

Abstract
An account is provided of the extraordinary features of buckminster fullerene cations and their chemistry that we discovered in our Ion Chemistry Laboratory at York University (Canada) during a ‘golden’ period of research in the early 1990s, just after C60 powder became available. We identified new chemical ways of C60 ionization and tracked novel chemistry of C60n+ as a function of charge state (n=1–3) with some 50 different reagent molecules. We found that multiple charges enhance reaction rates and diversify reaction products and mechanisms. Strong electrostatic interactions with reagent molecules were seen to reduce barriers to carbon surface bonding and charge-separation reactions, while intramolecular Coulomb repulsion appeared to localize charge on the surface or the substituent and so influence higher order chemistry, including ‘spindle’, ‘star’, ‘fuzzy ball’, ‘ball-and-chain’ and dimer ion formation. We introduced the notion of ‘apparent’ gas-phase acidity with measurements of proton-transfer reactions of multiply charged fullerene cations. We also explored the attachment of atomic metal cations to C60 and their subsequent reactions. All these findings were applied to the possible chemistry of fullerene cations in the interstellar medium with a focus on multiply charged fullerene ion formation and the intervention of fullerene cations in fullerene derivatization and molecular synthesis, with a view to their possible future detection.
This article is part of the themed issue ‘Fullerenes: past, present and future, celebrating the 30th anniversary of Buckminster Fullerene’.
Keywords: buckminster fullerene cations, electron transfer, charge state chemistry, extraterrestrial chemistry
1. Introduction
At the time of the dazzling news in 1985 of the mass-spectrometric observation of the buckminster fullerene cation C60+ by Sir Harry Kroto and his colleagues [1], my research group, in the Ion Chemistry Laboratory at York University in Canada, was uniquely poised to begin measurements of physico-chemical reactions of C60+ with atoms and molecules at room temperature. Our laboratory had just constructed a helium flow reactor with mass-spectrometric detection and coupled upstream to an electron-impact ionization source with ion selection using a quadrupole mass filter [2]. We were using this apparatus, known as a selected-ion flow tube (SIFT), routinely to measure kinetic properties of a large variety of chemical reactions between ions and molecules. Measurements with C60 became our focus in 1991 as soon as C60 powder became readily available [3] for vaporization and ionization and so began a journey of discovery and achievement beyond our wildest dreams. Two exceptional post-doctoral fellows had just joined my research group: Dr Reza Javahery, a very skilled experimenter from the UK, and Dr Simon Petrie, a splendid thinker and writer from Australia. Both fellows were very highly motivated and extremely talented, a powerful combination! We were able to explore many aspects of fullerene ions ranging from their fundamental physico-chemical properties to their chemistry as a function of charge state. Here, I provide a brief overview of some of the highlights of this early exciting period; our contributions in general have been surveyed previously in several review articles [4,5].
As was the case in Sir Kroto’s initial quest for carbon clusters and chains that led to the mass-spectrometric observation of C60+, our studies of ion–molecule reactions with C60+ were driven, in part, by our own fascination with interstellar and circumstellar chemistry. So, I will conclude this account with some of our early work in this field that now has taken on a new relevance with the very recent confirmation, in 2015, of the presence of C60+ in interstellar environments with a definitive spectroscopic assignment of two diffuse interstellar bands seen towards reddened stars [6] and the spectroscopic detection in 2010 of C60 and C70 in a young planetary nebula [7].
2. Chemical removal of electrons from C60
While ionization with photons and electrons quickly became the method of choice for the formation of fullerene cations in the laboratory once fullerene powder became available, we focused initially on chemical ways to remove electrons from C60. These early experiments showed that metastable atoms of He, Ne, Ar and Kr induce the loss of electrons from C60 in the gas phase at room temperature in chemi-ionization, also known as Penning ionization, reactions [8],
These metastable atoms all have energies that exceed the first ionization energy (IE) of C60. The mechanism of chemi-ionization can be understood in terms of the formation of an excited quasi-molecule (XC60)* that undergoes auto-ionization. We also found that C60 is readily ionized at room temperature in electron-transfer reactions with atomic ions that have a recombination energy (RE) larger than IE(C60) [9]. These include He+, Ne+, Ar+, D+ and Si+,
While exothermic dissociative electron transfer leading to the rupture of C60+ through loss of C2, for example, was not observed to compete with electron transfer under our experimental operating conditions, we did observe electron-transfer/electron-detachment reactions with X+=He+ and Ne+. These two atomic ions have recombination energies that exceed the double IE of C60,
This observation was surprising and an important result unprecedented in ion chemistry generally! The mechanism of this novel reaction is not known but may involve the initial transfer of a deeply embedded electron in C60 followed by an Auger emission of a second electron in a manner analogous to that which is known to occur when metals are exposed to slow beams of He+ ions. The occurrence of electron transfer/electron emission with C60 may therefore be a manifestation of the metallic character of C60 that arises from the ocean of electrons that surround its hollow cage of 60 carbon nuclei.
