

Abstract
In neurodegenerative diseases caused by misfolded proteins, including Huntington's disease (HD), the neuronal processes and terminals are particularly prone to the accumulation of misfolded proteins, leading to axonal and synaptic dysfunction. This compartment-dependent accumulation can result from either the altered transport of misfolded proteins or impaired protein degradation. Mutant huntingtin (mHtt), the HD protein, is known to affect intracellular transport and can be degraded by the proteasome and autophagy, but how mHtt accumulates in the neuronal processes, an early pathological event in the brains of HD patients, still remains unclear. Using an “optical pulse-chase” assay that can quantify protein degradation in specific subcellular regions, we found that neuronal mHtt is removed faster in the cell body than in neurites. Furthermore, mHtt is cleared more rapidly in astrocytes than in neurons. The ubiquitin-proteasome system plays a much bigger role than autophagy in degrading soluble mHtt via K48 ubiquitination in both the cytoplasm and processes of neurons and astrocytes. By injecting adenoviral vectors expressing mHtt into the mouse brain, we confirmed that mHtt is removed more slowly in neurites than in the cytoplasm of the cell body of neurons. Our findings provide evidence for the cell type- and compartment-dependent degradation of mHtt and explain why mHtt preferentially accumulates and aggregates in the neuropils of vulnerable neurons. In addition, our findings suggest that enhancing proteasomal activity could be an effective way to reduce the preferential accumulation of soluble mHtt in neuronal processes.
SIGNIFICANCE STATEMENT The clearance of misfolded proteins is key to preventing neurodegeneration in Huntington's disease, but how mutant huntingtin (mHtt) accumulates differentially in different cell types and subcellular regions remains unclear. We found mHtt is cleared slowly in neuronal processes compared with the cytoplasm and is cleared more efficiently in astrocytes than in neurons. Moreover, this compartment-dependent degradation of soluble mHtt is mediated primarily by the ubiquitin-proteasome system rather than autophagy. Our findings imply that enhancing proteasome activity could be an efficient way to clear soluble misfolded proteins in the neuronal processes.
Keywords: clearance, degradation, misfolding, neurodegeneration, polyglutamine, subcellular
Introduction
Huntington's disease (HD) is an autosomal dominant neurodegenerative disease caused by an expanded polyglutamine (polyQ) tract in the N-terminal region of mutant huntingtin (mHtt). Once the full-length mHtt is degraded by proteolysis, the polyQ expansion causes N-terminal Htt fragments to misfold and form aggregates (Ross and Tabrizi, 2011). Transgenic HD mouse models also indicate that it is N-terminal mHtt fragments with expanded polyQ repeats that can become misfolded, aggregated, and pathogenic (Davies et al., 1997; Schilling et al., 1999). Also, HD knock-in mice, which express full-length mHtt at the endogenous level, show that N-terminal mHtt preferentially forms aggregates in the processes of striatal neurons, which are those mostly affected in HD (Li et al., 2000). In fact, in the brains of HD patients, neuropil aggregates appear to be more abundant than nuclear aggregates in the early disease stage (DiFiglia et al., 1997; Gutekunst et al., 1999). Because neuropil aggregates result from the accumulation of misfolded proteins in neuronal process that can affect axonal transport and synaptic function (Milnerwood and Raymond, 2010; Millecamps and Julien, 2013) and suppression of neuropil aggregates can alleviate the neurological symptoms of HD mice (Wang et al., 2008a), it is important that we understand how mHtt accumulates preferentially in the neuronal processes.
The accumulation of misfolded proteins in the neuronal processes could be attributable to impaired protein clearance. The clearance of misfolded proteins in cells is primarily mediated by the ubiquitin-proteasome system (UPS) and autophagy (Ciechanover and Kwon, 2015); however, which of these clearance systems plays a larger role in removing mHtt in neuronal processes remains unclear. In addition, because mHtt can affect intracellular transport (Gunawardena et al., 2003; Gauthier et al., 2004; Trushina et al., 2004; Reddy and Shirendeb 2012; Wong and Holzbaur 2014), it is possible that mHtt accumulates in the elongated neuronal processes because of the adverse effect of the expanded polyQ on intracellular transport. Unfortunately, it is difficult to identify the causes for neuropil Htt accumulation using conventional biochemical or microscopic assays.
Recently, an “optical pulse-chase” assay was used to study the degradation of soluble mHtt in cultured neurons (Tsvetkov et al., 2013). This technique uses Dendra2, a photoconvertible fluorescent protein, to fuse to mHtt, so the decline of fluorescence after photoconversion represents degradation of the fused protein. In the present study, we used this technique to further study the degradation of mHtt in different subcellular compartments of neurons and glial cells in the brain. Our findings show that mHtt is cleared more slowly in neuronal processes than in the cell body and is also removed less efficiently from neurons than astrocytes. By inhibiting the UPS and autophagy, we found that the UPS plays a primary role in clearing soluble mHtt in the neuronal processes, suggesting that increasing proteasome activity could be an efficient way to clear soluble mHtt from the neuronal processes in HD brains.
Materials and Methods
Wild-type (wt) mice were maintained at the Emory University Animal facility. Both male and female pups from these mice were used for primary cultures, and both male and female adult mice at the age of 2 months were used for viral injection and brain slice preparation. This study was performed in strict accordance with the recommendations in the Guidelines for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Institutional Animal Care and Use Committee of Emory University (Permit 2002557).
Plasmids, antibodies, and reagents.
Htt-23Q and Htt-130Q were generated by subcloning N-terminal fragments of huntingtin (1–230 aa) containing 23Q or 130Q into pDendra2-N (Clontech) using SalI and ApaI cloning sites with a CMV promoter. For in vivo expression, Htt–Dendra2 fusion genes were subcloned into a pAAV–MCS vector (Cell Biolabs) with a synapsin-1 or GFAP promoter to generate adeno-associated virus (AAV-9). AAV-9 virus was generated by the Emory Viral Vector Core. Antibodies used were anti-huntingtin (rabbit or mouse EM48), anti-NeuN (ABN78; Millipore), anti-GFAP (MAB360; Millipore), anti-Dendra2 (TA180094; Origene), anti-LC3 (NB100-2220; Novus), anti-ubiquitin, K48-specific (05-1307; Millipore), and anti-β-actin (A5060; Sigma). Secondary antibodies were HRP-labeled donkey anti-mouse, donkey anti-rabbit, donkey anti-mouse Alexa Fluor 488 or 594, and donkey anti-rabbit Alexa Fluor 488 or 594 from Jackson ImmunoResearch. MG132, epoxomicin, and bafilomycin A (BFA) were purchased from Sigma, as were proteinase inhibitor cocktails.
Primary cell cultures.
Brains of postnatal (days 1–3) murine pups were used for culturing cortical astrocytes. After dissection, the cortex was subjected to 0.3 mg/ml papain digestion. The cell suspension flew through 70 μm nylon cell strainers (Thermo Fisher Scientific). Cells were plated onto Petri dishes; culture medium was replaced 24 h later and then once every 3 d thereafter. Microglia and oligodendrocytes were removed from cultures by shaking at DIV14. The remaining cells were detached with 0.25% trypsin and plated for the following experiments. For neuronal cultures, neurons were prepared from postnatal day 0 murine pups. Cortex or hippocampus was digested with 0.3 mg/ml papain. Cell suspension was filtered through 40 μm nylon cell strainers (Thermo Fisher Scientific) to remove debris. Neurons were cultured in Neurobasal-A medium supplemented with B27 and glutamine (Invitrogen). Half the culture medium was changed with fresh medium every 3 d. To reduce glial proliferation, cytosine was added to the cultures 3 d after plating. Cultured neurons at DIV3 and astrocytes at DIV21–DIV28 were used for the transfection of Htt–Dendra2 and were subjected to live imaging 24 h later or to Western blotting 40 h later.
