

Abstract
Motivation: It has been known that mature transfer RNAs (tRNAs) that are encoded in the nuclear genome give rise to short molecules, collectively known as tRNA fragments or tRFs. Recently, we reported that, in healthy individuals and in patients, tRFs are constitutive, arise from mitochondrial as well as from nuclear tRNAs, and have composition and abundances that depend on a person’s sex, population origin and race as well as on tissue, disease and disease subtype. Our findings as well as similar work by other groups highlight the importance of tRFs and presage an increase in the community’s interest in elucidating the roles of tRFs in health and disease.
Results: We created MINTbase, a web-based framework that serves the dual-purpose of being a content repository for tRFs and a tool for the interactive exploration of these newly discovered molecules. A key feature of MINTbase is that it deterministically and exhaustively enumerates all possible genomic locations where a sequence fragment can be found and indicates which fragments are exclusive to tRNA space, and thus can be considered as tRFs: this is a very important consideration given that the genomes of higher organisms are riddled with partial tRNA sequences and with tRNA-lookalikes whose aberrant transcripts can be mistaken for tRFs. MINTbase is extremely flexible and integrates and presents tRF information from multiple yet interconnected vantage points (‘vistas’). Vistas permit the user to interactively personalize the information that is returned and the manner in which it is displayed. MINTbase can report comparative information on how a tRF is distributed across all anticodon/amino acid combinations, provides alignments between a tRNA and multiple tRFs with which the user can interact, provides details on published studies that reported a tRF as expressed, etc. Importantly, we designed MINTbase to contain all possible tRFs that could ever be produced by mature tRNAs: this allows us to report on their genomic distributions, anticodon/amino acid properties, alignments, etc. while giving users the ability to at-will investigate candidate tRF molecules before embarking on focused experimental explorations. Lastly, we also introduce a new labeling scheme that is tRF-sequence-based and allows users to associate a tRF with a universally unique label (‘tRF-license plate’) that is independent of a genome assembly and does not require any brokering mechanism.
Availability and Implementation: MINTbase is freely accessible at http://cm.jefferson.edu/MINTbase/. Dataset submissions to MINTbase can be initiated at http://cm.jefferson.edu/MINTsubmit/.
Contact: isidore.rigoutsos@jefferson.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 Introduction
Transfer RNA (tRNA) molecules serve as the bridge between the messenger RNA that is being translated and the newly synthesized polypeptide chain, and, thus, are an integral part of the translation process, as described by the central dogma of biology (Barciszewska et al., 2016). Not surprisingly, tRNAs have long been considered to serve as housekeeping molecules.
In recent years, the use of next generation sequencing to investigate cellular transcriptomes has uncovered ample evidence that a tRNA can also be a source of a rich repertoire of non-coding RNAs (ncRNAs) (Gebetsberger and Polacek, 2013; Shigematsu et al., 2014; Sobala and Hutvagner, 2011). These derivative molecules are referred to as tRNA-derived fragments or tRFs, are generally short, correspond to segments of variable length and their functions are largely unknown. It is important to note that tRFs are not restricted to human biology; indeed, they have been reported in multiple organisms (Casas et al., 2015; Hirose et al., 2015; Karaiskos et al., 2015; Kumar et al., 2014).
Earlier work (Gebetsberger and Polacek, 2013; Kumar et al., 2014) recognized four structural types of molecules originating from the mature tRNA: 5′-halves, 3′-halves, 5′-tRFs and 3′-tRFs (Fig. 1). The 5′-halves and 3′-halves result from Angiogenin cleavage of the mature tRNA at the anticodon loop, and are typically 33 nucleotides (nt) in length. The 5′-tRFs and 3′-tRFs are of variable length, shorter than halves (≤ 30 nt) and originate from cleavage of the mature tRNA at the D and T arms, respectively.
Fig. 1.
