

Abstract
The neuromodulator acetylcholine (ACh) is crucial for several cognitive functions, such as perception, attention, and learning and memory. Whereas, in most cases, the cellular circuits or the specific neurons via which ACh exerts its cognitive effects remain unknown, it is known that auditory cortex (AC) neurons projecting from layer 5B (L5B) to the inferior colliculus, corticocollicular neurons, are required for cholinergic-mediated relearning of sound localization after occlusion of one ear. Therefore, elucidation of the effects of ACh on the excitability of corticocollicular neurons will bridge the cell-specific and cognitive properties of ACh. Because AC L5B contains another class of neurons that project to the contralateral cortex, corticocallosal neurons, to identify the cell-specific mechanisms that enable corticocollicular neurons to participate in sound localization relearning, we investigated the effects of ACh release on both L5B corticocallosal and corticocollicular neurons. Using in vitro electrophysiology and optogenetics in mouse brain slices, we found that ACh generated nicotinic ACh receptor (nAChR)-mediated depolarizing potentials and muscarinic ACh receptor (mAChR)-mediated hyperpolarizing potentials in AC L5B corticocallosal neurons. In corticocollicular neurons, ACh release also generated nAChR-mediated depolarizing potentials. However, in contrast to the mAChR-mediated hyperpolarizing potentials in corticocallosal neurons, ACh generated prolonged mAChR-mediated depolarizing potentials in corticocollicular neurons. These prolonged depolarizing potentials generated persistent firing in corticocollicular neurons, whereas corticocallosal neurons lacking mAChR-mediated depolarizing potentials did not show persistent firing. We propose that ACh-mediated persistent firing in corticocollicular neurons may represent a critical mechanism required for learning-induced plasticity in AC.
SIGNIFICANCE STATEMENT Acetylcholine (ACh) is crucial for cognitive functions. Whereas in most cases the cellular circuits or the specific neurons via which ACh exerts its cognitive effects remain unknown, it is known that auditory cortex (AC) corticocollicular neurons projecting from layer 5B to the inferior colliculus are required for cholinergic-mediated relearning of sound localization after occlusion of one ear. Therefore, elucidation of the effects of ACh on the excitability of corticocollicular neurons will bridge the cell-specific and cognitive properties of ACh. Our results suggest that cell-specific ACh-mediated persistent firing in corticocollicular neurons may represent a critical mechanism required for learning-induced plasticity in AC. Moreover, our results provide synaptic mechanisms via which ACh may mediate its effects on AC receptive fields.
Keywords: acetylcholine, auditory cortex, neuromodulation, persistent firing, synaptic excitability
Introduction
Acetylcholine (ACh) release in the cortex is crucial for perception, attention, learning, and memory (Bear and Singer, 1986; Everitt and Robbins, 1997; Kilgard and Merzenich, 1998a; Xiang et al., 1998; Weinberger, 2003; Froemke et al., 2007; Bajo et al., 2010; Hasselmo and Sarter, 2011; Leach et al., 2013; Wester and Contreras, 2013). As a result, the nucleus basalis (NB), which is the main source of cortical ACh (Lehmann et al., 1980; Mesulam et al., 1983), has been implicated in all of these cognitive aspects. Despite the importance of these cognitive functions, the specific cortical neuronal types mediating these functions, as well as the synaptic effects of ACh on these neurons, remain poorly understood.
In the auditory cortex (AC), pairing electrical stimulation of NB with an auditory stimulus induces stimulus-specific representational cortical plasticity and auditory memory (Kilgard and Merzenich, 1998a, 1998b; Bao et al., 2003; Weinberger et al., 2006; Froemke et al., 2007) and enhances discrimination learning (Reed et al., 2011). Moreover, cortical cholinergic input is required for normal perception of sound source location and experience-dependent plasticity involved in relearning sound localization after reversible occlusion of one ear (Leach et al., 2013). Recent studies revealed that the cholinergic- and experience-dependent plasticity involved in relearning sound localization after reversible occlusion of one ear is lost after specific elimination of corticocollicular neurons (Bajo et al., 2010), an AC layer 5B (L5B) neuronal type projecting to the inferior colliculus (IC). The knowledge of the specific neuronal population mediating learning-induced auditory plasticity, as well as the necessity of cortical ACh for this mechanism, provide an ideal model for determining the cell-specific synaptic mechanisms via which ACh enables sound localization.
Here, we investigated the effects of exogenous and endogenous ACh on the synaptic excitability of L5B corticocollicular neurons. However, L5B contains additional types of projection neurons, including corticocallosal neurons, a second major class of L5 neurons with axons projecting to the contralateral A1. Because recent studies in a variety of cortical areas, including the AC, revealed numerous differences in the physiological properties of pyramidal tract (PT) neurons, of which corticocollicular are a subtype, and intratelencephalic (IT) neurons, of which corticocallosal neurons are a subtype (for review, see Shepherd, 2013), we hypothesized that ACh may have cell-specific effects on the excitability of projection neurons. These cell-specific effects may be important for the cholinergic-mediated experience-dependent plasticity involved in relearning sound localization after plugging one ear (Bajo et al., 2010), as well as in the distinct role of PT and IT neurons in the delay period that occurs during movement (Li et al., 2015).
To study the effects of ACh on corticocollicular and L5B corticocallosal neurons, we used in vivo retrograde labeling, as well as in vitro electrophysiological methods combined with optogenetic activation of cholinergic fibers. We show that ACh has cell-specific effects on L5B projection neurons. Namely, ACh elicits nicotinic ACh receptor (nAChR)-mediated depolarizing potentials in both neuronal types, whereas ACh evokes muscarinic ACh receptor (mAChR)-mediated hyperpolarizing potentials in corticocallosal neurons, but long-lasting mAChR-mediated depolarizing potentials only in corticocollicular neurons. The long-lasting mAChR-mediated depolarizing potential generates persistent firing in corticocollicular neurons, which may be involved in top-down modulation of auditory learning.
Materials and Methods
Animals.
ICR mice (Harlan Laboratories) and Chat-ChR2-EYFP mice (The Jackson Laboratory) of either sex at age P22–P40 for microsphere injection and P24–P45 for recordings were used for experiments that examined the effect of endogenous release of ACh on corticocollicular and L5B corticocallosal neurons. All experimental procedures were approved by the Institutional Animal Care and Use Committee of the University of Pittsburgh.
Stereotaxic injections.
Mice were anesthetized with isofluorane (induction: 3% in O2, 0.6 L/min; maintenance: 50% of induction dose) and positioned in a stereotaxic frame (Kopf Instruments). Projection neurons in the AC were labeled retrogradely by injecting different colored fluorescent latex microspheres (Lumafluor) in the contralateral AC (in a small craniotomy drilled 4 mm posterior to bregma and 4 mm lateral, injection depth 1 mm) and the ipsilateral IC (1 mm posterior to lambda and 1 mm lateral, injection depth 0.75 mm). A volume of ∼ 0.1 μl microspheres was pressure injected (25 psi, 10–15 ms duration) from capillary pipettes (Drummond Scientific) with a Picospritzer (Parker–Hannifin). The injection volume was distributed between several sites along the injection depth so as to label the entire extent of the injection site. After injection, the pipette was held in the brain for 1.5 min before slowly withdrawing. The animals were allowed to recover for at least 48 h to allow time for retrograde transport of the tracers.
Slice electrophysiology.
Coronal slices (300 μm) containing AC were prepared from mice that had previously been injected with retrograde beads. The cutting solution (pH 7.35) contained the following (in mm): 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 0.5 CaCl2, 7 MgCl2, 7 dextrose, 205 sucrose, 1.3 ascorbic acid, and 3 sodium pyruvate (bubbled with 95% O2/5% CO2). The slices were transferred and incubated at 36°C in a holding chamber for 30 min. The holding chamber contained aCSF (pH 7.35) containing the following (in mm): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 2 CaCl2, 1 MgCl2, 10 glucose, 1.3 ascorbic acid, and 3 sodium pyruvate (bubbled with 95% O2/5% CO2). After incubation, the slices were stored at room temperature until the time of recording. Whole-cell recordings in voltage- and current-clamp modes were performed on slices bathed in carbogenated aCSF, which was identical to the incubating solution. The flow rate of the aCSF was ∼1.5 ml/min and its temperature was maintained at 32–34°C using an inline heating system (Warner Instruments). L5B of the AC was identified as the layer containing corticocollicular neurons (Games and Winer, 1988; Doucet et al., 2003; Slater et al., 2013; Joshi et al., 2015). Recordings were targeted to either green fluorescent corticocollicular neurons or red fluorescent corticocallosal neurons within L5B. Borosilicate pipettes (World Precision Instruments) were pulled into patch electrodes with 3–6 MΩ resistance (Sutter Instruments) and filled with a potassium-based intracellular solution composed of the following (in mm): 128 K-gluconate, 10 HEPES, 4 MgCl2, 4 Na2ATP, 0.3 Tris-GTP, 10 Tris phosphocreatine, 1 EGTA, and 3 sodium ascorbate. Data were sampled at 10 kHz and Bessel filtered at 4 kHz using an acquisition control software package Ephus (Suter et al., 2010). Pipette capacitance was compensated and series resistance for recordings was lower than 25 MΩ. Consistent with previous studies (Joshi et al., 2015), our results showed that L5B corticocallosal neurons have more hyperpolarized resting membrane potential (Vm) than corticocollicular neurons (corticocallosal Vm: −68.4 ± 0.5 mV, n = 41; corticocollicular Vm = −62.4 ± 3.5 mV, n = 41, p = 1e−8, t test). Therefore, before comparing cholinergic neurotransmission baseline membrane potential was maintained at −70 mV by injecting the required current when necessary.
To test for persistent firing, neurons were depolarized by a current injection to a membrane potential that was close to firing threshold. Firing frequency of the action potentials (APs) elicited in response to ACh release was plotted as a function of time starting from the stimulus onset (time, t = 0) to 15 s after the termination of the stimulus. Persistent firing was defined as the ability of the neuron to fire APs at least 5 s after the stimulus (t = 0) termination. AP threshold was measured in phase plane as the membrane potential at which the depolarization slope shows the first abrupt change (Δslope > 10 V/s). AP width was calculated as the full-width at the half-maximum amplitude of the AP (peak minus threshold). Input resistance, Ri, was calculated in voltage- or current-clamp mode by giving a −5 mV or −5 pA step, which resulted in transient current or voltage responses. In voltage-clamp mode, the difference between baseline and steady-state hyperpolarized current (ΔI) was used to calculate Ri using the following formula: Ri = −5 mV/ΔI − Rseries. In current-clamp mode, the difference in the steady-state voltage and baseline voltage (ΔV) was used to calculate Ri using the following formula: Ri = ΔV/−5 pA. The average Vm was calculated by holding the neuron in voltage-follower mode (current clamp at I = 0) immediately after breaking in and averaging the membrane potential over the next 20 s.
Pharmacology.