Stereotaxic injection of viral vectors.
Two-month-old mice were anesthetized with an intraperitoneal injection of avertin (0.5 mg/g). Their heads were placed and fixed in a David Kopf Instruments stereotaxic frame (model 1900) equipped with a digital manipulator and a UMP3-1 Ultra pump. Mice were kept deeply anesthetized as assessed by monitoring pinch withdrawal and respiration rate. Viral vector injections were given in the striatum (0.6 mm anterior to bregma, 2.0 mm lateral to the midline, and 3.5 mm ventral to dura) and motor cortex (1.0 mm anterior to bregma, 1.25 mm lateral to the midline, 0.8–1.0 mm ventral to dura). The injections were performed at a rate of 0.2 μl/min. The needle was left in place for 10 min after each injection to minimize upward flow of viral solution after raising the needle.
Photoconversion, imaging system, and data analysis.
Photoconversion of Dendra2 was performed with a Nikon A1R confocal live imaging system with a 60× objective lens. Transfected primary neurons and astrocytes, as well as acute brain slices, were placed in a chamber at 37°C, 5% CO2 during the imaging session. After transfection for 24 h, cultured hippocampal neurons or cortical astrocytes were subjected to live imaging. Approximately 2 weeks after viral injection, the brains were sliced with a vibratome (Leica) in cold artificial CSF. Subsequently, acute brain slices were allowed to recover for 30 min to 1 h in a 37°C and 5% CO2 incubator and then subjected to live imaging. During imaging sessions, brain slices were maintained in brain slice culture medium containing 50% MEM/HEPES (Gibco), 25% heat-inactivated horse serum (Gibco), 25% HBSS (Gibco), and 6.5 mg/ml glucose (Sigma), pH 7.2. UV light at 405 nm was used to activate Dendra2. To avoid UV damage to cells, the power and duration of UV irradiation was optimized. The photoconversion of Dendra2–Htt was performed on selected cell bodies and segments of processes in which no mHtt aggregates were visible and green fluorescence of inactive Dendra2 was seen. After photoconversion of green fluorescence to red fluorescence, images were taken at 10 min intervals. Decays of red fluorescence of active Dendra2 in the region of interest were used to measure degradation rates of Htt. Nikon Element software was used to quantify the red fluorescence in the region of interest. The intensity values were background subtracted. To avoid the influence of red fluorescence diffusion on degradation rates, the intensity value was normalized to that at 10 min after photoconversion, except for measuring degradation rates of Htt in cell bodies of cultured astrocytes, in which no diffusion of fluorescence was observed after photoconversion because target astrocytes did not have long processes.
Immunoprecipitation.
Primary cultures were harvested and lysed in ice-cold 0.5% Triton X-100/PBS solution with protease inhibitor cocktail and phosphatase inhibitors on ice. The lysates were centrifuged at 16,000 × g for 30 min. Protein concentrations were measured with BCA assay (Thermo Fisher Scientific). Total 300 μg samples were precleared with Protein A agarose beads (Sigma), and huntingtin proteins were immunoprecipitated by anti-Htt (mEM48) at 4°C overnight. Protein A agarose beads were added to capture the immunoprecipitates for 1 h at 4°C. Ice-cold lysis buffer was used to wash beads three times. Proteins from the immunoprecipitates and inputs were subjected to Western blotting.
Immunofluorescence staining.
Mice were anesthetized, perfused with fresh 4% paraformaldehyde in PBS, and postfixed overnight in the same fixative. Fixed brains were switched to 30% sucrose at 4°C. Mouse brains were sliced at 15 μm thickness with a cryostat at −20°C and then mounted onto gelatin-coated slides. The brain slices were blocked with 3% bovine serum albumin in PBS supplemented with 0.2% Triton X-100 for 30 min at room temperature. Slices were incubated with primary antibodies at 4°C overnight and washed. Fluor-conjugated secondary antibodies and nuclear dye Hoechst were added to the samples for staining. Images were acquired with an Imager Z1 microscope equipped with a 63× objective lens.
Western blot analysis.
Primary cultures were lysed in ice-cold RIPA buffer (50 mm Tris, pH 8.0, 150 mm NaCl, 1 mm EDTA, pH 8.0, 1 mm EGTA, pH 8.0, 0.1% SDS, 0.5% deoxycholate, and 1% Triton X-100) containing protease inhibitor cocktail and phosphatase inhibitors. The lysates were sonicated and subjected to SDS-PAGE. The proteins on the gel were transferred to a nitrocellulose membrane, which was then blocked with 5% milk/TBST for 1 h at room temperature. The blot was incubated with primary antibodies in 5% milk/TBST overnight at 4°C. After three washes in TBST, the blot was incubated with HRP-conjugated secondary antibodies in 5% milk/TBST for 1 h at room temperature. After three washes in TBST, ECL Prime (GE Healthcare) was used to detect immunoreactive bands on the blot.
Statistical analyses.
Unpaired two-tailed Student's t test and repeated-measures (RM) two-way ANOVA, followed by Bonferroni's post hoc test were performed with GraphPad Prism 6. Results are expressed as the means ± SEM. A p value < 0.05 was considered significant. The statistical significance level was set as follows: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Results
Soluble mHtt is cleared faster than wild-type Htt in the cell body
The photoconvertible fluorescent protein Dendra2 has been conjugated to Htt to examine the turnover of Htt, and this conjugation does not affect the aggregation or toxicity of mHtt (Tsvetkov et al., 2013). Indeed, mHtt with Dendra2 conjugation also formed aggregates in neurites as mHtt without Dendra2 and did not cause any obvious morphological difference in cultured neurons, suggesting that Dendra2 did not alter toxicity of mHtt (Fig. 1A,C). To evaluate the degradation rate of N-terminal Htt in different subcellular regions, we conjugated Dendra2 to the C terminus of N-terminal Htt (1–230 aa) with 23Q or 130Q (Fig. 1A). We transfected Htt–Dendra2, which is expressed under the CMV promoter, in cultured primary mouse hippocampal neurons and cortical astrocytes. Western blotting with anti-Htt (mEM48) and anti-Dendra2 showed the integrity of Htt–Dendra2 in cultured neurons and astrocytes (Fig. 1B). Thus, the proteolytic degradation of this fusion protein or decline in Dendra2 fluorescent signal reflects the degradation of Htt or its clearance in cells. Next, we used fluorescent microscopic imaging to evaluate the turnover of Htt–Dendra2. After activating Dendra2 to convert it to a red fluorescent protein within the region of cell bodies, the intensity of red fluorescence was quantified at different time points. We observed that a small fraction of red fluorescence promptly diffused into neurites immediately after photoconversion, and no such diffusion was seen at 10 min after photoconversion. This rapid diffusion may result from the intracellular transport of Htt. Thus, we started to measure red fluorescence intensity at 10 min after photoconversion to avoid diffusion effects on degradation rates. The fluorescence diffusion in astrocytes is negligible because they do not have long processes. Comparison of normal and mutant Htt red fluorescence intensities revealed faster degradation of mHtt (Htt-130Q) than wild-type Htt (Htt-23Q) in the cell bodies of neuronal and astrocytic cells (Fig. 1D–G, *p < 0.05, **p < 0.01).
Figure 1.