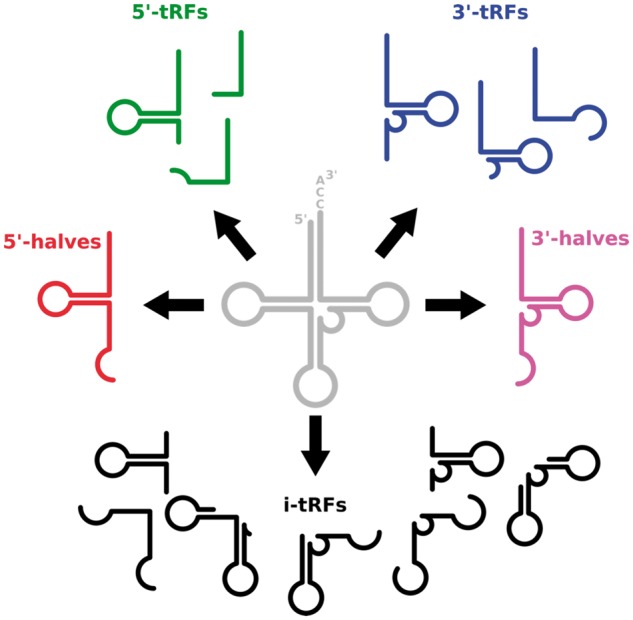
Structural types of tRNA fragments. This is a pictorial summary of the five structural categories of tRNA fragments that are now known to arise from mature tRNAs, both mitochondrially and nuclearly encoded ones
In recent work, we described a previously unreported fifth structural type of tRFs, the internal tRFs or i-tRFs (Telonis et al., 2015). The sequences of i-tRFs are wholly contained within the span of the mature tRNA sequence (Fig. 1). In terms of length, i-tRFs are atypical in that they can be as short as 5′-tRFs/3′-tRFs (e.g. 17-mers) and at least as long as 5′-halves/3′-halves (e.g. 33-mers) (Telonis et al., 2015). We also found that the 5′-tRF and 3′-tRF types have richer diversity than previously known, comprising molecules with many distinct and quantized lengths. In addition, we showed that the tRNAs of the mitochondrial genome (‘mitochondrially encoded’) are a very rich source of both halves and tRFs, just like the tRNAs that are encoded by the nuclear genome (‘nuclearly encoded’). Interestingly, the length distribution and other properties of all five structural types of fragments that are derived from mitochondrially encoded tRNAs differ from those of the fragments arising from nuclearly encoded tRNAs (Telonis et al., 2015).
In terms of function, tRFs are diverse. Not only has loading on Argonaute (Ago) and participation in transcriptional repression been associated with tRFs (Kumar et al., 2014; Shigematsu and Kirino, 2015; Sobala and Hutvagner, 2011; Telonis et al., 2015), but it is also a cell-type-dependent process as we reported in (Telonis et al., 2015). The regulation of translation initiation (Ivanov et al., 2011), the formation of stress granules (Emara et al., 2010), and the displacement of mRNAs from RNA-binding proteins by the newly discovered i-tRFs (Goodarzi et al., 2015) are also among the tRFs’ emerging roles. Recently, our group also showed that the structural type of tRNA-half can adopt multiple active instances that are distinguished by differences in their 3′-terminus modifications (e.g. a cyclic phosphate group instead of the typical hydroxyl), that the expression of these modified tRNA halves (‘SHOT-RNAs’) can depend on sex hormones, and that their silencing impedes cell proliferation (Honda et al., 2015).
Working with transcriptomes from human tissues, instead of cell lines, we were able to generate evidence of intriguing associations involving tRNA fragments. In particular, we showed that the expression profiles of 5′-halves, 3′-halves, 5′-tRFs, 3′-tRFs and i-tRFs from mitochondrially and nuclearly encoded tRNAs depend on tissue, tissue state, disease subtype and on an individual’s sex, population origin and race (Telonis et al., 2015). Moreover, we showed that these molecules are produced constitutively (Telonis et al., 2015).
