

Abstract
Stress and glucocorticoids stimulate the rapid mobilization of endocannabinoids in the basolateral amygdala (BLA). Cannabinoid receptors in the BLA contribute to anxiogenesis and fear-memory formation. We tested for rapid glucocorticoid-induced endocannabinoid regulation of synaptic inhibition in the rat BLA. Glucocorticoid application to amygdala slices elicited a rapid, nonreversible suppression of spontaneous, but not evoked, GABAergic synaptic currents in BLA principal neurons; the effect was also seen with a membrane-impermeant glucocorticoid, but not with intracellular glucocorticoid application, implicating a membrane-associated glucocorticoid receptor. The glucocorticoid suppression of GABA currents was not blocked by antagonists of nuclear corticosteroid receptors, or by inhibitors of gene transcription or protein synthesis, but was blocked by inhibiting postsynaptic G-protein activity, suggesting a postsynaptic nongenomic steroid signaling mechanism that stimulates the release of a retrograde messenger. The rapid glucocorticoid-induced suppression of inhibition was prevented by blocking CB1 receptors and 2-arachidonoylglycerol (2-AG) synthesis, and it was mimicked and occluded by CB1 receptor agonists, indicating it was mediated by the retrograde release of the endocannabinoid 2-AG. The rapid glucocorticoid effect in BLA neurons in vitro was occluded by prior in vivo acute stress-induced, or prior in vitro glucocorticoid-induced, release of endocannabinoid. Acute stress also caused an increase in anxiety-like behavior that was attenuated by blocking CB1 receptor activation and inhibiting 2-AG synthesis in the BLA. Together, these findings suggest that acute stress causes a long-lasting suppression of synaptic inhibition in BLA neurons via a membrane glucocorticoid receptor-induced release of 2-AG at GABA synapses, which contributes to stress-induced anxiogenesis.
SIGNIFICANCE STATEMENT We provide a cellular mechanism in the basolateral amygdala (BLA) for the rapid stress regulation of anxiogenesis in rats. We demonstrate a nongenomic glucocorticoid induction of long-lasting suppression of synaptic inhibition that is mediated by retrograde endocannabinoid release at GABA synapses. The rapid glucocorticoid-induced endocannabinoid suppression of synaptic inhibition is initiated by a membrane-associated glucocorticoid receptor in BLA principal neurons. We show that acute stress increases anxiety-like behavior via an endocannabinoid-dependent mechanism centered in the BLA. The stress-induced endocannabinoid modulation of synaptic transmission in the BLA contributes, therefore, to the stress regulation of anxiety, and may play a role in anxiety disorders of the amygdala.
Keywords: amygdala, cannabinoid, CB1, corticosteroid, GABA, glucocorticoid, LTD
Introduction
Stress plays an important role in the regulation of emotion and emotional-memory formation, generally increasing anxiety-like behavior (Cecchi et al., 2002) and facilitating the consolidation of emotionally arousing memories (Roozendaal, 2000). The amygdala occupies a central role in the neural circuitry responsible for emotions and emotional-memory formation. It has been associated with both the consolidation and extinction of fear memories (Roozendaal et al., 2009; Isogawa et al., 2013), processes directly related to stress-associated disorders, such as post-traumatic stress disorder (PTSD; Goldstein et al., 1996; Parsons and Ressler, 2013). The basolateral complex of the amygdala (BLA), comprising the basal, lateral, and accessory basal nuclei, stands out as a hub in the integration of different types of emotionally salient sensory and cognitive inputs that contribute to the generation of emotion and emotional memories (McGaugh, 2004; Phelps and LeDoux, 2005; Isogawa et al., 2013).
Glucocorticoids are the product of stress activation of the hypothalamic–pituitary–adrenal (HPA) axis. Stress-induced glucocorticoids promote emotional memory consolidation acutely via actions in the BLA (Roozendaal et al., 1999; Isogawa et al., 2013). Glucocorticoid actions also contribute to anxiety-related behaviors via activation of forebrain glucocorticoid receptors (Tronche et al., 1999; Boyle et al., 2006; Hartmann et al., 2016). The stress-induced facilitation of fear-memory consolidation (Roozendaal et al., 2009) is thought to be mediated by a rapid, nongenomic glucocorticoid action at GABA synapses within the BLA (Roozendaal et al., 2002, 2006).
Type I cannabinoid (CB1) receptors are highly expressed by GABAergic interneurons that make inhibitory synapses onto principal neurons in the BLA, where they act to suppress GABA release (Katona et al., 2001). Low-frequency stimulation in the BLA elicits a long-lasting suppression of inhibitory synaptic inputs to principal neurons, which requires endocannabinoid activation of CB1 receptors (Marsicano et al., 2002). Stress causes a decrease in anandamide (AEA) and an increase in 2-arachidonoylglycerol (2-AG) content in the BLA, whereas direct glucocorticoid actions in the BLA elicit increases in both AEA and 2-AG (Patel et al., 2005; Hill and McEwen, 2009; Hill and Tasker, 2012). Like glucocorticoids, endocannabinoids enhance fear-memory formation via actions in the BLA (Campolongo et al., 2009). Recent findings indicate that the facilitatory effect of glucocorticoids on fear-memory consolidation depend on endocannabinoid actions in the BLA, as they are blocked by inhibiting BLA CB1 receptors (Campolongo et al., 2009; Atsak et al., 2015).
Our previous studies demonstrated a rapid glucocorticoid-induced synthesis and retrograde release of the endocannabinoid 2-AG at glutamate synapses in the hypothalamus (Di et al., 2003, 2009), which contributes to the rapid glucocorticoid feedback inhibition of the HPA axis (Evanson et al., 2010). The cellular mechanisms in the BLA responsible for the rapid stress-induced glucocorticoid modulation of emotional-memory formation are not known. Here, we tested the hypothesis that glucocorticoids modulate GABAergic synaptic inputs to BLA neurons via rapid retrograde endocannabinoid release triggered by membrane-associated glucocorticoid receptors, and that stress-induced anxiety-like behavior is dependent on endocannabinoid signaling in the BLA.
Materials and Methods
Animals.
For electrophysiology experiments, male Wistar rats (4–6 weeks old; Charles River) were group housed on a 12 h light/dark schedule and allowed to acclimate to their surroundings for ≥4 d before experiments, and were used in accordance with Public Health Service guidelines and a protocol approved by the Tulane University Institutional Animal Care and Use Committee.
For behavior experiments, male Wistar rats weighing 225–275 g at the time of arrival were housed in groups of two on a 12 h light/dark schedule (lights off at 8:00 P.M.) and fed a standard rat diet (Purina Rat Chow, Ralston Purina) ad libitum except during experimental procedures. Animal procedures were approved by the Institutional Animal Care and Use Committee of the Louisiana State University Health Sciences Center and were used in accordance with National Institute of Health guidelines. Rats were given 1 week to acclimate to their surroundings and were handled daily before initiation of surgical or experimental procedures.
Slice preparation.
The morning of experiments, rats were anesthetized with ketamine/xylazine (80/5 mg/kg body weight, i.p.) and perfused with ice-cold modified artificial CSF (aCSF) containing the following (in mm): 260 sucrose, 3 KCl, 6 MgSO4, 1.4 NaH2PO4, 1 CaCl2, 11 glucose, 5 HEPES, bubbled with 100% O2. The brain was quickly removed and 3–4 coronal slices (300 μm) containing the BLA were sectioned on a vibratome in the same ice-cold, modified aCSF. Slices were placed in oxygenated modified aCSF at 32–34°C (30 min) and then transferred to a standard HEPES-based aCSF at room temperature (25°C), where they were held until recordings. The standard aCSF contained the following (in mm): 140 NaCl, 3 KCl, 1.3 MgSO4, 1.4 NaH2PO4, 2.4 CaCl2, 11 glucose, and 5 HEPES; pH was adjusted to 7.2–7.3 with NaOH (Di et al., 2009). For some electrophysiology experiments, rats were subjected to restraint stress for 30 min in a plastic decapitation cone immediately before decapitation and slice preparation.
Brain-slice electrophysiology.