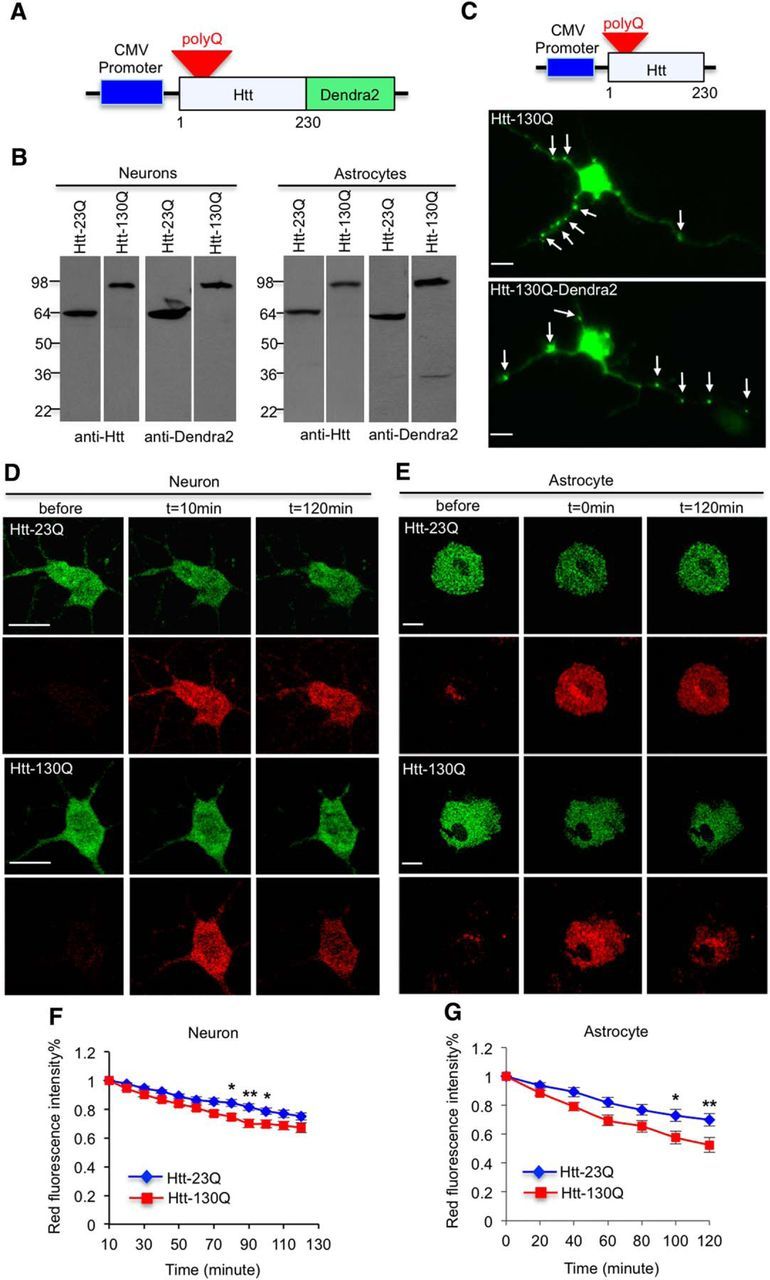
Soluble mHtt is promptly removed in the bodies of both neuronal and astrocytic cells. A, Schematic representation of Htt–Dendra2 plasmids. Dendra2 is conjugated to N-terminal (1–230 aa) Htt under the control of an exogenous promoter. In this figure, Htt–23Q and Htt–130Q are expressed by the CMV promoter. B, Western blotting analysis of transfected Htt–Dendra2 in cultured primary neurons and astrocytes. Antibodies to Htt (mEM48) and Dendra2 were used. C, Immunostaining with EM48 antibody showed that both mHtt and mHtt–Dendra2 fusion protein aggregated in processes of cultured neurons, and neuropil aggregates are indicated by arrow. D, E, mHtt (Htt–130Q) is degraded faster than wild-type Htt (Htt–23Q) in the cytoplasm of neuronal (D) and astrocytic (E) cell bodies (hippocampal neurons in D and cortical astrocytes in E). F, G, Reduction of red fluorescence intensity in D and E is quantified at indicated time points; n = 23 (23Q) and 21 (130Q) cells in F, n = 10 (23Q) and 14 (130Q) cells in G. For neurons, the red fluorescence intensity at 10 min was normalized to 1. For astrocytes, the red fluorescence intensity at 0 was normalized to 1. [*p < 0.05, **p < 0.01, two-way RM-ANOVA, followed by Bonferroni's post hoc test (factor 1, time; factor 2, genotype).] Error bars represent SEM. Scale bars, 10 μm.
mHtt is cleared slowly in neuronal processes
The advantage of using Htt–Dendra2 is that we can examine Htt turnover in neuronal processes. Thus, we activated Dendra2 in segments of processes and saw that a considerable amount of red fluorescence was stable in the activated regions, probably because of the association of Htt with microtubules because it interacts with many proteins and organelles (Hoffner et al., 2002; Harjes and Wanker, 2003; Trushina et al., 2004; Smith et al., 2009). Importantly, we found that Htt–130Q was cleared more slowly than Htt–23Q in neurites of cultured neurons (Fig. 2A,C, *p < 0.05, **p < 0.01), suggesting the impaired degradation of mHtt in this subcellular region. In contrast, in the processes of cultured astrocytes, mHtt (Htt–130Q) is still removed faster than normal Htt (Htt-23Q; Fig. 2B,D, ***p < 0.001, ****p < 0.0001). This difference indicates that neuronal cells and astrocytes have intrinsic differences in clearing mHtt in their processes.
Figure 2.

mHtt is slowly cleared in neuronal processes. A, Htt–130Q is degraded slower than Htt–23Q in processes of cultured hippocampal neurons; n = 13 (23Q) and 18 (130Q) cells. B, In the processes of cortical astrocytes, Htt–130Q is cleared more rapidly than Htt–23Q; n = 10 (23Q) and 11 (130Q) cells. C, D, Quantitative results for A and B showing that the degradation rate of mHtt in neuronal processes is slow. [*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; two-way RM-ANOVA, followed by Bonferroni's post hoc test (factor 1, time; factor 2, genotype).] Error bars represent SEM. Scale bars, 10 μm.
Soluble mHtt is more efficiently cleared by the UPS
We know that both proteasome and autophagy can degrade soluble mHtt (Waelter et al., 2001; Sarkar and Rubinsztein, 2008; Li et al., 2010); however, we know little about which quality control mechanism is primary for the removal of mHtt in processes. Therefore, we treated primary neurons or astrocytes with either 5 μm MG132 to inhibit the UPS or 100 nm BFA to inhibit autophagy. After adding these inhibitors to the culture medium, cells were subjected to live imaging. Again, without MG132 or BFA, normal Htt (Htt–23Q) is degraded much faster than mutant Htt (mHtt–130Q). After inhibiting the UPS by MG132, both Htt–23Q and mHtt–130Q were stable over the same period of time. We also used a specific proteasomal inhibitor, epoxomicin, and found that it also markedly suppressed the turnover of Htt–23Q and mHtt–130Q in neuronal processes. Interestingly, BFA treatment did not stabilize either Htt–23Q or Htt–130Q (Fig. 3, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, Con vs MG132; #p < 0.05, ##p < 0.01, ####p < 0.0001, Con vs Epoxomicin). These results suggest that soluble Htt in neuronal processes is degraded primarily by the UPS.
Figure 3.