Our discoveries of the existence of richer categories of 5′-/3′-tRFs and of the previously unrecognized i-tRFs, our finding that mitochondrially encoded tRNAs are also sources of tRNA fragments with profiles that differ distinctly from those of their nuclearly encoded counterparts as well as the numerous dependencies on tissue, disease and an individual’s attributes, which we uncovered, generate an acute need for an integrated and multi-faceted access to this knowledge. The current solutions, e.g. (Kumar et al., 2015), only partially meet this need while also offering limited search and user-interaction capabilities. Moreover, by considering short sequences (e.g. 14-nt, 15-nt, 16-nt, etc.) in a probabilistic search scheme whose performance is known to be influenced by the length of the query (Gusfield, 1997), instead of employing a deterministic and exhaustive approach such as the one we described in (Telonis et al., 2015), the current schemes increase the likelihood that aberrant transcripts will be misreported as tRFs (Kumar et al., 2014). Compounding these limitations are several facts that pertain to the idiosyncrasies of tRNA sequences and impose very concrete limitations on how one must go about identifying tRNA fragments among the reads of next generation sequencing datasets. As we detailed at length in (Telonis et al., 2015), and more recently in (Telonis et al., 2016), the mapping of such reads must be deterministic and exhaustive, should not permit replacements or insertions/deletions, and, most importantly, take into account the fact that the human genome is riddled with partial tRNA sequences and with tRNA-lookalikes (Telonis et al., 2014).
Here, we present a knowledge repository and interactive framework for the exploration of mi-tochondrial and n-uclear t-RFs (MINTbase). MINTbase recognizes that distinct isodecoders of the same anticodon comprise sequence segments that can differ ever so slightly from one another. Consequently, MINTbase does not group together instances of a structural type across isodecoders of the same anticodon (e.g. the 5′-halves from all AspGTC isodecoders): if we were to allow such a grouping, we would hinder the user’s ability to elucidate the differences of these distinct instances in terms of transcription and of genomic distribution. Instead, MINTbase treats each such tRF as a molecule that can be studied independently in its own right. MINTbase also introduces and adopts a second labeling scheme, one that associates each tRF with a unique label (‘tRF-license plate’) that is derived from the tRF’s nucleotide sequence. The uniqueness of the label ensures that a tRF-license plate corresponds to a unique nucleotide sequence. This new labeling approach complements the genome-centric labeling scheme that we introduced in (Telonis et al., 2015), and which is also used by MINTbase.
MINTbase comprehensively embraces this newly discovered complexity of the tRF biology, incorporates the latest discoveries in this field, brings about the various interconnections among tRFs and provides access to what is already known via goal-oriented displays while also remaining flexible enough to easily accommodate future discoveries. MINTbase is freely accessible at http://cm.jefferson.edu/MINTbase/. Codes for generating tRF-license plates and for converting them back to nucleotide sequences are available at https://cm.jefferson.edu/MINTcodes/.
2 Implementation
For the purposes of this presentation, we define as ‘tRF’, or ‘tRNA-derived fragment’, any sequence segment of mature tRNA with length between 16 and 50 nt inclusive (Telonis et al., 2015). This choice of length range for tRFs is dictated by the information that is currently available in the literature. Should subsequent studies provide evidence of tRFs that are longer than 50 nt, MINTbase can be trivially augmented to accommodate those as well. Also, integral to the definition of tRFs is the concomitant tRNA space. MINTbase makes use of our previously introduced definition of tRNA space (Telonis et al., 2015), which is the union of the nuclear and mitochondrial genomic loci that harbor tRNA genes plus the exact mitochondrial tRNA-lookalikes that are present in the nuclear genome and which we reported in (Telonis et al., 2014).
The MINTbase framework adopts a ‘tRF-centric’ approach to knowledge retrieval and exploratory access to the five types of molecules shown in (Fig. 1): 5′-tRFs, i-tRFs, 3′-tRFs, 5′-halves and 3′-halves. Under the hood, MINTbase integrates four kinds of information about these molecules:
Sequence information: this captures attributes of the molecule such as the corresponding sequence of nucleotides and the length of the sequence.
Expression information: this captures expression details such as the tissue type in which the tRF was reported expressed, the disease type, if applicable, PubMed identifier of the respective publication(s), unique identifier of the primary dataset, normalized expression in RPM (Reads Per Million), etc.
Parental tRNA (source) information: this captures knowledge about the fragment in relation to the mature tRNA from which it can arise, including the parent’s anticodon identity, the fragment’s structural type, the fragment’s starting position relative to the start of the parental tRNA, the fragment’s placement with respect to the D, T or anticodon loops of the parental tRNA, etc. On occasion, the parental tRNA cannot be determined unambiguously: in such cases, information about all possible parental tRNAs is included in MINTbase and reported to the user.
Genomic information: this captures global information about the fragment, including anticodon identity, chromosome and strand, genomic coordinates, etc.