Driven by our discovery of electron-transfer/electron-detachment reactions with C60, we also managed to observe (for the first time) the occurrence of double-electron transfer/electron detachment to yield C603+, but only with Ar2+. The mechanism in this case could involve the initial (highly exothermic) double-electron transfer to produce highly excited (C602+)* or Ar* as intermediates. C603+ formation could then follow either by auto-ionization of (C602+)* or by Penning ionization of C602+ by excited Ar* [10],
3. Chemistry of C60n+ as a function of charge state (n=1–3)
We found the removal of up to three electrons from C60 to be straightforward in the conventional electron-impact ion source that delivered ions, after mass selection, into our flow tube for kinetic studies of reactions with added gases. This ease of production of C60+, C602+ and C603+ prompted us to explore systematically the then unchartered chemistry of C60n+ as a function of charge state from n=1 to 3. Such investigations of reactivity were unprecedented in the greater world of gas-phase ion chemistry, in part because many multiply charged molecular ions explode by intramolecular Coulombic repulsion already in the second charge state. In sharp contrast, multiply charged fullerene cations are renowned for their stability against Coulomb explosion.
Our experiments with more than 30 different reagent atoms and molecules revealed that C60n+ carbocations are extensively reactive in the gas phase, and more so as the charge state increases, as is illustrated in figure 1. Charge made a big difference! New reaction pathways became accessible at higher degrees of ionization as the electron RE (C60n+) increases. The observed reaction channels included addition, dissociative addition, dissociative addition with charge separation, single-electron transfer, dissociative single-electron transfer and two-electron transfer
 |
Both the nature of the product channel and the rate of reaction were observed to be a function of the charge state of C60n+. Chemical pathways became increasingly pre-empted by electron transfer with increasing charge. We have discussed this trend qualitatively in terms of schematic potential-energy profiles that are crossed by single, double and dissociative electron-transfer potential-energy curves leading to a non-adiabatic transition to the transfer of an electron. Those C60n+/XY encounters that survive the electron-transfer crossings may undergo inelastic collisions with He buffer-gas atoms to produce stable adduct ions C60XYn+. Failing that, bond redisposition may occur by dissociative addition with or without charge separation. The opportunity for electron transfer increases with increasing charge state. In a sense, chemistry is increasingly pre-empted by physics with increasing charge state.
Figure 1.

Qualitative overview of the observed trend in the reactivity of C60n+ cations with charge state n. The predominant reaction channels that were observed are listed in the order of general importance. RE is the electron RE of the cation in eV.
(a). Electron transfer to C60n+ cations (n=1–3)
We have surveyed extensively the room temperature kinetics of electron transfer reactions with C60+, C602+ and C603+, initially to assess values for their electron recombination energies, namely the first three ionization energies of C60, with kinetic bracketing measurements of the occurrence or non-occurrence of single-electron transfer from molecules XY with known ionization energies [11],
In this way, IE(C60) was determined to be 7.61±0.11 eV. Single-electron transfer to C602+ and C603+ was not observed to occur with molecules with IE≥9.58 eV and ≥11.3 eV, respectively, and leads to two charged product ions. In these cases, Coulomb repulsion between the product ions, which can lead to energy barriers to electron transfer, must be taken into account,
 |
The energy barriers due to Coulomb repulsion were estimated to be 2.07 eV and 4.4 eV, respectively, assuming that electron transfer occurs at a separation of 7 Åwhen the reactants are in close proximity. This treatment leads to values for IE(C60+)=11.65 eV and for IE(C602+)=15.7 eV, in good agreement with the literature values.
To our great surprise, an unprecedented double-electron transfer was observed at room temperature between triply charged buckminster fullerene, C603+, and the polycyclic aromatic hydrocarbon (PAH) molecules anthracene, corranulene, benzo[rst]pentaphene and pyrene [12],
All of these PAH molecules have sufficiently low first and second ionization energies to make double-electron transfer thermodynamically and kinetically favourable. The mechanism of the two-electron transfer, whether concomitant or sequential, is not known. Two-electron transfer was observed to occur as a minor channel (less than 5%) in competition with single-electron transfer with anthracene, corannulene and pyrene and in almost equal amounts with benzo[rst]pentaphene, which requires the lowest energy for double ionization of these four PAH molecules.
Our rate coefficient measurements revealed an increase in the rate of electron transfer with increasing charge state in response to the increasing electrostatic interaction between reagents. The increase in the RE with increasing charge state leads to an increase in the exothermicity of electron transfer and this leads to the occurrence of dissociative electron transfer with C603+. For example, dissociative electron transfer was observed as the dominant channel with ethylamine to produce both CH2NH2+ and CH3+ along with C602+.
Electron transfer was observed to compete with adduct formation, more effectively in the reactions with C603+. Exothermicity appeared to play a role here too. We have examined the influence of exothermicity more closely and systematically for reactions of C602+ with a family of related reagent molecules with different ionization energies. Figure 2 illustrates the competition between electron transfer and adduct formation that we found for reactions with ammonia and several amines. Electron transfer was seen to dominate in reactions with amines having ionization energies below 8.5 eV while adduct formation dominated in reactions with amines having ionization energies above 8.5 eV. In comparison, while we observed the addition of ammonia to C603+, only electron transfer was seen with amines.
Figure 2.