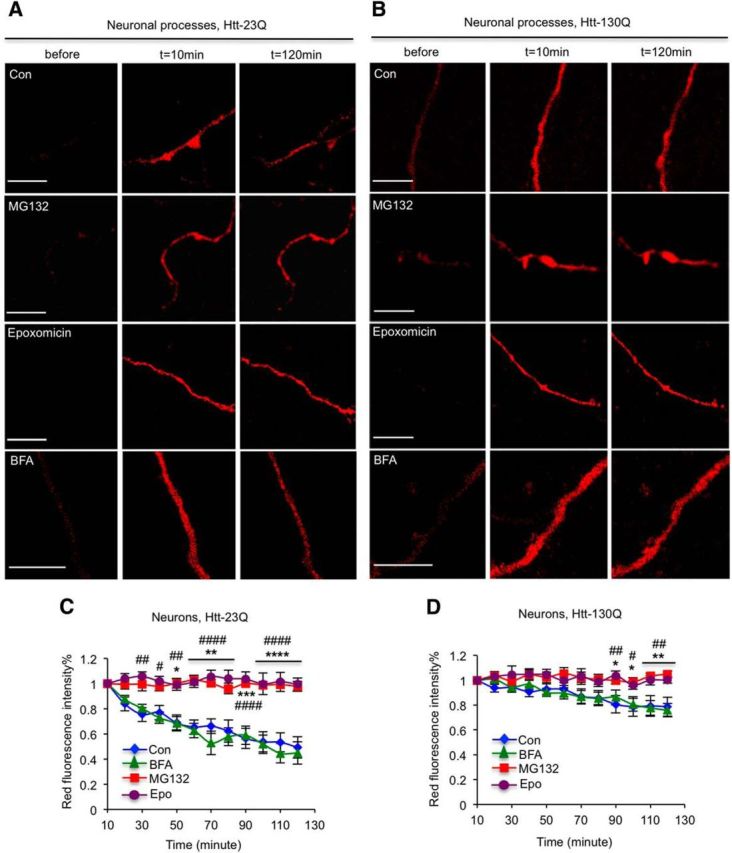
Proteasome plays a major role in clearing Htt in neuronal processes. A, B, Fluorescent images show the clearance of Htt–23Q (A) and Htt–130Q (B) in the processes of cultured hippocampal neurons. MG132 or epoxomicin stabilizes the levels of Htt–23Q and Htt–130Q in the neuronal process. n = 13 (Con), 8 (BFA), 5 (MG132), and 10 (epoxomicin) cells in A; n = 18 (Con), 8 (BFA), 9 (MG132), and 12 (epoxomicin) cells in B. C, D, Quantitative results of red fluorescent intensity of Htt–Dendra2 at different time points showing that MG132 (5 μm) and epoxomicin (100 nm), but not BFA (100 nm), can stabilize the levels of Htt–23Q and Htt–130Q in the neuronal processes. [*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, Con vs MG132; #p < 0.05, ##p < 0.01, ###p < 0.001, ####p < 0.0001, Con vs epoxomicin, two-way RM-ANOVA, followed by Bonferroni's post hoc test (factor 1, time; factor 2, treatment).] Error bars represent SEM. Scale bars, 10 μm. Con, Control.
To examine whether Htt in astrocytic processes is degraded by the UPS and autophagy, we treated Htt–130Q-transfected astrocytes in culture with MG132, epoxomicin, or BFA. Similarly, MG132 and epoxomicin, but not BFA, blocked the degradation of mHtt in astrocytes (Fig. 4, *p < 0.05, **p < 0.01, ***p < 0.001, Con vs MG132; #p < 0.05, ##p < 0.01, ###p < 0.001, ####p < 0.0001, Con vs. epoxomicin). Thus, in the processes of astrocytes, mHtt is also degraded primarily by the UPS.
Figure 4.
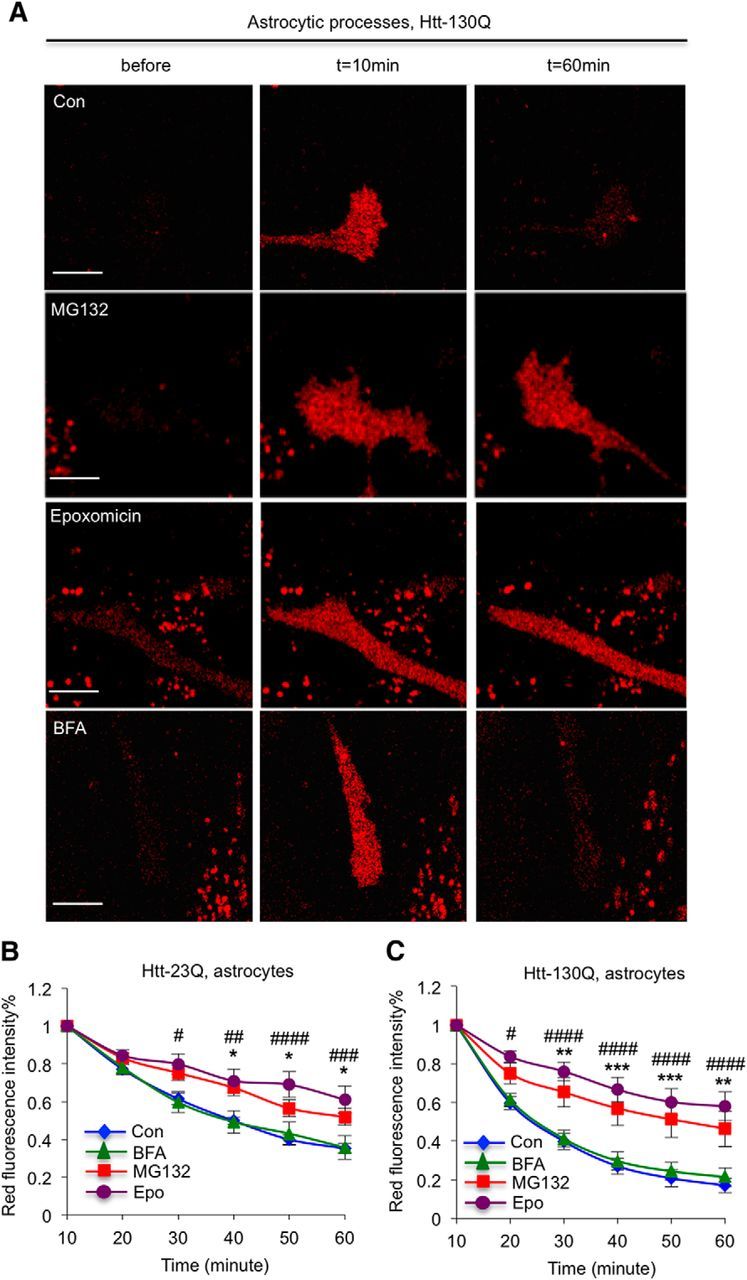
Proteasomal inhibitors also stabilize mHtt in the processes of cultured astrocytes. A, Fluorescent imaging showing that MG132 (5 μm) or epoxomicin (100 nm) treatment blocks the clearance of mHtt in the processes of cultured cortical astrocytes. B, C, Quantitative results of red fluorescence intensity of Htt–Dendra2 at different time points showing that MG132 and epoxomicin, but not BFA (100 nm), can stabilize the levels of Htt–23Q and Htt–130Q in processes of cultured astrocytes. n = 10 (Con), 10 (BFA), 15 (MG132), and 13 (epoxomicin) cells in B; n = 11 (Con), 10 (BFA), 12 (MG132), and 11 (epoxomicin) in C. [*p < 0.05, **p < 0.01, *** p < 0.001, Con vs MG132; #p < 0.05, ##p < 0.01, ###p < 0.001, ####p < 0.0001, Con vs epoxomicin, two-way RM-ANOVA, followed by Bonferroni's post hoc test (factor 1, time; factor 2, treatment).] Error bars represent SEM. Scale bars, 10 μm. Con, Control.
