

Abstract
Axon regeneration after spinal cord injury (SCI) fails due to neuron-intrinsic mechanisms and extracellular barriers including inflammation. microRNA (miR)-155–5p is a small, noncoding RNA that negatively regulates mRNA translation. In macrophages, miR-155-5p is induced by inflammatory stimuli and elicits a response that could be toxic after SCI. miR-155 may also independently alter expression of genes that regulate axon growth in neurons. Here, we hypothesized that miR-155 deletion would simultaneously improve axon growth and reduce neuroinflammation after SCI by acting on both neurons and macrophages. New data show that miR-155 deletion attenuates inflammatory signaling in macrophages, reduces macrophage-mediated neuron toxicity, and increases macrophage-elicited axon growth by ∼40% relative to control conditions. In addition, miR-155 deletion increases spontaneous axon growth from neurons; adult miR-155 KO dorsal root ganglion (DRG) neurons extend 44% longer neurites than WT neurons. In vivo, miR-155 deletion augments conditioning lesion-induced intraneuronal expression of SPRR1A, a regeneration-associated gene; ∼50% more injured KO DRG neurons expressed SPRR1A versus WT neurons. After dorsal column SCI, miR-155 KO mouse spinal cord has reduced neuroinflammation and increased peripheral conditioning-lesion-enhanced axon regeneration beyond the epicenter. Finally, in a model of spinal contusion injury, miR-155 deletion improves locomotor function at postinjury times corresponding with the arrival and maximal appearance of activated intraspinal macrophages. In miR-155 KO mice, improved locomotor function is associated with smaller contusion lesions and decreased accumulation of inflammatory macrophages. Collectively, these data indicate that miR-155 is a novel therapeutic target capable of simultaneously overcoming neuron-intrinsic and neuron-extrinsic barriers to repair after SCI.
SIGNIFICANCE STATEMENT Axon regeneration after spinal cord injury (SCI) fails due to neuron-intrinsic mechanisms and extracellular barriers, including inflammation. Here, new data show that deleting microRNA-155 (miR-155) affects both mechanisms and improves repair and functional recovery after SCI. Macrophages lacking miR-155 have altered inflammatory capacity, which enhances neuron survival and axon growth of cocultured neurons. In addition, independent of macrophages, adult miR-155 KO neurons show enhanced spontaneous axon growth. Using either spinal cord dorsal column crush or contusion injury models, miR-155 deletion improves indices of repair and recovery. Therefore, miR-155 has a dual role in regulating spinal cord repair and may be a novel therapeutic target for SCI and other CNS pathologies.
Keywords: axon regeneration, microRNA, neuroinflammation, neuroprotection, spinal cord injury
Introduction
Injured mammalian CNS axons fail to regenerate successfully. Both neuron-intrinsic and neuron-extrinsic mechanisms contribute to axon regeneration failure. After injury, CNS neurons fail to upregulate regeneration-associated genes (RAGs), those genes encoding proteins that promote cytoskeletal dynamics and growth-associated changes (Skene and Willard, 1981; Jacobson et al., 1986; Tetzlaff et al., 1991; Bonilla et al., 2002; Mason et al., 2002; Khazaei et al., 2014). Numerous neuron-extrinsic barriers also conspire to prevent efficient CNS growth and repair (McKeon et al., 1991; Bradbury et al., 2002; Brambilla et al., 2005; Donnelly and Popovich, 2008; Pineau et al., 2010; Alilain et al., 2011; Gaudet and Popovich, 2014). For example, inflammatory macrophages can cause axonal injury or inhibit axon regeneration after spinal cord injury (SCI) (Popovich et al., 1997; Kigerl et al., 2006; Kigerl et al., 2009; Beck et al., 2010; Prüss et al., 2011; Pool et al., 2012; Kroner et al., 2014). Therefore, therapies that boost intrinsic axon growth programs or that limit macrophage inflammatory signaling could improve spinal cord repair (Kobayashi et al., 1997; Popovich et al., 1999; Ramer et al., 2000; Liu et al., 2010; Sun et al., 2011; Bartus et al., 2014).
MicroRNAs (miRs) are ∼22-oligonucleotide sequences that target specific mRNAs for translational repression and/or cleavage. A key 6- to 7-oligonucleotide “seed sequence” in the miR binds a complementary sequence in the 3′-untranslated region of target mRNAs, thereby preventing translation (Bartel, 2009). The present study focuses on miR-155. The MIR155 host gene (MIR155HG) codes for both miR-155-5p and miR-155-3p and these sequences are conserved across species, including humans and rodents (Mashima, 2015). Whereas little is known about miR-155-3p, miR-155-5p has been studied extensively in inflammatory diseases and cancer. In the CNS, miR-155-5p can be found in microglia/macrophages (Tili et al., 2007; Cardoso et al., 2012), astrocytes (Tarassishin et al., 2011), and neurons (Herai et al., 2014). The therapeutic potential of targeting miR-155 is exemplified by the fact that miR-155 removal improves repair and recovery in other animal models of CNS pathology, including amyotrophic lateral sclerosis (Koval et al., 2013; Butovsky et al., 2015), multiple sclerosis (Murugaiyan et al., 2011; Moore et al., 2013), and central neuroinflammation (Lopez-Ramirez et al., 2014).
miR-155-5p modulates the stability and translation of mRNAs implicated in inflammation (Tili et al., 2007; Worm et al., 2009; Liu et al., 2011b; Fonken et al., 2016) and axon growth (Wang et al., 2013; Varendi et al., 2014). In macrophages and microglia, damage- or pathogen-associated molecular patterns bind toll-like receptors and elicit inflammatory signaling pathways (Graff et al., 2012; Freilich et al., 2013; Moore et al., 2013). Blocking miR-155-5p limits these inflammatory responses in macrophages (Cai et al., 2012; Nazari-Jahantigh et al., 2012; Jablonski et al., 2016) and microglia (Cardoso et al., 2012). Although the effects of miR-155-5p on axon growth are unclear, indirect evidence and in silico prediction models (Lewis et al., 2003; John et al., 2004; O'Connell et al., 2009; Liu et al., 2011b) suggest that miR-155-5p could bind various RAG-related mRNAs, which could restrict the synthesis of proteins that enhance axon growth.
Here, we tested the hypothesis that miR-155 deletion reduces inflammatory-mediated neurotoxicity and improves axon growth both by limiting activation of inflammatory signaling in macrophages and by increasing the expression of neuron-intrinsic RAGs. New data show that miR-155-deficient macrophages are less inflammatory and neurotoxic and promote axon growth through a contact- or proximity-dependent mechanism. In addition, deleting miR-155 in neurons augments intrinsic axon growth potential both in vitro and in vivo. Indeed, miR-155 deletion reduced macrophage/microglia inflammation by 46% with a concomitant increase in sensory axon growth beyond the lesion epicenter in an in vivo model of dorsal column SCI in which neuron-intrinsic axon growth potential was primed using a peripheral conditioning lesion. After SCI, miR-155 deletion reduces intraspinal inflammation, increases neuroprotection, and improves functional recovery. Together, these data implicate miR-155 as a novel regulator of destructive postinjury neuroinflammation and as a neuron-intrinsic “regeneration-inhibitory gene.”
Materials and Methods
Animals and surgery
All housing, surgery, and postoperative care conformed to guidelines set by The Ohio State University Institutional Animal Care and Use Committee. All animals were fed standard chow and filtered tap water ad libitum and maintained on a 12:12 light/dark cycle. miR-155 KO mice were a generous gift from Dr. Caroline Whitacre. All mice used were littermates generated from crossing heterozygote parents (heterozygotes were offspring of WT × miR-155 KO breeding pairs: C57BL/6J WT mice: stock 000664; KO mice: stock 007745, The Jackson Laboratory) (Thai et al., 2007; Gaudet et al., 2016). For experiments with WT and KO mice (all female), mice from both genotypes were randomly assigned to experimental groups and individuals involved in data analysis were blinded to group designations throughout all stages of the experiment, including animal care, behavior, tissue dissection, tissue sectioning, imaging, and data analysis. All mice were anesthetized with an intraperitoneal injection of ketamine (137.5 mg/kg; obtained from The Ohio State University Department of Pharmacy; JHP Pharmaceuticals) and xylazine (60 mg/kg; AnaSed, Lloyd Laboratories) and were pretreated with prophylactic antibiotics (gentamicin sulfate, 5 mg/kg, s.c., in 0.1 ml; Butler Schein). Postoperative animal care included daily administration of prophylactic gentamicin antibiotic and subcutaneous injection of Ringer's solution (2, 2, 1, 1, and 1 ml) for the first 5 d postinjury (dpi) to prevent dehydration. Bladders were voided manually twice daily. Animals were monitored daily for infection or signs of abnormal recovery. At the appropriate postinjury time points, mice were injected with an overdose of ketamine (300 mg/kg) and xylazine (150 mg/kg) before perfusion with 4% paraformaldehyde.
SCI.
Mice were treated with prophylactic antibiotics as above. A T9 laminectomy was performed before SCI. The periosteum, but not the dura, was removed for all surgeries. Animals were subjected to a moderately severe contusion injury (75 kDyn) using the Infinite Horizons impactor (Precision Systems and Instrumentation). Post hoc analysis of primary injury data revealed no significant differences in impact force between groups (WT force: 78 ± 1 kDyn, KO force: 76 ± 1 kDyn; p > 0.05). Postoperative animal care was completed as described above. To measure postinjury changes in miR-155-5p expression, 8- to 12-week-old female C57BL/6J mice (The Jackson Laboratory) were randomized into uninjured (sham surgery) control or spinal contusion injury groups. SCI mice survived 1, 3, 7, 14, or 28 dpi (n = 4 per group). For analysis of functional recovery and lesion morphometrics, a separate group of miR-155 KO mice and female WT control littermate mice were randomized into groups and received SCI as described above (n = 11 WT, n = 6 KO; 2–5 months old). Locomotor recovery was assessed before injury and at 1, 4, 7, 10, 14, 21, and 28 dpi using the Basso Mouse Scale (BMS) (Basso et al., 2006). At 28 dpi, mice were perfused and spinal cords were collected for morphometric and immunohistochemical analyses.
Spinal cord dorsal column injury with sciatic nerve-conditioning lesion.
Female miR-155 KO and WT littermate mice were used (n = 5 WT, n = 9 KO; 2–5 months old) in this experiment. Breeding produces unpredictable male/female and WT/KO ratios, resulting in unbalanced group sizes. To enhance the regenerative capacity of sensory axons in the gracile funiculus, mice received a bilateral sciatic nerve injury (i.e., “conditioning” lesion). Six days before dorsal column crush injury, sciatic nerves were exposed, tightly ligated using 7–0 silk suture (to prevent peripheral axon regeneration and maintain neuronal growth responses), and then transected distal to the suture. Animals received prophylactic gentamicin sulfate and 2 ml of subcutaneous Ringer's solution and were monitored daily.
Six days after sciatic nerve injury, animals received a C5 spinal dorsal column crush injury. Mice were anesthetized (see above) and the cervical spinal cord was exposed. By maneuvering the animal, a space between the C5 and C6 lamina was exposed without removing any vertebral lamina. Using a needle, a small transverse hole was cut in the dura. “Guide” holes were created by inserting no. 5 forceps at the spinal cord midline, held at a width of 0.9–1.0 mm with tips marked to a depth of 600 μm. Next, no. 55 forceps (labeled for depth) were inserted into the holes and closed for 3 × 10 s, thereby crushing both ascending sensory and descending corticospinal tract axons in the spinal cord dorsal column. While still anesthetized, mice received injections of adeno-associated virus 2 (AAV2)–green fluorescent protein (GFP) into both sciatic nerves (proximal to ligation) to label the central projections of sensory axons growing into/through the lesion site. Mice received ∼0.5 μl of AAV2-GFP into each sciatic nerve branch (2 branches per side, so 1 μl of AAV2-GFP per side) using a Hamilton syringe. In contrast to severe contusion injury (see below), all animals receiving dorsal column crush injury recover bladder function within 1 week. Regardless, animals were monitored daily beyond 1 week for abnormal patterns of recovery from surgery/injury. All mice were perfused at 32 dpi.
Tissue processing and immunohistochemistry
Under terminal anesthesia, mice were perfused intracardially with PBS (0.1 m, pH 7.4), followed by 4% paraformaldehyde in 0.1% phosphate buffer. Fixed spinal cords were placed in 4% paraformaldehyde overnight and then switched to 30% sucrose the next day before tissue blocking. To prepare tissues for cryosectioning, spinal cords and dorsal root ganglia (DRGs) were flash frozen in optimal cutting temperature compound. Spinal cord tissue (dorsal column injury epicenters) was sectioned horizontally at 12 μm and DRGs were sectioned at 10 μm, then slides were stored at −20°C. When in a single block, spinal cords from different time points or treatment groups were randomly distributed across blocks to ensure uniformity of staining. In addition, to ensure that all analyses were performed in a blinded manner, animal numbers were randomized; WT/KO mouse numbers were intermingled and indistinguishable without the code. For horizontal spinal cord sections, a PBS drop was applied to the slide immediately before picking up the section to prevent tissue folding and bubbles. For immunohistochemistry, slides were washed with 0.1 m PBS before applying a blocking solution (10% normal goat serum or 10% normal donkey serum, depending on secondary used) for 45 min. Primary antibodies (in 0.1 m PBS + 0.2% Triton-X) were applied overnight: rabbit anti-Iba1 (1:1000; 019-19741, WAKO), rat anti-CD86 (1:100; 553689, BD Biosciences), goat anti-CD206 (1:100; AF2535, R&D Systems), rabbit anti-GFP (1:500; A11122, Life Technologies), and chicken anti-GFP (1:500; GFP-1020, Aves). The next day, slides were washed 3 times with PBS and then the following secondary antibodies (1:500 in PBS/0.2% Triton X-100) with DAPI were added for 2 h: Alexa Fluor 546-conjugated goat anti-rat (A11081), goat-anti-rabbit (A11035), and donkey anti-goat (A11056); or 488-conjugated goat anti-rabbit (A11034) and goat anti-chicken (A11039) (all from Life Technologies). Slides were washed with PBS before coverslipping with Immu-Mount (9990402, Thermo Scientific).