Neurons in the BLA were selected for recording by their pyramidal-shaped cell body under infrared-differential interference contrast optics and whole-cell patch-clamp recordings were performed on a fixed-stage upright microscope (Olympus BXW51WI) at 30–32°C. Patch pipettes were pulled on a horizontal electrode puller (P97, Sutter Instrument) with a tip resistance of 3–4 MΩ, and were filled with either a high-[Cl−] and high-[Cs+] internal solution [containing the following (in mm): 120 CsCl, 2 MgCl2, 1 CaCl2, 11 EGTA, 2 Mg-ATP, 0.3 Na-GTP, and 30 HEPES] or a “normal”-[Cl−] internal solution [containing the following (in mm): 120 K-gluconate, 10 KCl, 1 NaCl, 1 MgCl2, 1 CaCl2, 10 EGTA, 2 Mg-ATP, 0.3 Na-GTP, and 10 HEPES]. All recordings were performed in voltage clamp using a Multiclamp 700A amplifier and pCLAMP 9 software (Molecular Devices). Recordings with an unstable input resistance or series resistance (i.e., that changed >20%) were not included in our analyses. IPSCs were recorded with the high-[Cl−] internal solution at a holding potential of −60 mV in the presence of the ionotropic glutamate receptor antagonists DNQX (20 μm) and AP-5 (40 μm). EPSCs were recorded with the normal-[Cl−] internal solution at a holding potential of −60 mV in the presence of the GABAA receptor antagonist bicuculline methiodide (10 μm). Tetrodotoxin (1 μm) was added to the perfusion medium to record miniature IPSCs (mIPSCs) and EPSCs (mEPSCs). Evoked IPSCs (eIPSCs) were elicited by constant current stimulation (Grass Technologies) using a bipolar stimulating electrode (FHC) placed in the external capsule. Recordings were low-pass filtered at 2 kHz, digitized at 5 kHz, and stored on a computer hard drive for off-line analysis. Miniature synaptic currents were analyzed for changes in mean frequency, amplitude, and decay time (defined as the time elapsed from the peak of the mIPSC to 63% decay to baseline) using the Minianalysis 6.0 program (Synaptosoft). Means were calculated from 3 min epochs recorded in the final 3 min of baseline activity and the final 3 min of the 10 min drug applications (Fig. 1A). The mIPSC and mEPSC data are reported in the text as group averages of the means of the 3 min epochs; they are illustrated in the figures as group means of the percentage change from baseline calculated in each cell. Evoked synaptic currents were analyzed for changes in mean amplitude.
Figure 1.

Rapid glucocorticoid induction of a long-lasting suppression of synaptic inhibition in BLA neurons. A, Timeline of the whole-cell recording experimental paradigm. The last 3 min of baseline and glucocorticoid application were used for normalization and calculation of percentage changes. B, mIPSC recording in control medium and DEX (1 μm) in a BLA neuron. DEX elicited a decrease in the frequency of mIPSCs compared with baseline. C, Mean values of mIPSC frequency, amplitude, and decay time with respect to baseline values (dashed line) following DEX (1 μm) and CORT (1 μm) application. D, Time course in a representative BLA neuron of relative changes in mIPSC frequency elicited by two successive applications of DEX. Each data point represents the mIPSC frequency over a 1 min period relative to baseline, designated by the dashed line. E, Concentration–response curves of the CORT-induced and DEX-induced decreases in mIPSC frequency relative to the maximal response (EC50: CORT, 350 nm; DEX, 384 nm). F, The mean amplitudes of eIPSCs (eIPSC Amp) recorded before (Basal) and after DEX (1 μm) application (DEX). **p < 0.01; numbers in parentheses in bar graphs represent numbers of BLA neurons included in each group.
Drug application.
Drugs used in brain-slice experiments included the following steroid ligands: water-soluble dexamethasone [(2-hydroxypropyl)-β-cyclodextrin-conjugated dexamethasone (DEX)] and corticosterone [(2-hydroxypropyl)-β-cyclodextrin-conjugated corticosterone (CORT)], DEX-bovine serum albumin (BSA) conjugate (Steraloids), spironolactone, and mifepristone (RU486); the following cannabinoid ligands: WIN55,212-2, AEA, and 2-AG (Tocris Bioscience); SR141716 (SR; National Institute of Mental Health Chemical Synthesis and Drug Supply Program); the nonhydrolyzable guanylyl nucleotide GDP-β-S; the selective inhibitor of eukaryotic protein synthesis cycloheximide (Tocris Bioscience); the gene transcription inhibitor actinomycin D (Tocris Bioscience); and the 11-β hydroxysteroid dehydrogenase type 1 inhibitor 2-methyl-1,2-di-3-pyridyl-1-propanone (metyrapone). All drugs, unless specified, were purchased from Sigma-Aldrich.
Intracranial injections.
Rats were implanted bilaterally with cannulae aimed at the BLA (coordinates: −2.1 mm anteroposterior, ±5.0 mm mediolateral, −8.0 mm dorsoventral) and allowed to recover for 5–7 d. They were then habituated to the intracerebral injection procedure by sham infusions for 4 consecutive days consisting of inserting injectors into cannulae without drug injection. On the day of testing, pairs of rats were removed from their home cages simultaneously during the dark cycle. Each rat was bilaterally infused in the BLA with either the CB1R antagonist SR (1 μg/0.5 μl/side) or vehicle (18:1:1 saline/ethanol/cremaphor solution) at a rate of 0.2 μl/min for 2.5 min, and injectors remained in the cannulae for 1 min after infusion to allow for diffusion. All rats were then returned to their home cage.
Restraint stress.
For electrophysiological experiments, rats were acutely stressed by placement in a rodent plastic decapitation cone (Kent Scientific) that was open-ended at the rostral end and closed caudally around the tail with a tie. They were immobilized for 30 min starting at 8:30–9:00 A.M., after which they were immediately killed by decapitation at 9:00–9:30 A.M. Control rats were cage mates of the stressed rats that were not subjected to restraint stress, but were also decapitated at ∼9:00 A.M. Some rats received metyrapone or vehicle injections subcutaneously 90 min before the start of the restraint stress.
Behavioral testing.
Control rats remained in their home cage for 45 min after intracranial drug infusion before being tested for anxiety-like behavior using the open-field test [black Plexiglas with grid floor; 72 cm (length) × 72 cm (width) × 36 cm (height)] or the elevated plus maze (four arms 10 cm wide × 50 cm long at right angles, elevated 60 cm from the floor; two arms with 40-cm-high walls and two arms with no walls). Rats in the stress group were removed from their home cage 15 min after infusion and were placed in clear Plexiglas restraint tubes for 30 min. Plexiglas restraint tubes were used in lieu of decapitation cones for the restraint stress here because the plastic tubes were less likely to dislodge the dental cement skull caps holding the intracranial cannulae. Following restraint (i.e., 45 min after infusion), rats were immediately tested in the open field or elevated plus maze for 300 s, then returned to their home cage. Unstressed control rats were tested at a parallel time point 45 min after infusion. Open-field and elevated plus-maze behavior was video recorded and scored later by an observer blind to the stress and drug-treatment conditions. Open-field tests were scored for time spent in the center versus periphery and for the number of center line crossings. Elevated plus-maze videos were scored for time spent in the open versus closed arms and for open-arm and closed-arm entries. Several days after testing, rats were bilaterally injected in the BLA with 0.5 μl of Evans blue dye under anesthesia and killed. Brains were then removed and flash frozen in 2-methylbutane. Brains were sectioned at 50 μm on a cryostat and stained with cresyl violet to determine the injection sites. Only rats with bilaterally accurate injection sites in the BLA were included in data analyses.
CORT assay.
Rats were decapitated and trunk blood was collected in Eppendorf tubes and allowed to clot at room temperature for 90 min. Blood samples were then centrifuged at 2000 × g for 15 min at room temperature and the serum fractions were carefully transferred to new Eppendorf tubes and stored at −20°C until analysis of CORT concentration. Radioimmunoassay of serum samples was performed by the Ligand Assay and Analysis Core Laboratory, Center for Research in Reproduction, University of Virginia [National Institute of Child Health and Human Development (Specialized Cooperative Centers Program in Reproductive and Infertility) Research Grant U54-HD28934].
Data analysis.
All data are expressed as means ± SEs. Statistical comparisons of electrophysiological data were performed using the Student's paired t test for within-group comparisons and the Student's unpaired t test for between-group comparisons. One-way repeated-measures ANOVAs were followed by post hoc pairwise comparisons with the Student–Newman–Keuls pairwise test where justified. Behavioral data were analyzed with two-way ANOVA with two between-subjects factors (stress × drug), followed by post hoc pairwise comparisons with the Student–Newman–Keuls test where justified. Non-normal data were square-root transformed before analysis. Statistical significance was set at p < 0.05 in all cases.
