes in the Ca2+-activated K+ channels of the coronary artery during left ventricular hypertrophy. Circ Res. 2003;93:541–547. doi: 10.1161/01.RES.0000090087.66390.F2. doi: 10.1161/01.RES.0000090087.66390.F2. [DOI] [PubMed] [Google Scholar]
- 4.Kim HK, Park WS, Warda M, Park SY, Ko EA, Kim MH, Jeong SH, Heo HJ, Choi TH, Hwang YW, Lee SI, Ko KS, Rhee BD, Kim N, Han J. Beta adrenergic overstimulation impaired vascular contractility via actin-cytoskeleton disorganization in rabbit cerebral artery. PLoS One. 2012;7:e43884. doi: 10.1371/journal.pone.0043884. doi: 10.1371/journal.pone.0043884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davel AP, Brum PC, Rossoni LV. Isoproterenol induces vascular oxidative stress and endothelial dysfunction via a Giα-coupled β2-adrenoceptor signaling pathway. PLoS One. 2014;9:e91877. doi: 10.1371/journal.pone.0091877. doi: 10.1371/journal.pone.0091877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oliveira EM, Krieger JE. Chronic beta-adrenoceptor stimulation and cardiac hypertrophy with no induction of circulating renin. Eur J Pharmacol. 2005;520:135–141. doi: 10.1016/j.ejphar.2005.07.026. doi: 10.1016/j.ejphar.2005.07.026. [DOI] [PubMed] [Google Scholar]
- 7.Zhang GX, Ohmori K, Nagai Y, Fujisawa Y, Nishiyama A, Abe Y, Kimura S. Role of AT1 receptor in isoproterenol-induced cardiac hypertrophy and oxidative stress in mice. J Mol Cell Cardiol. 2007;42:804–811. doi: 10.1016/j.yjmcc.2007.01.012. doi: 10.1016/j.yjmcc.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 8.Bos R, Mougenot N, Findji L, Médiani O, Vanhoutte PM, Lechat P. Inhibition of catecholamine-induced cardiac fibrosis by an aldosterone antagonist. J Cardiovasc Pharmacol. 2005;45:8–13. doi: 10.1097/00005344-200501000-00003. [DOI] [PubMed] [Google Scholar]
- 9.Martín-Fernández B, de las Heras N, Miana M, Ballesteros S, Valero-Muñoz M, Vassallo D, Davel AP, Rossoni LV, Cachofeiro V, Lahera V. Spironolactone prevents alterations associated with cardiac hypertrophy produced by isoproterenol in rats: involvement of serum- and glucocorticoid-regulated kinase type 1. Exp Physiol. 2012;97:710–718. doi: 10.1113/expphysiol.2011.063230. doi: 10.1113/expphysiol.2011.063230. [DOI] [PubMed] [Google Scholar]
- 10.Schiffrin EL, Park JB, Intengan HD, Touyz RM. Correction of arterial structure and endothelial dysfunction in human essential hypertension by the angiotensin receptor antagonist losartan. Circulation. 2000;101:1653–1659. doi: 10.1161/01.cir.101.14.1653. [DOI] [PubMed] [Google Scholar]
- 11.Moss ME, Jaffe IZ. Mineralocorticoid receptors in the pathophysiology of vascular inflammation and atherosclerosis. Front Endocrinol (Lausanne) 2015;6:153. doi: 10.3389/fendo.2015.00153. doi: 10.3389/fendo.2015.00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Briones AM, Nguyen Dinh Cat A, Callera GE, Yogi A, Burger D, He Y, Corrêa JW, Gagnon AM, Gomez-Sanchez CE, Gomez-Sanchez EP, Sorisky A, Ooi TC, Ruzicka M, Burns KD, Touyz RM. Adipocytes produce aldosterone through calcineurin-dependent signaling pathways: implications in diabetes mellitus-associated obesity and vascular dysfunction. Hypertension. 2012;59:1069–1078. doi: 10.1161/HYPERTENSIONAHA.111.190223. doi: 10.1161/HYPERTENSIONAHA.111.190223. [DOI] [PubMed] [Google Scholar]
- 13.Löhn M, Dubrovska G, Lauterbach B, Luft FC, Gollasch M, Sharma AM. Periadventitial fat releases a vascular relaxing factor. FASEB J. 2002;16:1057–1063. doi: 10.1096/fj.02-0024com. doi: 10.1096/fj.02-0024com. [DOI] [PubMed] [Google Scholar]