Image analysis
Images were collected using a Zeiss Axioplan 2 microscope. Confocal images were taken on an Olympus FV1000 filter confocal microscope. At the time of analysis, all researchers were blinded with respect to treatment group using random animal numbers that concealed genotype. Analysis of microscope images (fluorescent density, etc.) was performed using ImageJ or MetaMorph (Molecular Devices).
Lesion size: spinal contusion injury.
Cross-sections were cut every 200 μm rostral and caudal to epicenter, collected on slides, and double stained with Eriochrome cyanine (myelin) and anti-neurofilament antibodies. The epicenter for each mouse was defined as the spinal cord section with the least amount of spared myelin and axons. The border between intact and injured tissue (devoid of staining or exhibiting clear pathology) was identified and quantified at each level using the Cavalieri method, as described previously then were analyzed as described previously (Kigerl et al., 2006; Howard and Reed, 1998).
Iba1 and CD16/32 analyses in contused spinal cord.
Three to four images of both lesion epicenter and rostral dorsal columns (just rostral to frank lesion) were collected for each mouse with contusion SCI. Within these regions, computerized image analysis (MetaMorph) was used to threshold pixel area that was positively stained for Iba1 or CD16/32. These data were expressed as a percentage of the total region of interest (e.g., epicenter or dorsal column area in which image was captured).
DRG analyses: conditioning lesion and dorsal column injury study.
Three to four images per L4 DRG were captured for each marker for all DRG analyses. Results from each image were averaged for each animal. For assessing RAG expression, MetaMorph software was used to manually circle DRG neurons (>100 neurons per DRG per marker). DRG neuron diameter and intensity of ATF3/GFP or SPRR1A/GFP expression were recorded. The threshold of RAG or GFP positivity was defined by averaging the intensity of two slightly negative and two slightly positive neurons for each marker; the average of these four cells was the threshold of positivity for that image. The percentage of cells positive for each marker was then recorded. The size distribution of DRG neuron diameters was transformed using recursive translation, a stereological counting method that reconstructs cell populations based on a size distribution of analyzed profiles (Rose and Rohrlich, 1988; Ramer et al., 2001; McGraw et al., 2005). There were no significant differences in the neuron size distribution (or size distribution of cells expressing the markers) between genotypes (data not shown). Density of Iba1, CD86, and CD206 was examined by defining a region of interest within areas containing neurons. The image was thresholded for positive labeling and the result was expressed as a percentage of the total region of interest (Gaudet et al., 2015).
SCI epicenter Iba1 density analysis: conditioning lesion and dorsal column injury study.
For epicenter analysis, only spinal cords sectioned horizontally were used for analysis (n = 4 WT, n = 8 KO). Three to four images were captured for each epicenter and then a montage was created for Iba1 density analysis. Image montages consisted of 20× images stitched together (Adobe Photoshop 5.0) to allow accurate analysis of Iba1 density between 2000 μm rostral and 2000 μm caudal to epicenter. The epicenter was identified and labeled in each section. Using ImageJ software, Iba1 images were thresholded and then Iba1 density was measured systematically in 200 μm (rostral–caudal) by 100 μm (lateral) boxes centered on the midline (where axons also resided). To ensure that Iba1 measurements were relevant to axon plasticity, only sections containing axons were analyzed. Data from 200-μm-long regions are expressed as the middle of the region (e.g., a box extending from 0 to 200 μm is expressed as 100 μm in the figure).
Confirming complete injury of sensory axons in dorsal columns: conditioning lesion and dorsal column injury study.
Transverse spinal cord sections rostral (∼C2) and caudal (∼C8) to injury epicenter were examined for GFP immunoreactivity. The gracile fasciculus was identified; GFP+ axons were thresholded and the percentage GFP+ area was recorded (three images rostral and caudal were analyzed in each animal). No GFP+ axons were identified in the spinal cords of any mouse rostral to the injury (at the spinal C2 level), confirming that the C5 dorsal column crush injury successfully severed all ascending GFP+ gracile axons in all WT and KO animals.
Axon growth/dieback quantification: conditioning lesion and dorsal column injury study.
A montage of four to five images that encompassed the lesion was created for each animal. ImageJ was used for density analysis of GFP+ axons. Areas of autofluorescence (defined by conspicuous fluorescent spots/blobs that were clearly not axons present in all three channels, lacking axon morphology or a DAPI+ nucleus; commonly seen in lesion epicenters) were removed and then images were thresholded to identify GFP+ axons. Total GFP+ area was recorded in 200-μm-long boxes for each image and then added together for each animal. The GFP+ area was normalized within each animal by expressing GFP+ area as a percentage of maximal density (100%), which occurred caudal to epicenter in all animals. Data from 200-μm-long regions are expressed as the middle of the region (e.g., the box extending from 0 to 200 μm is expressed as 100 μm in the figure). For location of the farthest rostral axons, the location (vs epicenter) of 100+ of the most rostral axons was recorded. The 100 furthest rostral axons were averaged for each animal.
Cell culture and analysis
Bone-marrow-derived macrophage (BMDM) culture.
BMDM cultures were generated as described previously (Longbrake et al., 2007) from 2- to 5-month-old female WT or miR-155 KO mice. BMDMs were collected from both tibias and femurs using aseptic technique. Marrow cores were flushed into conical tubes using a syringe with a 26-gauge needle filled with DMEM (Life Technologies, 10313). Cell suspensions were centrifuged and the cells were resuspended in red blood cell lysis solution (0.15 m NH4Cl, 10 mm KHCO3, and 0.1 mm Na2EDTA, pH 7.4) for 3 min. After a final wash with medium, cells were plated and cultured in T-75 flasks (8–10 × 106 cells per flask) in 10 ml of DMEM supplemented with 20% L-cell-conditioned medium (sL929), 10% fetal bovine serum (FBS; Life Technologies, 16000-044), 1% Glutamax (Life Technologies, 35050-061), 1% HEPES (Sigma-Aldrich, H0887), 0.05% gentamicin (Life Technologies, 15710), and 0.001% -mercaptoethanol. sL929, which contains macrophage-colony-stimulating factor and is required to promote differentiation of bone marrow cells into macrophages (7–10 d; Burgess et al., 1985). The medium was replaced at 2, 4, and 6 d after culture. At 7 d after culture, macrophages were scraped and replated onto 24-well plates (cell lysates and media transfer) or 96-well plates (macrophage-neuron coculture) at a density of 106 cells/ml in DMEM supplemented with 10% FBS, 1% Glutamax and 1% gentamicin for 6–24 h. To elicit macrophage differentiation into M1 or M2 cells, 8 d after culture, macrophages were exposed to media alone, LPS (100 ng/ml; Sigma-Aldrich, L2880) plus IFN-γ (20 ng/ml; eBioscience, BMS326), or IL-4 (20 ng/ml; eBioscience, 14-80410-62), respectively, for 24–48 h.
Western blot.
Cells from WT and miR-155 KO mice were treated with media, IFN-γ/LPS, or IL-4. After 24 h stimulation, macrophages were washed once with PBS and then lysed and extracted using 100–150 μl of RIPA buffer (Pierce, 89900) containing protease inhibitors (Pierce, 87786). For each genotype, n = 6 mice were used. For Ym1 analysis, two M0 and two M1 samples were not analyzed due to patchy/uneven spots (e.g., due to air bubbles) on the exposed membrane. Sample protein concentrations were assessed using the Coomassie Plus Protein Assay Kit (Pierce, 23236). A total of 20 mg of protein from each sample was diluted in NuPAGE LDS sample buffer (Life Technologies, NP0008) before being loaded into a 4–12% Bis-Tris gel (Life Technologies, NP0336BOX) and run for 45 min at 200 V. Proteins were transferred to a nitrocellulose membrane (Bio-Rad, 162-01115) for 80 min at 30 V. The membrane was blocked with 5% bovine serum albumin (Fisher, BP9703-100) for 45 min and then incubated overnight at 4°C with primary antibody: mouse anti-iNOS (1:500; BD Biosciences, 610431); rabbit anti-Ym1 (1:500; Stem Cell Technologies, 01404); goat anti-arginase-1 (1:500; Santa Cruz Biotechnology, sc-18354); goat anti-CD206 (1:500; R&D Systems, AF2535); and rat anti-α-tubulin (1:80,000; Serotec MCA77G). The next day, the membrane was washed 3 times with PBS/Tween20 before incubating for 2 h at room temperature with secondary antibodies (all diluted 1:1000): horseradish peroxidase (HRP)-conjugated rabbit anti-goat (Jackson Immunoresearch, 305-035-003), goat anti-rat (Jackson Immunoresearch, 115-035-174), and goat anti-mouse (Jackson Immunoresearch, 112-035-003). Chemiluminescent HRP substrate (Pierce, 34078) was used to develop the blot, and the membrane was imaged and analyzed on a Kodak Image Station 4000MM PRO. Band densities were analyzed using ImageJ. For every marker, expression was normalized to its corresponding α-tubulin value and the mean of WT media-treated normalized values were set to 1.
DRG neuron culture.
DRG neurons from female WT and miR-155 KO mice were cultured as described previously (Gardiner et al., 2005). Wells were coated with 100 μl of poly-d-lysine (25 μg/ml; Sigma-Aldrich, P6407) and laminin (10 μg/ml; Life Technologies, 23017-015) diluted in deionized water. The next day, wells were washed three times with deionized water before adding neurons. After isolation, DRGs (from 2- to 5-month-old females) were placed in 0.125% collagenase at 37°C/5% CO2 for 80 min and then incubated in 0.25% trypsin for 20 min. After removing trypsin, 30% FBS was added to inactivate the enzyme; DRGs were washed twice with DMEM/F12 (Life Technologies, 21331-020) and then dissociated by trituration (∼15–20 cycles) in 1 ml of DMEM/F12. Dissociated cells were passed through a 70 μm filter, overlaid onto 2 ml of BSA, and then centrifuged (900 × g, 5 min). DRG neurons in the pellet were resuspended in “neuron medium”: DMEM/F12 with N2 supplement (Life Technologies, 17502-048), 0.25% Glutamax, and 0.05% gentamicin. Cells were counted and then plated at the appropriate density. Neurons cultured alone were plated at 700 cells/well in a 24-well plate (cross-point analysis; n = 3 WT and n = 4 KO mice) or a 96-well plate (neurite initiation and longest neurite analysis; n = 4 per genotype) for 24 h.
Macrophage and DRG neuron coculture.
In 4 separate cultures (n = 4 females per genotype, cells from individual mice grown separately for independent replicates), differentiated macrophages were plated in uncoated 96-well plates. At 8 d after culture, macrophages were stimulated with media or 2× M1/M2 factors (200 ng/ml LPS + 40 ng/ml IFN-γ; 40 ng/ml IL-4) in 100 μl of per well neuron media for 4–6 h. During this time, DRGs were isolated from adult female WT mice. Once DRG isolation and dissociation were completed, 1000 DRG neurons in 100 μl were plated into each well containing macrophages (thereby diluting M1/M2 factors to 1×). In some experiments, GFP-positive DRG neurons were plated and imaged at the beginning and end of culture to assess toxicity. Neurons were cultured at 37°C/5% CO2 for 48 h.
Macrophage-conditioned media transfer.
In 3 separate cultures (n = 3 per genotype; all in parallel with cocultures for direct comparison; cells from individual mice were grown in separate wells to maintain independent replicates), differentiated macrophages were cultured in 24-well plates and stimulated with neuron medium alone, LPS/IFN, or IL-4 beginning ∼6 h after plating. The next day, WT DRGs were isolated and plated in poly-d-lysine/laminin-coated 96-well plates at 700 neurons/well in 100 μl neuron medium. At 24 h after stimulation (∼40–60 min after neuron plating), media from WT and miR-155 KO macrophages were collected, centrifuged at 10,000 × g for 10 min to remove cells/debris, and then 100 μl of the medium was added to wells containing neurons. Cell lysates and surplus media were collected for protein analysis and Griess assays. Neurons were incubated for 48 h before fixation.
Neurite outgrowth and neurotoxicity assays.