Results
Rapid glucocorticoid induction of a long-lasting suppression of synaptic inhibition in BLA neurons
We tested for a rapid glucocorticoid modulation of GABA-mediated mIPSCs and eIPSCs in BLA neurons. In BLA neurons recorded in amygdala slices from 12 rats, glucocorticoid applied in the perfusion bath had no effect on the membrane holding current or input conductance, but suppressed mIPSCs beginning within 3–5 min and maximally within 5–7 min in 19 of 22 neurons recorded (86%). The synthetic glucocorticoid DEX (1 μm) induced a rapid decrease in the frequency of mIPSCs (from 1.4 ± 0.2 to 1.0 ± 0.1 Hz; p < 0.01; n = 12), but not the mIPSC amplitude (28.1 ± 3.4 vs 30.2 ± 4.6 pA; p = 0.4) or decay time (12.4 ± 1.0 vs 12.3 ± 1.2 ms; p = 0.9; Fig. 1A–D). The endogenous glucocorticoid CORT (1 μm) induced a similar rapid decrease in the mIPSC frequency (from 1.7 ± 0.2 to 1.4 ± 0.2 Hz; p < 0.01; n = 10), but had no effect on either the mIPSC amplitude (30.3 ± 2.7 vs 29.8 ± 2.3 pA; p = 0.43) or decay time (13.0 ± 1.3 vs 13.2 ± 1.4 ms; p = 0.58; Fig. 1C). This suggested a presynaptic site of modulation of GABA synapses by the glucocorticoids. The glucocorticoid effect was not reversible with 40–60 min of washout of the drug, and a second glucocorticoid application had no further effect on mIPSC frequency (Fig. 1D), indicating a long-lasting action of the glucocorticoid that occludes a subsequent response. Dose–response analyses of the rapid glucocorticoid response showed a threshold concentration of ∼10 nm for both CORT and DEX and a half-effective concentration (EC50) of 350 nm for CORT and 384 nm for DEX (Fig. 1E). These findings revealed, therefore, a rapid, nonreversible suppression of spontaneous inhibitory synaptic currents in BLA neurons by relatively high glucocorticoid concentrations (i.e., physiological stress levels). DEX (1 μm) had no effect on the amplitude of eIPSCs (Fig. 1F), suggesting that evoked GABA release was insensitive to the rapid glucocorticoid action, and that glucocorticoids modulate the spontaneous, but not the evoked, pool of GABA vesicles.
The glucocorticoid suppression of inhibition is mediated by a nongenomic mechanism
The rapid nature of the glucocorticoid modulation of GABAergic synaptic inputs to BLA neurons suggested that it was mediated by a membrane receptor-induced, nongenomic steroid action. To test this, we first determined whether the membrane–impermeant glucocorticoid conjugate DEX-BSA would retain the rapid suppressive effect of glucocorticoid on the mIPSC frequency. Application of DEX-BSA (5 μm) in the bath also decreased the mIPSC frequency (from 1.7 ± 0.2 to 1.2 ± 0.3 Hz, p < 0.01; n = 6; Fig. 2B), beginning within 3–5 min of exposure. This suggested a site of action of the glucocorticoid at the membrane instead of a site in the cytoplasm or nucleus. The membrane locus of the glucocorticoid receptor was supported by DEX (1 μm) application directly into the cytoplasm of the recorded cell via the patch pipette, which failed to elicit a significant change in mIPSC frequency (1.4 ± 0.2 vs 1.3 ± 0.2 Hz; p = 0.21, n = 6; Fig. 2B). We next tested whether the rapid glucocorticoid suppression of mIPSCs is sensitive to blockade of either the classical mineralocorticoid receptor with spironolactone (Young et al., 1998) or the classical glucocorticoid receptor with RU 486 (Agarwal et al., 1987). Blocking mineralocorticoid receptors with spironolactone (10 μm, n = 6) or glucocorticoid receptors with RU 486 (10 μm, n = 6) failed to inhibit the decrease in mIPSC frequency elicited by subsequent coapplication of DEX (1 μm; Fig. 2C). Together, these results suggest that the glucocorticoid suppression of inhibitory synaptic inputs to BLA neurons is not mediated by the intracellular mineralocorticoid or glucocorticoid receptor, but by activation of a membrane-associated receptor.
Figure 2.

The glucocorticoid effect was mediated by a membrane-initiated, nongenomic, G-protein-dependent signaling mechanism. A, The timeline of the whole-cell recording paradigm used for data presented in C and D, except that the antagonists in D were applied intracellularly via the patch pipette throughout the recordings. The timeline used for data presented in B was the same as that illustrated in Figure 1A. B, Bath application of the membrane-impermeant DEX-BSA conjugate maintained the steroid's suppressive effect on mIPSC frequency, whereas direct intracellular application of DEX via the patch pipette had no effect on mIPSC frequency. C, The DEX suppression of mIPSC frequency was not blocked by prior application of antagonists of the nuclear mineralocorticoid receptor spironolactone (Spiro+DEX) or the nuclear glucocorticoid receptor RU 486 (RU 486+DEX). The antagonists alone were without effect on the relative mIPSC frequency (Spiro and RU 486). D, Blockade of gene transcription and protein synthesis with intracellular application of actinomycin D (Act D) and cycloheximide (CHX), respectively, via the patch pipette did not block the DEX suppression of mIPSC frequency. Blockade of postsynaptic G-protein activity with intracellular application of GDP-β-S via the patch pipette abolished the decrease in mIPSC frequency induced by DEX. E, Time course of mean relative changes in mIPSC frequency elicited by two successive applications of DEX in the absence and presence of the protein synthesis inhibitor cycloheximide in the bath. Inhibiting protein synthesis did not block the maintenance of the glucocorticoid response, nor did it block the occlusion of the response to a second glucocorticoid application. *p < 0.05; **p < 0.01.
Next, to test whether the glucocorticoid effect on BLA neurons was mediated by a nongenomic mechanism, we applied via the patch pipette the gene transcription inhibitor actinomycin D and the protein synthesis inhibitor cycloheximide directly into the cytoplasm of the recorded cells. The rapid DEX (1 μm) reduction in mIPSC frequency was insensitive to gene transcription blockade with actinomycin D (10 μm; 1.9 ± 0.3 to 1.5 ± 0.2 Hz, p < 0.05, n = 5) and to blockade of protein synthesis with cycloheximide (10 μm; 1.8 ± 0.5 to 1.4 ± 0.4 Hz, p < 0.05, n = 5; Fig. 2D). Because the rapid glucocorticoid effect was mediated, therefore, by a membrane receptor-associated nongenomic signaling mechanism, we next tested for the postsynaptic G-protein dependence of the response with the intracellular application via the patch pipette of the G-protein blocker guanosine 5′-[β-thio]diphosphate (GDP-β-S; 0.5–1 mm). Postsynaptic GDP-β-S application (10 min) blocked the DEX-induced decrease in mIPSC frequency (1.6 ± 0.2 vs 1.7 ± 0.3 Hz, p = 0.25, n = 6; Fig. 2D), indicating that the rapid glucocorticoid suppression of GABAergic synaptic inputs to BLA neurons is mediated by a postsynaptic G-protein signaling mechanism, and implicating a retrograde messenger in the presynaptic glucocorticoid modulation of GABA release. Finally, we tested for the dependence of the long-term maintenance of the rapid glucocorticoid response on protein synthesis. The response to DEX (1 μm) did not reverse after >30 min of washout of the steroid with intracellular infusion of the protein synthesis inhibitor cycloheximide (10 μm; Fig. 2E). Therefore, like the induction of the glucocorticoid suppression of synaptic inhibition, the maintenance is not mediated by a protein synthesis-dependent mechanism. A second bath application of DEX (1 μm) with intracellular cycloheximide infusion also had no effect on mIPSC frequency (Fig. 2D), indicating that blocking protein translation did not prevent the occlusion of the second response.