- 14.Szasz T, Bomfim GF, Webb RC. The influence of perivascular adipose tissue on vascular homeostasis. Vasc Health Risk Manag. 2013;9:105–116. doi: 10.2147/VHRM.S33760. doi: 10.2147/VHRM.S33760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weston AH, Egner I, Dong Y, Porter EL, Heagerty AM, Edwards G. Stimulated release of a hyperpolarizing factor (ADHF) from mesenteric artery perivascular adipose tissue: involvement of myocyte BKCa channels and adiponectin. Br J Pharmacol. 2013;169:1500–1509. doi: 10.1111/bph.12157. doi: 10.1111/bph.12157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pritchard KA, Jr, Ackerman AW, Gross ER, Stepp DW, Shi Y, Fontana JT, Baker JE, Sessa WC. Heat shock protein 90 mediates the balance of nitric oxide and superoxide anion from endothelial nitric-oxide synthase. J Biol Chem. 2001;276:17621–17624. doi: 10.1074/jbc.C100084200. doi: 10.1074/jbc.C100084200. [DOI] [PubMed] [Google Scholar]
- 17.Xia N, Horke S, Habermeier A, Closs EI, Reifenberg G, Gericke A, Mikhed Y, Münzel T, Daiber A, Förstermann U, Li H. Uncoupling of endothelial nitric oxide synthase in perivascular adipose tissue of diet-induced obese mice. Arterioscler Thromb Vasc Biol. 2016;36:78–85. doi: 10.1161/ATVBAHA.115.306263. doi: 10.1161/ATVBAHA.115.306263. [DOI] [PubMed] [Google Scholar]
- 18.Pitt B, Poole-Wilson PA, Segal R, Martinez FA, Dickstein K, Camm AJ, Konstam MA, Riegger G, Klinger GH, Neaton J, Sharma D, Thiyagarajan B. Effect of losartan compared with captopril on mortality in patients with symptomatic heart failure: randomised trial–the Losartan Heart Failure Survival Study ELITE II. Lancet. 2000;355:1582–1587. doi: 10.1016/s0140-6736(00)02213-3. [DOI] [PubMed] [Google Scholar]
- 19.Pitt B, Remme W, Zannad F, Neaton J, Martinez F, Roniker B, Bittman R, Hurley S, Kleiman J, Gatlin M Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study Investigators. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med. 2003;348:1309–1321. doi: 10.1056/NEJMoa030207. doi: 10.1056/NEJMoa030207. [DOI] [PubMed] [Google Scholar]
- 20.Rossignol P, Ménard J, Fay R, Gustafsson F, Pitt B, Zannad F. Eplerenone survival benefits in heart failure patients post-myocardial infarction are independent from its diuretic and potassium-sparing effects. Insights from an EPHESUS (Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study) substudy. J Am Coll Cardiol. 2011;58:1958–1966. doi: 10.1016/j.jacc.2011.04.049. doi: 10.1016/j.jacc.2011.04.049. [DOI] [PubMed] [Google Scholar]
- 21.Leopold JA, Dam A, Maron BA, Scribner AW, Liao R, Handy DE, Stanton RC, Pitt B, Loscalzo J. Aldosterone impairs vascular reactivity by decreasing glucose-6-phosphate dehydrogenase activity. Nat Med. 2007;13:189–197. doi: 10.1038/nm1545. doi: 10.1038/nm1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mian KB, Martin W. Differential sensitivity of basal and acetylcholine-stimulated activity of nitric oxide to destruction by superoxide anion in rat aorta. Br J Pharmacol. 1995;115:993–1000. doi: 10.1111/j.1476-5381.1995.tb15909.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen W, Xiao H, Rizzo AN, Zhang W, Mai Y, Ye M. Endothelial nitric oxide synthase dimerization is regulated by heat shock protein 90 rather than by phosphorylation. PLoS One. 2014;9:e105479. doi: 10.1371/journal.pone.0105479. doi: 10.1371/journal.pone.0105479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Galigniana MD, Echeverría PC, Erlejman AG, Piwien-Pilipuk G. Role of molecular chaperones and TPR-domain proteins in the cytoplasmic transport of steroid receptors and their passage through the nuclear pore. Nucleus. 2010;1:299–308. doi: 10.4161/nucl.1.4.11743. doi: 10.4161/nucl.1.4.11743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sugiyama T, Yoshimoto T, Hirono Y, Suzuki N, Sakurada M, Tsuchiya K, Minami I, Iwashima F, Sakai H, Tateno T, Sato R, Hirata Y. Aldosterone increases osteopontin gene expression in rat endothelial cells. Biochem Biophys Res Commun. 2005;336:163–167. doi: 10.1016/j.bbrc.2005.08.056. doi: 10.1016/j.bbrc.2005.08.056. [DOI] [PubMed] [Google Scholar]
- 26.Fu GX, Xu CC, Zhong Y, Zhu DL, Gao PJ. Aldosterone-induced osteopontin expression in vascular smooth muscle cells involves MR, ERK, and p38 MAPK. Endocrine. 2012;42:676–683. doi: 10.1007/s12020-012-9675-2. doi: 10.1007/s12020-012-9675-2. [DOI] [PubMed] [Google Scholar]