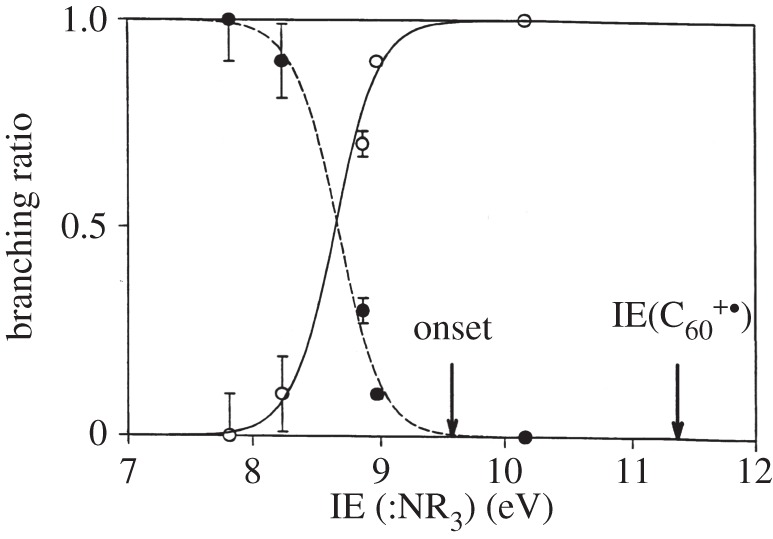
The competition between electron transfer (dotted line) and adduct formation (solid line) observed for reactions of C602+ with ammonia and amines in our SIFT apparatus at room temperature at a helium bath pressure of 0.35 Torr.
(b). Molecular interactions with C60+
We found that C60+ generally was quite unreactive under SIFT conditions: 50 non-polar, but also polar, inorganic and organic molecules were used as reagents. Electron transfer was rare because of the low electron RE of C60+. The few molecules that did add to C60+, presumably by chemical-bond formation with the carbonaceous surface, are summarized in figure 3. We found that C60+ can be derivatized with H atoms, strong nucleophiles such as ammonia and aliphatic amines and with molecules capable of Diels–Alder additions such as 1,3-cyclopentadiene, 1,3-cyclohexadiene, anthracene and corannulene. A reaction of C60+ with iron pentacarbonyl that produces C60Fe(CO)4++CO is the only example of a bimolecular derivatization reaction with C60+ that we observed. A remarkable trend in reactivity was seen for the reactions of C60+ with amines: ammonia added only slowly, but the rate of addition approached the collision rate with increasing alkyl substitution. This behaviour is consistent with a trend in inductive electron donation (the Lewis basicity of the amine) and a trend in the number of degrees of freedom in the intermediate adduct ion effective in energy dispersal.
Figure 3.

Overview of derivatization reactions of C60+ observed at 298 K in helium at 0.35 Torr. The assigned structures are speculative.
We envisaged chemical reactions with singly charged C60+ to proceed as follows. At long range, the charge on the isolated C60+ cation, initially delocalized over the entire C60 surface, will become localized due to electrostatic interaction with the induced and/or permanent dipole of the incoming molecule. Chemical attack at short separations should occur at the carbon site of a localized charge. For reaction to take place, a transition is required from largely sp2 to sp3 hybridization at the C-atom that becomes bonded. This requires surface deformation and so will involve an activation barrier that we estimated to be ca 15 kcal mol−1, thus accounting for the relatively low reactivity of C60+ at room temperature.
Our early investigations of addition reactions of the fullerene cations C56+, C58+ and C60+ with NH3 revealed an enhanced reactivity for the adjacent-pentagon fullerene ions of C56 and C58 relative to that of C60. We noted that adjacent pentagons enhance the curvature of the carbon surface and the sp3 character of selected C sites [13]. Further systematic studies of addition reactions of C56+, C58+, C60+, C70+, corannulene+ and coronene+ with cyclopentadiene and 1,3-cyclohexadiene indicated that the efficiency of bond formation with these two molecules also depends strongly on the curvature of the carbonaceous surface of the reacting cation [14]. This is illustrated in figure 4 for the Diels–Alder addition with cyclopentadiene that is faster with the smaller, more strained fullerene cations and immeasurably slow with the less-strained corannulene cation and the flat coronene cation. Thus, the relief of strain energy appeared to govern the rate of addition of molecules to carbonaceous cations.
Figure 4.

A correlation between reaction efficiency, kobs/kc, with the square of the pi orbital axis vector angle and the strain energy for addition reactions with cyclopentadiene at room temperature and a helium pressure of 0.35 Torr. kobs is the measured rate coefficient and kc is the collision rate coefficient, which is estimated to be 10−9 cm3 molecule−1 s−1. The dotted line defines the dependence of kobs on the strain energy, Estrain.
(c). Molecular interactions with C602+ and C603+
Increasing the charge state of the fullerene cation beyond +1 of course increases the electron RE of the fullerene cation as well as the electrostatic interaction between the ion/molecule reagents. So, C602+ was found to be much more reactive than C60+ in both electron transfer and chemical bonding. We attributed this enhanced reactivity to the higher RE of C602+ and the stronger electrostatic interaction between molecules and C602+ that may serve to overcome the energy barrier associated with the change in hybridization required at the site of bonding. Figure 5 summarizes derivatization reactions observed with C602+. We measured the chemistry of C602+ with H atoms, ammonia and aliphatic amines, nitriles, water, alcohols and ethers, aldehydes, ketones, carboxylic acids and esters, cyclic oxides and unsaturated hydrocarbons. Electron transfer was observed to be an important competitive channel for some of the derivatization reactions and, for molecules with a sufficiently low IE, became the only observed reaction channel. However, electron transfer with C602+ occurred in the presence of an activation barrier that arises from Coulomb repulsion between the charged product ions that were evident from an observed delayed onset for competing electron transfer at about 9.6 eV, well below RE(C602+) at 11.36 eV.