After 24 or 48 h, neurons (and macrophages, when present) were fixed in 4% paraformaldehyde (20–30 min) and stored in PBS. For imaging and analysis, rabbit anti-βIII-tubulin (Sigma-Aldrich, T2200) with Alexa Fluor 488-conjugated secondary antibodies (Life Technologies, A-11008) and DAPI were used. Neurite outgrowth was imaged (ThermoScientific, ArrayScan XTI) and then the length of the longest neurite, the percentage of cells bearing neurites, and cell survival were measured using MetaMorph. Thirty-five to 50 cells from each animal were examined for all analyses. For the proportion of cells with neurites, the number of cells with neurites per well was divided by the total number of cells per well (as defined by βIII-tubulin-positive cells with DAPI+ nuclei). For the longest neurite, 12–15 images per well were collected and then the longest neurite was measured. Sholl (branching) analysis was completed using MetaMorph, as described previously (Gensel et al., 2010), and the number of cross points at defined distances from the cell body was determined. For cell survival, the cells in the well were counted at initial culture (40 min after plating) and at fixation (48 h after plating); data ae expressed as percentage survival. Analysts were blinded using coded image names.
Statistics
Immunohistological, morphometric, and gene expression levels were analyzed using one- or two-way ANOVA, followed by Holm–Sidak post hoc tests. In experiments with two groups, a Student's t test (or nonparametric Mann–Whitney U test) was performed. SigmaPlot 12.0 (SPSS) was used to analyze these data. Spontaneous recovery of locomotor function, as measured using BMS scores, follows a nonlinear pattern. Therefore, a nonlinear model was used to fit a regression equation with the form of an exponential curve as we have described previously (Jones et al., 2002). In this case, we used the equation: BMS = α + βexp(γ * dpi) (Stevens, 1951), where α is the asymptotic value that BMS reaches, β is the change in BMS as dpi moves from 0 to ∞, and γ is the reduction (should be negative) in deviation of BMS from its asymptote for a given change in dpi. The BMS of left and right paws were averaged; one WT animal was removed based on Grubbs's method of detecting outliers. Average BMS scores were analyzed using a nonlinear mixed-effect model using the procedure of nlmixed in SAS 9.4. Results were considered significant when p < 0.05. All data, with the exception of BMS data, are plotted as mean ± SEM. For BMS data, the model fit showing mean group values and raw data are plotted together.
Results
Macrophage miR-155-5p is markedly increased by inflammatory stimuli
Macrophage and microglia effector functions are dictated by the signals that they receive in their microenvironment. Macrophages that contribute to secondary neurodegeneration or that inhibit axon growth/plasticity after SCI do so after exposure to inflammatory stimuli, either in vivo within the lesion microenvironment or using in vitro models (Popovich et al., 1999; Horn et al., 2008; Kigerl et al., 2009). To determine whether inflammatory signaling regulates miR-155-5p in macrophages, WT BMDMs were stimulated for 24 h with control medium or canonical inflammatory stimuli (IFN-γ + LPS). Expression of miR-155-5p increased ∼30-fold (H2 = 7.48; p < 0.01) compared with unstimulated macrophages (Fig. 1A). To confirm that miR-155-5p induction was not a generic response to cell activation, separate BMDMs were stimulated with IL-4, a cytokine that activates macrophages without eliciting neurotoxic effector functions (Stein et al., 1992; Kigerl et al., 2009; Miron et al., 2013). miR-155-5p was not increased in IL-4-activated BMDMs (Fig. 1A).
Figure 1.
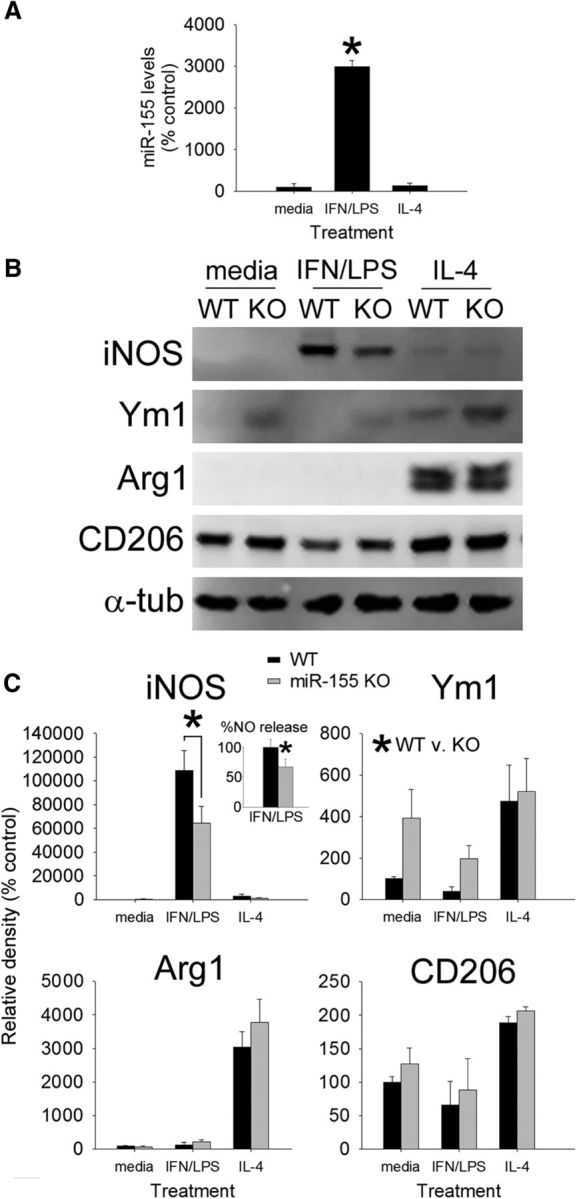
miR-155 is required for macrophages to acquire an inflammatory phenotype. A, BMDMs stimulated with IFN-γ + LPS increase their expression of miR-155 by ∼3000%. miR-155 expression was not increased by IL-4. B, C, WT and miR-155 KO macrophages were stimulated with media, IFN-γ + LPS, or IL-4 for 24 h (loading control: α-tubulin) and then cell lysates were collected to assess expression of canonical M1 (iNOS) and M2 (Ym1, Arg1, CD206) markers. Inset in C shows that, consistent with a significant reduction in iNOS, nitric oxide (NO) release was reduced (∼30%) in miR-155 KO macrophages after stimulation with IFN-γ + LPS. miR-155 KO macrophages also expressed higher levels of Ym1 (*p < 0.05; main effect of genotype). As expected, Arginase-1 and CD206 were significantly increased after activation with IL-4; however, expression of these M2 markers was not significantly different between genotypes. *p < 0.05 (ANOVA with Holm–Sidak post hoc test).
Deleting miR-155 reduces macrophage inflammatory signaling
To determine the effects of miR-155 deletion on macrophage phenotype and function, cell lysates were collected from WT and miR-155 KO macrophages activated as in Figure 2A. Western blotting for proteins that define inflammatory “M1” (iNOS) or alternatively activated “M2” (Ym1, Arginase-1, CD206) macrophages revealed expected changes: that is, WT M1 macrophages upregulated iNOS, and WT M2 macrophages upregulated Ym1, Arginase-1 (Arg1), and CD206 (Fig. 1C,D; data normalized to α-tubulin). Conversely, in miR-155 KO macrophages activated by inflammatory stimuli, iNOS expression and downstream release of nitric oxide were reduced 41% and 33%, respectively (Fig. 1C,D; F(1,35) = 4.58, p < 0.05; Holm–Sidak post hoc p = 0.001). In addition, miR-155 KO macrophages expressed higher levels of Ym1 compared with WT macrophages (F(1,25) = 4.42; p < 0.05; main effect of genotype; Fig. 1C,D). Other M2 markers, including Arg1 and CD206, were increased by IL-4 in both groups but were unaffected by miR-155 deletion. These data indicate that inflammatory signaling is impaired in miR-155 KO macrophages, but the absence of miR-155 does not enhance macrophage differentiation toward an M2 phenotype.
Figure 2.
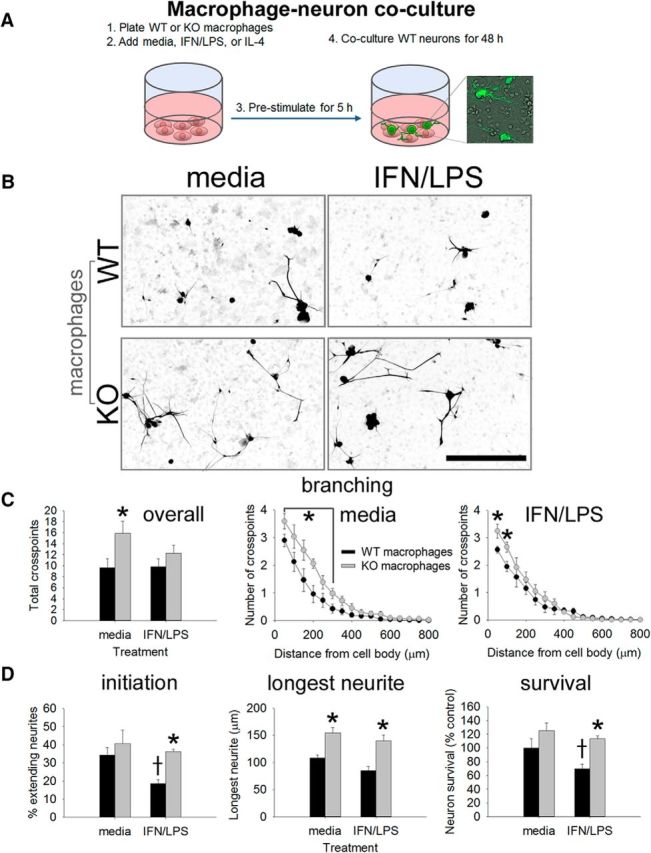
miR-155 KO macrophages enhance neurite outgrowth from adult DRG neurons in a coculture model. A, WT or miR-155 KO macrophages were stimulated with media (control) or IFN-γ + LPS for 5 h before adding WT DRG neurons (inset, GFP+ neurons growing on macrophages). B–D, At 48 h after initial culture, miR-155 KO macrophages were more supportive of neurite outgrowth. Sholl analysis showed that miR-155 KO macrophages enhanced neurite outgrowth, particularly in areas close to the cell body (C, overall total branching and individual media/IFN-γ + LPS Sholl analyses). D, miR-155 KO macrophages increased neurite initiation, the length of the longest neurite, and neuron survival. *p < 0.05 versus the appropriate WT control; †difference versus media-treated WT control. Scale bar, 200 μm.
miR-155 KO macrophages support axon growth without causing neurotoxicity
Inflammatory macrophages can kill neurons (Popovich et al., 1997; Kigerl et al., 2006; Kigerl et al., 2009; Beck et al., 2010; Prüss et al., 2011; Pool et al., 2012; Kroner et al., 2014). To determine whether reduced inflammatory signaling in miR-155 KO macrophages limits their ability to kill neurons while also enhancing neurite growth, WT and miR-155 KO macrophages were left untreated or were exposed to inflammatory stimuli as in Figure 1. Five hours later, WT adult DRG neurons were added to macrophage cultures (Fig. 2A). The percentage of neurons extending neurites, longest neurite, amount of branching, and neuron survival were quantified.
When in a KO macrophage environment, various indices of neurite growth were consistently increased compared with growth with WT macrophages. This effect was independent of whether macrophages had been exposed to “control” or “inflammatory” stimuli. When cocultured with miR-155 KO macrophages, neurite branching (F(1,23) = 10.04; p = 0.005), the percentage of neurons extending neurites (“neurite initiation”; F(1,23) = 19.59; p < 0.001) and the length of the longest neurite (F(1,23) = 11.62; p < 0.01) were consistently increased (Fig. 2B–D). Neuron survival also was enhanced in the presence of miR-155 KO macrophages (F(1,23) = 6.15; p < 0.05). The neuroprotective effect of deleting miR-155 in macrophages was particularly notable under inflammatory conditions (Fig. 2D). WT inflammatory macrophages were neurotoxic, which corroborates previous data from our laboratory (Kigerl et al., 2009). However, unlike those data, inflammatory macrophages did not enhance neurite growth. Apparent differences between studies may be explained by key variations in experimental design: the duration of cell culture was different and this dramatically affects neurite growth phenotype (Smith and Skene, 1997). In addition, neurons in these separate studies were exposed to macrophages/conditioned medium for different durations, which likely affects neurite growth dynamics and analyses.
To determine whether the growth-permissive environment created by miR-155 deletion was due to soluble factors released by macrophages, medium from WT or miR-155 KO macrophages was added to WT DRG neurons (Fig. 3). Regardless of stimulation protocol, neither WT nor KO macrophage conditioned media significantly affected neurite growth compared with control media (Fig. 3B,C). These data suggest that miR-155 KO macrophages support neurite outgrowth, and perhaps neuron survival, through contact (or close association) with neurons.
Figure 3.