Degradation of mHtt by the UPS via K48 ubiquitination
Although live imaging analysis led us to examine the degradation of mHtt in neuronal processes, quantitatively comparing the global effects of proteasomal and autophagic inhibitors on mHtt in neuronal and astrocytic cells is difficult. Thus, we performed Western blot analysis using antibodies to LC3 for detecting the production of LC3-II, an indicator of autophagy activation, because BFA increases LC3-II during its inhibition of autophagy (Myeku and Figueiredo-Pereira, 2011). As expected, BFA treatment markedly elevated the level of LC3-II, indicating that the doses and treatment of BFA we used indeed inhibited autophagic function (Fig. 5A,B); however, this BFA treatment apparently could not significantly increase levels of soluble normal Htt (Htt–23Q). Moreover, BFA treatment led to a small but significant increase in soluble mutant Htt (Htt–130Q) in neuronal and astrocytic cells. Compared with BFA, MG132 and epoxomicin remarkably increased both Htt–23Q and Htt–130Q in neuronal cells and astrocytes (Fig. 5, *p < 0.05, **p < 0.01, ***p < 0.01). These results support the live imaging results that soluble Htt is degraded primarily by the UPS.
Figure 5.
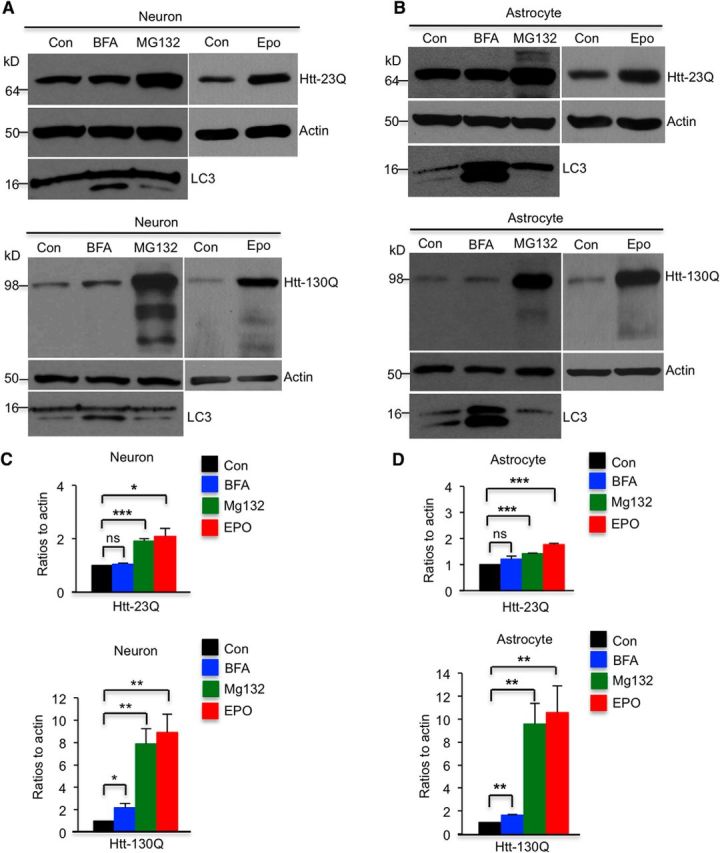
Western blot analysis of Htt–Dendra2 expression. A, B, Western blot analysis of cultured Htt-transfected cortical neurons (A) and astrocytes (B) after treatments with MG132 (5 μm), epoxomicin (100 nm), or BFA (100 nm). MG132 and epoxomicin have stronger effects on inhibiting clearance of Htt–130Q than BFA in neurons and astrocytes. C, D, Ratios of Htt–Dendra2 to actins from three independent experiments are presented. MG132, epoxomicin, or BFA treatment time is 16 h. Con, Control; Epo, epoxomicin. *p < 0.05, **p < 0.01, *** p < 0.001, unpaired two-tailed Student's t test. ns, Not significant. Error bars represent SEM.
To further investigate whether the degradation of Htt by the UPS is via K48 ubiquitination, we performed immunoprecipitation of Htt and then probed the immunoprecipitates with an anti-K48 antibody. We found that, in neuronal cells and astrocytes, both Htt–23Q and Htt–130Q are ubiquitinated via K48, but Htt–130Q is ubiquitinated via K48 to a much greater extent than Htt–23Q (Fig. 6A,B). Quantitative analysis of the ratio of ubiquitinated Htt to precipitated Htt verified that more mHtt is K48 ubiquitinated than normal Htt (Fig. 6C, *p < 0.05). Because K48 ubiquitination is important for protein degradation by the UPS (Pickart and Eddins, 2004), the result suggests that differential K48 ubiquitination may account for faster degradation of mHtt in neuronal cell bodies and astrocytes.
Figure 6.
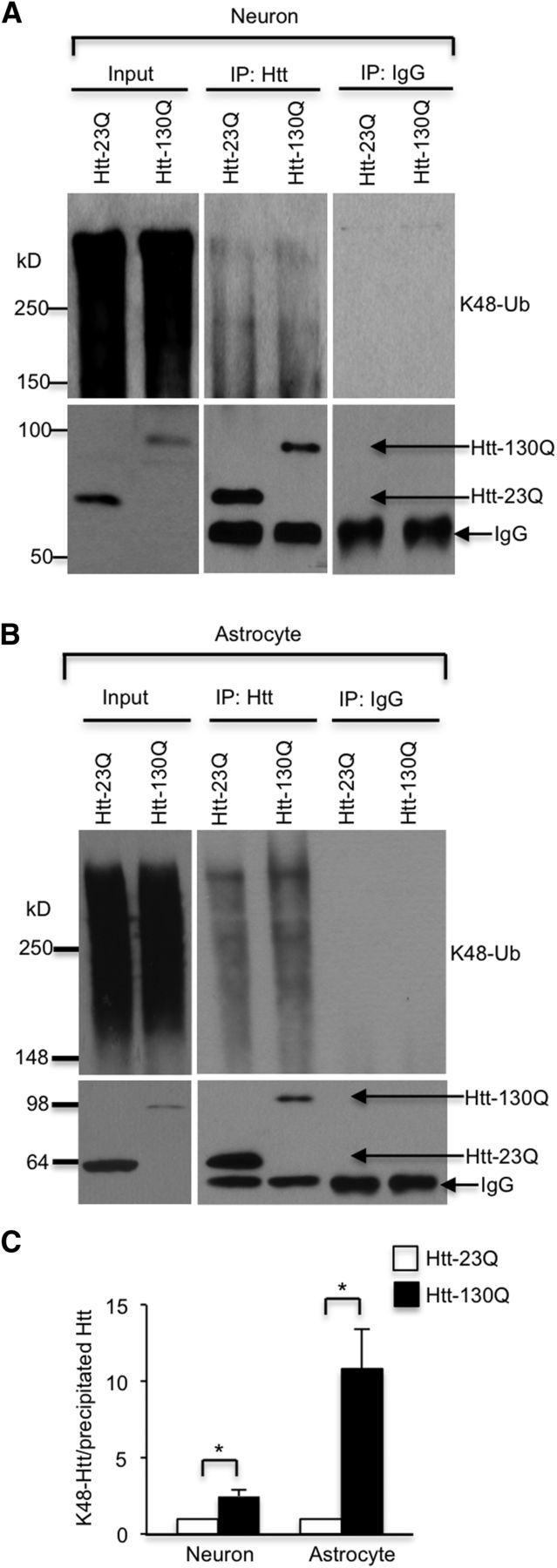
Western blot analysis of immunoprecipitated Htt. A, B, Transfected Htt was immunoprecipitated by anti-Htt antibody (EM48) from cultured cortical neurons (A) and astrocytes (B), and IgG was used as a control. The immunoprecipitates were probed with antibodies to Htt (EM48) or K48 ubiquitination. MG132 at 5 μm was used to inhibit proteasomal activity. C, Ratios of K48 ubiquitinated Htt to the immunoprecipitated Htt from three independent experiments are presented beneath the blots. *p < 0.05, unpaired two-tailed Student's t test. Error bars represent SEM.