A MINTbase user can access the available data from five distinct vantage points or ‘vistas’. The ‘Genomic Loci’ vista provides access to all possible genomic origins of the sought tRFs and their characteristics with reference to the source tRNA genes. The ‘RNA Molecule’ vista provides access to the distribution of a tRF across all tRNAs in the tRNA space. The ‘tRNA Alignment’ vista visualizes the tRF(s) in the sequence context of the parental tRNA. The ‘Expression’ vista provides information about the different datasets, tissues, diseases, etc. in which each tRF has been reported expressed, together with the corresponding PubMed identifier. Finally, the ‘tRF Summary’ vista summarizes all the information in MINTbase for each tRF individually in the form of a ‘record.’
Each vista can be accessed independently, and provides the user with a plethora of sorting options for reorganizing and further subselecting among the reported entries. Output data are presented as tables (‘genomic loci,’ ‘RNA molecule’ and ‘expression’ vistas), as a schematic (‘alignment’ vista), or in the form of records (‘summary’ vista) and include links that enable further exploration of the output at hand from the other vistas.
2.1 Searching MINTbase
To retrieve information from MINTbase the user is required to choose a vista, which decides the nature of the reported information and the organization of the output, and a genome assembly. The use of filters helps narrow the outcome to a specific subset of the tRF universe, if the user so desires: e.g. the user can ask to retrieve only the 5′-tRFs of all anticodons, only the 5′-tRFs of the AlaAGC or SerACT anticodons, all tRF types that originate from all the genomic instances of ValTAC, etc., or various combinations of such choices (Fig. 2). To ensure the effortless use of MINTbase, we accommodated multiple tRNA naming schemes and thus allow the use of different tRNA identifiers when composing a search. On-demand pop-up windows as well as help pages provide information at all stages of one’s interaction with MINTbase.
Fig. 2.

Search form of MINTbase. The user can select one of the five possible vistas then impose specific search criteria through specific selections and the available filters. The user can optionally work with only tRFs for which there is evidence of expression in the literature or with all tRFs that can potentially arise from a mature tRNA. Filters can include one or more of the structural types of a fragment, amino acid and anticodon, tRNA name, tRF nucleotide sequence, tRF name, chromosome, strand, etc
A novel key feature of MINTbase is that it provides the user with the ability to enumerate tRFs that have not been reported as yet in the literature as expressed. Since the tRNA sequence space of a genome is finite (Telonis et al., 2015), there exists an upper bound to the number of distinct tRF sequences that can ever be produced by mature tRNAs. The decision to incorporate all possible tRFs (and not only the ones that have been reported as expressed to date) stems from our previously reported findings that the tRF profiles in a given cell/tissue depend on multiple variables (Telonis et al., 2015). As the number of discovered dependencies is likely to increase it is reasonable to expect that at least some of these currently unreported tRFs will be discovered by subsequent studies to be expressed in some setting.
2.2 Interpreting a user’s request
Depending on the choice of vista, MINTbase will interpret the search parameters accordingly. As mentioned above and discussed in detail in (Telonis et al., 2015), there exist tRFs whose sequence is such that their parental anticodon gene cannot be determined unambiguously: imagine, e.g. that the user searches for all ‘5′-tRFs of the nuclearly encoded AlaAGC anticodon,’ which is denoted by AlaAGC (n) in MINTbase. It turns out that some of the 5′-tRFs of AlaAGC are also identically present in isodecoders for AlaCGC, AlaTGC, CysGCA and ValAAC. Consequently, when the user selects ‘RNA Molecule’, ‘Summary’ or the ‘Expression’ vista, MINTbase’s output will include information about this ambiguity, explicitly indicating those 5′-tRFs that exist in AlaAGC and in other anticodons. In other words, at all points of the user’s interaction with MINTbase, the user is reminded of the underlying ambiguity that is inherent to the biology of tRNAs and tRFs.
In the ‘Genomic Loci’ vista, if a user selects a specific anticodon, MINTbase’s output will not include the 5′-tRFs that have an identical nucleotide sequence but come from the isodecoder of a different anticodon: nonetheless, information about the inherent ambiguity will still be available and reported in a separate column of the output. In the ‘tRNA Alignment’ vista, the selection of a specific tRNA becomes mandatory and therefore a tRNA gene must be chosen. Finally, in the case of the ‘tRF Summary’ vista, the user is required to input a tRF, by providing its sequence (or its label).