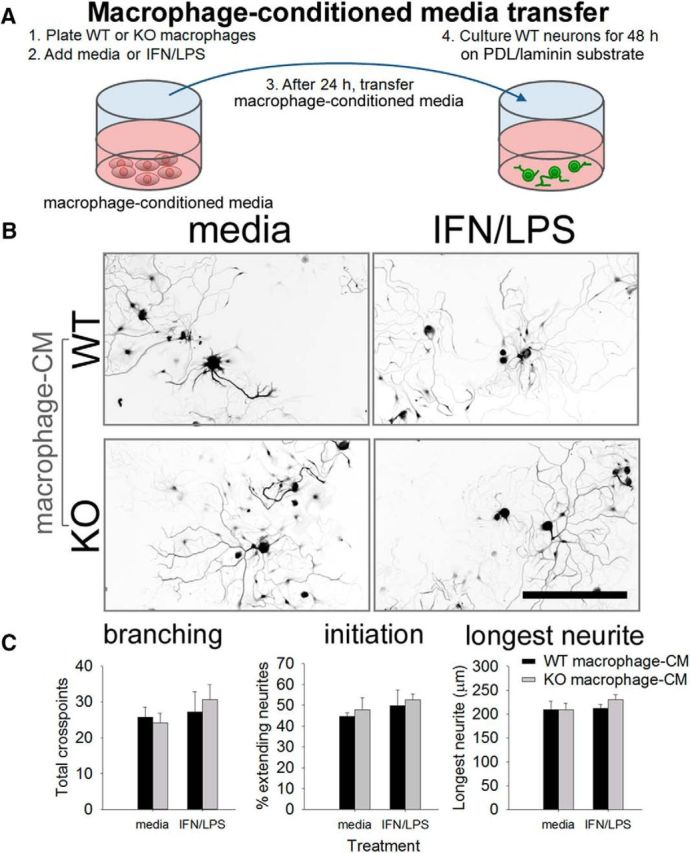
Secreted factors likely do not underlie KO-macrophage-enhanced neurite. Unlike cocultures, in which KO macrophages augment axon growth, transfer of macrophage-conditioned media (CM) from miR-155 KO macrophages does not enhance neurite outgrowth compared with WT macrophage-CM. A, DRG neurons were cultured in the presence of WT/miR-155 KO macrophage-CM for 48 h. B, C, miR-155 KO macrophage-CM (control or inflammatory) did not significantly enhance neurite branching, initiation, or length. Scale bar, 200 μm.
miR-155 as a neuron-intrinsic regeneration inhibitory gene
The above data indicate that miR-155 is essential for promoting the development of inflammatory macrophages and that these cells can limit various indices of axon growth. However, miR-155 could also affect neuron-intrinsic growth by targeting axon-growth-related mRNAs. To study the neuron-intrinsic effects of miR-155, spontaneous neurite outgrowth was measured in WT and miR-155 KO adult DRG neurons 24 h after plating them onto an axon growth-permissive poly-d-lysine/laminin substrate (Fig. 4). Compared with WT neurons, miR-155 KO neurons consistently exhibit more robust “arborizing” neurite growth (Fig. 4A,B; 27% more total cross points; t(5) = −2.65; p < 0.05). miR-155 KO neurons also extend longer neurites' the average longest neurite from KO neurons was 44% longer than from WT neurons (t(6) = 3.82; p < 0.01; Fig. 4C). In general, more miR-155 KO neurons extended neurites by 24 h (compared with WT), although this did not reach statistical significance (Fig. 4C). These data implicate miR-155 as an intrinsic inhibitor of axon growth.
Figure 4.

miR-155 KO neurons have greater intrinsic growth potential. A, Representative WT and miR-155 KO DRG neurons at 24 h after culture. B, C, Neurite outgrowth from adult miR-155 KO neurons is enhanced compared with WT neurons. miR-155 KO neurons had 27% more total cross points (B) and 44% longer neurites (C). The percentage of extending neurites was not significantly different between strains. *p < 0.05 versus WT control. Scale bar, 100 μm.
miR-155 deletion increases expression of axon growth-related proteins and reduces inflammation in DRGs after peripheral axotomy
The in vivo regenerative potential of injured DRG sensory axons, both the peripheral and central branches, is influenced by both neuron-intrinsic and neuron-extrinsic mechanisms (Lu and Richardson, 1991; Hellal et al., 2011; Sun et al., 2011; Kwon et al., 2013; Niemi et al., 2013). Given that miR-155 deletion improves both intrinsic and extrinsic mechanisms of axon growth in vitro (Figs. 1, 2, 3, 4), we next evaluated the effects of miR-155 deletion on postinjury expression of axon growth and inflammatory proteins in vivo using a model of spinal cord dorsal column injury. Dorsal column crush injuries create discrete zones of primary trauma that are ideal for studying cellular and molecular mechanisms of axon regeneration/plasticity (Kwon et al., 2002). Further, the dorsal column crush injury model enables accurate quantification of axon dieback and axon regeneration from this discrete injury border (Busch et al., 2011; Evans et al., 2014).
To enhance the regenerative potential of central branches of DRG sensory axons, WT and miR-155 KO mice received sciatic nerve conditioning lesions and then 6 d later were subjected to C5 spinal cord dorsal column crush injury (Fig. 5A). AAV2-GFP was injected into the sciatic nerves immediately after SCI to trace injured sensory axons from conditioned DRG neurons. Tissue was collected 32 dpi (38 d after conditioning lesion; Fig. 5A).
Figure 5.

After conditioning lesion and dorsal column injury, miR-155 KO DRGs show improved intrinsic growth responses of sensory neurons and reduced intraganglionic macrophage activation. A, Experimental model and timeline for studying DRG responses and sensory axon regeneration. B, A similar proportion of neurons expressed GFP and ATF3 in the DRG of WT and miR-155 KO mice; however, 44% more miR-155 KO neurons increased expression of the RAG SPRR1A. C, In WT DRGs, large, dense clusters of activated macrophages (arrowheads) accumulated in the DRG and surround a subset of DRG neurons (e.g., GFP-labeled neurons; arrows; also see inset). In miR-155 KO DRGs, macrophage density was reduced 28% and 47% fewer KO DRGs contained inflammatory foci. D, Both pro-inflammatory (CD86) and anti-inflammatory (CD206) markers were expressed in these lumbar DRGs. miR-155 KO DRGs reduced CD86 density by 45%. CD206 density was not different between strains. In all images, blue is nuclear DAPI stain. *p < 0.05 versus WT. Scale bars, 100 μm; inset scale bar, 25 μm.
SPRR1A and ATF3 proteins have been shown to enhance regeneration of injured DRG axons (Bonilla et al., 2002; Seijffers et al., 2007). Neither protein is detectable in uninjured DRG neurons, but both are upregulated in DRG neurons after conditioning lesion (Fig. 5B). Although ATF3 expression was increased to a similar extent in both WT and KO DRG neurons after conditioning lesion (∼20% of WT and KO neurons were ATF3+), significantly more (44%) KO DRG neurons expressed SPRR1A compared with WT DRG neurons (WT: 20%; KO 29%; t(12) = −2.29; p < 0.05). GFP expression was not significantly different between genotypes (∼20% of DRG neurons expressed GFP in both WT and KO mice), indicating that genotype had no effect on retrograde labeling with AAV2-GFP.
Peripheral nerve lesions elicit inflammation in the affected DRGs and macrophages are essential for effective regeneration of injured peripheral axons (Kwon et al., 2013). Accordingly, we evaluated macrophage responses in L4 DRGs of WT and miR-155 KO mice after sciatic nerve transection (Fig. 5C,D). Iba1+ macrophages accumulated in WT DRGs, including the formation of focal macrophage aggregates surrounding DRG neurons (arrowheads in Fig. 5C,D). In contrast, in miR-155 KO DRGs, Iba1+ macrophage density was reduced 28% (t(26) = 2.32; p < 0.05; Fig. 5C), with 47% fewer miR-155 KO DRGs containing Iba1+ inflammatory foci (7/10 WT DRGs had inflammatory foci, whereas 6/18 KO DRGs contained these foci). Macrophages in miR-155 KO DRGs also were less “inflammatory” based on the expression of canonical phenotypic markers (Fig. 5D); the inflammatory marker CD86 was reduced by 45% in KO DRGs (t(25) = 2.08; p < 0.05), with a corresponding decrease of ∼58% in the ratio of CD86:CD206 (inflammatory:anti-inflammatory markers; t(26) = 3.60; p < 0.001) compared with WT DRGs.
After SCI, miR-155 deletion reduces intraspinal inflammation and promotes the regeneration of sensory axons
We next determined whether miR-155 deletion also positively affects intraspinal inflammation and regeneration of sensory axons within the injured spinal cord. Consistent with the reduced inflammatory phenotype described in vitro and in DRGs after peripheral nerve lesion (Figs. 1, 5), miR-155 KO mice had reduced Iba1+ macrophage/microglia density in injured spinal cord dorsal columns (Fig. 6B,B′; F(1,247) = 10.53; p = 0.001; asterisks, Fig. 6D); Iba1+ density was reduced 46% compared with comparable regions in injured WT mouse spinal cord (t(10) = 2.46; p < 0.05; Fig. 6F).
Figure 6.

miR-155 deletion reduces intraspinal macrophage activation/accumulation in mice subjected to a peripheral conditioning lesion and spinal cord dorsal column crush injury. miR-155 KO spinal cord epicenters have reduced density of Iba1+ macrophages/microglia. Top, Overview of Iba1 immunoreactivity in horizontal sections of WT and miR-155 KO dorsal columns. A, A′, WT and miR-155 KO spinal cords had similar Iba1+ cell densities in rostral dorsal columns. Rostral is to the left; dashed line delineates injury epicenter. B, B′, graphs, miR-155 KO spinal cords had reduced Iba1+ macrophage/microglial density in the injury epicenter. C, C′, GFP+ axons and endbulbs localized near inflammatory Iba1+ cells. Note the more rostral location of activated macrophages that colocalize with regrowing axon tips in KO spinal cord. D, D′, Confocal images showing detail of Iba1+ cells and GFP+ axons at the rostral-most front of growing axons. E, In miR-155 KO mice, Iba1 density was significantly reduced compared with WT mice from 200 μm rostral to 400 μm caudal to injury epicenter. F, Compared with WT mice, Iba1+ cell density in the KO epicenter was reduced by 46% (quantified between 200 μm and −200 μm). *p < 0.05 versus WT control. R-C, Rostral–caudal axis; R-L, right–left (horizontal axis). Scale bars: top, alternating gray and white bars, each 400 μm; inset panels, 50 μm.
The dystrophic end bulbs of both WT and KO axons were closely associated with Iba1+ macrophages/microglia (Fig. 6C,C′,D,D′). To determine whether genotype-specific differences in intraspinal macrophages were associated with differences in regeneration or degeneration (“dieback”) of sensory axons, AAV2-GFP was used to trace axons from preconditioned DRG neurons into the dorsal columns (i.e., gracile fasciculus) of injured mouse spinal cord. In miR-155 KO spinal cords, GFP+ axon density was increased within 400 μm of the caudal pole of the lesion (F(9,119) = 18.15; p < 0.001; Fig. 7D,F). Closer to the lesion (∼0–200 μm), axon density increased further with 5-fold more GFP labeling in KO than WT spinal cords (WT: 8%; KO: 48%; post hoc p < 0.01); at 200–400 μm, KO axon density was 1.5-fold higher than WT density (WT: 22%; KO: 56%; post hoc p < 0.05). Therefore, the majority of axons undergo dieback after dorsal column injury (even with conditioning lesion); this postinjury axon dieback is reduced in miR-155 KO spinal cords.
Figure 7.

miR-155 KO axons show enhanced plasticity and regrowth after peripheral conditioning lesion and spinal cord dorsal column crush injury. Top, Overview of GFP immunoreactivity in horizontal sections of WT and miR-155 KO dorsal columns. A, A′, Axons at the regenerative front were located further rostral in KO spinal cords. B, B′, Areas containing axon growth cones or end bulbs in WT and KO mice. C, C′, Axons located in more caudal regions of the injury epicenter. D, Representative thresholded images of GFP+ axons illustrating the enhanced growth of miR-155 KO axons up to and beyond the injury site (dashed line). E, Translesional extension of the longest-growing axons is increased in miR-155 KO spinal cords. F, In miR-155 KO mice, GFP+ axon density was higher than in WT mice between 0 and −400 μm from epicenter, suggesting that KOs have improved axon growth and/or reduced dieback. *p < 0.05 versus WT control. R-C, Rostral–caudal axis; R-L, right–left (horizontal axis). Rostral is to the left; dashed lines identify injury epicenter. Scale bars: Top and D, gray and white bars, each 400 μm; A–C, 50 μm.
To differentiate between the effects of miR-155 deletion on axon dieback compared with axon regeneration, GFP+ axon density was measured through the lesion site into rostral spinal cord. In WT spinal cords, conditioning lesions promoted regeneration of a subset of axons through the lesion site, but none extended >400 μm rostral to the lesion (Fig. 7E). Conversely, axon regeneration was evident beyond this point in half of the miR-155 KO mice (n = 4/8), with axons extending as far as 1.4 mm past the lesion site (Fig. 7E). Overall, axons in miR-155 KO mice projected further into the rostral spinal cord (average axon location relative to the lesion epicenter: KO spinal cord, +11 μm; WT spinal cord, −482 μm; t(10) = −2.23; p = 0.05; Fig. 7E). These data show that dieback of injured sensory axons is reduced and axon regeneration is improved in miR-155 KO mice after conditioning lesion plus SCI.
miR-155 is upregulated in contused spinal cord; miR-155 deletion is neuroprotective and enhances recovery of locomotor function
To further evaluate the translational potential of manipulating miR-155 in vivo after SCI, we first measured postinjury changes in intraspinal miR-155-5p expression in a more clinically relevant spinal contusion injury model (Fig. 8A). In WT mice, miR-155-5p increased at 3 dpi (∼300% higher than uninjured; p < 0.05) and 14 dpi (∼400%; p < 0.01) and remained elevated for at least 6 weeks (42 dpi; >400%; p < 0.005).
Figure 8.