The glucocorticoid suppression of inhibition is mediated by a retrograde endocannabinoid messenger
That glucocorticoids decrease the mean mIPSC frequency without affecting either the mIPSC amplitude or decay kinetics suggests a presynaptic site of action, while the postsynaptic G-protein dependence suggests a postsynaptic G-protein signaling mechanism. In hypothalamic neuroendocrine cells, glucocorticoids rapidly suppress glutamatergic synaptic inputs by the retrograde release of the endocannabinoid 2-AG (Di et al., 2003, 2005, 2013). We tested, therefore, for the endocannabinoid dependence of the glucocorticoid-induced suppression of GABA release onto BLA neurons. Bath application of the CB1 receptor inverse agonist/antagonist SR (1 μm) blocked the decrease in mIPSC frequency caused by DEX (1 μm; 1.9 ± 0.3 Hz in DEX + SR vs 1.8 ± 0.2 Hz in SR; p = 0.72, n = 5; Fig. 3B). The DEX-induced decrease in mIPSC frequency was also blocked by tetrahydrolipstatin (THL, 10 μm), the selective inhibitor of the 2-AG synthetic enzyme diacylglycerol lipase (1.6 ± 0.2 Hz in THL vs 1.7 ± 0.3 Hz in DEX + THL; p = 0.50, n = 7; Fig. 3B). This suggests that the rapid glucocorticoid modulation of GABA synapses on BLA neurons is mediated by presynaptic CB1 activation via the retrograde release of 2-AG. No pharmacological inhibitor of AEA metabolism is currently available, so we were not able to eliminate entirely the possibility that AEA may contribute to the glucocorticoid-induced retrograde endocannabinoid signal; however, the complete block of the DEX-induced decrease in mIPSC frequency by THL suggests that the effect was mediated by 2-AG. The endocannabinoid signal was further supported by the observations that the DEX-induced decrease in mIPSC frequency was mimicked and occluded by bath application of high concentrations of the CB1 receptor agonists AEA (1 μm) and 2-AG (1 μm; Fig. 3C). To confirm the presynaptic site of the CB1 receptors, a CB1 receptor agonist was applied in the bath following postsynaptic blockade of the rapid glucocorticoid effect with the intracellular application of the G-protein antagonist GDP-β-S. Following bath application of DEX (1 μm), which was blocked with the postsynaptic G-protein blocker in the patch pipette, bath application of the high-affinity synthetic CB1 receptor agonist WIN55,212-2 (1 μm) caused a decrease in mIPSC frequency (1.9 ± 0.4 to 1.4 ± 0.3 Hz, p < 0.05; n = 5; Fig. 3C). The nongenomic nature of the sustained response to glucocorticoid and the fact that there was no response to a second application of glucocorticoid suggested that the maintenance of the glucocorticoid response may be mediated by the activation of a continuous release of endocannabinoid. We tested this by blocking CB1 receptors subsequent to the glucocorticoid response on mIPSCs. A 10 min application of SR (1 μm) reversed the DEX-induced decrease in the frequency of mIPSCs (Fig. 3E). This demonstrated that the sustained suppression of GABA synaptic inputs to the BLA neurons is caused by a glucocorticoid-induced continuous release of endocannabinoid. Together, these findings suggest that glucocorticoids trigger a rapid, sustained suppression of inhibitory inputs to BLA neurons by switching on a continuous retrograde release of 2-AG at GABA synapses.
Figure 3.

The rapid glucocorticoid suppression of synaptic inhibition is mediated by activation of the release of a retrograde endocannabinoid messenger. A, Timeline of the experimental paradigm that produced the data shown in B and C. B, The CB1 receptor antagonist SR and the inhibitor of 2-AG synthesis THL blocked the DEX-induced decrease in mIPSC frequency. C, The endocannabinoids 2-AG and AEA mimicked (AEA and 2-AG) and occluded (AEA+DEX and 2-AG+DEX) the rapid DEX-induced suppression of mIPSC frequency. Blocking postsynaptic G-protein activity with intracellular GDP-β-S application blocked the postsynaptic DEX-induced decrease in mIPSC frequency (DEX), but not that caused by presynaptic CB1 receptor activation with bath application of WIN55,212-2 (DEX+WIN). D, Time course of the mean relative changes in mIPSC frequency elicited by a single 10 min application of DEX (filled circles), and by a 10 min application of DEX followed by a 10 min application of the CB1 receptor antagonist SR (open circles). Blocking CB1 receptors reversed the glucocorticoid-induced suppression of mIPSC frequency. *p < 0.05; **p < 0.01.
Stress-induced endocannabinoid release and sustained suppression of inhibition
The previous experiments showed that a 10 min application of a relatively high glucocorticoid concentration induces a long-term suppression of synaptic inhibition in BLA neurons by triggering a continuous release of endocannabinoid at GABA synapses. We next tested whether acute stress-induced CORT secretion in vivo would have a similar long-lasting suppressive effect on synaptic inhibition in BLA neurons that could be detected subsequently in recordings in brain slices. We conducted whole-cell recordings in BLA neurons in amygdala slices from rats subjected to a 30 min restraint stress before being killed (Fig. 4A). Restraint stress increased the morning plasma CORT from a control level of 18.6 ± 3.5 ng/ml (n = 14) to 684.4 ± 49.7 ng/ml (n = 16; p < 0.01; Fig. 4B). In slices from unstressed rats, bath application of DEX (1 μm, 10 min) caused a reduction in mIPSC frequency in BLA neurons (from 1.3 ± 0.2 to 1.0 ± 0.2 Hz; p < 0.01, n = 5; Fig. 4C), as observed in the previous experiments. In contrast, in slices from stressed rats, DEX (1 μm) had no effect on mIPSC frequency (baseline: 1.8 ± 0.3 Hz; DEX: 1.7 ± 0.3 Hz; p = 0.63, n = 5; Fig. 4C). To determine whether the loss of the DEX-induced suppression of GABA inputs to BLA neurons following in vivo restraint stress was caused by endocannabinoid occlusion of the DEX effect, we tested for differences in CB1 receptor occupation at GABA synapses on BLA neurons in the slices from unstressed versus stressed rats. Bath application of the endocannabinoid 2-AG (1 μm) caused a decrease in mIPSC frequency in BLA neurons from unstressed rats (from 1.6 ± 0.4 to 1.2 ± 0.3 Hz; p < 0.05, n = 5), but this effect was lost in BLA neurons from stressed rats (baseline: 1.2 ± 0.4 Hz; 2-AG: 1.2 ± 0.3 Hz; p = 0.48, n = 9; Fig. 4C). Bath application of the CB1 receptor antagonist SR (1 μm) alone had no effect on mIPSC frequency in BLA neurons from unstressed rats (baseline: 1.7 ± 0.3 Hz; SR: 1.8 ± 0.3 Hz; p = 0.46, n = 6), but it caused a significant increase in the mIPSC frequency in BLA neurons from stressed rats (from 1.4 ± 0.2 to 1.7 ± 0.3 Hz; p < 0.05, n = 6; Fig. 4C). These findings suggested that the CB1 receptors at GABA synapses on BLA neurons were occupied by endogenously released cannabinoid in slices from stressed rats, but not in slices from unstressed control rats.
Figure 4.