2.3 Vista: genomic loci
This is the most detailed vista of the framework and provides a genome-wide overview of the available information (Fig. 3). Given the source ambiguity of some tRFs, in the general case, this vista’s output can comprise multiple rows that refer to all of the genomic instances of the same tRF sequence. Each of the optional search filters (Search by tRNA label, Search by fragment sequence, Search by fragment label) is applied at the genome level and allows the user to further sub-select among the currently reported results. The Genomic Loci vista is meant for those users who want to explore similarities among tRFs at the genome level or to study in detail the tRFs’ relative position with respect to the source tRNAs. The data are presented in a tabular format that contains one genomic instance per row.
Fig. 3.

Genomic loci vista. The output of this vista presents the user with information about the tRF type, the tRF’s nucleotide sequence, the corresponding amino acid and anticodon identity, the tRF's genomic coordinates (global) and the tRF’s coordinates within the parental tRNA (local). Also included is information on whether a fragment has been reported as expressed in the literature. The results are organized as a table. Menu bars above and below the table enable navigation in multi-page outputs and also give the user the ability to interact with the presented output: columns can be shown/hidden at will, the number of results shown per page can be modified and the generated results downloaded. The headers of several columns are interactive and allow the user to sort the corresponding column’s contents; a second click on a column’s header reverses the sorting order. Holding down the ‘shift’ key and clicking on multiple headers, allows for multi-sorting of the corresponding columns (in the order the headers were selected by the user). The sequence, expression and tRNA alignment columns provide links to the other four vistas of MINTbase
By default several features are reported for each tRF, including the tRF’s structural type, the tRF’s genome-centric and sequence-centric labels (see Subsection 2.9 for more information), the actual nucleotide sequence of the tRF, the identity of the corresponding amino acid and anticodon, chromosome and strand information, global genomic starting and ending coordinate, local starting and ending coordinates within the corresponding mature tRNA, the number of distinct anticodons that contain the tRF, the number of instances the tRF has among nuclear and mitochondrial tRNAs respectively, the number of instances the tRF has among the tRNA-lookalikes in the nuclear genome (Telonis et al. 2014), whether the tRF overlaps with the D-loop, whether it overlaps with the anticodon, etc. The user can selectively report any combination of additional tRF features, e.g. the global genomic coordinates of intronic endpoints (exon–exon junction spanning tRFs only), etc.
Several of the included features/columns are meant to remind the user of the idiosyncrasies of the tRNA sequence space, and in particular the parental tRNA ambiguity that characterizes many tRFs. Importantly, one of the included features pertains to whether the tRF’s sequence can also be found outside of the tRNA space: in such cases, the corresponding tRF should be treated with caution as it may be a potential false positive (i.e. a non-tRF).
The Genomic Loci vista is interconnected with all the other vistas allowing the fast exploration of different attributes of the currently ‘active list of results.’ For example, by clicking on the sequence of a tRF contained in the current output the user can open the ‘tRF Summary’ vista for this fragment. By clicking on the entry of the expression column, if labeled ‘yes,’ the user can retrieve information about the studies that reported the fragment as expressed whereas by clicking on the tRNA alignment column’s images the user can retrieve the alignment of the tRF with respect to its source tRNA.
2.4 Vista: RNA molecule
This vista is molecule-centric and presents a summary of the fragment’s basic characteristics (Fig. 4). The RNA Molecule vista is meant for those users who want to obtain basic level information of the tRF of interest and its potential origins in a summarized format.
Fig. 4.