The identity of the receptors mediating the responses elicited by the release of ACh on corticocollicular or L5B corticocallosal neurons was established by applying blockers of nAChRs and mAChRs (nAChR-mediated responses identified by the application of a mixture of nAChR blockers mecamylamine hydrochloride (5 μm) + hexamethonium bromide (50 μm), or dihydro-β-erythroidine (DHβE) (500 nm); mAChR-mediated responses identified by the application of atropine (1 μm). The blockers were applied for at least 10 min before assessing their effects on the evoked responses. All drugs were obtained from Sigma-Aldrich.
Puffing experiments.
After establishing whole-cell recording from corticocallosal or corticocollicular neurons, 100 μm ACh was puffed for 20 ms at 20 psi from a patch pipette placed 50 μm from the neuronal soma. The 100 μm ACh concentration was used because high concentrations of agonists (>100 μm ACh) affect nAChR-mediated responses by either desensitizing the receptors or causing an open channel block (Quick and Lester, 2002). Responses were identified if they were >2.5 SDs of the baseline noise level (noise levels were measured during a 100 ms timing period before the ACh puff) and were further analyzed.
Optogenetic simulation.
After establishing whole-cell recordings from corticocallosal or corticocollicular neurons, we used wide-field illumination (using a 40× objective) with a blue LED (470 nm at maximum intensity; Thor Laboratories) to activate ChR2-containing cholinergic axons. To assess the effects of endogenous release of ACh on A1 L5B corticocallosal neurons, stimulations ranged from a single 5 ms pulse of blue light to 10 or 60 pulses (5 ms each, 50 Hz). Responses were identified if they were >2.5 SDs of the baseline noise level (noise levels were measured during a 100 ms timing period before illumination) and were further analyzed.
Anatomy.
For anatomical visualization of EYFP-containing cholinergic axons among corticocollicular and L5B corticocallosal neurons, ChAT-ChR2-EYFP mice were injected with red fluorescent latex microspheres in the IC to label corticocollicular neurons and cholera toxin subunit B (CTB, far red emission) in the contralateral AC to label corticocallosal neurons. Mice were allowed to recover for 7 d and were subsequently perfused with 4% paraformaldehyde (PFA). The brains were extracted and postfixed in 4% PFA for 4 h, after which they were cryoprotected overnight in 25% sucrose solution maintained at 4°C. The brains were washed with phosphate buffer solution the next day and were sectioned on a microtome into 50-μm-thick sections containing the AC. The sections were mounted and imaged on an Olympus microscope with a 20× objective using standard filters for green, red, and far-red emissions. The acquired images were subsequently processed in ImageJ for brightness and contrast.
Statistical analysis.
Student's t tests were used for statistical comparisons between different population of neurons. Paired t tests were used for all statistical analyses to compare the effect of drug applications on responses generated by exogenous or endogenous release of ACh. In cases in which two drugs were added sequentially, a one-way ANOVA was used to examine the effect of each drug application on the response. Significance was reported if the p-value was found to be <0.05.
Results
Exogenous application of ACh generates distinct responses in AC L5B corticocallosal and corticocollicular neurons
To study the effect of ACh on AC L5B corticocallosal and corticocollicular neurons, AC L5B projection neurons were targeted for whole-cell recordings in current-clamp mode. Briefly, fluorescent microspheres were injected in the IC in vivo, whereas microspheres of different color were injected in the contralateral AC. In brain slices prepared 2–3 d later, corticocollicular and corticocallosal neurons were labeled selectively (Joshi et al., 2015). In agreement with previous studies, L5B of the AC was identified as the layer containing corticocollicular neurons (Games and Winer, 1988; Doucet et al., 2003; Slater et al., 2013; Joshi et al., 2015). In all of our experiments, recordings were targeted to either red fluorescent corticocollicular neurons or green fluorescent corticocallosal neurons within L5B.
In 23 of 43 corticocallosal neurons, puff application of 100 μm ACh onto their somata (20 psi, ∼50 μm from soma) generated a monophasic depolarizing potential (Fig. 1A1,A2 control black trace). This depolarizing potential was mediated by nAChRs because it was blocked by the application of a mixture of nAChR blockers (5 μm mecamylamine hydrochloride + 50 μm hexamethonium bromide; Fig. 1A1, orange trace, A3). Furthermore, this depolarizing potential was blocked by 500 nm DHβE, a selective antagonist of α4β2 nAChRs (Harvey and Luetje, 1996; Harvey et al., 1996), indicating that it was mediated by α4β2 (Fig. 1A2, magenta trace, A3). In three of 10 corticocallosal neurons, application of nAChR blockers eliminated the depolarizing potential and revealed a hyperpolarizing potential (Fig. 1B1, black trace = depolarizing potential, magenta trace = hyperpolarizing potential after application of DHβE). This hyperpolarizing potential was blocked by the application of 1 μm atropine (mAChR blocker; Fig. 1B1, green trace, B2), indicating that it was mediated by mAChRs.
Figure 1.

Effects of exogenous application of ACh on the membrane potential of L5B corticocallosal neurons. Baseline Vm was maintained at −70 mV by injecting the required current when necessary. A1, Representative example of a monophasic depolarizing potential before (Ctrl, black trace) and after application of a mixture of nAChR blockers (5 μm mecamylamine hydrochloride + 50 μm hexamethonium bromide, orange trace), in response to a single puff of 100 μm ACh (50 ms, 20 psi, 50 μm away from the soma, denoted by the black arrow), observed in a subset of corticocallosal neurons (23 of 43). A2, Representative example of a monophasic depolarizing potential before (Ctrl) and after application of 500 nm DHβE (selective antagonist of α4β2 subunit-containing nAChRs, magenta trace). A3, Average of the effects of the nAChR blocker mixture and DHβE on the amplitude of the monophasic depolarizing potential (normalized values: depolarizing potential amplitude in aCSF: 1.0; after nAChR blockers: −0.003 ± 0.02, n = 4, p = 0.0007; after DHβE: 0.03 ± 0.02; n = 3, p = 0.0002; non-normalized values: average depolarization response amplitude in aCSF: 16.71 ± 2.3 mV; after nAChR blockers: 0.018 ± 0.002 mV, n = 4, p = 0.0001; after DHβE: 0.2 ± 0.081 mV, n = 3, p = 0.16). B1, Representative example of hyperpolarizing potential before and after application of nAChR blocker mixture or DHβE on the monophasic depolarizing potential (black trace) observed in three of 10 neurons (black trace = depolarizing potential; DHβE magenta trace = hyperpolarizing potential). This hyperpolarizing potential was abolished by the addition of 1 μm atropine (green trace), a blocker of all mAChRs. B2, Average of the effect of atropine on the hyperpolarizing potential revealed in the presence of nAChR blockers (normalized values: hyperpolarizing amplitude in aCSF: 1.0; after atropine: −0.06 ± 0.07, n = 3, p = 0.004; non-normalized values: average hyperpolarization response amplitude after nAChR blockers: −2.74 ± 0.28 mV; after atropine: 0.16 ± 0.18 mV, n = 3, p = 0.01). C1, Representative example of a biphasic (depolarizing/hyperpolarizing) potential in response to a single puff of 100 μm ACh (Ctrl, black trace) observed in the rest of corticocallosal neurons (20 of 43). Sequential application of 1 μm atropine (green trace) and the mixture of nAChR blockers (orange trace) abolished the hyperpolarizing and depolarizing potential, respectively. C2, Average of the effect of atropine on the hyperpolarizing potential in the biphasic response. The amplitudes of the responses have been normalized to the amplitude of the hyperpolarizing potential in aCSF (normalized values: hyperpolarizing potential amplitude in aCSF: 1.0; after atropine: 0.10 ± 0.08, n = 6, p-value <0.0001; non-normalized values: average hyperpolarization response amplitude in aCSF: 1.31 ± 0.22 mV; after atropine: 0.09 ± 0.07 mV, n = 6, p = 0.004). C3, Average of the effect of atropine and the nAChR blocker mixture on the depolarizing potential in the biphasic response. The amplitudes of the responses have been normalized to amplitude of the depolarizing potential in aCSF (normalized values: depolarizing potential amplitude in aCSF: 1.0; after atropine: 1.09 ± 0.21, n = 6, p > 0.05; after the nAChR blocker mixture: −0.01 ± 0.02, n = 6, p < 0.01 vs aCSF, p < 0.01 vs atropine; non-normalized values: average depolarization response amplitude in aCSF: 5.05 ± 1.08 mV; after atropine: 5.25 ± 1.06, n = 6, p = 0.75; after nAChR blockers: −0.02 ± 0.08 mV, n = 6, p = 0.01).
In the rest of the recorded corticocallosal neurons (20 of 43), puffing ACh onto their somata generated biphasic responses consisting of a depolarizing potential followed by a hyperpolarizing potential (Fig. 1C1, black trace). Sequential application of 1 μm atropine and the nAChR blocker mixture abolished the hyperpolarizing and the depolarizing potential, respectively (Fig. 1C1, green trace = atropine, orange trace = nAChR blocker mixture, C2, C3). Together, our results show that exogenous application of ACh results in depolarizing and hyperpolarizing potentials in corticocallosal neurons, which are mediated by nAChRs and mAChRs, respectively.
Next, we studied the effect of exogenous ACh on corticocollicular neurons. In 18 of 28 corticocollicular neurons, puffing ACh onto their somata (20 psi, ∼50 μm from soma) generated a depolarizing potential (Fig. 2A1, A2, control black trace), which was mediated by nAChRs because it was blocked by the application of the nAChR blocker mixture (Fig. 2A1, orange trace, A3). Furthermore, the depolarizing potential was also blocked by 500 nm DHβE, indicating that it was mediated by α4β2 nAChRs (Fig. 2A2, magenta trace, A3). In two of 10 corticocollicular neurons, application of nAChR blockers eliminated the depolarizing potential and revealed a hyperpolarizing potential (Fig. 2B, black trace = depolarizing potential, magenta trace = hyperpolarizing potential after application of DHβE). These results suggest that application of ACh revealed similar monophasic potentials in corticocollicular and corticocallosal neurons.
Figure 2.