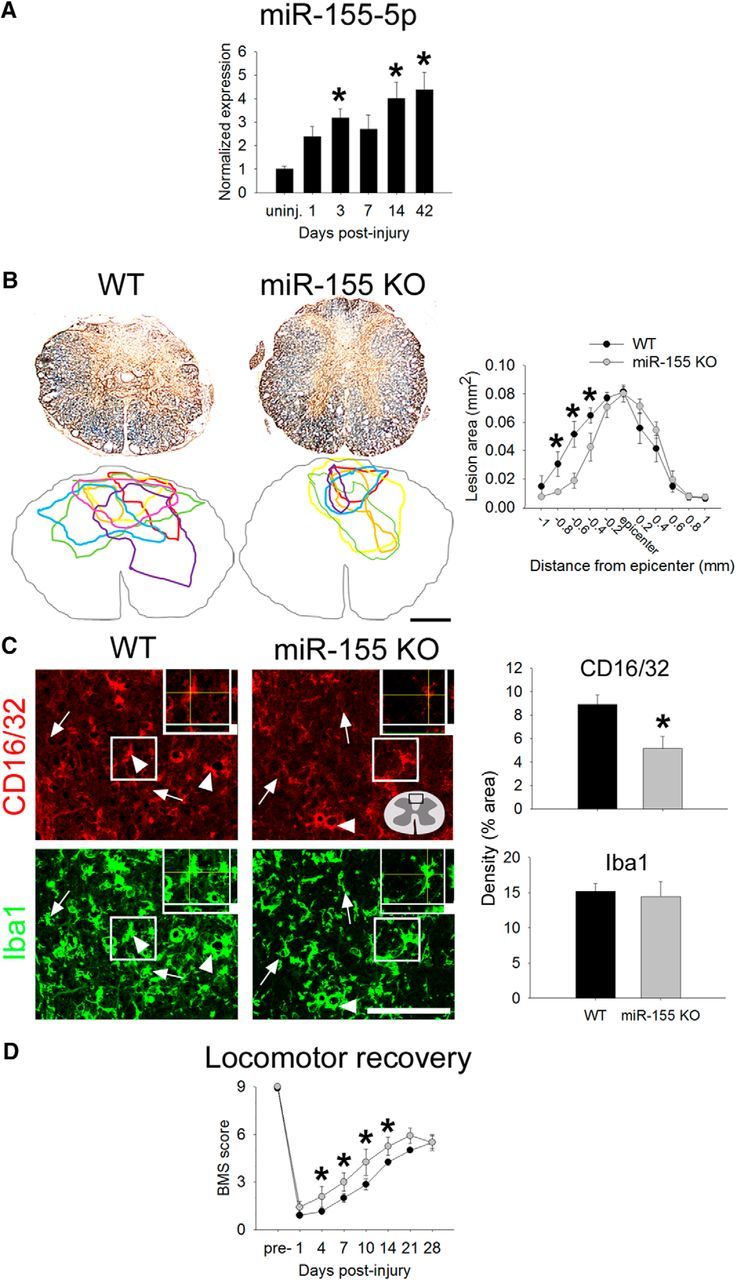
Contusion SCI increases expression of miR-155-5p in WT mice and miR-155 deletion promotes neuroprotection, reduces inflammatory macrophage accumulation, and improves spontaneous locomotor recovery. A, After SCI in WT mice, miR-155-5p expression increases over time after injury, reaching levels >300% above uninjured control by 42 dpi. B, Eriochrome and neurofilament were used to identify intact myelin and axons, respectively. miR-155 KO mice had significantly smaller lesions with the most notable changes evident rostral to injury epicenter: At 600 μm rostral to epicenter, miR-155 KO lesions were 64% smaller than WT lesions. Colored outlines represent lesion size in every animal examined for each genotype at 600 μm rostral to epicenter. C, Overall density of inflammatory CD16/32+ macrophages was reduced by 25% in rostral lesion extensions of miR-155 KO spinal cords, although overall Iba1+ macrophage density in this region was not different between KO and WT mice. D, Beginning at 4 dpi and extending until 14 dpi, spontaneous locomotor recovery was improved in miR-155 KO mice compared with WT mice. By 21 and 28 dpi, mice in both groups regained similar levels of locomotor function. Arrowheads, CD16/32+Iba1+ cells; arrows, CD16/32−Iba1+ cells. *p < 0.05 versus WT control. Scale bars: C, 100 μm; F, 50 μm; inset, 5 μm.
After SCI, a rapid and protracted increase in miR-155-5p in intraspinal macrophages, neurons/axons, and/or other CNS cells could affect intraspinal inflammation, lesion pathology, and recovery of function. The functional implications of miR-155 deletion were determined by comparing lesion pathology and locomotor recovery after spinal contusion injury in WT and miR-155 KO mice (Fig. 8B–F). Sparing of spinal cord tissue, especially in the rostral spinal cord, was enhanced in miR-155 KO mice after spinal contusion injury (Fig. 8B,C). Compared with injured WT spinal cords, lesion size in miR-155 KO mice was reduced throughout the rostral extent of the lesion, suggesting that miR-155 removal improved neuroprotection.
SCI-induced inflammatory macrophage/microglial reactions were also reduced in miR-155 KO mice, particularly in the spinal cord rostral to injury. In the contusion epicenter, immunoreactive densities of Iba1 (a pan-macrophage/microglia marker) and CD16/32 (an inflammatory marker expressed in macrophages/microglia) were increased in both genotypes, although there was a trend for reduced Iba1 density in the lesion epicenter of KO mice (WT: 40 ± 2% vs KO: 33 ± 3%; p = 0.07). Next, the dorsal columns rostral to injury epicenter were examined. Although the overall density of Iba1+ macrophages was not different between groups, CD16/32+ microglia/macrophage density was reduced >40% in KO mouse spinal cord rostral to SCI (WT: 8.9%, KO: 5.2%; p < 0.05; Fig. 8F). These data, which are similar to those described in vitro (Fig. 1) and in the spinal dorsal column crush injury model (Figs. 5, 6, 7), indicate that inflammatory signaling in macrophages is reduced by miR-155 deletion.
The enhanced neuroprotection in miR-155 KO mice was associated with improved locomotor recovery after contusion SCI. Using the BMS locomotor recovery scale, miR-155 KO mice had improved spontaneous recovery beginning at 4 dpi and extending until 14 dpi (compared with WT mice; p < 0.05 at all time points). By 21 and 28 dpi, both WT and miR-155 KO mice had regained similar levels of locomotor function (Fig. 8D,E); average BMS scores were ∼5.5 (some or mostly coordinated stepping with rotated paw position; Basso et al., 2006). Therefore, in the contusion lesion model, reduced post-SCI inflammation in miR-155 KO spinal cords is associated with significant neuroprotection and recovery of locomotor function.
Discussion
Here, we reveal a novel role for miR-155 in influencing CNS repair through concurrent regulation of inflammatory signaling and neuron-intrinsic inhibition of axon growth. In vitro, genetic deletion of miR-155 reduced inflammatory signaling in macrophages and improved their ability to support neuron survival and neurite growth. Neurons lacking miR-155 showed improved intrinsic axon growth capacity. Using a conditioning lesion plus dorsal column injury model, miR-155 KO spinal cords had reduced inflammation and improved axon growth in vivo. Similar benefits of miR-155 deletion were observed in mice subjected to a clinically relevant model of spinal contusion injury; miR-155 deletion improved tissue sparing and reduced inflammatory macrophage/microglial density, which likely contributed to improved locomotor recovery. Together, these data identify miR-155 as a potential therapeutic target to improve indices of repair after SCI.
Modifying macrophage-mediated neurotoxicity and repair in the injured spinal cord via miR-155 deletion
In the injured spinal cord, inflammatory stimuli persist indefinitely and likely exacerbate tissue pathology. Although understudied in the context of spinal cord inflammation, miR-155 has defined roles in eliciting inflammatory changes in macrophages. Therefore, the sustained increase in expression of miR-155-5p after SCI could implicate this miR as a robust molecular “ON” switch for propagating destructive intraspinal inflammation. We and others have shown that inflammatory signaling potently activates miR-155-5p in macrophages (Fig. 1; O'Connell et al., 2007; Ceppi et al., 2009; Cremer et al., 2009). For example, macrophages activated by extracellular damage- or pathogen-associated molecules (e.g., tenascin-C or LPS) increase their expression of miR-155-5p (O'Connell et al., 2007; Piccinini and Midwood, 2012). Increased miR-155-5p reduces the availability of anti-inflammatory RNAs (Wang et al., 2010; Martinez-Nunez et al., 2011), driving the differentiation of macrophages toward an inflammatory or “classically activated” M1 phenotype. During peripheral tissue trauma or infection, transient miR-155-5p-induced M1 macrophage polarization likely assists with tissue repair and pathogen removal (e.g., miR-155 KO mice are more susceptible to lung infection; Rodriguez et al., 2007); however, sustained miR-155 expression may cause chronic inflammation associated with diseases such as atherosclerosis (Nazari-Jahantigh et al., 2012) and arthritis (Kurowska-Stolarska et al., 2011). In the CNS, miR-155-5p is implicated in the onset or progression of amyotrophic lateral sclerosis (Koval et al., 2013) and multiple sclerosis (O'Connell et al., 2010; Murugaiyan et al., 2011; Moore et al., 2013).
The intracellular mechanisms by which miR-155-5p augments the inflammatory phenotype of macrophages are not clear, although several validated miR-155-5p mRNA targets encode anti-inflammatory mediators or immune-regulatory proteins, including Cebpb, Creb, Bcl6, Sfpi1 (PU.1), Il-13raI, Ship1, and Socs1 (Vigorito et al., 2007; Lu et al., 2009; O'Connell et al., 2009; Yin et al., 2010; Liu et al., 2011b; Martinez-Nunez et al., 2011; Nazari-Jahantigh et al., 2012; Table 1). Therefore, in response to inflammatory stimuli, preferential degradation of compensatory anti-inflammatory genes by miR-155 may polarize macrophages toward an inflammatory phenotype. Previously published data and the present data support this hypothesis. For example, cultured miR-155 KO macrophages produce lower levels of iNOS and the free radical nitric oxide, which likely reduces their oxidative and neurotoxic capacity (Fig. 1; also see Pearse et al., 2003; Xu et al., 2006; David and Kroner, 2011). At rest and in response to inflammatory stimuli, miR-155 KO macrophages also express higher mRNA and protein levels of the anti-inflammatory factor Ym1. These results suggest that KO macrophages' resting metabolism and functions are distinct from those of WT macrophages. Such differences could explain how miR-155 KO macrophages, activated by canonical inflammatory stimuli, enhance axon growth in vitro and in vivo after SCI while simultaneously showing reduced neurotoxicity.
Table 1.
miR-155-5p validated and predicted targets: a partial list
| Macrophages | Neurons | ||
|---|---|---|---|
| TFs controlling macrophage phenotype | TFs regulating neurite growth | ||
| C/EBPb | C/EBPb | ||
| Spi1 | Gene for PU.1 transcription factor | Creb1 | |
| Creb1 | Various eIFs | ||
| Bcl6 | Adenylate cyclases | ||
| Nr1h3 | LXRalpha gene | Growth factors/receptors | |
| Ppargc1b | PPAR, gamma, coactivator 1 beta gene (PGC1beta) | BDNF | |
| TNF signaling | Ntrk1 (TrkA) | ||
| Ikbkg | Inhibitor of kappaB kinase gamma gene | Gdnf | |
| Traf3 | TNF receptor-associated factor 3 gene | Gfra1 | |
| TGF signaling | IGF-2 | ||
| Igf1r | |||
| TGFbeta receptor 2 | Axon guidance factors/receptors | ||
| Tgf alpha | EphB4, B2 | ||
| Tgfbr1 | TGF, beta receptor 1 gene | Efna5 | Ephrin-A5 gene |
| Other, pro-inflammatory | Sema6c | ||
| IL-12 receptor beta 1 | Various RPTPs, not sigma | ||
| MMP-12 | Various MMPs, ADAMs, ADAM-TS | Bmpr integrin alpha 3, 4, 5, 6, 8, 9 | Various BMP and activin receptors |
| Ccr7 | |||
| Other, anti-inflammatory | Lama genes | Laminin beta and alpha | |
| Il-13RaI | Ntn3 | Netrin-3 gene | |
| Msr1 | Macrophage scavenger receptor 1 (Msr1) | Nrcam | NRCAM gene |
| Cd200r3 | CD200 receptor 3 gene | Tenascin-C | |
| CD200 | |||
| IL-4Ra | |||
| Mrc1, 2 | Mannose receptor, C type 1 gene (and 2) | ||
| Adpor1 | Adiponectin receptor 1 gene | ||
| Other, potentially important | |||
| Rheb | |||
| HMGB1 | |||
| SHIP1 | |||
| Socs1 | |||
| FGF-6 | |||
| P2rx7 | |||
| P2rx4 | |||
Genes in bold are validated miR-155 targets that could influence inflammation or neuroplasticity. Various databases, including targetscan.org, microrna.org, and mirbase.org, were used to generate lists of targets. These mRNAs have a sequence in the 3′-UTR that could be bound by the seed region of miR-155. Such binding could reduce translation of these mRNAs.
Sources: https://cm.jefferson.edu/rna22v1.0-mus_musculus/InputController?identifier=mmu-miR-155, http://www.targetscan.org/cgi-bin/targetscan/vert_61/targetscan.cgi?mirg=mmu-miR-155, http://www.microrna.org/microrna/getTargets.do?matureName=mmu-miR-155&startIndex=350&organism=10090, http://www.mirbase.org/cgi-bin/mirna_entry.pl?acc=MI0000177.