RNA molecule vista. The output of this vista presents the user with fragment level information. The results are again organized as a table with menu bars above and below the table enabling navigation and user-interaction with the data (see also text and caption of Fig. 3). As in the Genomic Loci vista, the column headers are interactive and permit single-column and multi-column sorting. The nucleotide sequence column links to the ‘Summary’ vista, the column listing the number of genomic loci links to the ‘Genomic Loci’ vista, and the expression column links to the ‘Expression’ vista
In this vista, the data is presented in a tabular format with each row containing a unique tRF sequence. Reported features include the actual nucleotide sequence of the tRF, the structural type of the tRF and the amino acid/anticodon combination corresponding to the parental tRNA gene, the total number of genomic instances that the tRF has in tRNA space, whether it is also present outside tRNA space, whether it has been reported as expressed in a tissue, and the tRF’s ‘license-plate.’ The latter is a unique sequence-centric label that simplifies referencing and bookkeeping as well as automatically serves as a lookup table for analyzing RNA-seq data. We discuss it at length in Subsection 2.9 below.
This vista explicitly captures the inherent ambiguity vis-à-vis a tRF’s parental tRNA gene, and is an important component of MINTbase. To this end, MINTbase’s output will group all of the amino acid/anticodon combinations that correspond to the same tRF sequence into a comma-separated list; e.g. see rows 4-6 and row 8 of Fig. 4. Doing so provides the user with a summary view of all the genomic instances of the tRF at hand and of the tRF’s distribution across the various amino acids and anticodons. Finally, the vista is interconnected with and allows a quick transition to the ‘Genomic Loci’ vista via the ‘number of genomic instances’ column and the ‘tRF Summary’ vista via the fragment sequence. It is also connected to the ‘Expression’ vista via the expression column.
2.5 Vista: tRNA alignment
This vista is mature-tRNA-centric and presents a summary of the parental tRNA gene’s repertoire of fragments (Fig. 5). The tRNA Alignment vista is meant for users who want to explore the tRF-generation potential of a tRNA across each of the five structural types or simply wish to visualize the tRF(s) of their choice. For the user-selected mature tRNA, this display allows the generation of an alignment of all tRFs, or only the selected ones, against the tRNA, one tRF per line.
Fig. 5.

tRNA Alignment vista. The output of this vista presents the user with information on how the tRFs relate to the parental tRNA. A counter at the top of the page lists the number of tRFs included on the page. The various tRFs are shown aligned to the parental tRNA, one tRF per line. The parental tRNA’s sequence remains floating as the page is scrolled up/down or right/left. tRFs can be selectively removed from the shown alignment (by clicking on the [×] beside them). Clicking on a fragment’s nucleotide sequence allows the user to transition to the Summary Vista for the corresponding fragment. Clicking on the parental tRNA’s genome-centric label will redirect the user to the UCSC Genome Browser and enable the user to view the tRNA in its genomic context. Clicking on ‘Undo,’ restores the output to its original version. Different colors indicate the location of relevant information such as D-loop, anticodon, T-loop, intron (if appropriate), etc. A legend at the top of the page describes the used color-coding
The generated output comprises explicit information about the mature tRNA’s secondary structure: specifically, the D-loop, anticodon loop and T-loop are marked with different colors on both the mature tRNA and the fragments, which facilitates tracking of these features across the fragments and enables a visual comparison of tRFs with one another and with the parental gene. Also marked with different colors are the anticodon triplet and the non-templated CCA tail. Individual fragments can be removed at will to allow juxtaposition of other fragments (e.g. see arrangement of Fig. 5); at any time, an already edited view can be restored to its initial state. Also, the intron of an intron-containing tRNA can be optionally hidden, an action that applies to both the shown mature tRNA and the shown fragments.
The links contained in the tRNA Alignment output page allow a quick transition to the ‘tRF Summary’ vista or a visualization of the parental tRNA’s genomic context via the UCSC Human Genome browser.
2.6 Vista: expression
This vista is fragment-centric and presents information about a tRF’s known expression status (Fig. 6). The information is reported in tabular form. If the search parameters retrieved multiple distinct tRFs from MINTbase, then the table will comprise as many rows as the number of tRFs: each row will list the sequence of the tRF and the number of datasets in which it is known to be expressed—clicking on the number of datasets produces a second tabular output that is tRF-specific. The detailed, tRF-centric output of the expression vista contains one row for each deep-sequencing dataset that is included in MINTbase and contains the tRF. The information of a given row includes columns that can be optionally hidden and include: the tRF’s normalized abundance (in RPM), the PubMed identifier of the primary publication that released the dataset to the public domain, the PubMed identifier of the publication that analyzed/reported the tRF, the tissue/cell type in which the tRF is expressed, a link to the public domain repository in which the original dataset can be found, a brief description of the dataset as found in this public repository, etc. The PubMed identifiers, the dataset identifier, the names of the submitter and corresponding authors, and the names of the corresponding institutions redirect the user to the NIH PubMed website, the public repository where the data is deposited, and the personal and institutional webpages respectively.