Effects of exogenous application of ACh on the membrane potential of corticocollicular neurons. Baseline Vm was maintained at −70 mV by injecting the required current when necessary. A1, Representative example of a monophasic depolarizing potential before (Ctrl, black trace) and after application of a mixture of nAChR blockers in response to a single puff of 100 μm ACh observed in a subset of corticocollicular neurons (18 of 28). A2, Representative example of a monophasic depolarizing potential before (Ctrl) and after application of 500 nm DHβE (magenta trace). A3, Average of the effects of the nAChR blocker mixture and DHβE on the monophasic depolarizing potential (normalized values: depolarizing potential amplitude in aCSF: 1.0; after nAChR blockers: 0.05 ± 0.11, n = 4, p = 0.003; after DHβE: −0.01 ± 0.13; n = 4, p = 0.004; non-normalized values: average depolarization response amplitude in aCSF: 2.91 ± 0.81 mV; after nAChR blockers: 0.14 ± 0.33 mV, n = 4, p = 0.04; after DHβE: −0.07 ± 0.44 mV, n = 4, p = 0.09). B, Representative example of a hyperpolarizing potential after application of DHβE on the monophasic depolarizing potential, observed in 2 of 10 neurons (black trace = depolarizing potential; DHβE magenta trace = hyperpolarizing potential). C1, Representative example of two-peak depolarizing potential (black trace, peak 1 and peak 2) in response to a single puff of 100 μm ACh observed in the rest of the corticocollicular neurons (10 of 28). C2, Sequential application of the mixture of nAChR blockers (orange trace) and 1 μm atropine (green trace) abolished peak 1 and peak 2, respectively (control same as in C1 but in a different time scale). C3, Average of the effects of the nAChR blocker mixture on peak 1 of the two-peak depolarizing potential. The amplitudes of the responses have been normalized to amplitude of peak 1 in aCSF (normalized values: peak 1 amplitude in aCSF: 1.0; peak after nAChR blocker mixture: −0.07 ± 0.02, n = 3, p = 0.0003; non-normalized values: average depolarization peak 1 amplitude in aCSF: 2.18 ± 0.02 mV; after nAChR blockers: −0.16 ± 0.03 mV, n = 3, p = 0.0001). C4, Average of the effects of the nAChR blocker mixture and atropine on peak 2 of the two-peak depolarizing potential. The amplitudes of the responses have been normalized to amplitude of peak 2 in aCSF (normalized values: peak 2 amplitude in aCSF: 1.0; after the nAChR blocker mixture: 1.47 ± 0.27, n = 3, p-value >0.05; after atropine: −0.02 ± 0.03, n = 3, p < 0.01 vs aCSF, p < 0.02 vs nAChR blocker mixture; non-normalized values: average depolarization peak 2 amplitude in aCSF: 0.96 ± 0.02 mV after nAChR blockers: 1.20 ± 0.41 mV, n = 3, p = 0.13; after atropine: −0.05 ± 0.03 mV, n = 3, p = 0.006). C5, Average of the effect of atropine on the hyperpolarizing potential, revealed in the presence of nAChR blockers (normalized values: average hyperpolarizing amplitude in aCSF + nAChR blockers: 1.0; after atropine: −0.02 ± 0.03, n = 3, p = 0.033; non-normalized values: average hyperpolarizing response amplitude after nAChR blockers: 0.76 ± 0.22; after atropine: 0.02 ± 0.03, n = 3, p = 0.024).
In 10 of 28 corticocollicular neurons, exogenous application of ACh revealed a distinct response. Namely, puffing of ACh generated a two-peak (peak 1 and peak 2) depolarizing potential (Fig. 2C1, black trace). Sequential application of nAChR blockers and 1 μm atropine (mAChR blocker) abolished peak 1 and peak 2, respectively (Fig. 2C2, orange trace = nAChR blocker mixture, green trace = atropine, C3, C4). The application of nAChR blockers also revealed a hyperpolarizing response, which was blocked by subsequent application of atropine (Fig. 2C5). These results suggest that peak 1 is mediated by nAChRs, whereas the slower peak 2 and the hyperpolarizing response are mediated by mAChRs. Together, our results show that the exogenous ACh caused nAChR-mediated depolarizing potentials, which were similar between L5B corticocallosal and corticocollicular neurons. Conversely, exogenous ACh activated mAChRs, which caused hyperpolarizing potentials in corticocallosal neurons, but delayed, long-lasting depolarizing and hyperpolarizing potentials in corticocollicular neurons.
Endogenous release of ACh evokes distinct responses in corticocollicular and corticocallosal neurons
Next, we determined whether endogenous release of ACh on AC L5B corticocallosal and corticocollicular neurons has actions similar to those of exogenous ACh. To investigate the effect of endogenous ACh, we used the ChAT-ChR2-EYFP mouse line, which expresses the light-activated cation channel channelrhodopsin (ChR2) selectively in cholinergic axons (Zhao et al., 2011). The ChAT-ChR2-EYFP mouse line carries several copies of the vesicular ACh transporter gene (VAChT), which leads to overexpression of functional VAChT and to a threefold increase in ACh release in these mice compared with control mice (Kolisnyk et al., 2013). Although this VAChT overexpression may contribute to cellular outputs that differ from the outputs due to normal levels of ACh release, we used this mouse cell line because it can reveal cell-specific effects of endogenous ACh release between L5B corticocollicular and corticocallosal neurons and further test the validity of the differential effect of exogenous ACh application on corticocollicular and L5B corticocallosal excitability.
To confirm the presence of ChR2-EYFP fibers among AC L5B corticocallosal and corticocollicular neurons, we performed in vivo injections of fluorescent retrograde tracers in ChAT-ChR2-EYFP mice. Small volumes of red fluorescent microspheres (red emission) were injected in the IC, whereas CTB (far red emission) was injected in the contralateral AC (Fig. 3A). Mice were perfused 1 week later and then their brains were cryoprotected and subsequently sectioned into 50-μm-thick AC-containing sections. Epifluorescence imaging revealed labeled corticocollicular neurons in L5B of the AC (Fig. 3B), whereas CTB-labeled corticocallosal neurons were present in L5B and other layers of the AC (Fig. 3C). EYFP-containing green cholinergic axons were also present in L5B AC (Fig. 3D). An overlay of the three separate images (Fig. 3B–D) revealed an intermingled population of AC L5B corticocollicular and corticocallosal neurons (red and blue) among green cholinergic axons (Fig. 3E), thus confirming the presence of cholinergic axons expressing ChR2 in AC L5B.
Figure 3.

ChR2-EYFP fibers among AC L5B corticocallosal and corticocollicular neurons in the ChAT-ChR2-EYFP mouse line. A, Labeling of corticocollicular and corticocallosal neurons with fluorescent tracers. Projection neurons in the AC were labeled by injecting different colored retrograde tracers in the contralateral AC (choleratoxin to label corticocallosal neurons) and the ipsilateral IC (red fluorescent microspheres to label corticocollicular neurons). B, A 20× epifluorescence image showing labeled corticocollicular neurons in L5B of the AC. C, A 20× epifluorescence image showing labeled corticocallosal neurons in L5B and other layers of the AC. D, A 20× epifluorescence image showing green cholinergic axons in L5B of AC. E. Merged image (B–D combined) showing intermingled population of corticocallosal and corticocollicular neurons among green cholinergic axons in L5B of AC.
To assess the effects of endogenous release of ACh on AC L5B corticocallosal neurons, we used wide-field illumination of the slice with a blue LED (λ = 470 nm) to activate ChR2-containing cholinergic terminals and evoke ACh release. In 12 of 21 corticocallosal neurons, endogenous release of ACh by stimulation with a single pulse of blue light (pulse width = 5 ms) generated a monophasic depolarizing potential (Fig. 4A1, control black trace), which was similar to the monophasic depolarizing potential that we observed with exogenous ACh. To assess the pharmacology of these responses, we stimulated with 10 pulses of blue light (at 50 Hz) because the responses were more robust. This monophasic depolarizing potential was mediated by nAChRs because it was blocked by the application of nAChR blockers (Fig. 4A2, orange trace, A4). Furthermore, the depolarizing potential was also blocked by 500 nm DHβE, indicating that it was mediated by α4β2 nAChRs (Fig. 4A3, magenta trace, A4).
Figure 4.

Effects of optogenetic stimulation of cholinergic fibers on the membrane potential of L5B corticocallosal neurons. Baseline Vm was maintained at −70 mV by injecting the required current when necessary. A1, Representative example of a monophasic depolarizing potential (Ctrl, black trace) in response to optogenetic stimulation with a single pulse (λ = 470 nm, pulse width 5 ms, denoted by the blue vertical bar) observed in a subset of corticocallosal neurons (12 of 21). A2, Representative example of a monophasic depolarizing potential before (Ctrl, black trace) and after application of the nAChR mixture blocker (orange trace). A3, Representative example of a monophasic depolarizing potential before (Ctrl, black trace) and after application of 500 nm DHβE (magenta trace). A4, Average of the effects of nAChR blocker mixture and DHβE on amplitude of the monophasic depolarizing potential (normalized values: depolarizing potential amplitude in aCSF: 1.0; after nAChR blockers: 0.01 ± 0.04, n = 3, p = 0.001; after DHβE: 0.03 ± 0.04; n = 5, p < 0.0001; non-normalized values: average depolarization response amplitude in aCSF: 3.81 ± 1.01 mV; after nAChR blockers: −0.04 ± 0.13 mV, n = 3, p = 0.01; after DHβE: −0.10 ± 0.18 mV, n = 5, p = 0.04). B1, Representative example of biphasic (depolarizing/hyperpolarizing) potential in response to a single pulse observed in another subset of corticocallosal neurons (4 of 21). B2, Sequential application of 1 μm atropine (green trace) and the mixture of nAChR blockers (orange trace) abolished the hyperpolarizing and depolarizing potential, respectively. B3, Average of the effect of atropine on the amplitude of the hyperpolarizing potential of the biphasic response. The amplitudes of the responses have been normalized to amplitude of the hyperpolarizing potential in aCSF (normalized values: hyperpolarizing potential amplitude in aCSF: 1.0 ± 0.0; after atropine: −0.05 ± 0.07, n = 3, p = 0.008; non-normalized values: average hyperpolarization response amplitude in aCSF: 0.74 ± 0.05 mV; after atropine: −0.04 ± 0.05 mV, n = 3, p = 0.03). B4, Average of the effects of the nAChR blocker mixture on the amplitude of the depolarizing potential of the biphasic response. The amplitudes of the responses have been normalized to the amplitude of the depolarizing potential in aCSF (normalized values: depolarizing potential amplitude in aCSF: 1.0; after atropine: 2.07 ± 0.15, n = 3, p < 0.01; after the nAChR, n = 3, p = 0.01; non-normalized values: average depolarization response amplitude in aCSF: 0.65 ± 0.08 mV; after atropine: 1.32 ± 0.14 mV, n = 3, p = 0.04; after nAChR blockers: −0.08 ± 0.02 mV, n = 3, p = 0.01). C1, Representative example of a monophasic hyperpolarizing potential in response to optogenetic stimulation with a single pulse observed in another subset of corticocallosal neurons (5 of 21). C2, Hyperpolarizing potential is abolished by the addition of 1 μm atropine (green trace). C3, Average of the effect of atropine on the amplitude of the hyperpolarizing potential inhibitory responses (normalized values: hyperpolarizing potential amplitude in aCSF: 1.0 ± 0.0; after atropine: 0.12 ± 0.08; n = 3, p = 0.008; non-normalized values: average hyperpolarization response amplitude in aCSF: 1.52 ± 0.25 mV; after atropine: 0.14 ± 0.06 mV, n = 3, p = 0.04).