Although miR-155 KO macrophages enhanced in vitro axon growth, miR-155 KO DRGs and spinal cords had reduced postinjury macrophage density that still correlated with improved axon growth dynamics. Because inflammatory signaling is reduced in miR-155 KO macrophages (Fig. 1; O'Connell et al., 2007) and microglia (Cardoso et al., 2012; Butovsky et al., 2015), these KO cells likely release fewer cytokines and chemokines that induce proliferation and recruitment of microglia/monocytes after nervous system trauma. This could explain the reduced postinjury density and inflammatory phenotype of macrophages in KO DRGs and spinal cords (Figs. 5, 6, 7, 8). Deleting miR-155 in macrophages also could influence inflammation-induced expression of integrins, netrins, semaphorins or other neuroimmune regulatory proteins (e.g., CD200/CD200L, CX3CL1/CX3CR1) that strongly affect axon guidance and neuronal function (Togari et al., 2000; Popovich and Longbrake, 2008; Schmidt and Moore, 2013; Ramkhelawon et al., 2014). Therefore, manipulating miR-155 (and its downstream signaling) has complex effects on the microenvironment created by activated macrophages. miR-155 deletion likely alters the secretory profile of macrophages; however, this would not explain how miR-155 KO macrophages improve axon growth in vitro and in vivo. Otherwise, transfer of macrophage-conditioned medium would have been as effective as coculture in promoting neurite outgrowth (Fig. 2, 3). Instead, miR-155 deletion in macrophages likely alters the expression of adhesion and/or extracellular matrix molecules, creating a microenvironment that is less toxic and more permissive for axon growth.
Improving intrinsic neuron growth capacity via miR-155 deletion
Novel data in this report also implicate miR-155 as a neuron-intrinsic regulator of axon growth. Using dissociated adult sensory neurons from miR-155 KO mice, we found that spontaneous axon growth was enhanced in vitro. Similarly, sensory axons in the injured spinal cord of miR-155 KO mice regenerated more effectively than injured WT axons. A peripheral conditioning lesion (Richardson and Issa, 1984; Neumann and Woolf, 1999) before dorsal column injury was used to boost the regenerative potential of sensory axons in the spinal cord of both WT and KO mice. Using this model, deletion of miR-155 significantly augmented the regenerative response of axons (Fig. 5, 6, 7).
Low intrinsic neuron growth potential contributes to CNS axon regeneration failure (Ye and Houle, 1997; Plunet et al., 2002; Liu et al., 2011a; Tom et al., 2013). Unlike injured peripherally projecting neurons, which upregulate genes that coordinate axon regeneration (RAGs; e.g., ATF3, SPRR1A, GAP-43, CAP-23, α-tubulin; Miller et al., 1989; Bomze et al., 2001; Bonilla et al., 2002; Seijffers et al., 2007), injured CNS neurons do not initiate intrinsic growth programs. miR-155-5p may prevent or reduce axon growth by restricting the availability of specific RAG mRNAs, including Cebpb and Creb (Nadeau et al., 2005; Table 1). Interestingly, these same genes can also limit inflammatory macrophage activation. Other potential miR-155-5p targets that enhance axon growth include BDNF (Varendi et al., 2014) and Rheb/mTOR pathway mRNAs (Roitbak et al., 2011; Wang et al., 2013). Together, these data raise the possibility that miR-155 is a “regeneration-inhibitory gene” in adult neurons that could be targeted to improve intrinsic axon growth capacity.
Conclusions
Because miRs simultaneously regulate the expression/translation of hundreds of mRNAs, strategies designed to upregulate or downregulate their expression may provide unique control over coincident mechanisms of CNS pathology and repair. Here, we reveal a novel role for miR-155 in restricting endogenous spinal cord repair through detrimental activation of inflammatory signaling and neuron-intrinsic axon growth inhibition. Future research could identify factors that regulate miR-155 expression, how miR-155 KO macrophages affect other CNS cells (glia, endothelia, etc.), and whether other effector functions regulated by miR-155 are detrimental after SCI. Indeed, the immunomodulatory and neuroprotective effects of miR-155 deletion may also benefit SCI-induced neuropathic pain or autonomic dysfunction (Gris et al., 2004; Detloff et al., 2008; Schwab et al., 2014; Allison and Ditor, 2015; Tan et al., 2015; Brommer et al., 2016; Offiah et al., 2016). Further studies may also incorporate male mice because miR-155 may have different effects on cell metabolism and host physiology in males and females (Sipski et al., 2004; Furlan et al., 2005; Swartz et al., 2007; Luchetti et al., 2010; Gaudet et al., 2016).
Our data identify a dual role for miR-155 after SCI: miR-155 both activates inflammatory programs in macrophages and limits neuron-intrinsic axon growth capacity. Therefore, postinjury removal of miR-155 could simultaneously improve macrophage and neuron responses to enhance neuroprotection and repair.
Footnotes
This work was supported by the Canadian Institutes of Health (Research Postdoctoral Fellowship to A.D.G.), the Ray W. Poppleton Endowment (P.G.P.), the National Institutes of Health (Grant R21NS081413 to P.G.P. and M.G.d.A.), and the International Foundation for Research in Paraplegia (Grant P129 to P.G.P. and M.G.d.A.). We thank Kristina Kigerl for assisting with laboratory equipment, Laura Fonken for editorial assistance, and Lori Hudson for helping with mouse colonies and genotyping.
The authors declare no competing financial interests.
References
- Alilain WJ, Horn KP, Hu H, Dick TE, Silver J. Functional regeneration of respiratory pathways after spinal cord injury. Nature. 2011;475:196–200. doi: 10.1038/nature10199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allison DJ, Ditor DS. Immune dysfunction and chronic inflammation following spinal cord injury. Spinal Cord. 2015;53:14–18. doi: 10.1038/sc.2014.184. [DOI] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartus K, James ND, Didangelos A, Bosch KD, Verhaagen J, Yáñez-Muñoz RJ, Rogers JH, Schneider BL, Muir EM, Bradbury EJ. Large-scale chondroitin sulfate proteoglycan digestion with chondroitinase gene therapy leads to reduced pathology and modulates macrophage phenotype following spinal cord contusion injury. J Neurosci. 2014;34:4822–4836. doi: 10.1523/JNEUROSCI.4369-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso DM, Fisher LC, Anderson AJ, Jakeman LB, McTigue DM, Popovich PG. Basso Mouse Scale for locomotion detects differences in recovery after spinal cord injury in five common mouse strains. J Neurotrauma. 2006;23:635–659. doi: 10.1089/neu.2006.23.635. [DOI] [PubMed] [Google Scholar]
- Beck KD, Nguyen HX, Galvan MD, Salazar DL, Woodruff TM, Anderson AJ. Quantitative analysis of cellular inflammation after traumatic spinal cord injury: evidence for a multiphasic inflammatory response in the acute to chronic environment. Brain. 2010;133:433–447. doi: 10.1093/brain/awp322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomze HM, Bulsara KR, Iskandar BJ, Caroni P, Skene JH. Spinal axon regeneration evoked by replacing two growth cone proteins in adult neurons. Nat Neurosci. 2001;4:38–43. doi: 10.1038/82881. [DOI] [PubMed] [Google Scholar]
- Bonilla IE, Tanabe K, Strittmatter SM. Small proline-rich repeat protein 1A is expressed by axotomized neurons and promotes axonal outgrowth. J Neurosci. 2002;22:1303–1315. doi: 10.1523/JNEUROSCI.22-04-01303.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradbury EJ, Moon LD, Popat RJ, King VR, Bennett GS, Patel PN, Fawcett JW, McMahon SB. Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature. 2002;416:636–640. doi: 10.1038/416636a. [DOI] [PubMed] [Google Scholar]
- Brambilla R, Bracchi-Ricard V, Hu WH, Frydel B, Bramwell A, Karmally S, Green EJ, Bethea JR. Inhibition of astroglial nuclear factor kappaB reduces inflammation and improves functional recovery after spinal cord injury. J Exp Med. 2005;202:145–156. doi: 10.1084/jem.20041918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brommer B, Engel O, Kopp MA, Watzlawick R, Muller S, Prüss H, Chen Y, DeVivo MJ, Finkenstaedt FW, Dirnagl U, Liebscher T, Meisel A, Schwab JM. Spinal cord injury-induced immune deficiency syndrome enhances infection susceptibility dependent on lesion level. Brain. 2016;139:692–707. doi: 10.1093/brain/awv375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess AW, Metcalf D, Kozka IJ, Simpson RJ, Vairo G, Hamilton JA, Nice EC. Purification of two forms of colony-stimulating factor from mouse L-cell-conditioned medium. J Biol Chem. 1985;260:16004–16011. [PubMed] [Google Scholar]
- Busch SA, Hamilton JA, Horn KP, Cuascut FX, Cutrone R, Lehman N, Deans RJ, Ting AE, Mays RW, Silver J. Multipotent adult progenitor cells prevent macrophage-mediated axonal dieback and promote regrowth after spinal cord injury. J Neurosci. 2011;31:944–953. doi: 10.1523/JNEUROSCI.3566-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butovsky O, Jedrychowski MP, Cialic R, Krasemann S, Murugaiyan G, Fanek Z, Greco DJ, Wu PM, Doykan CE, Kiner O, Lawson RJ, Frosch MP, Pochet N, Fatimy RE, Krichevsky AM, Gygi SP, Lassmann H, Berry J, Cudkowicz ME, Weiner HL. Targeting miR-155 restores abnormal microglia and attenuates disease in SOD1 mice. Ann Neurol. 2015;77:75–99. doi: 10.1002/ana.24304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Yin Y, Li N, Zhu D, Zhang J, Zhang CY, Zen K. Re-polarization of tumor-associated macrophages to pro-inflammatory M1 macrophages by microRNA-155. J Mol Cell Biol. 2012;4:341–343. doi: 10.1093/jmcb/mjs044. [DOI] [PubMed] [Google Scholar]
- Cardoso AL, Guedes JR, Pereira de Almeida L, Pedroso de Lima MC. miR-155 modulates microglia-mediated immune response by down-regulating SOCS-1 and promoting cytokine and nitric oxide production. Immunology. 2012;135:73–88. doi: 10.1111/j.1365-2567.2011.03514.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceppi M, Pereira PM, Dunand-Sauthier I, Barras E, Reith W, Santos MA, Pierre P. MicroRNA-155 modulates the interleukin-1 signaling pathway in activated human monocyte-derived dendritic cells. Proc Natl Acad Sci U S A. 2009;106:2735–2740. doi: 10.1073/pnas.0811073106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremer TJ, Ravneberg DH, Clay CD, Piper-Hunter MG, Marsh CB, Elton TS, Gunn JS, Amer A, Kanneganti TD, Schlesinger LS, Butchar JP, Tridandapani S. miR-155 induction by F. novicida but not the virulent F. tularensis results in SHIP down-regulation and enhanced pro-inflammatory cytokine response. PLoS One. 2009;4:e8508. doi: 10.1371/journal.pone.0008508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David S, Kroner A. Repertoire of microglial and macrophage responses after spinal cord injury. Nat Rev Neurosci. 2011;12:388–399. doi: 10.1038/nrn3053. [DOI] [PubMed] [Google Scholar]
- Detloff MR, Fisher LC, McGaughy V, Longbrake EE, Popovich PG, Basso DM. Remote activation of microglia and pro-inflammatory cytokines predict the onset and severity of below-level neuropathic pain after spinal cord injury in rats. Exp Neurol. 2008;212:337–347. doi: 10.1016/j.expneurol.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly DJ, Popovich PG. Inflammation and its role in neuroprotection, axonal regeneration and functional recovery after spinal cord injury. Exp Neurol. 2008;209:378–388. doi: 10.1016/j.expneurol.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans TA, Barkauskas DS, Myers JT, Hare EG, You JQ, Ransohoff RM, Huang AY, Silver J. High-resolution intravital imaging reveals that blood-derived macrophages but not resident microglia facilitate secondary axonal dieback in traumatic spinal cord injury. Exp Neurol. 2014;254:109–120. doi: 10.1016/j.expneurol.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonken LK, Gaudet AD, Gaier KR, Nelson RJ, Popovich PG. MicroRNA-155 deletion reduces anxiety- and depressive-like behaviors in mice. Psychoneuroendocrinology. 2016;63:362–369. doi: 10.1016/j.psyneuen.2015.10.019. [DOI] [PubMed] [Google Scholar]
