

Abstract
Summary: MOCAT2 is a software pipeline for metagenomic sequence assembly and gene prediction with novel features for taxonomic and functional abundance profiling. The automated generation and efficient annotation of non-redundant reference catalogs by propagating pre-computed assignments from 18 databases covering various functional categories allows for fast and comprehensive functional characterization of metagenomes.
Availability and Implementation: MOCAT2 is implemented in Perl 5 and Python 2.7, designed for 64-bit UNIX systems and offers support for high-performance computer usage via LSF, PBS or SGE queuing systems; source code is freely available under the GPL3 license at http://mocat.embl.de.
Contact: bork@embl.de
Supplementary information: Supplementary data are available at Bioinformatics online.
1 Introduction
Metagenomics has enabled large-scale studies investigating the structure, function and diversity of microbial communities. The computational analysis of samples, often totaling many gigabases of sequence data, usually involves mapping reads to taxonomic and functional reference databases (which may require the de novo assembly of predicted genes), and subsequent abundance profiling. Whereas taxonomic profiling methodology has matured recently (Segata et al., 2013; Sunagawa et al., 2013), functional profiling still remains challenging due to the difficulties in assigning functions to millions of reads from metagenomes. Moreover, current metagenomic pipelines (Abubucker et al., 2012; Bose et al., 2015; Edwards et al., 2012; Glass et al., 2010; Huson et al., 2011; Lingner et al., 2011; Markowitz et al., 2008; Meinicke, 2015; Glass et al., 2010; Huson et al., 2011; Lingner et al., 2011; Abubucker et al., 2012; Edwards et al., 2012; Bose et al., 2015; Silva et al., 2015) for functional annotation and/or profiling mainly implement metabolic pathway or protein domain databases (Segata et al., 2013) such as KEGG (Kanehisa et al., 2014), SEED (Overbeek et al., 2014) or Pfam (Finn et al., 2014). Here, we present metagenomic analysis toolkit version 2 (MOCAT2), which was developed to enable functional profiling of metagenomes based on a much wider range and diversity of functional gene annotations. Its features are compared to existing tools in Supplementary Table S1.
2 The MOCAT2 pipeline
The metagenomic analysis toolkit (MOCAT) (Kultima et al., 2012) proceeds through the following steps: raw sequence reads are quality-filtered and subsequently assembled into longer contigs, on which open reading frames are predicted (Fig. 1).
Fig. 1.
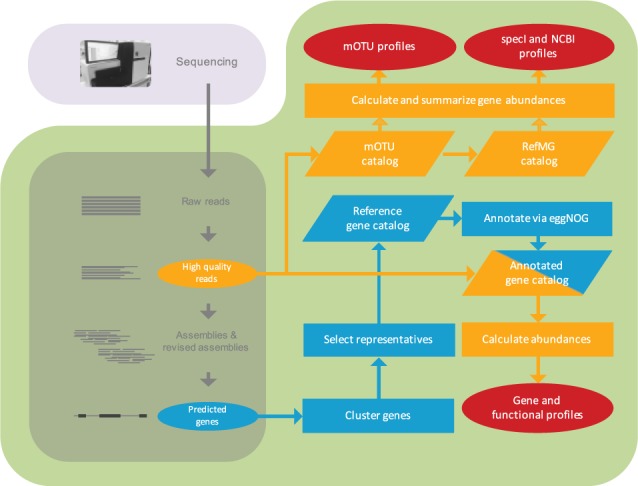
The MOCAT2 pipeline. Read quality control, assembly and gene prediction represent the original MOCAT pipeline (dark green box). Blue path: Genes are clustered into reference gene catalogs, which are functionally annotated. Orange path: To quantify functional composition, reads are mapped to the annotated gene catalog and summarized over the respective annotation categories. Taxonomic profiles (mOTU, specI and NCBI) are generated by mapping reads to mOTU and reference marker gene (RefMG) catalogs
Its main extensions in MOCAT2 enable comprehensive functional profiling, in addition to the eggNOG database, by integrating 18 publicly available resources that cover diverse functional properties (Table 1). The databases were selected to include large, widely used protein databases, as well as ones targeting specific functional categories (Supplementary Text). Each database has been filtered for relevance, for example from the eukaryote-centered database DrugBank only the genes with bacterial homologs were extracted.