In four of 21 corticocallosal neurons, endogenous release of ACh with a single pulse of blue light generated biphasic responses: a depolarizing potential followed by a hyperpolarizing potential (Fig. 4B1, control black trace). Sequential application of 1 μm atropine and nAChR blockers abolished the hyperpolarizing and depolarizing potential, respectively, showing that the hyperpolarizing phase is mediated mAChRs and the depolarizing phase is mediated by nAChRs (Fig. 4B2, control black trace = depolarization/hyperpolarization, green trace = after atropine, orange trace = after nAChR blockers; B3, summary of the effect of atropine on the hyperpolarizing potential; B4, summary of the effect of atropine and nAChR blockers on the depolarizing potential). Finally, in five of 21 corticocallosal neurons, endogenous release of ACh by optogenetic stimulation with a single pulse of blue light generated a hyperpolarizing potential (Fig. 4C1, control black trace). This hyperpolarizing potential was mediated by mAChRs because it was abolished by the application of 1 μm atropine (Fig. 4C2, control black trace = hyperpolarizing potential, green trace = atropine, C3). Although we did not observe monophasic hyperpolarizing potentials after extracellular ACh application, together, our results indicate that exogenous and endogenous ACh had similar effects on the excitability of L5B corticocallosal neurons.
Next, we studied the effect of endogenous ACh on the excitability of corticocollicular neurons. In five of 13 corticocollicular neurons, one or 10 pulses of blue light elicited monophasic depolarizing responses (Fig. 5A1). Because corticocollicular neurons consistently gave responses to 60 pulses delivered at 50 Hz, we used this stimulation protocol for assessing the response of corticocollicular neurons to endogenous ACh. Under these conditions, in seven of 13 corticocollicular neurons, endogenous release of ACh generated a monophasic depolarizing potential, which was similar to the monophasic depolarizing potential that we observed with exogenous ACh (Fig. 5A2, control black trace). This depolarizing potential was mediated by α4β2 nAChRs because it was blocked by DHβE (Fig. 5A2, magenta trace, A3). In one of five corticocollicular neurons that showed a depolarizing potential, application of nAChR blockers eliminated the depolarizing potential and revealed a hyperpolarizing potential, which was blocked by the application of atropine (Fig. 5B), indicating that it was mediated by mAChRs. These results show that the responses due to endogenous ACh resemble the responses obtained with exogenous application of ACh.
Figure 5.

Effects of optogenetic stimulation of cholinergic fibers on the membrane potential of corticocollicular neurons. Baseline Vm was maintained at −70 mV by injecting the required current when necessary. A1, Representative example of a monophasic depolarizing potential before (Ctrl, black trace) in response to optogenetic stimulation with 10 pulses (λ = 470 nm, pulse width 5 ms @ 50 Hz), observed in a subset of corticocollicular neurons (5 of 13). A2, Representative example of a monophasic depolarizing potential before (Ctrl, black trace) and after (magenta trace) application of DHβE in response to optogenetic stimulation with 60 pulses. A3, Average of the effects of the nAChR blocker mixture and DHβE on the amplitude of the monophasic depolarizing potential (normalized values: depolarizing potential amplitude in aCSF: 1.0; after nAChR blockers and DHβE: 0.05 ± 0.12, n = 3, p = 0.003; non-normalized values: average depolarization response amplitude in aCSF: 2.91 ± 1.20 mV; after nAChR blockers: 0.36 ± 0.27 mV, n = 3, p = 0.02). B, Representative example of a hyperpolarizing potential after application of the nAChR blocker mixture on the monophasic depolarizing potential (black trace = depolarizing potential; orange trace = hyperpolarizing potential after the nAChR mixture blocker) elicited in one of five neurons. This hyperpolarizing potential was abolished by the addition of atropine (green trace). C1, Representative example of a broad depolarizing potential in response to optogenetic stimulation with 60 pulses observed in the rest of the corticocollicular neurons (6 of 13). Representative example showing that application of atropine changed the kinetics of the broad depolarizing potential to resemble that of a monophasic depolarizing potential (green trace) and that subsequent application of nAChR blockers abolished the remaining response (orange trace). C2, Average of the effect of atropine and nAChR blockers on the amplitude of the narrow depolarizing potentials. To quantify the atropine effect on the narrow depolarizing potential, we measured the latency of the narrow peak amplitude (331 ± 32 ms) after atropine application and used the corresponding latency as the time point to measure the amplitude in control condition and after nAChR blocker application (normalized values: narrow depolarization amplitude in aCSF: 1.0; after atropine: 2.79 ± 0.56, n = 4, p = 0.014; after nAChR blockers: −0.17 ± 0.12, n = 4, p = 0.007; non-normalized values: average narrow depolarization response amplitude in aCSF: 1.47 ± 0.27; after atropine: 3.83 ± 0.54, n = 4, p = 0.01; after nAChR blockers: −0.23 ± 0.15 mV, n = 4, p = 0.007). C3, Average of the effect of atropine on the amplitude of the broad depolarizing potentials. To quantify the atropine effect on the broad depolarizing potential, we used as control the membrane depolarization of the black trace by averaging 500 ms of membrane potential starting at 50 ms after the green trace reached baseline value in C1. (normalized values: depolarizing potential amplitude in aCSF: 1.0; after atropine: 0.12 ± 0.09; n = 4, p = 0.002; non-normalized values: average broad depolarization response amplitude in aCSF: 0.91 ± 0.29 mV; after atropine: 0.10 ± 0.04 mV, n = 4, p = 0.04).
In six of 13 corticocollicular neurons, photostimulation of cholinergic fibers elicited a broader depolarizing potential (Fig. 5C1, black trace). This broad depolarizing potential was reminiscent of peak 2 of the two-peak depolarizing potential obtained in response to exogenous ACh application. Because peak 2 of the two-peak depolarizing potential was mediated by mAChRs (Fig. 2C1,C2), we tested whether the broad depolarizing potential was also mediated by mAChRs. Indeed, application of 1 μm atropine revealed a narrower depolarizing potential (Fig. 5C1, green trace, C3), which was subsequently eliminated by application of nAChR blockers (Fig. 5C1, orange trace, C2). Together, these results suggest that endogenous ACh, like exogenous ACh, generates nAChR- and mAChR-mediated depolarizing potentials in corticocollicular neurons and the mAChR-mediated depolarizing potentials are long-lasting.
No evidence for subdivision of corticocollicular neurons based on the variability of their intrinsic properties
Because approximately half of the recorded corticocollicular neurons displayed long-lasting depolarizing potentials after exogenous or endogenous ACh application, we tested whether these neurons comprised a distinct subgroup within the corticocollicular neuronal population. Because corticocollicular and corticocallosal L5B neurons in mouse AC display distinct dendritic morphology and distinct intrinsic properties such as resting membrane potential, input resistance, AP threshold, and AP width (Joshi et al., 2015), we tested whether differences in these properties are associated with the ability of a subpopulation of corticocollicular neurons to generate long-lasting mAChR-mediated depolarizing potentials. Our results showed that the average input resistance, resting membrane potential, AP width, and AP threshold were not different between the corticocollicular neurons displaying long-lasting depolarization (broad) and the corticocollicular not displaying long-lasting depolarization (narrow; Fig. 6A1–A4). This finding is inconsistent with potential further subdivision of the corticollicular neuronal population. To further validate the lack of subdivision, we tested for potential correlations between the variability of these intrinsic parameters and the amplitude of the narrow and broad ACh-mediated responses. Because all corticocollicular neurons displayed fast onset (rapid) cholinergic depolarization, we tested whether there is any correlation between the amplitude of the early depolarization and the observed variability in intrinsic properties. Aside from an expected correlation between the variability of input resistance and the amplitude (1st amplitude) of the early cholinergic response in the neurons not displaying the long-lasting depolarization (Fig. 6B1, black, p = 0.007), the variability of the intrinsic properties of either group of corticocollicular neurons was not correlated with the amplitude of the early cholinergic depolarization (Fig. 6B1–B4). Furthermore, for the corticocollicular neurons displaying the long-lasting depolarization, we observed no significant correlation between the amplitude of this response (2nd amplitude) and the variability of the intrinsic properties of these neurons (Fig. 6C1–C4). Combined with our previous study showing homogeneity in the dendritic morphology of the population of corticocollicular neurons (Joshi et al., 2015), our results suggest that the variability in the intrinsic properties and morphology of corticocollicular neurons is not correlated with the presence or absence of the long-lasting cholinergic depolarization that we observed.
Figure 6.

There was no evidence for distinct subgroups of corticocollicular neurons based on variability in intrinsic properties. A1, Input resistance was not different between corticocollicular neurons displaying broad depolarization (gray) and those without (black; Ri: narrow depolarization: 211.9 ± 22.6 MΩ, n = 23; broad depolarization = 184 ± 17.1 MΩ, n = 15; p = 0.32). A2, Resting membrane potential (Vm) was not different between corticocollicular neurons displaying and neurons not displaying broad depolarization (Vm: narrow depolarization = −63.1 ± 5.0 mV, n = 22; broad depolarization = −61.4 ± 4.4 mV, n = 15; p = 0.26). A3, AP width was not different between corticocollicular neurons displaying and neurons not displaying broad depolarization (AP width: narrow depolarization = 1.94 ± 0.33 ms, n = 8; broad depolarization = 1.75 ± 0.25 ms, n = 10; p = 0.08). A4, AP threshold was not different between corticocollicular neurons displaying and neurons not displaying broad depolarization (narrow depolarization = −44 ± 7.5 mV, n = 8; broad depolarization = −41.3 ± 5.8 mV, n = 10; p = 0.09). B1, Relationship of the variability of the amplitude of the narrow depolarization (first amp.) and Ri in corticocollicular neurons (narrow depolarization: R2 = 0.55, n = 23, p = 0.007; broad depolarization: R2 = 0.20, n = 13, p = 0.50). B2, Relationship of the variability of the amplitude of the narrow depolarization and Vm in corticocollicular neurons (narrow depolarization: R2 = 0.10, n = 19, p = 0.68; broad depolarization: R2 = 0.20, n = 13, p = 0.50). B3, Relationship of the variability of the amplitude of the narrow depolarization and AP width in corticocollicular neurons (narrow depolarization: R2 = 0.27, n = 8, p = 0.57; broad depolarization: R2 = 0.05, n = 10, p = 0.88). B4, Relationship of the variability of amplitude of the narrow depolarization and AP threshold in corticocollicular neurons (narrow depolarization: R2 = 0.10, n = 8, p = 0.67; broad depolarization: R2 = 0.41, n = 10, p = 0.28). C1, Relationship of the variability of the amplitude of the broad depolarization (second amp.) and Ri in corticocollicular neurons (broad depolarization: R2 = 0.53, n = 13, p = 0.15). C2, Relationship of the variability of the amplitude of the broad depolarization and Vm in corticocollicular neurons (broad depolarization: R2 = 0.46, n = 13, p = 0.12). C3, Relationship of the variability of the amplitude of the broad depolarization and AP width in corticocollicular neurons (broad depolarization: R2 = 0.43, n = 10, p = 0.25). C4, Relationship of the variability of the amplitude of the broad depolarization and AP threshold in corticocollicular neurons (broad depolarization: R2 = 0.38, n = 10, p = 0.31).