- Freilich RW, Woodbury ME, Ikezu T. Integrated expression profiles of mRNA and miRNA in polarized primary murine microglia. PLoS One. 2013;8:e79416. doi: 10.1371/journal.pone.0079416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furlan JC, Krassioukov AV, Fehlings MG. The effects of gender on clinical and neurological outcomes after acute cervical spinal cord injury. J Neurotrauma. 2005;22:368–381. doi: 10.1089/neu.2005.22.368. [DOI] [PubMed] [Google Scholar]
- Gardiner NJ, Fernyhough P, Tomlinson DR, Mayer U, von der Mark H, Streuli CH. Alpha7 integrin mediates neurite outgrowth of distinct populations of adult sensory neurons. Mol Cell Neurosci. 2005;28:229–240. doi: 10.1016/j.mcn.2004.08.017. [DOI] [PubMed] [Google Scholar]
- Gaudet AD, Popovich PG. Extracellular matrix regulation of inflammation in the healthy and injured spinal cord. Exp Neurol. 2014;258:24–34. doi: 10.1016/j.expneurol.2013.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudet AD, Sweet DR, Polinski NK, Guan Z, Popovich PG. Galectin-1 in injured rat spinal cord: Implications for macrophage phagocytosis and neural repair. Mol Cell Neurosci. 2015;64:84–94. doi: 10.1016/j.mcn.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudet AD, Fonken LK, Gushchina LV, Aubrecht TG, Maurya SK, Periasamy M, Nelson RJ, Popovich PG. miR-155 deletion in female mice prevents diet-induced obesity. Sci Rep. 2016;6:22862. doi: 10.1038/srep22862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gensel JC, Schonberg DL, Alexander JK, McTigue DM, Popovich PG. Semi-automated Sholl analysis for quantifying changes in growth and differentiation of neurons and glia. J Neurosci Methods. 2010;190:71–79. doi: 10.1016/j.jneumeth.2010.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graff JW, Dickson AM, Clay G, McCaffrey AP, Wilson ME. Identifying functional microRNAs in macrophages with polarized phenotypes. J Biol Chem. 2012;287:21816–21825. doi: 10.1074/jbc.M111.327031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gris D, Marsh DR, Oatway MA, Chen Y, Hamilton EF, Dekaban GA, Weaver LC. Transient blockade of the CD11d/CD18 integrin reduces secondary damage after spinal cord injury, improving sensory, autonomic, and motor function. J Neurosci. 2004;24:4043–4051. doi: 10.1523/JNEUROSCI.5343-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellal F, Hurtado A, Ruschel J, Flynn KC, Laskowski CJ, Umlauf M, Kapitein LC, Strikis D, Lemmon V, Bixby J, Hoogenraad CC, Bradke F. Microtubule stabilization reduces scarring and causes axon regeneration after spinal cord injury. Science. 2011;331:928–931. doi: 10.1126/science.1201148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herai RR, Stefanacci L, Hrvoj-Mihic B, Chailangkarn T, Hanson K, Semendeferi K, Muotri AR. Micro RNA detection in long-term fixed tissue of cortical glutamatergic pyramidal neurons after targeted laser-capture neuroanatomical microdissection. J Neurosci Methods. 2014;235:76–82. doi: 10.1016/j.jneumeth.2014.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn KP, Busch SA, Hawthorne AL, van Rooijen N, Silver J. Another barrier to regeneration in the CNS: activated macrophages induce extensive retraction of dystrophic axons through direct physical interactions. J Neurosci. 2008;28:9330–9341. doi: 10.1523/JNEUROSCI.2488-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard CV, Reed MG. Unbiased stereology: three-dimensional measurement in microscopy. New York: Springer; 1998. [Google Scholar]
- Jablonski KA, Gaudet AD, Amici SA, Popovich PG, Guerau-de-Arellano M. Control of the inflammatory macrophage transcriptional signature by miR-155. PLOS ONE. 2016 doi: 10.1371/journal.pone.0159724. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson RD, Virág I, Skene JH. A protein associated with axon growth, GAP-43, is widely distributed and developmentally regulated in rat CNS. J Neurosci. 1986;6:1843–1855. doi: 10.1523/JNEUROSCI.06-06-01843.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John B, Enright AJ, Aravin A, Tuschl T, Sander C, Marks DS. Human MicroRNA targets. PLoS Biol. 2004;2:e363. doi: 10.1371/journal.pbio.0020363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones TB, Basso DM, Sodhi A, Pan JZ, Hart RP, MacCallum RC, Lee S, Whitacre CC, Popovich PG. Pathological CNS autoimmune disease triggered by traumatic spinal cord injury: implications for autoimmune vaccine therapy. J Neurosci. 2002;22:2690–2700. doi: 10.1523/JNEUROSCI.22-07-02690.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khazaei MR, Girouard MP, Alchini R, Ong Tone S, Shimada T, Bechstedt S, Cowan M, Guillet D, Wiseman PW, Brouhard G, Cloutier JF, Fournier AE. Collapsin response mediator protein 4 regulates growth cone dynamics through the actin and microtubule cytoskeleton. J Biol Chem. 2014;289:30133–30143. doi: 10.1074/jbc.M114.570440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kigerl KA, McGaughy VM, Popovich PG. Comparative analysis of lesion development and intraspinal inflammation in four strains of mice following spinal contusion injury. J Comp Neurol. 2006;494:578–594. doi: 10.1002/cne.20827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci. 2009;29:13435–13444. doi: 10.1523/JNEUROSCI.3257-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi NR, Fan DP, Giehl KM, Bedard AM, Wiegand SJ, Tetzlaff W. BDNF and NT-4/5 prevent atrophy of rat rubrospinal neurons after cervical axotomy, stimulate GAP-43 and Talpha1-tubulin mRNA expression, and promote axonal regeneration. J Neurosci. 1997;17:9583–9595. doi: 10.1523/JNEUROSCI.17-24-09583.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koval ED, Shaner C, Zhang P, du Maine X, Fischer K, Tay J, Chau BN, Wu GF, Miller TM. Method for widespread microRNA-155 inhibition prolongs survival in ALS-model mice. Hum Mol Genet. 2013;22:4127–4135. doi: 10.1093/hmg/ddt261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroner A, Greenhalgh AD, Zarruk JG, Passos Dos Santos R, Gaestel M, David S. TNF and increased intracellular iron alter macrophage polarization to a detrimental M1 phenotype in the injured spinal cord. Neuron. 2014;83:1098–1116. doi: 10.1016/j.neuron.2014.07.027. [DOI] [PubMed] [Google Scholar]
- Kurowska-Stolarska M, Alivernini S, Ballantine LE, Asquith DL, Millar NL, Gilchrist DS, Reilly J, Ierna M, Fraser AR, Stolarski B, McSharry C, Hueber AJ, Baxter D, Hunter J, Gay S, Liew FY, McInnes IB. MicroRNA-155 as a pro-inflammatory regulator in clinical and experimental arthritis. Proc Natl Acad Sci U S A. 2011;108:11193–11198. doi: 10.1073/pnas.1019536108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon BK, Oxland TR, Tetzlaff W. Animal models used in spinal cord regeneration research. Spine. 2002;27:1504–1510. doi: 10.1097/00007632-200207150-00005. [DOI] [PubMed] [Google Scholar]
- Kwon MJ, Kim J, Shin H, Jeong SR, Kang YM, Choi JY, Hwang DH, Kim BG. Contribution of macrophages to enhanced regenerative capacity of dorsal root ganglia sensory neurons by conditioning injury. J Neurosci. 2013;33:15095–15108. doi: 10.1523/JNEUROSCI.0278-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003;115:787–798. doi: 10.1016/S0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- Liu K, Lu Y, Lee JK, Samara R, Willenberg R, Sears-Kraxberger I, Tedeschi A, Park KK, Jin D, Cai B, Xu B, Connolly L, Steward O, Zheng B, He Z. PTEN deletion enhances the regenerative ability of adult corticospinal neurons. Nat Neurosci. 2010;13:1075–1081. doi: 10.1038/nn.2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Tedeschi A, Park KK, He Z. Neuronal intrinsic mechanisms of axon regeneration. Annu Rev Neurosci. 2011a;34:131–152. doi: 10.1146/annurev-neuro-061010-113723. [DOI] [PubMed] [Google Scholar]
- Liu S, Yang Y, Wu J. TNFalpha-induced up-regulation of miR-155 inhibits adipogenesis by down-regulating early adipogenic transcription factors. Biochem Biophys Res Commun. 2011b;414:618–624. doi: 10.1016/j.bbrc.2011.09.131. [DOI] [PubMed] [Google Scholar]
- Longbrake EE, Lai W, Ankeny DP, Popovich PG. Characterization and modeling of monocyte-derived macrophages after spinal cord injury. J Neurochem. 2007;102:1083–1094. doi: 10.1111/j.1471-4159.2007.04617.x. [DOI] [PubMed] [Google Scholar]
- Lopez-Ramirez MA, Wu D, Pryce G, Simpson JE, Reijerkerk A, King-Robson J, Kay O, de Vries HE, Hirst MC, Sharrack B, Baker D, Male DK, Michael GJ, Romero IA. MicroRNA-155 negatively affects blood-brain barrier function during neuroinflammation. FASEB J. 2014;28:2551–2565. doi: 10.1096/fj.13-248880. [DOI] [PubMed] [Google Scholar]
- Lu LF, Thai TH, Calado DP, Chaudhry A, Kubo M, Tanaka K, Loeb GB, Lee H, Yoshimura A, Rajewsky K, Rudensky AY. Foxp3-dependent microRNA155 confers competitive fitness to regulatory T cells by targeting SOCS1 protein. Immunity. 2009;30:80–91. doi: 10.1016/j.immuni.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Richardson PM. Inflammation near the nerve cell body enhances axonal regeneration. J Neurosci. 1991;11:972–978. doi: 10.1523/JNEUROSCI.11-04-00972.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchetti S, Beck KD, Galvan MD, Silva R, Cummings BJ, Anderson AJ. Comparison of immunopathology and locomotor recovery in C57BL/6, BUB/BnJ, and NOD-SCID mice after contusion spinal cord injury. J Neurotrauma. 2010;27:411–421. doi: 10.1089/neu.2009.0930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Nunez RT, Louafi F, Sanchez-Elsner T. The interleukin 13 (IL-13) pathway in human macrophages is modulated by microRNA-155 via direct targeting of interleukin 13 receptor alpha1 (IL13Ralpha1) J Biol Chem. 2011;286:1786–1794. doi: 10.1074/jbc.M110.169367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashima R. Physiological roles of miR-155. Immunology. 2015;145:323–333. doi: 10.1111/imm.12468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason MR, Lieberman AR, Grenningloh G, Anderson PN. Transcriptional upregulation of SCG10 and CAP-23 is correlated with regeneration of the axons of peripheral and central neurons in vivo. Mol Cell Neurosci. 2002;20:595–615. doi: 10.1006/mcne.2002.1140. [DOI] [PubMed] [Google Scholar]
- McGraw J, Gaudet AD, Oschipok LW, Steeves JD, Poirier F, Tetzlaff W, Ramer MS. Altered primary afferent anatomy and reduced thermal sensitivity in mice lacking galectin-1. Pain. 2005;114:7–18. doi: 10.1016/j.pain.2004.10.009. [DOI] [PubMed] [Google Scholar]
- McKeon RJ, Schreiber RC, Rudge JS, Silver J. Reduction of neurite outgrowth in a model of glial scarring following CNS injury is correlated with the expression of inhibitory molecules on reactive astrocytes. J Neurosci. 1991;11:3398–3411. doi: 10.1523/JNEUROSCI.11-11-03398.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller FD, Tetzlaff W, Bisby MA, Fawcett JW, Milner RJ. Rapid induction of the major embryonic alpha-tubulin mRNA, T alpha 1, during nerve regeneration in adult rats. J Neurosci. 1989;9:1452–1463. doi: 10.1523/JNEUROSCI.09-04-01452.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miron VE, Boyd A, Zhao JW, Yuen TJ, Ruckh JM, Shadrach JL, van Wijngaarden P, Wagers AJ, Williams A, Franklin RJ, ffrench-Constant C. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci. 2013;16:1211–1218. doi: 10.1038/nn.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore CS, Rao VT, Durafourt BA, Bedell BJ, Ludwin SK, Bar-Or A, Antel JP. miR-155 as a multiple sclerosis-relevant regulator of myeloid cell polarization. Ann Neurol. 2013;74:709–720. doi: 10.1002/ana.23967. [DOI] [PubMed] [Google Scholar]
- Murugaiyan G, Beynon V, Mittal A, Joller N, Weiner HL. Silencing microRNA-155 ameliorates experimental autoimmune encephalomyelitis. J Immunol. 2011;187:2213–2221. doi: 10.4049/jimmunol.1003952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadeau S, Hein P, Fernandes KJ, Peterson AC, Miller FD. A transcriptional role for C/EBP beta in the neuronal response to axonal injury. Mol Cell Neurosci. 2005;29:525–535. doi: 10.1016/j.mcn.2005.04.004. [DOI] [PubMed] [Google Scholar]
- Nazari-Jahantigh M, Wei Y, Noels H, Akhtar S, Zhou Z, Koenen RR, Heyll K, Gremse F, Kiessling F, Grommes J, Weber C, Schober A. MicroRNA-155 promotes atherosclerosis by repressing Bcl6 in macrophages. J Clin Invest. 2012;122:4190–4202. doi: 10.1172/JCI61716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann S, Woolf CJ. Regeneration of dorsal column fibers into and beyond the lesion site following adult spinal cord injury. Neuron. 1999;23:83–91. doi: 10.1016/S0896-6273(00)80755-2. [DOI] [PubMed] [Google Scholar]