Figure 5.

Overview of derivatization reactions of C602+ observed at 298 K in helium at 0.35 Torr. The assigned structures are speculative.
With C603+, addition was generally observed to be faster and more efficient than the corresponding addition to C602+ and was seen only with H atoms, ammonia, nitriles, an aldehyde, and a ketone, an ester, ethylene oxide and some unsaturated hydrocarbons. Electron transfer, now more exothermic because of the higher RE, became more competitive and even dissociative, as mentioned earlier. Also, bimolecular reactions formally involving hydride or hydroxide transfer, which we have called dissociative addition reactions, became more effective. This was the case with water, some alcohols and some alkanes.
Halide transfer from HCl and HBr also was observed [15]. HCl reacts both by Cl− transfer to produce, remarkably, a free proton as the second cation product (88%) and by hydride transfer (12%). HBr also reacts by halide transfer, but the competing channel in this case is dissociative electron transfer,
 |
Bimolecular chloride transfer was also observed with CH3Cl, CH2Cl2, CDCl3 and CCl4, but not with Cl2 and not for C602+ and C60+. Apparently, the extra electrostatic interaction with C603+ is decisive for the dechlorination of these molecules. On the other hand, chlorinated ethylenes reacted almost exclusively by rapid electron transfer with C603+, but exhibited addition and double chlorination as well.
4. Higher order reactions: spindle, star, fuzzy ball, ball-and-chain and dimer ring formation
We were able to observe a variety of higher order reactions of fullerene cations, especially when they were multiply charged. The large number of carbon atoms available on the surface of these ions and the possible localization of charge by intramolecular Coulomb repulsion when multiply charged in principle provide extensive opportunities for higher order chemistry both on and away from the surface of the fullerene. We have talked about ‘spindle’, ‘star’, ‘fuzzy’ ball, ‘ball-and-chain’ and dimer ring formation in our publications. In a sense, chemistry and physics operate in concert here as Coulomb repulsion between charges drives intramolecular charge transport that enhances charge localization on the fullerene surface, or on a substituent, and so enhances the chemical reactivity of the bare or derivatized cation.
Extensive surface derivatization of C60 cations was observed with H atoms [16,17]. While the first three charge states of C60n+ were seen to be unreactive towards molecular hydrogen at room temperature, extensive hydrogenation was achieved under SIFT conditions in He buffer gas at 0.35 Torr by sequential addition of hydrogen atoms,
The lower limits obtained for the effective bimolecular rate coefficient for the primary addition step were 0.10, 0.30 and 1.0×10−9 cm3 molecule−1 s−1 for singly, doubly and triply charged cations, respectively. Up to 10 additions were observed for each of the first three charge states with the maximum number of additions being constrained experimentally only by the hydrogen-atom density experimentally available. The mechanisms for these addition reactions are most intriguing as they may involve alternate formation of distonic ions as intermediates in the hydrogenation of the carbon skeleton, the formation of a hydrogen ‘fuzzy ball’. The SIFT experiments were not able to exclude the occurrence of the formation of molecular hydrogen in secondary reactions, which can be exothermic given the high H–H bond energy
The sequence of H atom addition and H atom elimination would be quite remarkable in that it would lead to the recombination of H atoms with the C60n+ cation acting as a (surface) catalyst.
Selected molecules also exhibited extensive sequential addition to fullerene cations. The most remarkable example is shown in figure 6, in which rapid sequential addition, presumably on the carbonaceous surface, is clearly visible.
Figure 6.

Data recorded for the sequential addition of methyl isocyanide to C603+ in helium buffer gas at 0.35 Torr and 294 K.
C602+ was observed to react with methyl nitrite in sequential bimolecular reactions, leading to the transfer of at least 15 methoxy groups to the fullerene surface. The addition kinetics showed periodicities: odd-numbered adduct ions reacted faster, presumably because of their radical nature,
By contrast, with other molecules, the number of charges on the C60n+ appeared to govern the number of additions. Aliphatic nitriles, RCN, in particular were seen to be effective in attaching ‘handles’ to produce the single, double (spindle) and triple (star) adducts with singly, doubly and triply charged C60n+, respectively. The derivatization can be attributed to donation of an electron pair from the electronegative nitrogen atom of the nitrile to the site of charge on the surface of the fullerene cation forming a dative bond between the nitrogen atom and a carbon atom on the fullerene shell with concomitant charge propagation to the nitrogen atom.
The observed addition of pyridine was also instructive [18]. Again, the number of molecules added was observed to be equal to the number of charges on C60n+, but, in this case, we were able to perform collision-induced dissociation (CID) experiments with the adduct ions that revealed the presence of isomers for C60(C5H5N)22+ and C60(C5H5N)33+ in which the pyridine molecules add either 1 to each charge site (2 or 3, respectively) or disproportionately to one or two charge sites, e.g. all three of the pyridine molecules are attached to one charge site in a ‘ball-and-chain’-type structure in C60(C5H5N)33+ (figure 7). The influence of Coulomb repulsion appeared to propagate charge to the first substituent and so direct a second addition to that charge site.
Figure 7.

Isomers of C60(C5H5N)33+ identified with CID. (a) ‘Star’ structure; (c) ‘ball-and-chain’ structure.