Muscarinic AChRs mediate persistent firing in corticocollicular neurons
Our results show that release of ACh onto AC L5B pyramidal neurons resulted in distinct responses in corticocollicular and L5B corticocallosal neurons. The most prominent difference was seen in a subset of corticocollicular neurons, which exhibited a fast nicotinic depolarizing potential and a prolonged muscarinic depolarizing potential upon the release of ACh (Fig. 5C1). Such a prolonged depolarizing potential may cause persistent firing in response to transient suprathreshold stimulus. Because cholinergic activation leads to persistent firing in cortical neurons, which is associated with mnemonic and learning tasks (Haj-Dahmane and Andrade, 1996; Egorov et al., 2002; Egorov et al., 2006; Fransén et al., 2006; Gulledge et al., 2009; Dembrow et al., 2010; Hedrick and Waters, 2015), we hypothesized that corticocollicular neurons displaying prolonged mAChR-mediated depolarizing potentials will display persistent firing in response to endogenous ACh release or exogenous ACh application.
Consistent with our hypothesis, puffing or endogenous release of ACh onto corticocallosal neurons with monophasic depolarizing potentials or biphasic (depolarizing/hyperpolarizing potentials) responses (Fig. 7A1,B1,C1), when held at subthreshold but close to threshold potential, elicited transient firing, but failed to elicit any persistent firing (Fig. 7A2,B2,C2). Plots of the firing frequency as a function of time indicated that corticocallosal neurons fired APs only during the stimulus or immediately after stimulus termination (Fig. 7A3,B3,C3). Note that even stimulation with 60 pulses did not induce persistent firing in corticocallosal neurons (Fig. 7C4–C6). Similar results were obtained from exogenous application (Fig. 8A1–A3) or endogenous release of ACh (Fig. 8B1–B3) onto corticocollicular neurons exhibiting monophasic depolarizing potentials. However, the subset of corticocollicular neurons exhibiting two-peak depolarizing potentials or broad depolarizing potentials (Fig. 9A1,B1) showed persistent firing in response to either exogenous or endogenous ACh, respectively (Fig. 9A2,B2). Plots of the firing frequency as a function of time indicated that corticocollicular neurons that exhibited two-peak depolarizing potentials or broad depolarizing potentials fired APs for >10 s after the termination of the stimulus (Fig. 9A3,B3). Persistent firing was abolished upon application of atropine, suggesting that mAChRs are crucial for the persistent firing of corticocollicular neurons (Fig. 9A4–A5, B4–B5). Finally, the intrinsic properties, such as AP threshold and AP width, did not change from the onset of firing and during the spike train (Fig. 9C1–C2), suggesting that mAChRs promote persistent firing without affecting the intrinsic AP properties. This finding suggests that ACh is capable of converting AC neurons projecting to the IC into a “persistent activity” mode, whereas intracortically projection neurons do not enter this mode. The persistent firing may be essential for the ACh-dependent, learning-induced plasticity mediated by corticocollicular neurons (Bajo et al., 2010; Leach et al., 2013).
Figure 7.
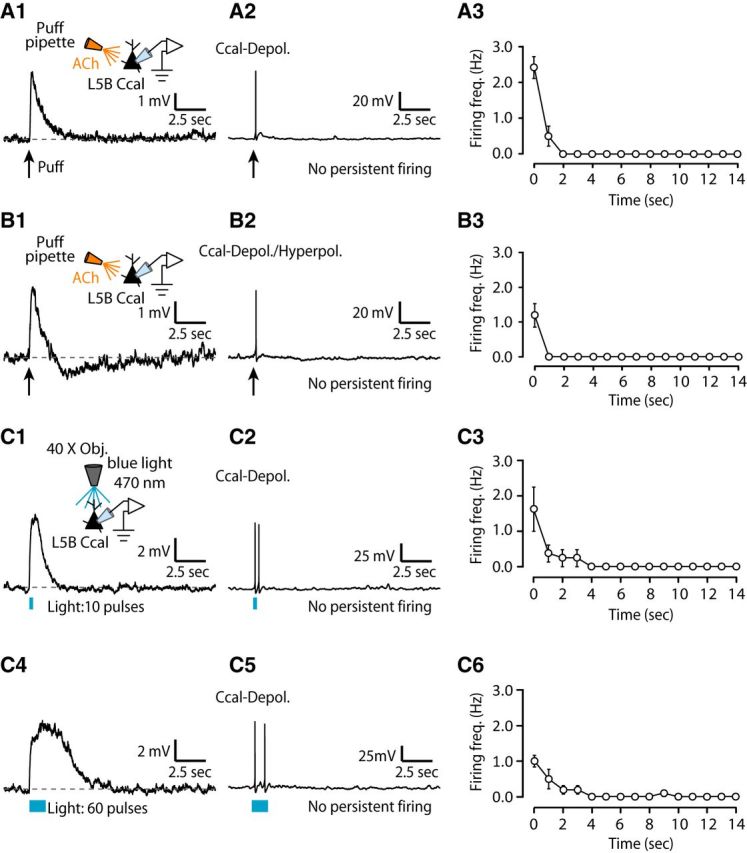
Lack of persistent firing in L5B corticocallosal neurons. A1, Representative example of a corticocallosal neuron held at ∼−70 mV, which responds with a monophasic depolarizing potential to a single puff of 100 μm ACh, denoted by the black arrow. A2, Same neuron as in A1, when held at subthreshold but closer to threshold potential, fires transiently in response to a puff of ACh. This transient spiking was observed in seven of seven corticocallosal neurons with monophasic depolarizing potentials. A3, Average firing frequency quantified for 15 s starting at the time of stimulus onset (t = 0) for APs as in A2 (n = 7). B1, An example of a corticocallosal neuron held at ∼−70 mV, which responds with a biphasic potential to a puff of 100 μm ACh. B2, Same neuron as in B1, when held at subthreshold but closer to threshold potential, fires transiently in response to a puff of ACh. This transient spiking was observed in five of five corticocallosal neurons with biphasic responses. B3, Average firing frequency quantified for 15 s starting at the time of stimulus onset (t = 0) for APs as in B2 (n = 5). C1, Representative example of a corticocallosal neuron held at ∼−70 mV, which responds with a monophasic depolarizing potential in response to optogenetic stimulation with 10 pulses of blue light (λ = 470 nm, pulse width = 5 ms @ 50 Hz). C2, Same neuron as in C1, when held at subthreshold but closer to threshold potential, fires transiently in response to the same optogenetic stimulation used in C1. This transient spiking was observed in four of four corticocallosal neurons with monophasic depolarizing potentials. C3, Average firing frequency quantified for 15 s starting at the time of stimulus onset (t = 0) for APs as in C2 (n = 4). C4, Same corticocallosal neuron as in C1 responds with a broader monophasic depolarizing potential in response to optogenetic stimulation with 60 pulses of blue light (λ = 470 nm, pulse width = 5 ms at 50 Hz). C5, Same neuron as in C4, when held at subthreshold but closer to threshold potential, fires transiently in response to the same optogenetic stimulation used in C4. C6, Average firing frequency quantified for 15 s starting at the time of stimulus onset (t = 0) for APs as in C5 (n = 5).
Figure 8.

Lack of persistent firing in L5B corticocollicular neurons exhibiting ACh-evoked monophasic depolarizing potentials. A1, Representative example of a corticocollicular neuron held at ∼−70 mV, which responds with a monophasic depolarizing potential to a puff of 100 μm ACh. A2, Same neuron as in A1, when held at subthreshold but closer to threshold potential, fires transiently in response to a puff of ACh. This transient spiking was observed in five of five corticocollicular neurons exhibiting monophasic depolarizing potentials. A3, Average firing frequency quantified for 15 s starting at the time of stimulus onset (t = 0) for APs as in A2 (n = 5). B1, Representative example of a corticocollicular neuron held at ∼−70 mV, which responds with a monophasic depolarizing potential to the release of endogenous ACh by optogenetic stimulation with 60 pulses of blue light. B2, Same neuron as in B1, when held close to its AP threshold, fires transiently in response to the same optogenetic stimulation used in B1. This transient spiking was observed in three of three corticocollicular neuron with a monophasic depolarizing potentials. B3, Average firing frequency quantified for 15 s starting at the time of stimulus onset (t = 0) for APs as in B2 (n = 3).
Figure 9.
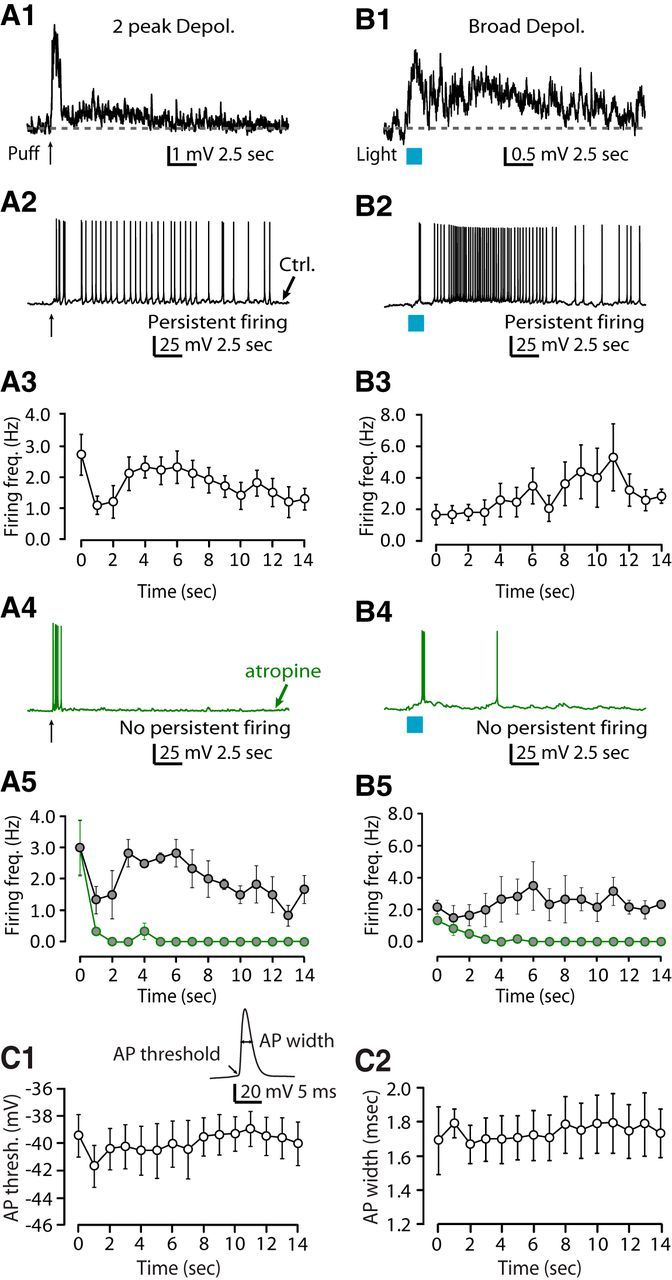
Persistent firing in L5B corticocollicular neurons exhibiting ACh-evoked two-peak, broad depolarizing potentials depends on mAChRs. A1, Representative example of a corticocollicular neuron held at ∼−70 mV, which responds with a two-peak depolarizing potential to a puff of ACh. A2, Same neuron as in A1, when held at subthreshold but closer to threshold potential, fires persistently in response to a puff of ACh. This persistent firing was observed in five of five corticocollicular neurons with two-peak depolarizing potentials. A3, Average firing frequency quantified for 15 s starting at the time of stimulus onset (t = 0) for trains of APs as generated in A2 (n = 5). A4, After the application of 1 μm atropine, the same neuron as in A1 and A2 fails to fire persistently in response to a puff of ACh. This effect of atropine was seen in three of three persistently firing corticocollicular neurons with two-peak depolarizing potentials. A5, Average firing frequency quantified for 15 s starting at the time of stimulus onset, before and after the application of atropine (average firing frequency for the last 5 s (t = 10–14) in control: 1.52 ± 0.34; in atropine: 0.00 ± 0.00, n = 3, p < 0.05). B1, Representative example of a corticocollicular neuron held at ∼−70 mV, which responds with a broad depolarizing in response to optogenetic stimulation with 60 pulses of blue light. B2, Same neuron as in B1, when held at subthreshold but closer to threshold potential, fires persistently in response to the same optogenetic stimulation used in B1. This persistent firing was observed in four of four corticocollicular neurons with broad depolarizing potentials. B3, Average firing frequency quantified for 15 s starting at the time of stimulus onset for trains of APs as generated in B2 (n = 4). B4, After the application of 1 μm atropine, the same neuron as in B1 and B2 fails to fire persistently in response to the same optogenetic stimulation used in B1 and B2. This effect of atropine was seen in three of three persistently firing corticocollicular neurons with broad depolarizing potentials. B5, Average firing frequency quantified for 15 s starting at the time of stimulus onset before and after the application of atropine (average firing frequency for the last 5 s in control: 2.36 ± 0.54; in atropine: 0.00 ± 0.00, n = 3, p < 0.05). C1, C2, AP properties during persistent firing. Persistent firing was evoked in response to optogenetic stimulation as in Figure 8B2. Inset shows representative AP waveform. C1, Average of the AP threshold plotted as a function of time during persistent firing (average AP threshold during stimulus (t = 0–2): −40.63 ± 1.55, average AP threshold during the last five seconds of the train (t = 10–14): −39.62 ± 1.36, n = 4, p = 0.16). C2, Average of the AP width plotted as a function of time during persistent firing (average AP width during stimulus (t = 0–2): 1.70 ± 0.12, average AP width during the last 5 s (t = 10–14): 1.77 ± 0.16, n = 4, p = 0.22).