Degradation of mHtt is compartment dependent in neurons in the mouse brain
To explore whether mHtt is differentially cleared in the cell body and processes in the brain, we generated AAV-9 viral vectors that express Htt–Dendra2 under the control of the neuronal promoter synapsin-1 (syn–Htt) or the glial promoter GFAP (GFAP–Htt; Fig. 7A). Stereotaxic injection of these viral vectors allows for the selective expression of Htt in neurons or astrocytes. Therefore, we injected syn–wt–Htt or syn–mHtt into the striatum of 2-month-old wild-type mice (Fig. 7B). Fluorescence imaging demonstrated restricted expression of the injected AAV viruses in the mouse striatum (Fig. 7B). Double immunofluorescent staining showed that syn–mHtt is distributed in NeuN-positive cells but not GFAP-positive glial cells (Fig. 7C). Also, syn–mHtt is present in the neuronal process or accumulates in the nuclei to form aggregates (Fig. 7D).
Figure 7.

Construction of AAV vectors to express Htt–Dendra2 selectively in neurons and astrocytes in the mouse brain. A, Schematic representation of AAV plasmids. The specific expression of Htt is controlled by the synapsin-1 or GFAP promoter. B, Immunostaining confirms the selective expression of Htt–Dendra2 in neurons and astrocytes in the mouse brain after stereotaxic injection for 1–2 weeks. C, Immunostaining of fixed brain sections of mice injected with AAV–Htt–Dendra2. Double immunostaining with antibodies to Htt, the neuronal marker protein NeuN, and astrocytic marker protein GFAP. The result shows that synapsin promoter-driven mHtt (syn–mHtt) is selectively distributed in NeuN-positive neuronal cells. D, mHtt forms aggregates in the neuropil (arrow) and nuclei (arrowhead) of striatal neurons in the mouse brain. Scale bars, 20 μm.
We next sectioned brain slices from the injected mouse striatum and used fluorescence microcopy to examine the degradation of syn–Htt. Compared with normal Htt (syn–wt–Htt), syn–mHtt in the brain slices showed faster degradation in the neuronal cell body and slower degradation in neuronal processes (Fig. 8A–C, *p < 0.05, **p < 0.01, ***p < 0.001). In the cell body of astrocytes in the brain slices, GFAP–mHtt is also degraded faster than GFAP–wt–Htt (Fig. 8D,E, **p < 0.01, ***p < 0.001). These findings are consistent with what we have observed in cultured cells and demonstrate that mHtt is indeed differentially degraded in the cell body and processes of neurons. Furthermore, we saw faster degradation of mHtt in astrocytes than neurons in the striatum of brain slices (Fig. 8A,D), suggesting that the ability of cell body and processes to clear polyQ proteins may depend on cell types, in which the UPS and autophagy, as well as proteases, could function differentially in different types of cells. It is well established that striatal projection neurons are most affected by HD. To compare the degradation rates of mHtt between cortical and striatal neurons, we injected AAV–mHtt–Dendra2 into the striatum and motor cortex and found that cortical neurons were able to clear mHtt faster than striatal neurons (Fig. 8F,G, *p < 0.05). This finding suggests that cell-autonomous mechanisms contribute, at least in part, to the vulnerability of striatal neurons in HD. Together, our results from brain slices not only support compartment-dependent degradation of mHtt in neurons but also show distinct capacity of clearing mHtt in different types of brain cells.
Figure 8.
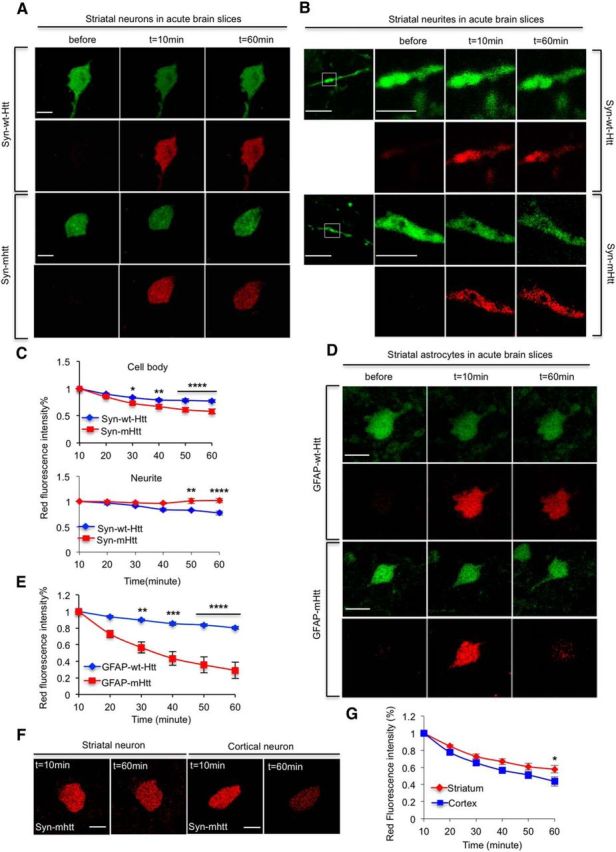
Expanded polyQ promotes the clearance of soluble Htt in the cell bodies but impairs the clearance of Htt in neurites in the mouse brain. A, B, mHtt expressed under the synapsin promoter is cleared faster than wild-type Htt (wt–Htt) in neuronal cell bodies (A) in brain slices. In B, mHtt is more stable in neuronal process than wt–Htt. C, Quantitative results of decline in mHtt and wt–Htt. n = 13 (wt–Htt) and 15 (mHtt) cells for cell bodies. n = 4 (wt–Htt) and 3 (mHtt) cells for neurites. D, E, mHtt is removed more promptly than wt–Htt in astrocytic cells in the mouse brain slices. D shows representative fluorescent images, and E shows quantitative data for the clearance of wt–Htt and mHtt. n = 7 (wt–Htt) and 11 (mHtt) cells. F, G, cortical neurons cleared mHtt faster than striatal neurons. F shows representative fluorescent images, and the statistical result is shown in G, n = 15 (striatal neurons) and 13 (cortical motor neurons). [*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, two-way RM-ANOVA, followed by Bonferroni's post hoc test (factor 1, time; factor 2, genotype for C and E; factor 1, time; factor 2, neuronal type for G).] Error bars represent SEM. Scale bars: 10 μm; for enlargements in B, 3 μm.
Discussion
In this study, we used an optical pulse-chase assay with Dendra2 to show for the first time that mHtt is differentially cleared in the body and processes of neuronal cells. Many previous studies have used GFP-conjugated mHtt to investigate the distribution and toxicity of expanded polyQ proteins. Our results demonstrated that Dendra2, a GFP derivative, did not change the pattern of formation of mHtt aggregates in neuropils and did not cause obvious morphological alterations in transfected neurons. Although it requires additional studies to determine whether the conjugation of GFP or Dendra2 can modify mHtt function or toxicity, Dendra2 conjugation allowed us to investigate the turnover of Htt in live cells. Using Dendra2–Htt, we also showed that mHtt is degraded faster in astrocytes than in neuronal cells. These findings explain why mHtt accumulates preferentially in neuronal processes, a subcellular site whose function is important for neuronal interaction and is vulnerable in many neurodegenerative diseases at the early stages (Li et al., 2001; Fischer and Glass, 2007; Volpicelli-Daley et al., 2014).