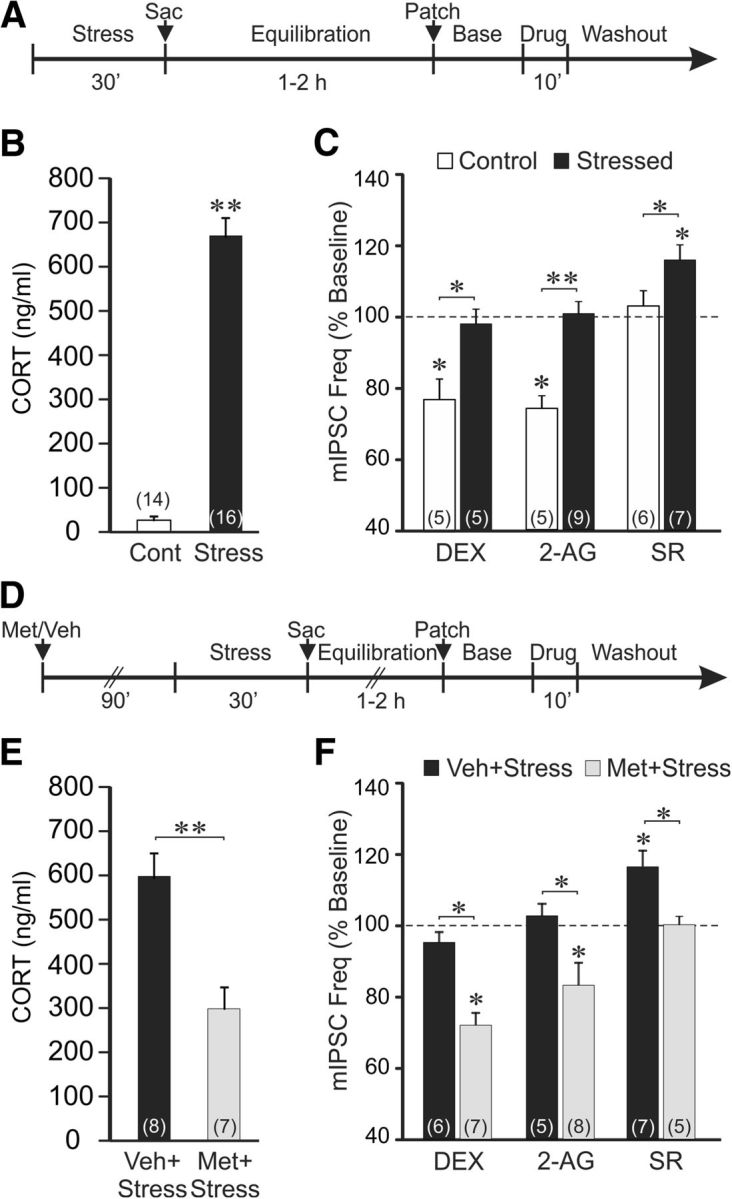
Acute stress causes a long-lasting suppression of synaptic inhibition in BLA neurons via glucocorticoid-dependent endocannabinoid activation of CB1 receptors. A, Timeline of experimental paradigm used to generate data presented in C. B, Trunk blood serum CORT levels measured in control (Cont) and stressed rats (Stress). Exposure to a 30 min restraint stress increased the serum CORT level. C, Effects of DEX and CB1 receptor ligands on mean normalized mIPSC frequency in BLA neurons from unstressed (white bars, Control) and stressed rats (black bars, Stressed). DEX and 2-AG both caused decreases in mIPSC frequency in BLA neurons from unstressed control rats, but had no effect on mIPSC frequency in BLA neurons from stressed rats. Blocking CB1 receptors with SR had no effect on mIPSC frequency in BLA neurons from unstressed control rats, but caused an increase in mIPSC frequency in BLA neurons from stressed rats. These findings suggested CB1 receptor occupation at GABA synapses following stress. D, Timeline of experimental paradigm used to generate data presented in F. E, Trunk blood serum CORT levels measured in restraint-stressed animals injected intraperitoneally with vehicle or the glucocorticoid synthesis inhibitor metyrapone. Compared with vehicle-injected rats (Veh+Stress), metyrapone pretreatment reduced the acute stress-induced circulating CORT (Met+Stress) by ∼50%. F, Effects of DEX and CB1 receptor ligands on mean normalized mIPSC frequency in BLA neurons from vehicle-injected stressed rats (black bars, Veh+Stress) and metyrapone-injected stressed rats (gray bars, Met+Stress). As in the uninjected stressed rats, BLA neurons from vehicle-injected stressed rats showed no effect of DEX and 2-AG on mIPSC frequency, and an increase in mIPSC frequency with SR. Metyrapone suppression of stress-induced glucocorticoid synthesis restored the DEX-induced and 2-AG-induced decreases in mIPSC frequency, and abolished the SR-induced increase in mIPSC frequency. This suggested the CB1 receptor occupation at GABA synapses following stress was prevented by reducing stress-induced CORT synthesis. *p < 0.05; **p < 0.01.
To determine whether the endocannabinoid occupation of CB1 receptors at GABA synapses on BLA neurons in slices from stressed rats was dependent on glucocorticoids released during the acute stress, we tested the effect of blockade of CORT synthesis during the stress exposure with the 11-β-HSD inhibitor metyrapone (Roozendaal et al., 1996; Blundell et al., 2011). Metyrapone applied intraperitoneally (50 mg/kg) 90 min before the start of the restraint stress (Fig. 4D) resulted in a ∼50% decrease in serum CORT compared with the vehicle control group (vehicle: 597.4 ± 54.5 ng/ml; metyrapone: 297.4 ± 47.9 ng/ml; n = 7, 8, p < 0.01; Fig. 4E). In BLA neurons from stressed rats pretreated with metyrapone, but not vehicle, the DEX-induced decrease in mIPSC frequency was restored (from 1.6 ± 0.3 to 1.2 ± 0.2 Hz; p < 0.05, n = 6; Fig. 4F). In BLA neurons from stressed rats treated with metyrapone, but not vehicle, the 2-AG-induced decrease in mIPSC frequency was also restored (from 1.5 ± 0.3 to 1.2 ± 0.2 Hz; p < 0.05, n = 8) and the SR-induced increase in mIPSC frequency was lost (baseline: 1.6 ± 0.4 Hz; SR: 1.6 ± 0.5 Hz; p = 0.47, n = 5; Fig. 4F). These findings together suggest that acute restraint stress in vivo leads to a glucocorticoid-induced sustained release of endocannabinoid and a continuous occupation of CB1 receptors at GABA synapses that causes the suppression of inhibitory synaptic inputs to BLA neurons for up to several hours in vitro. Consistent with this, we found that mIPSCs recorded in BLA principal neurons in slices from stressed rats showed a trend toward a lower baseline frequency than mIPSCs recorded in slices from unstressed rats (unstressed: 1.7 ± 0.2 Hz, n = 62; stressed: 1.3 ± 0.2 Hz, n = 21; p = 0.06).
DEX (1 μm) had no effect on the amplitude of eIPSCs recorded in BLA neurons in slices from stressed rats (p = 0.65, n = 16). The CB1 receptor antagonist SR (1 μm) also had no effect on the amplitude of eIPSCs in slices from stressed rats (p = 0.5, n = 10), further suggesting that the sustained endocannabinoid release elicited by stress-induced glucocorticoid actions did not modulate evoked GABA responses.
Stress causes anxiety-like behavior via endocannabinoid release in the BLA
A sustained suppression of synaptic inhibition in BLA neurons should shift the excitation–inhibition balance in the BLA toward excitation, which could alter the anxiogenic output of the BLA. We tested this hypothesis using the open-field and elevated plus-maze tests of anxiety-like behavior in restraint-stressed and unstressed rats that received bilateral injections in the BLA of vehicle or antagonists of endocannabinoid signaling.
Rats were injected in the BLA with the CB1 receptor inverse agonist/antagonist SR (1 μg/0.5 μl) or vehicle 15 min before restraint stress, or no stress, and were then placed in the open field 5 min later to test for anxiety-like behavior (i.e., time in center) and locomotor activity (i.e., line crossings). A two-way ANOVA revealed a main effect of stress (F(1,32) = 15.5, p < 0.001), no main effect of drug (F(1,32) = 0.34, p = 0.86), and a significant stress × drug interaction (F(1,32) = 33.7, p = 0.001) on time in center of the open field (Fig. 5A). Restraint stress reduced time spent in the center of the open field (i.e., increased anxiety-like behavior) in rats that received intra-BLA vehicle injections (p < 0.05), but not in rats that received intra-BLA injections of the CB1 antagonist SR. Blocking CB1 receptors with SR injection in control rats reduced time in the center of the open field (p < 0.05; i.e., increased anxiety-like behavior), which revealed an endocannabinoid tone in the BLA that was anxiolytic (Fig. 5A). We also analyzed line crossings in the open field after intra-BLA infusions of SR. A two-way ANOVA revealed a main effect of stress (F(1,32) = 36.6, p < 0.001), but no effect of drug (p > 0.05), and no stress × drug interaction effect (p > 0.05) on line crossings in the open field.
Figure 5.

Stress increases anxiety-like behavior via 2-AG signaling in the BLA. A, Percentage of total time (5 min) spent in the center of the open field by rats pretreated with either vehicle (Veh) or the CB1 receptor antagonist SR in the BLA and exposed to restraint stress (30 min; Stress) or returned to the home cage (Control). Stress reduced the percentage of time spent in the center, indicating an increase in anxiety-like behavior, in rats that received bilateral vehicle injections in the BLA, but not injections of SR. Intra-BLA blockade of CB1 receptors in unstressed control rats led to a decrease in percentage time spent in the center, indicating an anxiolytic tone exerted by CB1 receptor activation in the BLA. B, Percentage of total time (5 min) spent in the center of the open field by rats pretreated with either vehicle (Veh) or the 2-AG synthesis inhibitor THL in the BLA and exposed to restraint stress (30 min; Stress) or returned to the home cage (Control). Stress reduced the percentage of time spent in the center in rats that received bilateral vehicle injections in the BLA, and this effect was abolished by intra-BLA blockade of 2-AG synthesis. Intra-BLA blockade of 2-AG synthesis had no effect on center time in unstressed control rats, suggesting that the anxiogenic effect of blocking CB1 receptors was not dependent on 2-AG synthesis. C, Percentage of total time (5 min) spent in the open arms of the elevated plus maze by rats pretreated with either vehicle (Veh) or THL in the BLA and exposed to restraint stress (30 min; Stress) or returned to the home cage (Control). D, Number of entries into the open arms of the elevated plus maze by rats pretreated with vehicle (Veh) or THL in the BLA and undergoing stress or no stress (Control). The open-arm time and open-arm entries in the elevated plus maze revealed a similar pattern to that observed in the open field, but did not show a significant effect of drug or stress. **p < 0.01 vs Control/Veh; #p < 0.05 vs Stress/Veh.