Discussion
To assess the effects of ACh on the excitability of L5B projection neurons, we used in vivo retrogradely fluorescent labeling to label corticocollicular and L5B corticocallosal neurons selectively, single-cell electrophysiology, exogenous application of ACh, and selective stimulation of cholinergic fibers. Whereas exogenous and endogenous ACh generated fast nAChR-mediated depolarizing potentials in corticocollicular and corticocallosal neurons, ACh release generated mAChR-mediated hyperpolarizing potentials in corticocallosal neurons, but long-lasting mAChR-mediated depolarizing potentials in corticocollicular neurons. The long-lasting mAChR-mediated depolarizing potentials were crucial for the persistent firing observed selectively in corticocollicular neurons, which may be involved in auditory learning.
ACh-mediated persistent firing in corticocollicular neurons: roles and mechanisms
ACh-mediated neuronal modulation causes persistent firing in several neocortical areas and in the substancia nigra pars compacta and subthalamic nucleus (Andrade, 1991; Haj-Dahmane and Andrade, 1996; Egorov et al., 2002; Yamashita and Isa, 2003a, 2003b; Egorov et al., 2006; Fransén et al., 2006; Dembrow et al., 2010; Hedrick and Waters, 2015). In these brain areas, persistent firing of selective neuronal populations is a proposed cellular mechanism underlying learning and mnemonic functions, such as working memory (Wang, 2001; Hasselmo and Stern, 2006; Barak and Tsodyks, 2014). Our results show that cholinergic activation generates persistent firing in a subset of corticocollicular neurons, which display prolonged mAChR-mediated depolarizing potentials. Because these neurons are crucial for cholinergic-mediated auditory learning of sound localization (Bajo et al., 2010; Leach et al., 2013), we propose that persistent firing in corticocollicular neurons is a critical mechanism for auditory learning. Moreover, we predict that mAChR blockers will block this form of auditory learning.
ACh-mediated persistent firing in other brain areas relies either on mAChR-mediated enhancement of postsynaptic Ca2+ and enhancement of afterdepolarizing potentials generated by a Ca2+ -activated nonselective cation current, or voltage-dependent increase in input resistance mediated by a reduction in afterhyperpolarization potentials, or muscarinic mediated inhibition of M-type (KCNQ) potassium channels, or activation of nAChRs and elevations in postsynaptic Ca2+ (McCormick and Prince, 1985; McCormick and Prince, 1986; McCormick and Williamson, 1989; Haj-Dahmane and Andrade, 1999; Egorov et al., 2002; Yamashita and Isa, 2003a, 2003b; Delmas and Brown, 2005; Egorov et al., 2006; Zhang and Séguéla, 2010; Rahman and Berger, 2011; Hedrick and Waters, 2015). Our results show that the persistent firing activity of corticocollicular neurons in response to ACh release is mediated by mAChRs. Whereas our studies did not evaluate the role of postsynaptic Ca2+, because mAChR activation does not affect the spiking properties of corticocollicular neurons and because the time course of persistent firing matches the time course of the mAChR-mediated prolonged depolarization, we propose that it is this depolarization that generates persistent firing lasting for ∼10–20 s after stimulus termination. Our studies did not assess whether this depolarization generates persistent firing in a cell autonomous manner; however, our findings support an additional mechanism for generating persistent firing in cortical neurons.
Cell-specific cholinergic neuromodulation of AC L5B pyramidal neurons promotes tonic activity in corticocollicular neurons and phasic activity in corticocallosal neurons
AC L5B corticocallosal and corticocollicular neurons fall in the broader category of IT and PT neurons, respectively. PT neurons project to subcortical targets, whereas IT neurons project to the contralateral cortex. Studies in different cortical areas showed that PT and IT type neurons differ not only in their projection targets, but also display differences in their anatomical (Gao and Zheng, 2004; Dembrow et al., 2010; Sun et al., 2013), intrinsic (Dembrow et al., 2010; Sheets et al., 2011; Slater et al., 2013; Suter et al., 2013; Joshi et al., 2015), and synaptic/circuit properties (Morishima and Kawaguchi, 2006; Anderson et al., 2010; Dembrow et al., 2010; Shepherd, 2013; Lee et al., 2014; Joshi et al., 2015). These multiple projection-specific cellular and synaptic mechanisms collectively promote dichotomous activity in L5 neurons, leading to sustained (tonic) responses in corticocollicular neurons and transient (phasic) responses in corticocallosal neurons. This hypothesis is further validated by the dichotomous responses of PT and IT neurons to sound in vivo (Sun et al., 2013). Our results, which are consistent with differential effects of neuromodulatory systems on PT and IT neurons (Dembrow et al., 2010; Avesar and Gulledge, 2012; Gee et al., 2012), further contribute to the dichotomous phasic/tonic activity in L5 projection neurons because ACh promotes persistent firing in PT, but not in IT, neurons.
The distinct responses of PT and IT neurons to cholinergic modulation suggest that they subserve distinct functions. This is consistent with recent findings showing that, during motor planning and movement, behavior activity with a contralateral population bias arises specifically in PT, but not in IT, neurons (Li et al., 2015). The ability of PT neurons to undergo persistent firing beyond their stimulus input makes them good candidates to contribute to persistent activity that occurs during the delay period in movement planning. Indeed, recent findings show that population activity in PT neurons appears and persists for hundreds of milliseconds before movement onset (Li et al., 2015).
Limitations of the ChAT-ChR2-EYFP mouse line used in our studies
The majority of cholinergic axons in neocortex originate from somata of cholinergic neurons in the basal forebrain, primarily in NB, with a minority originating from cholinergic interneurons and other nuclei within the basal forebrain complex, such as the medial septum (Bigl et al., 1982). ChR2-YFP observed in neocortex in ChAT-ChR2-EYFP mice was expressed mostly by projections from NB and also by local cholinergic interneurons (Zhao et al., 2011). It is therefore impossible to stimulate selectively the projection from NB in these mice, so ACh released from cholinergic interneurons also contributes to the observed responses.
Whereas neither exogenous application of ACh nor optogenetic stimulation of cholinergic fibers matches endogenous ACh release levels (Kolisnyk et al., 2013), both approaches are consistent with a cell-specific effect of cholinergic modulation on L5B corticocollicular and corticocallosal neurons. Moreover, both approaches resulted in consistent findings on the effects of ACh on L5B corticocollicular and corticocallosal neurons. One notable difference between the effects of exogenous application of ACh and optogenetic stimulation of endogenous ACh is the lack of nicotinic responses in a subset of corticocallosal neurons in response to optogenetic stimulation. This difference could be due to desensitization of nAChRs in ChAT-ChR2-EYFP mice due the enhanced cholinergic tone observed in these mice (Kolisnyk et al., 2013).
nAChR- and mAChR-mediated responses in L5 cortical pyramidal neurons
Several studies have used exogenous ACh and one study has used endogenous ACh (Hedrick and Waters, 2015) to study cholinergic neuromodulation in L5 cortical neurons. Exogenous and endogenous ACh causes mAChR-mediated hyperpolarizing and depolarizing responses, as well as facilitation and inhibition in L5 principal neurons of different cortices (Metherate et al., 1992; Gulledge and Stuart, 2005; Gulledge et al., 2007; Hedrick and Waters, 2015). Overall, these responses are consistent with mAChRs mediating slow depolarizing potentials observed in corticocollicular neurons and hyperpolarizing potentials in corticocallosal neurons in L5B AC. However, the biphasic response seen in AC L5 corticocallosal neurons is unique to the AC. In AC L5 corticocallosal neurons biphasic responses consisted of a depolarization followed a hyperpolarization, which were mediated by nAChRs and mAChRs, respectively, whereas in the somatosensory cortex, biphasic responses consisted of a hyperpolarization followed by a depolarization, which were both mediated by mAChRs (Gulledge and Stuart, 2005).
Because we observed the effects of long-lasting depolarization in corticocollicular neurons at different membrane potentials (e.g., long-lasting depolarization at ∼−70 mV and persistent firing near AP threshold in Figs. 7, 8), we suggest that this response is a feature of some corticocollicular neurons and not, for example, a voltage-dependent property of all corticocollicular neurons. In addition, because we never observed this long-lasting mAChR-mediated depolarization in corticocallosal neurons at any membrane potential, this result further supports the notion that this response is specific to only some corticollicular neurons. Although we did not identify any correlations between the variability in the intrinsic properties of corticocollicular with the presence of the long-lasting muscarinic depolarization, our results do not exclude the influence of cell-specific factors mediating this differential response. For example, in layer 2/3 of the AC, a long-lasting depolarization after muscarinic activation is mediated by M1 receptors (Aramakis et al., 1999). Therefore, potential differential expression of muscarinic receptor subtypes within the corticollicular neuronal population could explain the lack of the long-lasting mAChR-mediated depolarization in a subset of corticollicular neurons. However, further studies are needed to elucidate the mAChR subtypes underlying the hyperpolarization and long-lasting depolarization observed in corticocallosal and corticollicular neurons, respectively.