Table 1.
Databases from which functional properties are obtained
| Proteins | Coverage | Precision | Recall | Reference | |
|---|---|---|---|---|---|
| Protein domains and families | |||||
| eggNOG | 7 449 593 | 100 | 100 | 100 | Huerta-Cepas et al. (2015) |
| Pfam | 16 230* | 87 | 90 | 94 | Finn et al. (2014) |
| Superfamily | 15 438* | 93 | 89 | 94 | Gough et al. (2001) |
| (Metabolic) pathways | |||||
| KEGG | 7 423 864 | 98 | 93 | 93 | Kanehisa et al. (2014) |
| MetaCyc | 388 782 | 100 | 89 | 94 | Caspi et al. (2014) |
| SEED | 4 247 700 | 99 | 94 | 94 | Overbeek et al. (2014) |
| Antibiotic resistance | |||||
| ARDB | 25 360 | 89 | 99 | 88 | Liu and Pop (2009) |
| CARD | 2 820 | 100 | 81 | 93 | McArthur et al. (2013) |
| Resfams | 123* | 80 | 94 | 94 | Gibson et al. (2014) |
| Virulence factors | |||||
| MvirDB | 29 357 | 100 | 95 | 93 | Zhou et al. (2007) |
| PATRIC | 2 194 475 | 93 | 93 | 93 | Mao et al. (2015) |
| vFam | 29 655 | 35 | 99 | 86 | Skewes-Cox et al. (2014) |
| VFDB | 1 627 380 | 86 | 89 | 91 | Chen et al. (2012) |
| Victors | 3 329 893 | 91 | 92 | 94 | Mao et al. (2015) |
| Complex carbohydrate metabolism | |||||
| dbCAN | 333* | 76 | 99 | 99 | Yin et al. (2012) |
| Bacterial drug targets and exotoxins | |||||
| DBETH | 228 | 100 | 99 | 86 | Chakraborty et al. (2012) |
| DrugBank | 3 899 | 99 | 88 | 94 | Knox et al. (2011) |
| Mobile genetic elements | |||||
| ICEberg | 13 984 | 98 | 79 | 91 | Bi et al. (2012) |
| Prophages | 119 183 | 95 | 88 | 91 | Waller et al. (2014) |
Coverage of each database in percent, e.g., of the 18 202 orthologous groups in KEGG (KO), 17 773 (98%) are covered and thus propagated by the eggNOG database. Coverage, precision and recall are given as percentages.
*Number of hidden Markov models (HMMs), whereby one HMM can hit several proteins and several HMMs can map to one protein.
To avoid the computational burden of mapping reads to multiple databases, predicted genes are first clustered using CD-HIT (Huang et al., 2010) into a non-redundant gene set, called a reference gene catalog (Qin et al., 2010). Next, this gene catalog is mapped to the eggNOG database with wide taxonomic coverage of orthologous groups, to which sequence annotations from other databases have been pre-computed so that functional information from multiple databases can be transferred efficiently to the catalog. This indirect annotation methodology not only provides a 10-fold speed up compared to directly mapping to each database separately, but also enables annotations of short genes, which would otherwise be missed (Supplementary Figure Fig. S1). For computational efficiency MOCAT2 uses DIAMOND (Buchfink et al., 2014) in the annotation step. Combined, these features yield a more than 1400-fold annotation speedup over a conventional BLAST-based annotation pipeline (Supplementary Text). Users can either create and annotate their own gene catalogs de novo, or use pre-computed and pre-annotated reference gene catalogs for the human gut and skin, mouse gut, or the ocean (Li et al., 2014; Oh et al., 2014; Sunagawa et al., 2015; Xiao et al., 2015).