We observed multiple molecular derivatization consistent with ‘ball-and-chain’ formation for reactions of multiply charged fullerenes, with monomers well known from traditional polymer chemistry. The monomers 1,3-butadiene, ethylene oxide, 1-butene, allene and propyne, ethylene and acetylene, vinyl chloride, 1,1-dichloroethylene and trichloroethylene all have been found to polymerize with C602+, C702+ or C603+. Separate measurements of the CID of the polymerized cations  ,
,  ,
,  and
and  clearly pointed towards growth by ball-and-chain propagation,
clearly pointed towards growth by ball-and-chain propagation,
 |
The chemistry of C602+ with allene was particularly interesting because the sequential addition kinetics showed a striking periodicity in reactivity; the even-numbered adducts react about 10 times faster than the odd-numbered adducts, and a striking periodicity in stability was evident from the CID experiments. We proposed a mechanism that involves alternating formation of even-numbered charge-localized acyclic allyl cations that are more reactive and odd-numbered charge-delocalized cyclic allyl cations that are less reactive [19]. Our proposed structure is shown in figure 8.
Figure 8.

Semi-empirical MP3 structure calculated for C60(allene)72+.
Several reactions of derivatized fullerene trications that we observed with cyanide molecules involve the complete transfer of the ionized molecule with concomitant formation of the corresponding dimer cation. Termolecular polymerization is pre-empted by bimolecular dimer cation formation. We measured such ‘associative ionization’ reactions with R=CH2CH, CH2CHCH2, CN and CH2CN,
These charge-separation reactions are fast and appear to be driven by the stability of the product dimer cation, possibly involving the formation of a ‘dimer ring’.
The most striking example involves cyanoacetylene and the doubly charged fullerene cation which we investigated in considerable detail and for which dimer ring formation was established unequivocally [20],
CID experiments with the dimer cation (HC3N)2+, together with computations of the energies and structures of various isomers of (HC3N)2+, have pointed to the formation of a dicyanocyclobutadiene cation rather than a solvated dimer cation of the type that we were able to form directly by association of ionized cyanoacetylene with neutral cyanoacetylene (in the absence of C60) and that dissociates quite differently. This revealed a role for C602+ akin to a surface that chemisorbs the first molecule and then promotes formation of free dimers with the next molecule. We have called this a ‘molecular dock mechanism’, because the first reagent molecule is docked to the charged fullerene as seen in figure 9.
Figure 9.

‘Molecular dock mechanism’ proposed for the 2+2 cyclization of cyanoacetylene assisted by C602+.
5. Proton-transfer reactions: the notion of ‘apparent’ gas-phase acidity
The reaction of C603+ with C3H8 provided us with a route to the production of the doubly charged C60H2+ cation and thus allowed, what were probably the first, quantitative measurements of the kinetics of proton transfer from a multiply charged cation [21],
Figure 10 shows the results of systematic studies of rates of proton transfer from C60H2+ to various molecules X with known gas-phase basicities, GB(X), in the range from about 95 to 105 kcal mol−1. Proton transfer becomes exoergic when the gas-phase acidity GA(C60H2+)<GB(X), but the onset for proton transfer in figure 10 is delayed. This observation led us to introduce the concept of an apparent gas-phase acidity for C60H2+,  , which is different from the true gas-phase acidity by δ, the Coulomb repulsion associated with the charge separation involved in the deprotonation reaction [22],
, which is different from the true gas-phase acidity by δ, the Coulomb repulsion associated with the charge separation involved in the deprotonation reaction [22],
The onset for proton transfer shown in figure 10 indicates a value for GAapp(C60H2+)=166±4 kcal mol−1. We estimated δ to be 42±4 kcal mol−1 so that GA(C60H2+) becomes equal to 124±8 kcal mol−1. The notion of apparent gas-phase acidity, first introduced by us for the occurrence of deprotonation reactions of C60H2+, has since been adopted in the description of the acidities of polycations of biomolecules that had been generated by electrospray ionization only a few years earlier.
Figure 10.
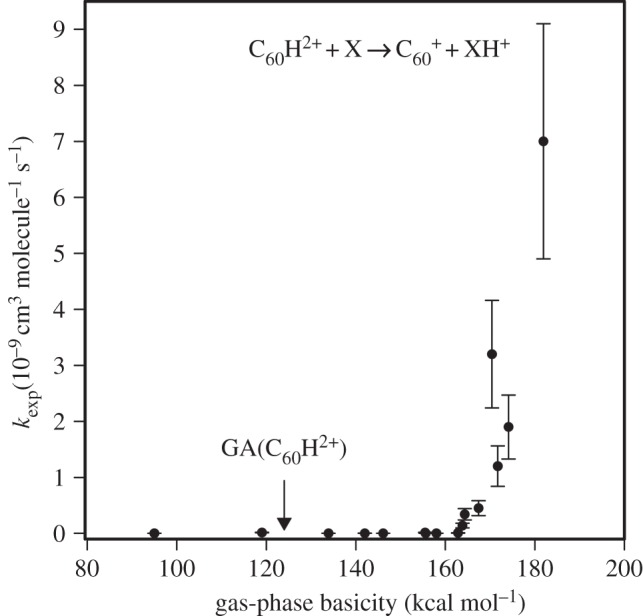
Observed variation of the rate coefficient for proton transfer with the gas-phase basicity of X, GB(X), for proton-transfer reactions of the type indicated.  at the onset of proton transfer.
at the onset of proton transfer.