- 27.Jia G, Habibi J, Aroor AR, Martinez-Lemus LA, DeMarco VG, Ramirez-Perez FI, Sun Z, Hayden MR, Meininger GA, Mueller KB, Jaffe IZ, Sowers JR. Endothelial mineralocorticoid receptor mediates diet-induced aortic stiffness in females. Circ Res. 2016;118:935–943. doi: 10.1161/CIRCRESAHA.115.308269. doi: 10.1161/CIRCRESAHA.115.308269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Callera GE, Montezano AC, Yogi A, Tostes RC, He Y, Schiffrin EL, Touyz RM. c-Src-dependent nongenomic signaling responses to aldosterone are increased in vascular myocytes from spontaneously hypertensive rats. Hypertension. 2005;46:1032–1038. doi: 10.1161/01.HYP.0000176588.51027.35. doi: 10.1161/01.HYP.0000176588.51027.35. [DOI] [PubMed] [Google Scholar]
- 29.Farman N, Rafestin-Oblin ME. Multiple aspects of mineralocorticoid selectivity. Am J Physiol Renal Physiol. 2001;280:F181–F192. doi: 10.1152/ajprenal.2001.280.2.F181. [DOI] [PubMed] [Google Scholar]
- 30.Funder JW. Mineralocorticoid receptor activation and oxidative stress. Hypertension. 2007;50:840–841. doi: 10.1161/HYPERTENSIONAHA.107.098012. doi: 10.1161/HYPERTENSIONAHA.107.098012. [DOI] [PubMed] [Google Scholar]
- 31.Moniri NH, Daaka Y. Agonist-stimulated reactive oxygen species formation regulates beta2-adrenergic receptor signal transduction. Biochem Pharmacol. 2007;74:64–73. doi: 10.1016/j.bcp.2007.03.016. doi: 10.1016/j.bcp.2007.03.016. [DOI] [PubMed] [Google Scholar]
- 32.Ayuzawa N, Nagase M, Ueda K, Nishimoto M, Kawarazaki W, Marumo T, Aiba A, Sakurai T, Shindo T, Fujita T. Rac1-mediated activation of mineralocorticoid receptor in pressure overload-induced cardiac injury. Hypertension. 2016;67:99–106. doi: 10.1161/HYPERTENSIONAHA.115.06054. doi: 10.1161/HYPERTENSIONAHA.115.06054. [DOI] [PubMed] [Google Scholar]
- 33.Liu Y, Mladinov D, Pietrusz JL, Usa K, Liang M. Glucocorticoid response elements and 11 beta-hydroxysteroid dehydrogenases in the regulation of endothelial nitric oxide synthase expression. Cardiovasc Res. 2009;81:140–147. doi: 10.1093/cvr/cvn231. doi: 10.1093/cvr/cvn231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.White CI, Jansen MA, McGregor K, Mylonas KJ, Richardson RV, Thomson A, Moran CM, Seckl JR, Walker BR, Chapman KE, Gray GA. Cardiomyocyte and vascular smooth muscle-independent 11β-hydroxysteroid dehydrogenase 1 amplifies infarct expansion, hypertrophy, and the development of heart failure after myocardial infarction in male mice. Endocrinology. 2016;157:346–357. doi: 10.1210/en.2015-1630. doi: 10.1210/en.2015-1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jaffe IZ, Mendelsohn ME. Angiotensin II and aldosterone regulate gene transcription via functional mineralocortocoid receptors in human coronary artery smooth muscle cells. Circ Res. 2005;96:643–650. doi: 10.1161/01.RES.0000159937.05502.d1. doi: 10.1161/01.RES.0000159937.05502.d1. [DOI] [PubMed] [Google Scholar]
- 36.Caprio M, Newfell BG, la Sala A, Baur W, Fabbri A, Rosano G, Mendelsohn ME, Jaffe IZ. Functional mineralocorticoid receptors in human vascular endothelial cells regulate intercellular adhesion molecule-1 expression and promote leukocyte adhesion. Circ Res. 2008;102:1359–1367. doi: 10.1161/CIRCRESAHA.108.174235. doi: 10.1161/CIRCRESAHA.108.174235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bene NC, Alcaide P, Wortis HH, Jaffe IZ. Mineralocorticoid receptors in immune cells: emerging role in cardiovascular disease. Steroids. 2014;91:38–45. doi: 10.1016/j.steroids.2014.04.005. doi: 10.1016/j.steroids.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Caprio M, Fève B, Claës A, Viengchareun S, Lombès M, Zennaro MC. Pivotal role of the mineralocorticoid receptor in corticosteroid-induced adipogenesis. FASEB J. 2007;21:2185–2194. doi: 10.1096/fj.06-7970com. doi: 10.1096/fj.06-7970com. [DOI] [PubMed] [Google Scholar]