Finally, to quantify functional composition, reads from each sample are mapped to the annotated gene catalog and summarized over the respective annotation categories (Fig. 1).
MOCAT2 now also offers several approaches for taxonomic profiling, all of which are based on mapping reads to a benchmarked set of single copy marker genes (Fig. 1). Taxonomic abundance estimates are calculated not only for different NCBI taxonomic levels, but also for species clusters defined based on molecular sequence identity (specI; Mende et al., 2013) and species that currently lack sequenced reference genomes based on metagenomic operational taxonomic units (mOTU; Sunagawa et al., 2013).
3 Annotation and profiling benchmarks
As complex functional annotation based on 18 databases via indirect propagation of eggNOG annotations is conceptually new, we benchmarked the (indirect) MOCAT2 annotations and functional profiles (Supplementary Table S2 and Supplementary Text).
First, we compared the indirect annotations to the direct ones (generated using the annotation tool of each individual database or recommended pipeline and cutoffs) for >65 million genes from five diverse datasets (precision and recall are listed in Table 1).
Next, using data from (Zeller et al., 2014) we compared the direct annotations to ones produced by COGNIZER and UProC (Bose et al., 2015; Meinicke et al., 2015), two recently developed annotation tools integrating multiple databases. In our tests, MOCAT2 annotations were either similar to, or more accurate, than those of COGNIZER and UProC (Supplementary Table S3).
Finally, the functional abundance profiles obtained using the indirect MOCAT2 annotations were very similar to those obtained using the direct method (Spearman = 0.95; n = 1300).
4 Conclusions
MOCAT2 is a software pipeline for metagenomics using state of the art assembly, annotation as well as taxonomic and functional profiling approaches in this fast moving field. Generating and annotating gene catalogs with precomputed assignments to a large selection of functional databases allows for comprehensive and efficient functional profiling of complex microbial communities. MOCAT2 thus enables such analysis at an extent far beyond what other tools currently offer and is scalable to the anticipated deluge of metagenomic data from diverse sources.
Supplementary Material
Acknowledgements
We wish to thank Bernd Klaus for valuable feedback on statistical analyses and Alison Waller for kindly providing the prophages database.
Funding
This work was supported by the European Research Council CancerBiome project [grant number 268985], the International Human Microbiome Standards project [grant number HEALTH-2010-261376] and the MetaCardis project [grant number HEALTH-2012-305312]. S.S.L. is the recipient of an Australian Postgraduate Award and EMBL Australia International PhD Fellowship.
Conflict of Interest: none declared.
References
- Abubucker S. et al. (2012) Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS Comput. Biol., 8, e1002358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi D. et al. (2012) ICEberg: a web-based resource for integrative and conjugative elements found in Bacteria. Nucleic Acids Res., 40, D621–D626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose T. et al. (2015) COGNIZER: a framework for functional annotation of metagenomic datasets. PLoS One, 10, e0142102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchfink B. et al. (2014) Fast and sensitive protein alignment using DIAMOND. Nat. Methods, 12, 59–60. [DOI] [PubMed] [Google Scholar]
- Caspi R. et al. (2014) The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res., 42, D459–D471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty A. et al. (2012) DBETH: a database of bacterial exotoxins for human. Nucleic Acids Res., 40, D615–D620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L. et al. (2012) VFDB 2012 update: Toward the genetic diversity and molecular evolution of bacterial virulence factors. Nucleic Acids Res., 40, 641–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards R.A. et al. (2012) Real Time Metagenomics: Using k-mers to annotate metagenomes. Bioinformatics, 28, 3316–3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn R.D. et al. (2014) Pfam: the protein families database. Nucleic Acids Res., 42, D222–D230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson M.K. et al. (2014) Improved annotation of antibiotic resistance determinants reveals microbial resistomes cluster by ecology. ISME J., 9, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass E.M. et al. (2010) Using the metagenomics RAST server (MG-RAST) for analyzing shotgun metagenomes. Cold Spring Harb. Protoc., 2010, 1–10. [DOI] [PubMed] [Google Scholar]