- Niemi JP, DeFrancesco-Lisowitz A, Roldán-Hernandez L, Lindborg JA, Mandell D, Zigmond RE. A critical role for macrophages near axotomized neuronal cell bodies in stimulating nerve regeneration. J Neurosci. 2013;33:16236–16248. doi: 10.1523/JNEUROSCI.3319-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc Natl Acad Sci U S A. 2007;104:1604–1609. doi: 10.1073/pnas.0610731104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell RM, Chaudhuri AA, Rao DS, Baltimore D. Inositol phosphatase SHIP1 is a primary target of miR-155. Proc Natl Acad Sci U S A. 2009;106:7113–7118. doi: 10.1073/pnas.0902636106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell RM, Kahn D, Gibson WS, Round JL, Scholz RL, Chaudhuri AA, Kahn ME, Rao DS, Baltimore D. MicroRNA-155 promotes autoimmune inflammation by enhancing inflammatory T cell development. Immunity. 2010;33:607–619. doi: 10.1016/j.immuni.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Offiah I, Didangelos A, Dawes J, Cartwright R, Khullar V, Bradbury EJ, O'Sullivan S, Williams D, Chessell IP, Pallas K, Graham G, O'Reilly BA, McMahon SB. The expression of inflammatory mediators in bladder pain syndrome. Eur Urol. 2016;70:283–290. doi: 10.1016/j.eururo.2016.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearse DD, Chatzipanteli K, Marcillo AE, Bunge MB, Dietrich WD. Comparison of iNOS inhibition by antisense and pharmacological inhibitors after spinal cord injury. J Neuropathol Exp Neurol. 2003;62:1096–1107. doi: 10.1093/jnen/62.11.1096. [DOI] [PubMed] [Google Scholar]
- Piccinini AM, Midwood KS. Endogenous control of immunity against infection: tenascin-C regulates TLR4-mediated inflammation via microRNA-155. Cell Rep. 2012;2:914–926. doi: 10.1016/j.celrep.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineau I, Sun L, Bastien D, Lacroix S. Astrocytes initiate inflammation in the injured mouse spinal cord by promoting the entry of neutrophils and inflammatory monocytes in an IL-1 receptor/MyD88-dependent fashion. Brain Behav Immun. 2010;24:540–553. doi: 10.1016/j.bbi.2009.11.007. [DOI] [PubMed] [Google Scholar]
- Plunet W, Kwon BK, Tetzlaff W. Promoting axonal regeneration in the central nervous system by enhancing the cell body response to axotomy. J Neurosci Res. 2002;68:1–6. doi: 10.1002/jnr.10176. [DOI] [PubMed] [Google Scholar]
- Pool M, Rambaldi I, Darlington PJ, Wright MC, Fournier AE, Bar-Or A. Neurite outgrowth is differentially impacted by distinct immune cell subsets. Mol Cell Neurosci. 2012;49:68–76. doi: 10.1016/j.mcn.2011.09.004. [DOI] [PubMed] [Google Scholar]
- Popovich PG, Longbrake EE. Can the immune system be harnessed to repair the CNS? Nat Rev Neurosci. 2008;9:481–493. doi: 10.1038/nrn2398. [DOI] [PubMed] [Google Scholar]
- Popovich PG, Wei P, Stokes BT. Cellular inflammatory response after spinal cord injury in Sprague-Dawley and Lewis rats. J Comp Neurol. 1997;377:443–464. doi: 10.1002/(SICI)1096-9861(19970120)377:3%3C443::AID-CNE10%3E3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Popovich PG, Guan Z, Wei P, Huitinga I, van Rooijen N, Stokes BT. Depletion of hematogenous macrophages promotes partial hindlimb recovery and neuroanatomical repair after experimental spinal cord injury. Exp Neurol. 1999;158:351–365. doi: 10.1006/exnr.1999.7118. [DOI] [PubMed] [Google Scholar]
- Prüss H, Kopp MA, Brommer B, Gatzemeier N, Laginha I, Dirnagl U, Schwab JM. Non-resolving aspects of acute inflammation after spinal cord injury (SCI): indices and resolution plateau. Brain Pathol. 2011;21:652–660. doi: 10.1111/j.1750-3639.2011.00488.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramer MS, Priestley JV, McMahon SB. Functional regeneration of sensory axons into the adult spinal cord. Nature. 2000;403:312–316. doi: 10.1038/35002084. [DOI] [PubMed] [Google Scholar]
- Ramer MS, Duraisingam I, Priestley JV, McMahon SB. Two-tiered inhibition of axon regeneration at the dorsal root entry zone. J Neurosci. 2001;21:2651–2660. doi: 10.1523/JNEUROSCI.21-08-02651.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramkhelawon B, Hennessy EJ, Ménager M, Ray TD, Sheedy FJ, Hutchison S, Wanschel A, Oldebeken S, Geoffrion M, Spiro W, Miller G, McPherson R, Rayner KJ, Moore KJ. Netrin-1 promotes adipose tissue macrophage retention and insulin resistance in obesity. Nat Med. 2014;20:377–384. doi: 10.1038/nm.3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson PM, Issa VM. Peripheral injury enhances central regeneration of primary sensory neurones. Nature. 1984;309:791–793. doi: 10.1038/309791a0. [DOI] [PubMed] [Google Scholar]
- Rodriguez A, Vigorito E, Clare S, Warren MV, Couttet P, Soond DR, van Dongen S, Grocock RJ, Das PP, Miska EA, Vetrie D, Okkenhaug K, Enright AJ, Dougan G, Turner M, Bradley A. Requirement of bic/microRNA-155 for normal immune function. Science. 2007;316:608–611. doi: 10.1126/science.1139253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roitbak T, Bragina O, Padilla JL, Pickett GG. The role of microRNAs in neural stem cell-supported endothelial morphogenesis. Vasc Cell. 2011;3:25. doi: 10.1186/2045-824X-3-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose RD, Rohrlich D. Counting sectioned cells via mathematical reconstruction. J Comp Neurol. 1988;272:365–386. [PubMed] [Google Scholar]
- Schmidt AM, Moore KJ. The Semaphorin 3E/PlexinD1 axis regulates macrophage inflammation in obesity. Cell Metab. 2013;18:461–462. doi: 10.1016/j.cmet.2013.09.011. [DOI] [PubMed] [Google Scholar]
- Schwab JM, Zhang Y, Kopp MA, Brommer B, Popovich PG. The paradox of chronic neuroinflammation, systemic immune suppression, autoimmunity after traumatic chronic spinal cord injury. Exp Neurol. 2014;258:121–129. doi: 10.1016/j.expneurol.2014.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seijffers R, Mills CD, Woolf CJ. ATF3 increases the intrinsic growth state of DRG neurons to enhance peripheral nerve regeneration. J Neurosci. 2007;27:7911–7920. doi: 10.1523/JNEUROSCI.5313-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipski ML, Jackson AB, Gómez-Marin O, Estores I, Stein A. Effects of gender on neurologic and functional recovery after spinal cord injury. Arch Phys Med Rehabil. 2004;85:1826–1836. doi: 10.1016/j.apmr.2004.04.031. [DOI] [PubMed] [Google Scholar]
- Skene JH, Willard M. Axonally transported proteins associated with axon growth in rabbit central and peripheral nervous systems. J Cell Biol. 1981;89:96–103. doi: 10.1083/jcb.89.1.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DS, Skene JH. A transcription-dependent switch controls competence of adult neurons for distinct modes of axon growth. J Neurosci. 1997;17:646–658. doi: 10.1523/JNEUROSCI.17-02-00646.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein M, Keshav S, Harris N, Gordon S. Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J Exp Med. 1992;176:287–292. doi: 10.1084/jem.176.1.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens WL. Asymptotic regression. Biometrics. 1951;7:247–267. doi: 10.2307/3001809. [DOI] [Google Scholar]
- Sun F, Park KK, Belin S, Wang D, Lu T, Chen G, Zhang K, Yeung C, Feng G, Yankner BA, He Z. Sustained axon regeneration induced by co-deletion of PTEN and SOCS3. Nature. 2011;480:372–375. doi: 10.1038/nature10594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartz KR, Fee DB, Joy KM, Roberts KN, Sun S, Scheff NN, Wilson ME, Scheff SW. Gender differences in spinal cord injury are not estrogen-dependent. J Neurotrauma. 2007;24:473–480. doi: 10.1089/neu.2006.0167. [DOI] [PubMed] [Google Scholar]
- Tan Y, Yang J, Xiang K, Tan Q, Guo Q. Suppression of microRNA-155 attenuates neuropathic pain by regulating SOCS1 signalling pathway. Neurochem Res. 2015;40:550–560. doi: 10.1007/s11064-014-1500-2. [DOI] [PubMed] [Google Scholar]
- Tarassishin L, Loudig O, Bauman A, Shafit-Zagardo B, Suh HS, Lee SC. Interferon regulatory factor 3 inhibits astrocyte inflammatory gene expression through suppression of the pro-inflammatory miR-155 and miR-155*. Glia. 2011;59:1911–1922. doi: 10.1002/glia.21233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetzlaff W, Alexander SW, Miller FD, Bisby MA. Response of facial and rubrospinal neurons to axotomy: changes in mRNA expression for cytoskeletal proteins and GAP-43. J Neurosci. 1991;11:2528–2544. doi: 10.1523/JNEUROSCI.11-08-02528.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thai TH, Calado DP, Casola S, Ansel KM, Xiao C, Xue Y, Murphy A, Frendewey D, Valenzuela D, Kutok JL, Schmidt-Supprian M, Rajewsky N, Yancopoulos G, Rao A, Rajewsky K. Regulation of the germinal center response by microRNA-155. Science. 2007;316:604–608. doi: 10.1126/science.1141229. [DOI] [PubMed] [Google Scholar]
- Tili E, Michaille JJ, Cimino A, Costinean S, Dumitru CD, Adair B, Fabbri M, Alder H, Liu CG, Calin GA, Croce CM. Modulation of miR-155 and miR-125b levels following lipopolysaccharide/TNF-alpha stimulation and their possible roles in regulating the response to endotoxin shock. J Immunol. 2007;179:5082–5089. doi: 10.4049/jimmunol.179.8.5082. [DOI] [PubMed] [Google Scholar]
- Togari A, Mogi M, Arai M, Yamamoto S, Koshihara Y. Expression of mRNA for axon guidance molecules, such as semaphorin-III, netrins and neurotrophins, in human osteoblasts and osteoclasts. Brain Res. 2000;878:204–209. doi: 10.1016/S0006-8993(00)02700-1. [DOI] [PubMed] [Google Scholar]
- Tom VJ, Sandrow-Feinberg HR, Miller K, Domitrovich C, Bouyer J, Zhukareva V, Klaw MC, Lemay MA, Houlé JD. Exogenous BDNF enhances the integration of chronically injured axons that regenerate through a peripheral nerve grafted into a chondroitinase-treated spinal cord injury site. Exp Neurol. 2013;239:91–100. doi: 10.1016/j.expneurol.2012.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varendi K, Kumar A, Härma MA, Andressoo JO. miR-1, miR-10b, miR-155, and miR-191 are novel regulators of BDNF. Cell Mol Life Sci. 2014;71:4443–4456. doi: 10.1007/s00018-014-1628-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigorito E, Perks KL, Abreu-Goodger C, Bunting S, Xiang Z, Kohlhaas S, Das PP, Miska EA, Rodriguez A, Bradley A, Smith KG, Rada C, Enright AJ, Toellner KM, Maclennan IC, Turner M. microRNA-155 regulates the generation of immunoglobulin class-switched plasma cells. Immunity. 2007;27:847–859. doi: 10.1016/j.immuni.2007.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Yang K, Zhou L, Minhaowu Wu Y, Zhu M, Lai X, Chen T, Feng L, Li M, Huang C, Zhong Q, Huang X. MicroRNA-155 promotes autophagy to eliminate intracellular mycobacteria by targeting Rheb. PLoS Pathog. 2013;9:e1003697. doi: 10.1371/journal.ppat.1003697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Hou J, Lin L, Wang C, Liu X, Li D, Ma F, Wang Z, Cao X. Inducible microRNA-155 feedback promotes type I IFN signaling in antiviral innate immunity by targeting suppressor of cytokine signaling 1. J Immunol. 2010;185:6226–6233. doi: 10.4049/jimmunol.1000491. [DOI] [PubMed] [Google Scholar]
- Worm J, Stenvang J, Petri A, Frederiksen KS, Obad S, Elmén J, Hedtjärn M, Straarup EM, Hansen JB, Kauppinen S. Silencing of microRNA-155 in mice during acute inflammatory response leads to derepression of c/ebp Beta and down-regulation of G-CSF. Nucleic Acids Res. 2009;37:5784–5792. doi: 10.1093/nar/gkp577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Wang BR, Wang X, Kuang F, Duan XL, Jiao XY, Ju G. ERK1/2 and p38 mitogen-activated protein kinase mediate iNOS-induced spinal neuron degeneration after acute traumatic spinal cord injury. Life Sci. 2006;79:1895–1905. doi: 10.1016/j.lfs.2006.06.023. [DOI] [PubMed] [Google Scholar]
- Ye JH, Houle JD. Treatment of the chronically injured spinal cord with neurotrophic factors can promote axonal regeneration from supraspinal neurons. Exp Neurol. 1997;143:70–81. doi: 10.1006/exnr.1996.6353. [DOI] [PubMed] [Google Scholar]
- Yin Q, Wang X, Fewell C, Cameron J, Zhu H, Baddoo M, Lin Z, Flemington EK. MicroRNA miR-155 inhibits bone morphogenetic protein (BMP) signaling and BMP-mediated Epstein-Barr virus reactivation. J Virol. 2010;84:6318–6327. doi: 10.1128/JVI.00635-10. [DOI] [PMC free article] [PubMed] [Google Scholar]