Further studies of proton transfer from derivatized multiply charged C60 cations became possible when it was observed that a hydrogen-containing molecule could be docked onto C602+ or C603+ and then be allowed to react with a second molecule of its kind [22,23],
 |
This was achieved for 16 different molecules docked on C602+ and 12 different molecules docked on C603+. Proton transfer was the major, often only, secondary reaction for XH=ammonia, amines, alcohols, ketones, carboxylic acids, and esters and hydrogen cyanide. In these cases, the observed occurrence or non-occurrence of proton transfer from the adduct ion allowed the estimation of its apparent gas-phase acidity.
6. C60 as a ligand surface for atomic metal cations: fullerometallic ion chemistry
Our ability to readily generate atomic Fe+ cations by electron impact on ferrocene provided the opportunity to explore the bonding of Fe+ to C60 and the ensuing chemistry of the adduct ion. C60 provides a flawless carbonaceous surface for the chemisorption of Fe+; ensuing chemistry of Fe+ attached to C60 therefore will mimic surface chemistry.
A comparative study of reactions of Fe+ coordinated to the π-donating ligands C2H4, c-C5H5, C6H6 and C60 with N2O and CO. There was an observed match in the ligation chemistry of Fe+ (addition of up to four CO molecules) for C60 only with C2H4 and a similar addition and oxidation chemistry with N2O to release O2. This suggests that C60 acts as a two-electron donor and forms (η2–C60)Fe+ with Fe+ [24].
With this information in hand, we undertook a systematic study of the chemical reactivity of (η2–C60)Fe+ with the small molecules D2, N2, CO2, CH4, C2H2, C2H4, SO2, C6D6, NH3, H2O and CO, for which only ligation was observed (which was absent in the absence of Fe), as well as with N2O and O2, which showed more appealing chemistry. Especially noteworthy is the rapid reaction with O2 that eliminates FeO2 that needs to be the inserted isomer OFeO according to thermodynamics,
 |
When preceded by the collisional or radiative attachment of Fe to C60+, the reaction with O2 provides a catalytic pathway for the reaction of Fe with O2 to produce OFeO with C60+ acting as the catalytic surface [25].
More recently, we also explored the attachment of C60 to the atomic cations Mo+ (transition metal) and Sr+ (non-transition metal) that we could generate within an inductively coupled plasma. We were able to observe the ‘packing’ of these two atomic cations with up to four C60 molecules (within the available mass range of our mass spectrometer) by sequential ligation reactions to form M+(C60)4, shown in figure 11 [26].
Figure 11.

Proposed structure for M+(C60)4. Bonding is characterized by η6 interaction of the metal with the C60 ligands (see lower left) and by η2-to-η2 bonding of the ligands to one another (see left-most pair of ligands).
7. Extraterrestrial buckminster fullerene ion chemistry
We first addressed the implications of the results of our fullerene ion SIFT measurements for the chemistry occurring within diffuse and dense interstellar clouds and in circumstellar envelopes in 1992/1993 when the actual presence of C60+ in these environments was not yet established [27,28].
In this early work, we emphasized the possibility of formation of C602+ from C60 by electron transfer/electron detachment with He+, the kinetics of the associations and reactions of C60+ and C602+ with radicals and small molecules, the kinetics of proton transfer from derivatized fullerene dications and the existence of activation energy barriers towards many electron and hydride transfer reactions. Several properties of the fullerenes themselves, and their cations, were also considered, including difficulties inherent in the dissociative electron/ion recombination of C60+ and C602+, the formation of C60+ and C602+ by sequential cosmic ray and UV photoionization of C60, the possibility of fullerene cages acting as ‘traps’ for cosmic rays, and the possible role of neutral and ionized fullerenes as models for interstellar dust grains in surface recombination of atomic hydrogen and other species.
We considered the possible detection of derivatized fullerenes as signatures of fullerene cations that have undergone derivatization by chemical reactions (with H atoms, ammonia, amines and saturated hydrocarbons) with subsequent neutralization by electron/ion recombination and with due regard to the resilience of these derivatized fullerenes to photodissociation in diffuse and dense interstellar clouds. As a supplement, we also drew attention to the possible role for charge-separation reactions of interstellar fullerene dications in the chemical evolution of cold, dense interstellar clouds. Such charge-separation reactions are not only efficient but also can be highly exothermic and partition a large fraction of the exothermicity into the translational excitation of the light ion/neutral product. The fullerene cations can serve as ‘cannons’ launching fast ions and atoms/molecules into the interstellar medium. These internally cold, highly kinetically excited cations/neutrals may then serve as a driving force for the subsequent occurrence of kinetically unfavourable reactions, for example, hydrogen-atom abstraction reactions with the dominant dense-cloud species H2 [29].
In a later discourse in 2000, we extended our treatment of the chemical processing of fullerenes in various astrophysical environments to include fullerene anions, triply charged fullerene cations and other known interstellar molecules, including PAHs [30]. We discussed in more detail the prospects for the neutralization of derivatized fullerene cations by proton transfer and electron/ion recombination. Our overview of the chemistry of C602+ is provided in figure 12.
Figure 12.

Schematic of chemical pathways that we have proposed that can be initiated by doubly charged fullerenes in interstellar or circumstellar environments.