Although we did not investigate the AChR subunit subtypes and the signaling mechanisms mediating the mAChR-evoked potentials, based on studies in other brain regions, we propose that the hyperpolarizing potentials in corticocallosal neurons could be either mediated by activation of either the Gi-coupled M2/M4 receptors or the Gq-coupled M1 receptor that triggers IP3-mediated increase in intracellular Ca2+ and subsequent activation of SK potassium channels (Newberry and Priestley, 1987; Gulledge and Stuart, 2005; Gulledge et al., 2007; Eggermann and Feldmeyer, 2009; Gulledge et al., 2009). In corticollicular neurons, the long-lasting depolarization is potentially mediated by a direct activation of M1 and/or M3 receptors on corticocollicular neurons, which may in turn inhibit KCNQ potassium channels (Delmas and Brown, 2005), or by activation of M1-mediated enhancement of NMDA responses through an IP3-dependent pathway (Aramakis et al., 1999).
Exogenous application or endogenous release of ACh also generates nAChR-mediated responses in L5 pyramidal neurons somatosensory, prefrontal, and motor cortices (Zolles et al., 2009; Poorthuis et al., 2013; Hedrick and Waters, 2015). Nicotinic AChR-mediated depolarizing responses in L5 prefrontal cortex are mediated by α7-subunit-containing nAChRs (Poorthuis et al., 2013), whereas depolarizing responses in L5 somatosensory cortex are mediated by α7-subunit-containing and α4-subunit-containing nAChRs (Zolles et al., 2009). In L5B of the AC, exogenous and endogenous ACh generates nAChR-mediated depolarizing potentials in both corticocallosal and corticocollicular neurons, which are mediated by α4-subunit-containing nAChRs, presumably α4β2 nAChRs. Our results are similar to results obtained from motor cortex, which showed that the α4-subunit-containing nAChR-mediated depolarizing potentials in L5 pyramidal neurons (Zolles et al., 2009).
Cellular mechanisms underlying system-level effects of ACh in AC
The hypothesized combined effect of mAChRs and nAChRs on AC receptive fields is to reduce receptive field width and to enhance responsiveness within the sharpened receptive field (Metherate, 2011). Several in vivo studies have shown that stimulation of NB enhances, via mAChRs, afferent responses in AC evoked by thalamic (Metherate et al., 1992; Metherate and Ashe, 1993) or acoustic (Edeline et al., 1994; Chen and Yan, 2007) stimulation. This result is consistent with the mAChR-mediated prolonged depolarizing potentials that we observed in corticocollicular neurons. Moreover, sharpening of receptive fields by mAChRs is consistent with the biphasic and inhibitory potentials that we observed in L5B corticocallosal neurons. Although previous studies have shown that nAChRs enhance responsiveness via presynaptic regulation of thalamocortical transmission (Metherate, 2004), our results add an additional mechanism that can enhance responsiveness: the nAChR-mediated depolarizing potentials observed in corticocollicular and L5B corticocallosal neurons also contribute ACh-mediated enhanced responsiveness of AC receptive fields. Together, our results are consistent with in vivo studies revealing sharpening and enhancement of AC receptive fields by ACh and, importantly, provide cellular and synaptic mechanisms via which ACh mediates its effects on AC receptive fields.
Footnotes
This work was supported by National Institutes of Health (Grant DC013272 to T.T.). We thank Dr. Brian Davis for help with anatomical experiments.
The authors declare no competing financial interests.
References
- Anderson CT, Sheets PL, Kiritani T, Shepherd GM. Sublayer-specific microcircuits of corticospinal and corticostriatal neurons in motor cortex. Nat Neurosci. 2010;13:739–744. doi: 10.1038/nn.2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade R. Cell excitation enhances muscarinic cholinergic responses in rat association cortex. Brain Res. 1991;548:81–93. doi: 10.1016/0006-8993(91)91109-E. [DOI] [PubMed] [Google Scholar]
- Aramakis VB, Bandrowski AE, Ashe JH. Role of muscarinic receptos, G-proteins and intracellular messengers in muscarinic modulation of NMDA receptor-mediated synaptic transmission. Synapse. 1999;32:262–275. doi: 10.1002/(SICI)1098-2396(19990615)32:4<262::AID-SYN3>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Avesar D, Gulledge AT. Selective serotonergic excitation of callosal projection neurons. Front Neural Circuits. 2012;6:12. doi: 10.3389/fncir.2012.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajo VM, Nodal FR, Moore DR, King AJ. The descending corticocollicular pathway mediates learning-induced auditory plasticity. Nat Neurosci. 2010;13:253–260. doi: 10.1038/nn.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao S, Chang EF, Davis JD, Gobeske KT, Merzenich MM. Progressive degradation and subsequent refinement of acoustic representations in the adult auditory cortex. J Neurosci. 2003;23:10765–10775. doi: 10.1523/JNEUROSCI.23-34-10765.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barak O, Tsodyks M. Working models of working memory. Curr Opin Neurobiol. 2014;25:20–24. doi: 10.1016/j.conb.2013.10.008. [DOI] [PubMed] [Google Scholar]
- Bear MF, Singer W. Modulation of visual cortical plasticity by acetylcholine and noradrenaline. Nature. 1986;320:172–176. doi: 10.1038/320172a0. [DOI] [PubMed] [Google Scholar]
- Bigl V, Woolf NJ, Butcher LL. Cholinergic projections from the basal forebrain to frontal, parietal, temporal, occipital, and cingulate cortices: a combined fluorescent tracer and acetylcholinesterase analysis. Brain Res Bull. 1982;8:727–749. doi: 10.1016/0361-9230(82)90101-0. [DOI] [PubMed] [Google Scholar]
- Chen G, Yan J. Cholinergic modulation incorporated with a tone presentation induces frequency-specific threshold decreases in the auditory cortex of the mouse. Eur J Neurosci. 2007;25:1793–1803. doi: 10.1111/j.1460-9568.2007.05432.x. [DOI] [PubMed] [Google Scholar]
- Delmas P, Brown DA. Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat Rev Neurosci. 2005;6:850–862. doi: 10.1038/nrn1785. [DOI] [PubMed] [Google Scholar]
- Dembrow NC, Chitwood RA, Johnston D. Projection-specific neuromodulation of medial prefrontal cortex neurons. J Neurosci. 2010;30:16922–16937. doi: 10.1523/JNEUROSCI.3644-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doucet JR, Molavi DL, Ryugo DK. The source of corticocollicular and corticobulbar projections in area Te1 of the rat. Exp Brain Res. 2003;153:461–466. doi: 10.1007/s00221-003-1604-4. [DOI] [PubMed] [Google Scholar]
- Edeline JM, Hars B, Maho C, Hennevin E. Transient and prolonged facilitation of tone-evoked responses induced by basal forebrain stimulations in the rat auditory cortex. Exp Brain Res. 1994;97:373–386. doi: 10.1007/BF00241531. [DOI] [PubMed] [Google Scholar]
- Eggermann E, Feldmeyer D. Cholinergic filtering in the recurrent excitatory microcircuit of cortical layer 4. Proc Natl Acad Sci U S A. 2009;106:11753–11758. doi: 10.1073/pnas.0810062106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egorov AV, Hamam BN, Fransén E, Hasselmo ME, Alonso AA. Graded persistent activity in entorhinal cortex neurons. Nature. 2002;420:173–178. doi: 10.1038/nature01171. [DOI] [PubMed] [Google Scholar]
- Egorov AV, Unsicker K, von Bohlen und Halbach O. Muscarinic control of graded persistent activity in lateral amygdala neurons. Eur J Neurosci. 2006;24:3183–3194. doi: 10.1111/j.1460-9568.2006.05200.x. [DOI] [PubMed] [Google Scholar]
- Everitt BJ, Robbins TW. Central cholinergic systems and cognition. Annu Rev Psychol. 1997;48:649–684. doi: 10.1146/annurev.psych.48.1.649. [DOI] [PubMed] [Google Scholar]
- Fransén E, Tahvildari B, Egorov AV, Hasselmo ME, Alonso AA. Mechanism of graded persistent cellular activity of entorhinal cortex layer v neurons. Neuron. 2006;49:735–746. doi: 10.1016/j.neuron.2006.01.036. [DOI] [PubMed] [Google Scholar]
- Froemke RC, Merzenich MM, Schreiner CE. A synaptic memory trace for cortical receptive field plasticity. Nature. 2007;450:425–429. doi: 10.1038/nature06289. [DOI] [PubMed] [Google Scholar]
- Games KD, Winer JA. Layer V in rat auditory cortex: projections to the inferior colliculus and contralateral cortex. Hear Res. 1988;34:1–25. doi: 10.1016/0378-5955(88)90047-0. [DOI] [PubMed] [Google Scholar]
- Gao WJ, Zheng ZH. Target-specific differences in somatodendritic morphology of layer V pyramidal neurons in rat motor cortex. J Comp Neurol. 2004;476:174–185. doi: 10.1002/cne.20224. [DOI] [PubMed] [Google Scholar]
- Gee S, Ellwood I, Patel T, Luongo F, Deisseroth K, Sohal VS. Synaptic activity unmasks dopamine D2 receptor modulation of a specific class of layer V pyramidal neurons in prefrontal cortex. J Neurosci. 2012;32:4959–4971. doi: 10.1523/JNEUROSCI.5835-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulledge AT, Stuart GJ. Cholinergic inhibition of neocortical pyramidal neurons. J Neurosci. 2005;25:10308–10320. doi: 10.1523/JNEUROSCI.2697-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulledge AT, Park SB, Kawaguchi Y, Stuart GJ. Heterogeneity of phasic cholinergic signaling in neocortical neurons. J Neurophysiol. 2007;97:2215–2229. doi: 10.1152/jn.00493.2006. [DOI] [PubMed] [Google Scholar]
- Gulledge AT, Bucci DJ, Zhang SS, Matsui M, Yeh HH. M1 receptors mediate cholinergic modulation of excitability in neocortical pyramidal neurons. J Neurosci. 2009;29:9888–9902. doi: 10.1523/JNEUROSCI.1366-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haj-Dahmane S, Andrade R. Muscarinic activation of a voltage-dependent cation nonselective current in rat association cortex. J Neurosci. 1996;16:3848–3861. doi: 10.1523/JNEUROSCI.16-12-03848.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haj-Dahmane S, Andrade R. Muscarinic receptors regulate two different calcium-dependent non-selective cation currents in rat prefrontal cortex. Eur J Neurosci. 1999;11:1973–1980. doi: 10.1046/j.1460-9568.1999.00612.x. [DOI] [PubMed] [Google Scholar]