Fig. 6.
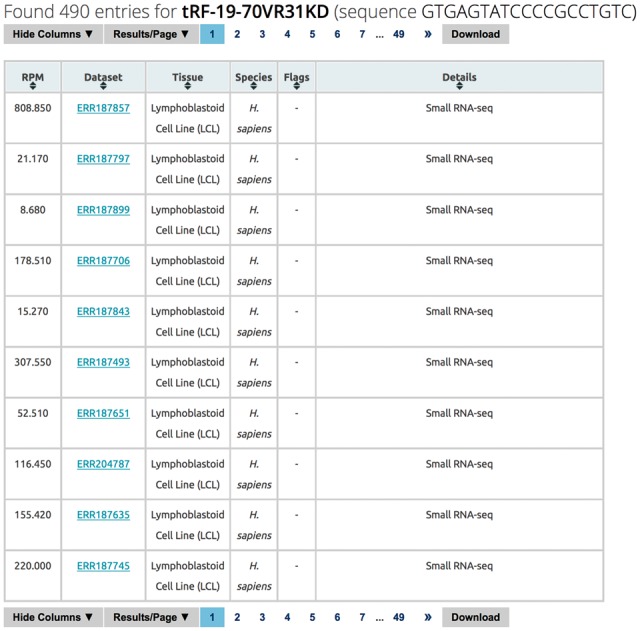
Expression vista. The above is an instance of the detailed tRF-centric layer of this vista. For the user-selected fragment, the user is presented with information about the tRF’s expression across the MINTbase datasets in which it is expressed. Menu bars above and below the table enable navigation and user-interaction with the presented data. Columns can be hidden at will, the number of results shown per page can be modified, and the generated results downloaded. The table’s headers are interactive and allow the user to sort the columns’ contents. In the above example, only a few of the many possible columns are shown (‘unhidden’)
MINTbase is meant to serve as a repository for tRFs across various tissues and cell types. With that in mind, we also provide an automated mechanism that allows users to submit their datasets to MINTbase (described below). This should help MINTbase grow quickly. In the meantime, we have pre-populated MINTbase with expressed tRFs from 832 deep-sequencing datasets that include: (i) the dataset reported in one of the first studies to describe tRFs using next generation sequencing (Cole et al., 2009); (ii) the dataset from the study of (Honda et al., 2015); (iii) 28 datasets from the study of (Selitsky et al., 2015)—tRFs are listed in Supplementary Table 1; and (iv) the 802 datasets that were analyzed in (Telonis et al., 2015) and which were deposited in the public domain as part of three earlier studies (Lappalainen et al., 2013; Pillai et al., 2014; The Cancer Genome Atlas Network, 2012).
2.7 Vista: tRF summary
This vista provides a tRF-specific record page that summarizes basic information about the tRF (Fig. 7). This serves not only as a reference for each molecule but also as a starting point for exploring the genomic and molecular characteristics of a tRF (e.g. its type, the potential tRNA gene sources, the number of instances in tRNA space, etc.) across the available vistas. The power of this vista also lies on the fact that a tRF’s record lists the number of public datasets in which the tRF is found expressed.
Fig. 7.

tRF summary vista. The output of this vista presents basic information about each tRF that is in MINTbase. It includes the tRF’s license plate (the unique sequence-centric identifier introduced by MINTbase—see text), a list of genome-centric labels as discussed in the text, the number of the tRF’s genomic instances, its structural type, whether the tRF appears only inside the tRNA space, a list of all possible parental tRNAs with the tRF underlined in each and the number of datasets currently in MINTbase in which the tRF is expressed
2.8 Filtering, sorting, downloading and printing
All five vistas were implemented in a way that offers easy navigation, selection of the maximum number of rows listed per page, selection of the type of features included in each row (some vistas), and the sorting of the presented information based on the contents of one or more columns that the user can select on-the-fly. Furthermore, the active list of results can be sub-selected based on user-selected values for a variety of attributes (all vistas). Criteria that the user can impose on the reported output include one or more of: chromosome identifier, strand, window of genomic coordinates, structural type of a tRF, amino acid, anticodon, fragment sequence, fragment name, etc. Multiple user-selected filters are combined using a Boolean AND operation. Such combinations of conjunctive filters can help the user ‘zoom in’ on tRFs that exhibit characteristics of interest, which can then be explored further by transitioning to one of the other vistas. The user can download the list of active results (Genomic Loci, RNA Molecule and Expression vistas), print an alignment (tRNA Alignment vista), or print a tRF record (tRF Summary vista) as needed.