We next tested the effect of the 2-AG synthesis inhibitor THL on stress-induced anxiety-like behavior in the open field. Rats received bilateral intra-BLA THL (10 μg/0.5 μl) injections 15 min before placement in the restraint tube. A two-way ANOVA revealed a main effect of stress (F(1,27) = 55.7, p < 0.001), a main effect of drug (F(1,27) = 7.5, p = 0.011), and a significant stress × drug interaction (F(1,27) = 9.02, p = 0.006) on time in the center of the open field (Fig. 5B). Restraint stress reduced time spent in the center of the open field (i.e., increased anxiety-like behavior) in rats that received intra-BLA vehicle injections (p < 0.05), but not in rats that received intra-BLA injections of THL. Intra-BLA THL significantly rescued stress-induced anxiety-like behavior (p < 0.05), but unlike the CB1 receptor antagonist, intra-BLA THL infusion had no effect on anxiety-like behavior in stress-naive controls (Fig. 5B). This indicates that the endocannabinoid anxiolytic tone revealed with CB1 receptor blockade (Fig. 5A) is not mediated by 2-AG release, and may be due, therefore, to the tonic release of AEA or another endocannabinoid. We also analyzed line crossings in the open field after intra-BLA infusions of THL. A two-way ANOVA revealed a main effect of stress (F(1,27) = 17.2, p < 0.001), but no effect of drug (p > 0.05), and no stress × drug interaction effect (p > 0.05) on line crossings in the open field.
We next tested the effect of the 2-AG synthesis inhibitor THL on stress-induced anxiety-like behavior in the elevated plus maze. Rats received bilateral intra-BLA THL (10 μg/0.5 μl) injections 15 min before placement in the restraint tube. A two-way ANOVA revealed no main effect of stress or drug, and no stress × drug interaction effect (p > 0.05 in all cases) on time spent in the open arms or open-arm entries in the elevated plus maze (Fig. 5C,D). There was also no main effect of stress or drug, and no stress × drug interaction effect (p > 0.05) on closed-arm entries in the elevated plus maze, indicating no effect on locomotor activity.
Discussion
Our findings demonstrate a long-lasting suppression of spontaneous inhibitory synaptic inputs to BLA principal neurons via retrograde endocannabinoid release stimulated by rapid stress-induced glucocorticoid signaling. The rapid glucocorticoid actions were nongenomic and mediated by the activation of a membrane-associated receptor, and were long lasting due to the sustained release of endocannabinoid, which occluded any further glucocorticoid suppression of synaptic inhibition. Glucocorticoids were without effect on evoked inhibitory synaptic inputs to the BLA neurons, suggesting that the glucocorticoid-induced endocannabinoid release modulates spontaneous, but not evoked, release of GABA at inhibitory synapses on BLA principal neurons.
The glucocorticoid suppression of basal inhibition was triggered by a membrane-associated glucocorticoid receptor. The decrease in mIPSCs was rapid in onset, was seen with a membrane-impermeant glucocorticoid, but not with intracellular glucocorticoid application, and was insensitive to antagonists of nuclear glucocorticoid and mineralocorticoid receptors and to inhibitors of gene transcription and protein synthesis. However, it was blocked by inhibiting postsynaptic G-protein signaling, implicating a postsynaptic nongenomic, membrane-associated receptor and G-protein signaling mechanism like that seen at glutamate synapses on hypothalamic neuroendocrine (Di et al., 2003, 2009) and presympathetic neurons (Boychuk et al., 2013). Duvarci and Paré (2007) reported a suppression of evoked IPSCs in BLA neurons with a concentration of CORT (100 nm) that was near-threshold in our analysis. However, the glucocorticoid effect in that study required a 1–2 h delay, suggesting a delayed transcriptional action of the steroid. Consistent with our data showing lack of rapid CORT effect on evoked GABA release, Patel et al. (2009) found that a single restraint stress failed to alter depolarization-induced endocannabinoid release at GABA synapses, suggesting no effect of stress-induced endocannabinoid release on evoked IPSCs in BLA principal neurons.
This mechanism of rapid glucocorticoid modulation of inhibitory synapses differs from the rapid corticosteroid modulation of excitatory synapses in the BLA, where CORT induced a rapid increase in glutamate release via a presynaptic mineralocorticoid receptor-dependent mechanism (Karst et al., 2010). The CORT facilitation of glutamate release was also nonreversible, but unlike the suppression of GABA release, the sustained facilitation of glutamate release was transcription-dependent, suggesting a genomic mechanism (Karst et al., 2010). The mineralocorticoid receptor dependence of the rapid corticosteroid facilitation of glutamate release in the BLA and hippocampus (Karst et al., 2005, 2010) suggests that multiple membrane-associated corticosteroid receptors mediate the rapid corticosteroid effects at different synapses in the brain (Di et al., 2013; Tasker and Joëls, 2015).
The long-lasting glucocorticoid suppression of spontaneous synaptic inhibition was mediated by the retrograde release of the endocannabinoid 2-AG, since the effect was blocked by a CB1 receptor antagonist and by a selective inhibitor of the 2-AG synthetic enzyme diacylglycerol lipase. CB1 receptors are highly expressed in the BLA, and are concentrated at presynaptic GABAergic terminals that synapse onto BLA principal neurons (Katona et al., 2001). The glucocorticoid effect was nonreversible, its maintenance did not require protein synthesis, and a second glucocorticoid application, or glucocorticoid application following prior in vivo acute stress, was without effect on GABA synaptic currents. The maintenance of the glucocorticoid-induced suppression of inhibition was due to a sustained release of 2-AG and occupation of CB1 receptors because (1) a CB1 receptor antagonist applied following the glucocorticoid application reversed the steroid effect, and (2) CB1 receptors were occupied by glucocorticoid-dependent endocannabinoid release in slices from stressed, but not unstressed, rats. The endocannabinoid release following restraint stress in vivo was dependent on glucocorticoid secretion, since it was reversed when glucocorticoid synthesis was inhibited with metyrapone. That a 50% reduction, and not a complete block, of the stress-induced CORT secretion (i.e., from ∼600 to ∼300 ng/ml) was sufficient to reverse the effect of stress on the in vitro glucocorticoid response is consistent with the high concentration of CORT required for the rapid effect in BLA neurons. This suggests that the normal circadian rhythm of CORT secretion in the blood may not have the same effect, since it would not be expected to reach the level we measured following stress in the presence of metyrapone.
The glucocorticoid suppression of inhibition was long-lasting. Thus, restraint stress in vivo, and glucocorticoid application in vitro, induced a sustained release of endocannabinoid and suppression of synaptic inhibition in BLA neurons for hours. This implies a glucocorticoid-induced molecular mechanism that serves as a switch that turns on endocannabinoid production at GABA synapses. It remains to be determined how the glucocorticoid-induced endocannabinoid synthesis at GABA synapses in the BLA is maintained and how it is turned off.
Acute stress and/or systemic corticosteroids cause a rapid modulation of endocannabinoid levels in the BLA (Hill and McEwen, 2009) and other stress-related brain regions, such as the hippocampus, the prefrontal cortex, and the hypothalamus (Hill et al., 2010a). The two endogenous cannabinoids, AEA and 2-AG, possess distinct metabolic pathways (Ahn et al., 2008; Di Marzo, 2008) and may serve different physiological functions (Hill and Tasker, 2012; Luchicchi and Pistis, 2012). In the BLA, AEA levels are decreased and 2-AG levels are increased during stress, the former possibly playing a permissive role in stress activation and the latter contributing to the feedback effects of glucocorticoids on these stress circuits (Evanson et al., 2010; Hill and Tasker, 2012). Our electrophysiological findings here in the BLA and in the hypothalamus are consistent with this, as 2-AG mediates the glucocorticoid modulation of inhibitory synapses on BLA neurons and excitatory synapses on hypothalamic neuroendocrine cells (Di et al., 2013).