- Harvey SC, Luetje CW. Determinants of competetive antagonist sensitivity on neuronal nicotinic receptor beta subunits. J Neurosci. 1996;16:3798–3806. doi: 10.1523/JNEUROSCI.16-12-03798.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey SC, Maddox FN, Luetje CW. Multiple determinants of dihydro-beta-erythroidine sensitivity on rat neuronal nicotinic receptor alpha subunits. J Neurochem. 1996;67:1953–1959. doi: 10.1046/j.1471-4159.1996.67051953.x. [DOI] [PubMed] [Google Scholar]
- Hasselmo ME, Sarter M. Modes and models of forebrain cholinergic neuromodulation of cognition. Neuropsychopharmacology. 2011;36:52–73. doi: 10.1038/npp.2010.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasselmo ME, Stern CE. Mechanisms underlying working memory for novel information. Trends Cogn Sci. 2006;10:487–493. doi: 10.1016/j.tics.2006.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrick T, Waters J. Acetylcholine excites neocortical pyramidal neurons via nicotinic receptors. J Neurophysiol. 2015;113:2195–2209. doi: 10.1152/jn.00716.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi A, Middleton JW, Anderson CT, Borges K, Suter BA, Shepherd GM, Tzounopoulos T. Cell-specific activity-dependent fractionation of layer 2/3–>5B excitatory signaling in mouse auditory cortex. J Neurosci. 2015;35:3112–3123. doi: 10.1523/JNEUROSCI.0836-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilgard MP, Merzenich MM. Cortical map reorganization enabled by nucleus basalis activity. Science. 1998a;279:1714–1718. doi: 10.1126/science.279.5357.1714. [DOI] [PubMed] [Google Scholar]
- Kilgard MP, Merzenich MM. Plasticity of temporal information processing in the primary auditory cortex. Nat Neurosci. 1998b;1:727–731. doi: 10.1038/3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolisnyk B, Guzman MS, Raulic S, Fan J, Magalhães AC, Feng G, Gros R, Prado VF, Prado MA. ChAT-ChR2-EYFP mice have enhanced motor endurance but show deficits in attention and several additional cognitive domains. J Neurosci. 2013;33:10427–10438. doi: 10.1523/JNEUROSCI.0395-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach ND, Nodal FR, Cordery PM, King AJ, Bajo VM. Cortical cholinergic input is required for normal auditory perception and experience-dependent plasticity in adult ferrets. J Neurosci. 2013;33:6659–6671. doi: 10.1523/JNEUROSCI.5039-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AT, Gee SM, Vogt D, Patel T, Rubenstein JL, Sohal VS. Pyramidal neurons in prefrontal cortex receive subtype-specific forms of excitation and inhibition. Neuron. 2014;81:61–68. doi: 10.1016/j.neuron.2013.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann J, Nagy JI, Atmadia S, Fibiger HC. The nucleus basalis magnocellularis: the origin of a cholinergic projection to the neocortex of the rat. Neuroscience. 1980;5:1161–1174. doi: 10.1016/0306-4522(80)90195-5. [DOI] [PubMed] [Google Scholar]
- Li N, Chen TW, Guo ZV, Gerfen CR, Svoboda K. A motor cortex circuit for motor planning and movement. Nature. 2015;519:51–56. doi: 10.1038/nature14178. [DOI] [PubMed] [Google Scholar]
- McCormick DA, Prince DA. Two types of muscarinic response to acetylcholine in mammalian cortical neurons. Proc Natl Acad Sci U S A. 1985;82:6344–6348. doi: 10.1073/pnas.82.18.6344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick DA, Prince DA. Mechanisms of action of acetylcholine in the guinea-pig cerebral cortex in vitro. J Physiol. 1986;375:169–194. doi: 10.1113/jphysiol.1986.sp016112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick DA, Williamson A. Convergence and divergence of neurotransmitter action in human cerebral cortex. Proc Natl Acad Sci U S A. 1989;86:8098–8102. doi: 10.1073/pnas.86.20.8098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesulam MM, Mufson EJ, Levey AI, Wainer BH. Cholinergic innervation of cortex by the basal forebrain: cytochemistry and cortical connections of the septal area, diagonal band nuclei, nucleus basalis (substantia innominata), and hypothalamus in the rhesus monkey. J Comp Neurol. 1983;214:170–197. doi: 10.1002/cne.902140206. [DOI] [PubMed] [Google Scholar]
- Metherate R. Nicotinic acetylcholine receptors in sensory cortex. Learn Mem. 2004;11:50–59. doi: 10.1101/lm.69904. [DOI] [PubMed] [Google Scholar]
- Metherate R. Functional connectivity and cholinergic modulation in auditory cortex. Neurosci Biobehav Rev. 2011;35:2058–2063. doi: 10.1016/j.neubiorev.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metherate R, Ashe JH. Nucleus basalis stimulation facilitates thalamocortical synaptic transmission in the rat auditory cortex. Synapse. 1993;14:132–143. doi: 10.1002/syn.890140206. [DOI] [PubMed] [Google Scholar]
- Metherate R, Cox CL, Ashe JH. Cellular bases of neocortical activation: modulation of neural oscillations by the nucleus basalis and endogenous acetylcholine. J Neurosci. 1992;12:4701–4711. doi: 10.1523/JNEUROSCI.12-12-04701.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishima M, Kawaguchi Y. Recurrent connection patterns of corticostriatal pyramidal cells in frontal cortex. J Neurosci. 2006;26:4394–4405. doi: 10.1523/JNEUROSCI.0252-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newberry NR, Priestley T. Pharmacological differences between two muscarinic responses of the rat superior cervical ganglion in vitro. Br J Pharmacol. 1987;92:817–826. doi: 10.1111/j.1476-5381.1987.tb11386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poorthuis RB, Bloem B, Schak B, Wester J, de Kock CP, Mansvelder HD. Layer-specific modulation of the prefrontal cortex by nicotinic acetylcholine receptors. Cereb Cortex. 2013;23:148–161. doi: 10.1093/cercor/bhr390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quick MW, Lester RA. Desensitization of neuronal nicotinic receptors. J Neurobiol. 2002;53:457–478. doi: 10.1002/neu.10109. [DOI] [PubMed] [Google Scholar]
- Rahman J, Berger T. Persistent activity in layer 5 pyramidal neurons following cholinergic activation of mouse primary cortices. Eur J Neurosci. 2011;34:22–30. doi: 10.1111/j.1460-9568.2011.07736.x. [DOI] [PubMed] [Google Scholar]
- Reed A, Riley J, Carraway R, Carrasco A, Perez C, Jakkamsetti V, Kilgard MP. Cortical map plasticity improves learning but is not necessary for improved performance. Neuron. 2011;70:121–131. doi: 10.1016/j.neuron.2011.02.038. [DOI] [PubMed] [Google Scholar]
- Sheets PL, Suter BA, Kiritani T, Chan CS, Surmeier DJ, Shepherd GM. Corticospinal-specific HCN expression in mouse motor cortex: I(h)-dependent synaptic integration as a candidate microcircuit mechanism involved in motor control. J Neurophysiol. 2011;106:2216–2231. doi: 10.1152/jn.00232.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd GM. Corticostriatal connectivity and its role in disease. Nat Rev Neurosci. 2013;14:278–291. doi: 10.1038/nrn3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater BJ, Willis AM, Llano DA. Evidence for layer-specific differences in auditory corticocollicular neurons. Neuroscience. 2013;229:144–154. doi: 10.1016/j.neuroscience.2012.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun YJ, Kim YJ, Ibrahim LA, Tao HW, Zhang LI. Synaptic mechanisms underlying functional dichotomy between intrinsic-bursting and regular-spiking neurons in auditory cortical layer 5. J Neurosci. 2013;33:5326–5339. doi: 10.1523/JNEUROSCI.4810-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suter BA, O'Connor T, Iyer V, Petreanu LT, Hooks BM, Kiritani T, Svoboda K, Shepherd GM. Ephus: multipurpose data acquisition software for neuroscience experiments. Front Neural Circuits. 2010;4:100. doi: 10.3389/fncir.2010.00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suter BA, Migliore M, Shepherd GM. Intrinsic electrophysiology of mouse corticospinal neurons: a class-specific triad of spike-related properties. Cereb Cortex. 2013;23:1965–1977. doi: 10.1093/cercor/bhs184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XJ. Synaptic reverberation underlying mnemonic persistent activity. Trends Neurosci. 2001;24:455–463. doi: 10.1016/S0166-2236(00)01868-3. [DOI] [PubMed] [Google Scholar]
- Weinberger NM. The nucleus basalis and memory codes: auditory cortical plasticity and the induction of specific, associative behavioral memory. Neurobiol Learn Mem. 2003;80:268–284. doi: 10.1016/S1074-7427(03)00072-8. [DOI] [PubMed] [Google Scholar]
- Weinberger NM, Miasnikov AA, Chen JC. The level of cholinergic nucleus basalis activation controls the specificity of auditory associative memory. Neurobiol Learn Mem. 2006;86:270–285. doi: 10.1016/j.nlm.2006.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wester JC, Contreras D. Differential modulation of spontaneous and evoked thalamocortical network activity by acetylcholine level in vitro. J Neurosci. 2013;33:17951–17966. doi: 10.1523/JNEUROSCI.1644-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Z, Huguenard JR, Prince DA. Cholinergic switching within neocortical inhibitory networks. Science. 1998;281:985–988. doi: 10.1126/science.281.5379.985. [DOI] [PubMed] [Google Scholar]
- Yamashita T, Isa T. Ca2+-dependent inward current induced by nicotinic receptor activation depends on Ca2+/calmodulin-CaMKII pathway in dopamine neurons. Neurosci Res. 2003a;47:225–232. doi: 10.1016/S0168-0102(03)00201-3. [DOI] [PubMed] [Google Scholar]
- Yamashita T, Isa T. Fulfenamic acid sensitive, Ca(2+)-dependent inward current induced by nicotinic acetylcholine receptors in dopamine neurons. Neurosci Res. 2003b;46:463–473. doi: 10.1016/S0168-0102(03)00128-7. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Séguéla P. Metabotropic induction of persistent activity in layers II/III of anterior cingulate cortex. Cereb Cortex. 2010;20:2948–2957. doi: 10.1093/cercor/bhq043. [DOI] [PubMed] [Google Scholar]
- Zhao S, Ting JT, Atallah HE, Qiu L, Tan J, Gloss B, Augustine GJ, Deisseroth K, Luo M, Graybiel AM, Feng G. Cell type-specific channelrhodopsin-2 transgenic mice for optogenetic dissection of neural circuitry function. Nat Methods. 2011;8:745–752. doi: 10.1038/nmeth.1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolles G, Wagner E, Lampert A, Sutor B. Functional expression of nicotinic acetylcholine receptors in rat neocortical layer 5 pyramidal cells. Cereb Cortex. 2009;19:1079–1091. doi: 10.1093/cercor/bhn158. [DOI] [PubMed] [Google Scholar]