Not included in figure 12 are higher order reactions that may lead to spindle, star, fuzzy balls, ball-and-chain and dimer ion formation. Except for dimer ion formation, these all require radiative stabilization to occur and propagate, as third-body collisions, with He atoms in our experiment, are essentially absent in the interstellar medium. On the other hand, dimer ion formation by associative ionization occurs in a rapid, bimolecular fashion after the first molecule has ‘docked’. The molecular dock mechanism that we proposed for the formation of the derivatized cyclobutadiene isomer of (HC3N)2+• is also expected to allow the formation of other derivatized cyclobutadienes from higher homologues of cyanoacetylene that are known to be present in the interstellar medium. Perhaps such ring formation can occur more generally with any carbonaceous molecule or dust particle in the interstellar medium that can carry multiple charges and provide an atomic site for initial covalent bonding,
After the very recent unambiguous detection of C60+ [6], we now await detections of derivatized fullerene cations, and perhaps dications, and hope that this review and our earlier detailed discourse will provide some guidance. The abundance of fullerene trications is almost certainly too low for the reaction chemistry of these ions to leave a detectable imprint upon the chemical evolution of the interstellar medium.
Acknowledgements
Special thanks go to Dr Simon Petrie and Dr Reza Javahery, both post-doctoral fellows in my research group, who were an invaluable part and driving force of this research effort.
Competing interests
I have no competing interests.
Funding
I thank the Natural Sciences and Engineering Research Council of Canada for the continuous financial support of this research and the Canada Council for a Killam Research Fellowship (1991–1993).
References
- 1.Kroto HW, Heath JR, O’Brian SC, Curl RF, Smalley RE. 1985. C60: Buckminsterfullerene. Nature 318, 162–163. ( 10.1038/318162a0) [DOI] [Google Scholar]
- 2.Mackay GI, Vlachos GD, Bohme DK, Schiff HI. 1980. Studies of reactions involving C2H+X ions with HCN using a modified selected ion flow tube. J. Mass Spectrom. Ion Phys. 36, 259–270. ( 10.1016/0020-7381(80)85059-5) [DOI] [Google Scholar]
- 3.Krätschmer W, Lamb LD, Fostiropoulis K, Huffman DR. 1990. Solid C60: a new form of carbon. Nature 347, 354–358. ( 10.1038/347354a0) [DOI] [Google Scholar]
- 4.Böhme DK. 1999. 1998 J.C. Polanyi Award Lecture. Fullerene ions in the gas phase: chemistry as a function of charge state. Can. J. Chem. 77, 1453–1464. ( 10.1139/v99-158) [DOI] [Google Scholar]
- 5.Bohme DK. 2009. Buckminsterfullerene cations: new dimensions in gas-phase ion chemistry. Mass Spectrom. Rev. 28, 672–693. ( 10.1002/mas.20227) [DOI] [PubMed] [Google Scholar]
- 6.Campbell EK, Holz M, Gerlich D, Maier JP. 2015. Laboratory conformation of C+60 as the carrier of two interstellar bands. Nature 523, 323–324. ( 10.1038/nature14566) [DOI] [PubMed] [Google Scholar]
- 7.Cami J, Bernard-Salas J, Peeters E, Malek SE. 2010. Detection of C60 and C70 in a young planetary nebula. Science 329, 1180–1182. ( 10.1126/science.1192035) [DOI] [PubMed] [Google Scholar]
- 8.Javahery G, Petrie S, Wang J, Wang X, Bohme DK. 1993. Penning ionisation of fullerenes: reactions of C60 and C70 with metastable atoms of the rare gases He, Ne, Ar and Kr. Int. J. Mass Spectrom. Ion Processes 125, R13–R15. ( 10.1016/0168-1176(93)80021-6) [DOI] [Google Scholar]
- 9.Javahery G, Petrie S, Wang J, Bohme DK. 1992. Fullerene cation and dication production by novel thermal-energy reactions of He+, Ne+, and Ar+ with C60. Chem. Phys. Lett. 195, 7–10. ( 10.1016/0009-2614(92)85901-L) [DOI] [Google Scholar]
- 10.Blagojevic V, Petrie S, Bohme DK. 2004. Reaction of Ar2+ with C60 to produce C603+: first observation of double electron-transfer ionisation? Int. J. Mass Spectrom. 233, 33–37. ( 10.1016/j.ijms.2003.10.015) [DOI] [Google Scholar]
- 11.Bohme DK. 1994. Electron transfer reactions with buckminsterfullerene, C60, in the gas phase. Int. Rev. Phys. Chem. 13, 163–185. ( 10.1080/01442359409353293) [DOI] [Google Scholar]
- 12.Javahery G, Becker H, Petrie S, Cheng P-C, Schwarz H, Scott LT, Bohme DK. 1993. Unprecedented double-electron transfer to a triply-charged cation: reactions of C603+ with anthracene, corannulene, benzo[rst]pentaphene and pyrene. Org. Mass Spectrom. 28, 1005–1008. ( 10.1002/oms.1210281006) [DOI] [Google Scholar]