The glucocorticoid-induced endocannabinoid suppression of GABA inputs to BLA neurons provides a potential cellular mechanism for the stress-induced increases in anxiety-like behavior and modulation of fear-memory formation (Roozendaal et al., 2009, Cecchi et al., 2002). Endocannabinoids in the BLA enhance fear-memory consolidation, and stress facilitates fear-memory consolidation via a CB1 receptor-dependent mechanism in the BLA (Campolongo et al., 2009). Rapid glucocorticoid-induced endocannabinoid suppression of basal synaptic inhibition should alter the excitation–inhibition balance in the BLA and increase BLA principal neuron excitability. Our finding that a CB1 receptor antagonist blocks the acute stress-induced increase in anxiety-like behavior suggests that the stress-induced decrease in inhibition in the BLA contributes to increased anxiety-like behavior. Glucocorticoids also rapidly facilitate excitatory synaptic inputs to BLA neurons, which should further enhance BLA neuron excitability, although this is not endocannabinoid-dependent (Karst et al., 2010), and may not, therefore, contribute to the rapid, endocannabinoid-dependent facilitatory effect of glucocorticoids on anxiety-like behavior (Patel et al., 2005; our findings) and fear-memory consolidation (Campolongo et al., 2009). A recent study suggests that glucocorticoid modulation of excitatory synaptic circuits in the BLA, while critical for fear learning, does not mediate stress-induced increases in anxiety-like behavior (Hartmann et al., 2016).
The findings here and by others (Patel et al., 2005) that blocking CB1 receptors in the BLA increases anxiety-like behavior in unstressed animals indicate a basal inhibitory endocannabinoid tone on BLA anxiogenic output. Given the anxiety-like behavior caused by CB1 receptor blockade, but not by blocking 2-AG synthesis, and the lack of effect of CB1 antagonist on basal IPSC frequency in our experiments, the endocannabinoid tone is likely to be mediated by AEA actions in glutamate circuits. The decrease in AEA levels in the BLA mediated by increased fatty acid amide hydrolase with stress exposure supports an AEA tone in the BLA (Hill et al., 2009; Hill and Tasker, 2012; Gray et al., 2015). We failed to detect a tonic endocannabinoid regulation of miniature EPSCs in BLA neurons with application of the CB1 receptor antagonist SR (1 μm; n = 12, data not shown); however, we cannot rule out the possibility that the endocannabinoid tone may be upstream in the BLA glutamate circuits, or that it may have been lost during the slice preparation.
The glucocorticoid-induced suppression of inhibitory synaptic inputs to BLA principal neurons described here, and the glucocorticoid-induced facilitation of excitatory inputs described by Karst et al. (2010), are likely to play a crucial role in the limbic regulation of stress and fear. Because the BLA is an important locus for encoding and regulating stress-related emotion and emotional-memory information, this rapid glucocorticoid–endocannabinoid signaling provides a potential mechanism for stress regulation of anxiety and emotional memories, as well as a potential target for the treatment of traumatic stress disorders, such as PTSD. Glucocorticoids increase endocannabinoid synthesis in multiple stress-related brain regions (Di et al., 2005; Malcher-Lopes et al., 2006; Hill and McEwen, 2009; Hill et al., 2010b), such that the functional interaction between glucocorticoids and endocannabinoids described here in the BLA, like that described in the hypothalamus (Di et al., 2003), may represent a generalized cellular mechanism of nongenomic glucocorticoid regulation throughout stress circuits in the brain (Tasker and Herman, 2011).
Footnotes
This work was supported by National Institutes of Health Grants R21 MH090453 (J.G.T.), R01 MH104373 (J.G.T.), R00 AA018400 (N.W.G.), and R01 AA023305 (N.W.G.); the Catherine and Hunter Pierson Chair in Neuroscience (J.G.T.); and a Tulane Research Bridge Grant (J.G.T.).
The authors declare no competing financial interests.
References
- Agarwal MK, Hainque B, Moustaid N, Lazer G. Glucocorticoid antagonists. FEBS Lett. 1987;217:221–226. doi: 10.1016/0014-5793(87)80667-1. [DOI] [PubMed] [Google Scholar]
- Ahn K, McKinney MK, Cravatt BF. Enzymatic pathways that regulate endocannabinoid signaling in the nervous system. Chem Rev. 2008;108:1687–1707. doi: 10.1021/cr0782067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atsak P, Hauer D, Campolongo P, Schelling G, Fornari RV, Roozendaal B. Endocannabinoid signaling within the basolateral amygdala integrates multiple stress hormone effects on memory consolidation. Neuropsychopharmacology. 2015;40:1485–1494. doi: 10.1038/npp.2014.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blundell J, Blaiss CA, Lagace DC, Eisch AJ, Powell CM. Block of glucocorticoid synthesis during re-activation inhibits extinction of an established fear memory. Neurobiol Learn Mem. 2011;95:453–460. doi: 10.1016/j.nlm.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boychuk CR, Zsombok A, Tasker JG, Smith BN. Rapid glucocorticoid-induced activation of TRP and CB1 receptors causes biphasic modulation of glutamate release in gastric-related hypothalamic preautonomic neurons. Front Neurosci. 2013;7:3. doi: 10.3389/fnins.2013.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle MP, Kolber BJ, Vogt SK, Wozniak DF, Muglia LJ. Forebrain glucocorticoid receptors modulate anxiety-associated locomotor activation and adrenal responsiveness. J Neurosci. 2006;26:1971–1978. doi: 10.1523/JNEUROSCI.2173-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campolongo P, Roozendaal B, Trezza V, Hauer D, Schelling G, McGaugh JL, Cuomo V. Endocannabinoids in the rat basolateral amygdala enhance memory consolidation and enable glucocorticoid modulation of memory. Proc Natl Acad Sci U S A. 2009;106:4888–4893. doi: 10.1073/pnas.0900835106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cecchi M, Khoshbouei H, Morilak DA. Modulatory effects of norepinephrine, acting on alpha 1 receptors in the central nucleus of the amygdala, on behavioral and neuroendocrine responses to acute immobilization stress. Neuropharmacology. 2002;43:1139–1147. doi: 10.1016/S0028-3908(02)00292-7. [DOI] [PubMed] [Google Scholar]
- Di S, Malcher-Lopes R, Halmos KC, Tasker JG. Nongenomic glucocorticoid inhibition via endocannabinoid release in the hypothalamus: a fast feedback mechanism. J Neurosci. 2003;23:4850–4857. doi: 10.1523/JNEUROSCI.23-12-04850.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di S, Malcher-Lopes R, Marcheselli VL, Bazan NG, Tasker JG. Rapid glucocorticoid-mediated endocannabinoid release and opposing regulation of glutamate and gamma-aminobutyric acid inputs to hypothalamic magnocellular neurons. Endocrinology. 2005;146:4292–4301. doi: 10.1210/en.2005-0610. [DOI] [PubMed] [Google Scholar]
- Di S, Maxson MM, Franco A, Tasker JG. Glucocorticoids regulate glutamate and GABA synapse-specific retrograde transmission via divergent nongenomic signaling pathways. J Neurosci. 2009;29:393–401. doi: 10.1523/JNEUROSCI.4546-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di S, Popescu IR, Tasker JG. Glial control of endocannabinoid heterosynaptic modulation in hypothalamic magnocellular neuroendocrine cells. J Neurosci. 2013;33:18331–18342. doi: 10.1523/JNEUROSCI.2971-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marzo V. Endocannabinoids: synthesis and degradation. Rev Physiol Biochem Pharmacol. 2008;160:1–24. doi: 10.1007/112_0505. [DOI] [PubMed] [Google Scholar]