- Gough J. et al. (2001) Assignment of homology to genome sequences using a library of hidden Markov models that represent all proteins of known structure. J. Mol. Biol., 313, 903–919. [DOI] [PubMed] [Google Scholar]
- Huang Y. et al. (2010) CD-HIT Suite: a web server for clustering and comparing biological sequences. Bioinformatics, 26, 680–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta-Cepas J. et al. (2015) eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res, 44, D286–D293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson D.H. et al. (2011) Integrative analysis of environmental sequences using MEGAN4. Genome Res., 21, 1552–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M. et al. (2014) Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res., 42, D199–D205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knox C. et al. (2011) DrugBank 3.0: a comprehensive resource for ‘omics’ research on drugs. Nucleic Acids Res., 39, D1035–D1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kultima J.R. et al. (2012) MOCAT: a metagenomics assembly and gene prediction toolkit. PLoS One, 7, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J. et al. (2014) An integrated catalog of reference genes in the human gut microbiome. Nat. Biotechnol., 32, 834–841. [DOI] [PubMed] [Google Scholar]
- Lingner T. et al. (2011) CoMet – a web server for comparative functional profiling of metagenomes. Nucleic Acids Res., 39, 1–6, doi:10.1093/nar/gkr388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B., Pop M. (2009) ARDB – antibiotic resistance genes database. Nucleic Acids Res., 37, D443–D447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao C. et al. (2015) Curation, integration and visualization of bacterial virulence factors in PATRIC. Bioinformatics, 31, 252–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markowitz V.M. et al. (2008) IMG/M: a data management and analysis system for metagenomes. Nucleic Acids Res., 36, D534–D538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McArthur A.G. et al. (2013) The comprehensive antibiotic resistance database. Antimicrob. Agents Chemother., 57, 3348–3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinicke P. (2015) UProC: tools for ultra-fast protein domain classification. Bioinformatics, 31, 1382–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mende D.R. et al. (2013) Accurate and universal delineation of prokaryotic species. Nat. Methods, 10, 881–884. [DOI] [PubMed] [Google Scholar]
- Oh J. et al. (2014) Biogeography and individuality shape function in the human skin metagenome. Nature, 514, 59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overbeek R. et al. (2014) The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res., 42, D206–D214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J. et al. (2010) A human gut microbial gene catalogue established by metagenomic sequencing. Nature, 464, 59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segata N. et al. (2013) Computational meta’omics for microbial community studies. Mol. Syst. Biol., 9, 666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva G.G.Z. et al. (2015) SUPER-FOCUS: a tool for agile functional analysis of shotgun metagenomic data. Bioinformatics, 32, 354–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skewes-Cox P. et al. (2014) Profile hidden markov models for the detection of viruses within metagenomic sequence data. PLoS One, 9, e105067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunagawa S. et al. (2013) Metagenomic species profiling using universal phylogenetic marker genes. Nat. Methods, 10, 1196–1199. [DOI] [PubMed] [Google Scholar]
- Sunagawa S. et al. (2015) Structure and function of the global ocean microbiome. Science, 348, 1–10. [DOI] [PubMed] [Google Scholar]
- Waller A.S. et al. (2014) Classification and quantification of bacteriophage taxa in human gut metagenomes. ISME J., 8, 1391–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao L. et al. (2015) A catalog of the mouse gut metagenome. Nat. Biotechnol., 33, 1103–1108. [DOI] [PubMed] [Google Scholar]
- Yin Y. et al. (2012) DbCAN: a web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res., 40, W445–W451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeller G. et al. (2014) Potential of fecal microbiota for early-stage detection of colorectal cancer. Mol. Syst. Biol., 10, 766–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C.E. et al. (2007) MvirDB – a microbial database of protein toxins, virulence factors and antibiotic resistance genes for bio-defence applications. Nucleic Acids Res, 35, 391–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.