The accumulation of misfolded proteins in neuronal cells is a prerequisite for their toxicity (Sherman and Goldberg, 2001; Goldberg, 2003). Thus, the clearance of misfolded proteins is key to preventing their neurotoxicity. Although mHtt is ubiquitously expressed in all types of cells and is distributed widely in different subcellular regions, why this disease protein can preferentially accumulate in the neuronal process and form neuropil aggregates, which appear more abundant than nuclear Htt aggregates in HD patient brains at early disease stages, remains unclear (DiFiglia et al., 1997; Gutekunst et al., 1999). We already know that Htt plays roles in a broad range of cellular functions (Zuccato et al. 2010). One of its important functions is to regulate intracellular trafficking (Gunawardena et al., 2003; Gauthier et al., 2004; Trushina et al., 2004; Wong and Holzbaur 2014). Consistent with this function, mHtt can associate with microtubules and affect microtubule-dependent transport (Trushina et al., 2004; Smith et al., 2009; Reddy and Shirendeb 2012). Because microtubule-dependent transport is essential for maintaining the function and integrity of long neuronal processes, it is possible that mHtt may affect its transport along the processes, leading to its accumulation and aggregation in the neuropil. We also know that mHtt can be cleared by the UPS and autophagy (Sarkar and Rubinsztein et al., 2008; Li et al., 2010). However, sorting out the mechanism for the accumulation of mHtt in neuronal processes is a challenge, because conventional biochemical assays and microcopy cannot distinguish the causes for the accumulation of mHtt in neuronal processes.
Our studies provide evidence for the slow degradation of mHtt in neuronal processes. First, the property of photoconversion of green Dendra2 to red fluorescence allowed us to examine the half-life of mHtt located in a defined region in neuronal processes. By examining this half-life and comparing normal and mutant Htt, we found that the decline in mHtt or its degradation is slower in the neuronal process than in the cell body. Second, we also performed pharmacological experiments to block the function of the UPS and autophagy and found that the UPS plays a more important role in clearing soluble mHtt. Third, we examined the decline of mHtt in the mouse brain by expressing viral Htt–Dendra2. This study also verified that mHtt is cleared slowly in neuronal processes in the context of neuron–glia interactions.
Using optical pulse-chase, Tsvetkov et al. (2013) observed a faster degradation of exon1 mHtt (1–67 aa) than its wild-type form in the soma of cultured striatal and cortical neurons. In our study, we examined the degradation of a larger N-terminal Htt fragment (1–230 aa), whose toxicity has been confirmed in the soma and processes of hippocampal neurons and astrocytes in transgenic HD mice (Bradford et al., 2009; Huang et al., 2015). In the finding by Tsvetkov et al. (2013), exon1 mHtt is cleared mainly by autophagy, whereas our finding show that the larger N-terminal mHtt fragment (1–230 aa) is more efficiently degraded by the UPS. These differences suggest that the length of N-terminal htt and protein context can influence the degradation of Htt in cells by different proteolytic machineries. Also, different methodologies, such as differences in types of cultured neurons examined, culturing conditions, and drug treatments used, may lead to different results. For example, we examined healthy cells in culture in which the UPS may play a more important role in removing Htt than autophagy. By comparing the UPS versus autophagy in removing mHtt in neuronal processes under the same experimental conditions, we found that the UPS plays a more important role in removing mHtt in neuronal processes. This finding is consistent with the age-dependent decline in the UPS in the brain (Chondrogianni et al., 2015) and explains why, in the aged HD brain, reduced UPS activity promotes the accumulation of mHtt in neuronal processes or neuropil. In support of this idea, astrocytes, which can clear mHtt more efficiently than neurons, are found to have higher UPS activities than neurons (Tydlacka et al., 2008).
Understanding how mHtt preferentially accumulates in neuronal processes will help us to find effective strategies to treat HD. This is because, in many neurodegenerative diseases, including Alzheimer's disease, Parkinson's disease, and amyotrophic lateral sclerosis, neuronal process degeneration and dysfunction are the early pathological events (Li et al., 2001; Fischer and Glass, 2007; Volpicelli-Daley et al., 2014). In HD knock-in mice that express full-length mHtt, neuropil aggregates appear to occur at the earlier HD stage (Li et al., 2001), further underscoring the important role of neuropil mHtt in HD pathology (DiFiglia et al., 1997; Gutekunst et al., 1999). The accumulated mHtt in neuronal processes can affect intracellular transport and synaptic function (Trushina et al., 2004; Xu et al., 2013). Although mHtt does not impair the overall proteasomal function in brain homogenates (Bett et al., 2009), it can locally affect the proteasomal activity in axonal terminals (Wang et al., 2008b). Our findings suggest that improving proteasomal function would be an effective way to alleviate the neuropathology caused by neuropil mHtt. Moreover, we show the feasibility of using optical pulse-chase to study protein turnover at subcellular levels, which could be expanded to other neurodegenerative disease proteins, providing new insights into their pathogenesis and neuropathology at subcellular levels.
Footnotes
This work was supported by National Institutes of Health Grants AG19206 and NS041449 (X.-J.L.) and AG031153 and NS045016 (S.L.), and the National Natural Science Foundation of China (91332206 to X.J.L.). We thank the Integrated Cellular Imaging Core at Emory University for the use of imaging facilities and Cheryl Strauss for critical reading of this manuscript.
The authors declare no competing financial interests.
References
- Bett JS, Cook C, Petrucelli L, Bates GP. The ubiquitin-proteasome reporter GFPu does not accumulate in neurons of the R6/2 transgenic mouse model of Huntington's disease. PLoS One. 2009;4:e5128. doi: 10.1371/journal.pone.0005128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford J, Shin JY, Roberts M, Wang CE, Li XJ, Li S. Expression of mutant huntingtin in mouse brain astrocytes causes age-dependent neurological symptoms. Proc Natl Acad Sci U S A. 2009;106:22480–22485. doi: 10.1073/pnas.0911503106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chondrogianni N, Voutetakis K, Kapetanou M, Delitsikou V, Papaevgeniou N, Sakellari M, Lefaki M, Filippopoulou K, Gonos ES. Proteasome activation: an innovative promising approach for delaying aging and retarding age-related diseases. Ageing Res Rev. 2015;23:37–55. doi: 10.1016/j.arr.2014.12.003. [DOI] [PubMed] [Google Scholar]
- Ciechanover A, Kwon YT. Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Exp Mol Med. 2015;47:e147. doi: 10.1038/emm.2014.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537–548. doi: 10.1016/S0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- Fischer LR, Glass JD. Axonal degeneration in motor neuron disease. Neurodegener Dis. 2007;4:431–442. doi: 10.1159/000107704. [DOI] [PubMed] [Google Scholar]
- Gauthier LR, Charrin BC, Borrell-Pagès M, Dompierre JP, Rangone H, Cordelières FP, De Mey J, MacDonald ME, Lessmann V, Humbert S, Saudou F. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118:127–138. doi: 10.1016/j.cell.2004.06.018. [DOI] [PubMed] [Google Scholar]
- Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature. 2003;426:895–899. doi: 10.1038/nature02263. [DOI] [PubMed] [Google Scholar]
- Gunawardena S, Her LS, Brusch RG, Laymon RA, Niesman IR, Gordesky-Gold B, Sintasath L, Bonini NM, Goldstein LS. Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron. 2003;40:25–40. doi: 10.1016/S0896-6273(03)00594-4. [DOI] [PubMed] [Google Scholar]
- Gutekunst CA, Li SH, Yi H, Mulroy JS, Kuemmerle S, Jones R, Rye D, Ferrante RJ, Hersch SM, Li XJ. Nuclear and neuropil aggregates in Huntington's disease: relationship to neuropathology. J Neurosci. 1999;19:2522–2534. doi: 10.1523/JNEUROSCI.19-07-02522.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harjes P, Wanker EE. The hunt for huntingtin function: interaction partners tell many different stories. Trends Biochem Sci. 2003;28:425–433. doi: 10.1016/S0968-0004(03)00168-3. [DOI] [PubMed] [Google Scholar]
- Hoffner G, Kahlem P, Djian P. Perinuclear localization of huntingtin as a consequence of its binding to microtubules through an interaction with beta-tubulin: relevance to Huntington's disease. J Cell Sci. 2002;115:941–948. doi: 10.1242/jcs.115.5.941. [DOI] [PubMed] [Google Scholar]
- Huang B, Wei W, Wang G, Gaertig MA, Feng Y, Wang W, Li XJ, Li S. Mutant huntingtin downregulates myelin regulatory factor-mediated myelin gene expression and affects mature oligodendrocytes. Neuron. 2015;85:1212–1226. doi: 10.1016/j.neuron.2015.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Li SH, Johnston H, Shelbourne PF, Li XJ. Amino-terminal fragments of mutant huntingtin show selective accumulation in striatal neurons and synaptic toxicity. Nat Genet. 2000;25:385–389. doi: 10.1038/78054. [DOI] [PubMed] [Google Scholar]
- Li H, Li SH, Yu ZX, Shelbourne P, Li XJ. Huntingtin aggregate-associated axonal degeneration is an early pathological event in Huntington's disease mice. J Neurosci. 2001;21:8473–8481. doi: 10.1523/JNEUROSCI.21-21-08473.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Wang CE, Huang S, Xu X, Li XJ, Li H, Li S. Inhibiting the ubiquitin-proteasome system leads to preferential accumulation of toxic N-terminal mutant huntingtin fragments. Hum Mol Genet. 2010;19:2445–2455. doi: 10.1093/hmg/ddq127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millecamps S, Julien JP. Axonal transport deficits and neurodegenerative diseases. Nat Rev Neurosci. 2013;14:161–176. doi: 10.1038/nrn3380. [DOI] [PubMed] [Google Scholar]
- Milnerwood AJ, Raymond LA. Early synaptic pathophysiology in neurodegeneration: insights from Huntington's disease. Trends Neurosci. 2010;33:513–523. doi: 10.1016/j.tins.2010.08.002. [DOI] [PubMed] [Google Scholar]
- Myeku N, Figueiredo-Pereira ME. Dynamics of the degradation of ubiquitinated proteins by proteasomes and autophagy: association with sequestosome 1/p62. J Biol Chem. 2011;286:22426–22440. doi: 10.1074/jbc.M110.149252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickart CM, Eddins MJ. Ubiquitin: structures, functions, mechanisms. Biochim Biophys Acta. 2004;1695:55–72. doi: 10.1016/j.bbamcr.2004.09.019. [DOI] [PubMed] [Google Scholar]
- Reddy PH, Shirendeb UP. Mutant huntingtin, abnormal mitochondrial dynamics, defective axonal transport of mitochondria, and selective synaptic degeneration in Huntington's disease. Biochim Biophys Acta. 2012;1822:101–110. doi: 10.1016/j.bbadis.2011.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross CA, Tabrizi SJ. Huntington's disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 2011;10:83–98. doi: 10.1016/S1474-4422(10)70245-3. [DOI] [PubMed] [Google Scholar]
- Sarkar S, Rubinsztein DC. Huntington's disease: degradation of mutant huntingtin by autophagy. FEBS J. 2008;275:4263–4270. doi: 10.1111/j.1742-4658.2008.06562.x. [DOI] [PubMed] [Google Scholar]
- Schilling G, Becher MW, Sharp AH, Jinnah HA, Duan K, Kotzuk JA, Slunt HH, Ratovitski T, Cooper JK, Jenkins NA, Copeland NG, Price DL, Ross CA, Borchelt DR. Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of huntingtin. Hum Mol Genet. 1999;8:397–407. doi: 10.1093/hmg/8.3.397. [DOI] [PubMed] [Google Scholar]
- Sherman MY, Goldberg AL. Cellular defenses against unfolded proteins: a cell biologist thinks about neurodegenerative diseases. Neuron. 2001;29:15–32. doi: 10.1016/S0896-6273(01)00177-5. [DOI] [PubMed] [Google Scholar]
- Smith R, Bacos K, Fedele V, Soulet D, Walz HA, Obermüller S, Lindqvist A, Björkqvist M, Klein P, Onnerfjord P, Brundin P, Mulder H, Li JY. Mutant huntingtin interacts with beta-tubulin and disrupts vesicular transport and insulin secretion. Hum Mol Genet. 2009;18:3942–3954. doi: 10.1093/hmg/ddp336. [DOI] [PubMed] [Google Scholar]
- Trushina E, Dyer RB, Badger JD, 2nd, Ure D, Eide L, Tran DD, Vrieze BT, Legendre-Guillemin V, McPherson PS, Mandavilli BS, Van Houten B, Zeitlin S, McNiven M, Aebersold R, Hayden M, Parisi JE, Seeberg E, Dragatsis I, Doyle K, Bender A, et al. Mutant huntingtin impairs axonal trafficking in mammalian neurons in vivo and in vitro. Mol Cell Biol. 2004;24:8195–8209. doi: 10.1128/MCB.24.18.8195-8209.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsvetkov AS, Arrasate M, Barmada S, Ando DM, Sharma P, Shaby BA, Finkbeiner S. Proteostasis of polyglutamine varies among neurons and predicts neurodegeneration. Nat Chem Biol. 2013;9:586–592. doi: 10.1038/nchembio.1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tydlacka S, Wang CE, Wang X, Li S, Li XJ. Differential activities of the ubiquitin-proteasome system in neurons versus glia may account for the preferential accumulation of misfolded proteins in neurons. J Neurosci. 2008;28:13285–13295. doi: 10.1523/JNEUROSCI.4393-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpicelli-Daley LA, Gamble KL, Schultheiss CE, Riddle DM, West AB, Lee VM. Formation of α-synuclein Lewy neurite-like aggregates in axons impedes the transport of distinct endosomes. Mol Biol Cell. 2014;25:4010–4023. doi: 10.1091/mbc.E14-02-0741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waelter S, Boeddrich A, Lurz R, Scherzinger E, Lueder G, Lehrach H, Wanker EE. Accumulation of mutant huntingtin fragments in aggresome-like inclusion bodies as a result of insufficient protein degradation. Mol Biol Cell. 2001;12:1393–1407. doi: 10.1091/mbc.12.5.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CE, Zhou H, McGuire JR, Cerullo V, Lee B, Li SH, Li XJ. Suppression of neuropil aggregates and neurological symptoms by an intracellular antibody implicates the cytoplasmic toxicity of mutant huntingtin. J Cell Biol. 2008a;181:803–816. doi: 10.1083/jcb.200710158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Wang CE, Orr A, Tydlacka S, Li SH, Li XJ. Impaired ubiquitin-proteasome system activity in the synapses of Huntington's disease mice. J Cell Biol. 2008b;180:1177–1189. doi: 10.1083/jcb.200709080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong YC, Holzbaur EL. The regulation of autophagosome dynamics by huntingtin and HAP1 is disrupted by expression of mutant huntingtin, leading to defective cargo degradation. J Neurosci. 2014;34:1293–1305. doi: 10.1523/JNEUROSCI.1870-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Huang S, Song M, Wang CE, Yan S, Liu X, Gaertig MA, Yu SP, Li H, Li S, Li XJ. Synaptic mutant huntingtin inhibits synapsin-1 phosphorylation and causes neurological symptoms. J Cell Biol. 2013;202:1123–1138. doi: 10.1083/jcb.201303146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccato C, Valenza M, Cattaneo E. Molecular mechanisms and potential therapeutical targets in Huntington's disease. Physiol Rev. 2010;90:905–981. doi: 10.1152/physrev.00041.2009. [DOI] [PubMed] [Google Scholar]