- 13.Petrie S, Bohme DK. 1993. Enhanced reactivity of fullerene cations containing adjacent pentagons. Nature 365, 426–429. ( 10.1038/365426a0) [DOI] [Google Scholar]
- 14.Becker H, Scott LT, Bohme DK. 1997. The influence of surface strain on the chemical reactivity of fullerene ions: addition reactions with cyclopentadiene and 1,3-cyclohexadiene. Int. J. Mass Spectrom. Ion Process. 167/168, 519–524. ( 10.1016/S0168-1176(97)00097-9) [DOI] [Google Scholar]
- 15.Baranov V, Bohme DK. 1996. Proton elimination in charge-separation reactions with hydrogen halides driven by chemical-bond formation with triply-charged C60 cations. Chem. Phys. Lett. 258, 203–206. ( 10.1016/0009-2614(96)00642-2) [DOI] [Google Scholar]
- 16.Petrie S, Javahery G, Wang J, Bohme DK. 1992. Hydrogenation of fullerene cations in the gas phase: reactions of fullerene cations and dications with atomic and molecular hydrogen. J. Am. Chem. Soc. 114, 6268–6269. ( 10.1021/ja00041a068) [DOI] [Google Scholar]
- 17.Petrie S, Becker H, Baranov VI, Bohme DK. 1995. Repeated addition of atomic hydrogen to fullerene cations, dications and trications. Int. J. Mass Spectrom. Ion Process. 145, 79–88. ( 10.1016/0168-1176(95)04172-H) [DOI] [Google Scholar]
- 18.Sun J, Bohme DK. 1998. Gas-phase reactions of fullerene cations C60x+ (x=1–3) with pyridine and pyrrole: formation of ‘ball-and-chain’ and ‘spindle’ isomers and their interconversion. Int. J. Mass Spectrom. Ion Process. 179/180, 267–275. ( 10.1016/S1387-3806(98)14104-0) [DOI] [Google Scholar]
- 19.Baranov V, Wang J, Javahery G, Hopkinson AC, Bohme DK. 1997. Fullerene dications and trications as initiators in the das-phase ‘ball-and-chain’ polymerization of allene and propyne: observation of a remarkable periodicity in chain growth with allene. J. Am. Chem. Soc. 119, 2040–2049. ( 10.1021/ja961359j) [DOI] [Google Scholar]
- 20.Milburn RK, Hopkinson AC, Bohme DK. 1999. Dimer cations of cyanoacetylene: theoretical isomers and their laboratory production in the absence and presence of C602+. J. Phys. Chem. 103, 7528–7534. ( 10.1021/jp991656l) [DOI] [Google Scholar]
- 21.Petrie S, Javahery G, Wincel H, Bohme DK. 1993. Proton transfer from a fullerene dication: bracketing the gas-phase acidity of C60H⋅2+. J. Am. Chem. Soc. 115, 6290–6294. ( 10.1021/ja00067a050) [DOI] [Google Scholar]
- 22.Petrie S, Javahery G, Bohme DK. 1993. First steps towards a gas-phase acidity ladder for derivatized fullerene dications. Int. J. Mass Spectrom. Ion Process. 124, 145–156. ( 10.1016/0168-1176(93)80005-Y) [DOI] [Google Scholar]
- 23.Petrie S, Bohme DK. 1998. Proton transfer reactions of derivatized fullerene trications. J. Am. Soc. Mass Spectrom. 9, 114–120. ( 10.1016/S1044-0305(97)00234-1) [DOI] [Google Scholar]
- 24.Baranov VI, Bohme DK. 1995. Reactions of Fe+ coordinated to the π-donating ligands C2H4, c-C5H5, C6H6 and C60 with N2O and CO: probing the bonding in (C60)Fe+. Int. J. Mass Spectrom. Ion Process. 149/150, 543–555. ( 10.1016/0168-1176(95)04287-U) [DOI] [Google Scholar]
- 25.Caraiman D, Koyanagi G, Scott LT, Preda DV, Bohme DK. 2001. Fullerometallic ion chemistry: reactions of C60Fe+ and C20H10Fe+ in the gas phase. J. Am. Chem. Soc. 123, 8573–8582. ( 10.1021/ja0104857) [DOI] [PubMed] [Google Scholar]
- 26.Koyanagi GK, Xu J, Bohme DK. 2008. Packing atomic metal cations with C60: mass spectrometric observation of M+(C60)n with M=Sr and Mo and n=0 to 4. Chem. Phys. Lett. 450, 228–231. ( 10.1016/j.cplett.2007.11.040) [DOI] [Google Scholar]
- 27.Bohme DK. 1992. PAH and fullerene ions and ion/molecule reactions in interstellar and circumstellar chemistry. Chem. Rev. 92, 1487–1508. ( 10.1021/cr00015a002) [DOI] [Google Scholar]
- 28.Petrie S, Bohme DK. 1993. Experimental results for ion-molecule reactions of fullerenes: implications for interstellar and circumstellar chemistry. Astron. Astrophys. 271, 662–674. [Google Scholar]
- 29.Petrie S, Bohme DK. 1994. Formation and charge-separation reactions of large molecular dications within interstellar clouds: consequences for cold cloud chemical evolution. Mon. Not. Roy. Astron. Soc. 268, 103–108. ( 10.1093/mnras/268.1.103) [DOI] [Google Scholar]
- 30.Petrie S, Bohme DK. 2000. Laboratory studies of ion/molecule reactions of fullerenes: chemical derivatization of fullerenes within dense interstellar clouds and circumstellar shells. Astrophys. J. 540, 869–885. ( 10.1086/309346) [DOI] [Google Scholar]