- Duvarci S, Paré D. Glucocorticoids enhance the excitability of principal basolateral amygdala neurons. J Neurosci. 2007;27:4482–4491. doi: 10.1523/JNEUROSCI.0680-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evanson NK, Tasker JG, Hill MN, Hillard CJ, Herman JP. Fast feedback inhibition of the HPA axis by glucocorticoids is mediated by endocannabinoid signaling. Endocrinology. 2010;151:4811–4819. doi: 10.1210/en.2010-0285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein LE, Rasmusson AM, Bunney BS, Roth RH. Role of the amygdala in the coordination of behavioral, neuroendocrine, and prefrontal cortical monoamine responses to psychological stress in the rat. J Neurosci. 1996;16:4787–4798. doi: 10.1523/JNEUROSCI.16-15-04787.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray JM, Vecchiarelli HA, Morena M, Lee TT, Hermanson DJ, Kim AB, McLaughlin RJ, Hassan KI, Kühne C, Wotjak CT, Deussing JM, Patel S, Hill MN. Corticotropin-releasing hormone drives anandamide hydrolysis in the amygdala to promote anxiety. J Neurosci. 2015;35:3879–3892. doi: 10.1523/JNEUROSCI.2737-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann J, Dedic N, Pöhlmann ML, Häusl A, Karst H, Engelhardt C, Westerholz S, Wagner KV, Labermaier C, Hoeijmakers L, Kertokarijo M, Chen A, Joëls M, Deussing JM, Schmidt MV. Forebrain glutamatergic, but not GABAergic, neurons mediate anxiogenic effects of the glucocorticoid receptor. Mol Psychiatry. 2016 doi: 10.1038/mp.2016.87. Advance online publication. Retrieved July 1, 2016. [DOI] [PubMed] [Google Scholar]
- Hill MN, McEwen BS. Endocannabinoids: the silent partner of glucocorticoids in the synapse. Proc Natl Acad Sci U S A. 2009;106:4579–4580. doi: 10.1073/pnas.0901519106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MN, Tasker JG. Endocannabinoid signaling, glucocorticoid-mediated negative feedback, and regulation of the hypothalamic-pituitary-adrenal axis. Neuroscience. 2012;204:5–16. doi: 10.1016/j.neuroscience.2011.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MN, McLaughlin RJ, Morrish AC, Viau V, Floresco SB, Hillard CJ, Gorzalka BB. Suppression of amygdalar endocannabinoid signaling by stress contributes to activation of the hypothalamic-pituitary-adrenal axis. Neuropsychopharmacology. 2009;34:2733–2745. doi: 10.1038/npp.2009.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MN, Karatsoreos IN, Hillard CJ, McEwen BS. Rapid elevations in limbic endocannabinoid content by glucocorticoid hormones in vivo. Psychoneuroendocrinology. 2010a;35:1333–1338. doi: 10.1016/j.psyneuen.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MN, Patel S, Campolongo P, Tasker JG, Wotjak CT, Bains JS. Functional interactions between stress and the endocannabinoid system: from synaptic signaling to behavioral output. J Neurosci. 2010b;30:14980–14986. doi: 10.1523/JNEUROSCI.4283-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isogawa K, Bush DE, LeDoux JE. Contrasting effects of pretraining, post-training, and pretesting infusions of corticotropin-releasing factor into the lateral amygdala: attenuation of fear memory formation but facilitation of its expression. Biol Psychiatry. 2013;73:353–359. doi: 10.1016/j.biopsych.2012.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karst H, Berger S, Turiault M, Tronche F, Schütz G, Joëls M. Mineralocorticoid receptors are indispensable for nongenomic modulation of hippocampal glutamate transmission by corticosterone. Proc Natl Acad Sci U S A. 2005;102:19204–19207. doi: 10.1073/pnas.0507572102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karst H, Berger S, Erdmann G, Schütz G, Joëls M. Metaplasticity of amygdalar responses to the stress hormone corticosterone. Proc Natl Acad Sci U S A. 2010;107:14449–14454. doi: 10.1073/pnas.0914381107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katona I, Rancz EA, Acsady L, Ledent C, Mackie K, Hajos N, Freund TF. Distribution of CB1 cannabinoid receptors in the amygdala and their role in the control of GABAergic transmission. J Neurosci. 2001;21:9506–9518. doi: 10.1523/JNEUROSCI.21-23-09506.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchicchi A, Pistis M. Anandamide and 2-arachidonoylglycerol: pharmacological properties, functional features, and emerging specificities of the two major endocannabinoids. Mol Neurobiol. 2012;46:374–392. doi: 10.1007/s12035-012-8299-0. [DOI] [PubMed] [Google Scholar]
- Malcher-Lopes R, Di S, Marcheselli VS, Weng FJ, Stuart CT, Bazan NG, Tasker JG. Opposing crosstalk between leptin and glucocorticoids rapidly modulates synaptic excitation via endocannabinoid release. J Neurosci. 2006;26:6643–6650. doi: 10.1523/JNEUROSCI.5126-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsicano G, Wotjak CT, Azad SC, Bisogno T, Rammes G, Cascio MG, Hermann H, Tang J, Hofmann C, Zieglgänsberger W, Di Marzo V, Lutz B. The endogenous cannabinoid system controls extinction of aversive memories. Nature. 2002;418:530–534. doi: 10.1038/nature00839. [DOI] [PubMed] [Google Scholar]
- McGaugh JL. The amygdala modulates the consolidation of memories of emotionally arousing experiences. Annu Rev Neurosci. 2004;27:1–28. doi: 10.1146/annurev.neuro.27.070203.144157. [DOI] [PubMed] [Google Scholar]
- Parsons RG, Ressler KJ. Implications of memory modulation for post-traumatic stress and fear disorders. Nat Neurosci. 2013;16:146–153. doi: 10.1038/nn.3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S, Roelke CT, Rademacher DJ, Hillard CJ. Inhibition of restraint stress-induced neural and behavioural activation by endogenous cannabinoid signalling. Eur J Neurosci. 2005;21:1057–1069. doi: 10.1111/j.1460-9568.2005.03916.x. [DOI] [PubMed] [Google Scholar]
- Patel S, Kingsley PJ, Mackie K, Marnett LJ, Winder DG. Repeated homotypic stress elevates 2-arachidonoylglycerol levels and enhances short-term endocannabinoid signaling at inhibitory synapses in basolateral amygdala. Neuropsychopharmacology. 2009;34:2699–2709. doi: 10.1038/npp.2009.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelps EA, LeDoux JE. Contributions of the amygdala to emotion processing: from animal models to human behavior. Neuron. 2005;48:175–187. doi: 10.1016/j.neuron.2005.09.025. [DOI] [PubMed] [Google Scholar]
- Roozendaal B. 1999 Curt P. Richter award. Glucocorticoids and the regulation of memory consolidation. Psychoneuroendocrinology. 2000;25:213–238. doi: 10.1016/S0306-4530(99)00058-X. [DOI] [PubMed] [Google Scholar]
- Roozendaal B, Bohus B, McGaugh JL. Dose-dependent suppression of adrenocortical activity with metyrapone: effects on emotion and memory. Psychoneuroendocrinology. 1996;21:681–693. doi: 10.1016/S0306-4530(96)00028-5. [DOI] [PubMed] [Google Scholar]
- Roozendaal B, Nguyen BT, Power AE, McGaugh JL. Basolateral amygdala noradrenergic influence enables enhancement of memory consolidation induced by hippocampal glucocorticoid receptor activation. Proc Natl Acad Sci U S A. 1999;96:11642–11647. doi: 10.1073/pnas.96.20.11642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roozendaal B, Quirarte GL, McGaugh JL. Glucocorticoids interact with the basolateral amygdala beta-adrenoceptor–cAMP/cAMP/PKA system in influencing memory consolidation. Eur J Neurosci. 2002;15:553–560. doi: 10.1046/j.0953-816x.2001.01876.x. [DOI] [PubMed] [Google Scholar]
- Roozendaal B, Okuda S, Van der Zee EA, McGaugh JL. Glucocorticoid enhancement of memory requires arousal-induced noradrenergic activation in the basolateral amygdala. Proc Natl Acad Sci U S A. 2006;103:6741–6746. doi: 10.1073/pnas.0601874103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roozendaal B, McReynolds JR, Van der Zee EA, Lee S, McGaugh JL, McIntyre CK. Glucocorticoid effects on memory consolidation depend on functional interactions between the medial prefrontal cortex and basolateral amygdala. J Neurosci. 2009;29:14299–14308. doi: 10.1523/JNEUROSCI.3626-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasker JG, Herman JP. Mechanisms of rapid glucocorticoid feedback inhibition of the hypothalamic-pituitary-adrenal axis. Stress. 2011;14:398–406. doi: 10.3109/10253890.2011.586446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasker JG, Joëls M. The synaptic physiology of the central nervous system response to stress. In: Russell JA, Shipston MJ, editors. International neuroendocrine federation masterclass in neuroendocrinology series: neuroendocrinology of stress. Hoboken, NJ: Wiley-Blackwell; 2015. pp. 43–70. [Google Scholar]
- Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R, Schütz G. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet. 1999;23:99–103. doi: 10.1038/12703. [DOI] [PubMed] [Google Scholar]
- Young EA, Lopez JF, Murphy-Weinberg V, Watson SJ, Akil H. The role of mineralcoticorid receptors in hypothalamic-pituitary-adrenal axis regulation in humans. J Clin Endocrinol Metab. 1998;83:3339–3345. doi: 10.1210/jcem.83.9.5077. [DOI] [PubMed] [Google Scholar]