2.9 Sequence-centric tRF-license plates
In Telonis et al. (2015), we introduced a genome-centric labeling scheme. This scheme, which is also used in MINTbase, comprises two components separated by the ‘@’ symbol. The first component is the tRNA name listed in gtRNAdb (Chan and Lowe, 2009). The second component consists of the tRF’s starting position, its ending position, its length, the number of distinct anticodons and the number of instances within tRNA space.
Considering that a given tRF can appear in different isodecoders of the same anticodon, in different anticodons, or, in the mature tRNAs of different organisms (Telonis et al., 2015) we wanted to also acknowledge and emphasize this ‘motif-like,’ i.e. recurrent, character exhibited by some tRFs. To this end, we designed a labeling scheme that is decoupled from the genomic location of a tRF, and, thus, independent of any given genome assembly, a design approach that meshes well with the tRF-centric character of MINTbase. Equally importantly, this new scheme enables any user to employ our provided codes to generate a unique label for their tRF(s) of interest without requiring a brokering framework such as the one currently employed by, e.g. miRBase (Kozomara and Griffiths-Jones, 2011). We emphasize that use of these codes is optional. Nonetheless, the codes provide users with the ability to generate a unique label that can be used as a short-hand in a manuscript, to look-up entries in MINTbase, to compare one’s data with data reported by other research groups, etc. Any tRF sequence can be mapped to a single and unique label (the tRF’s ‘license plate’), and any license plate can be mapped back always to the same tRF sequence. Different researchers who independently discover the same tRF in different contexts will independently assign the same label to this common tRF. We stress that assigning a label to a tRF can be done without the requirement of a central brokering system—as long as researchers use the provided codes, the resulting tRF labels will remain consistent. It is our hope that this scheme will help speed up tRF research. Importantly, allowing researchers at different institutions to generate the same unambiguous and unique label for the same tRF sequence should facilitate comparisons of a given tRF’s behavior in different tissues, under different conditions, across different publications, etc.
The labeling system is essentially a remapping of a tRF’s nucleotide sequence from a base-4 system (A, C, G, T) to a base-32 system that uses the following alphanumeric symbols: B, D, 0, E, F, 1, H, I, 2, J, K, 3, L, M, 4, N, O, 5, P, Q, 6, R, S, 7, U, V, 8, W, X, 9, Y, Z. By symmetrically interweaving letters and numbers, the resulting tRF label resembles an automobile’s license plate. Note that the new encoding does not use any of A, C, G or T; this is intentional and ensures that the tRF license plates do not contain any of the letters found in the tRF’s DNA sequence.
Let us use the i-tRF CTGTCACGCGGGAGACCGGGGTT from AspGTC to demonstrate this mapping scheme. First, we segment the tRF into consecutive, non-overlapping 5-mers; for tRFs whose length is not a multiple of 5, the last tuple will be between 1 and 4 nt long. The above tRF will give us CTGTC|ACGCG|GGAGA|CCGGG|GTT. We map each 5-mer to a pair of symbols from the above alphabet using standard base-32 mapping; this will result in the following four pairs: NM|EH|62|3K. The leftover suffix string can have four instances of length 1, 16 instances of length 2, 64 instances of length 3 and 256 instances of length 4: we sort these 340 base-4 strings in lexicographic order and map the first 32 to symbols B through Z (see above list) and the remaining 308 to pairs DB through KQ. In our example, the trailing GTT will map to 0E. To avoid mistaking symbol pairs representing a leftover suffix from symbol pairs representing 5-mers, we include the length of the original tRF in the license plate: using this information we can unambiguously decode a tRF-license plate back into the original nucleotide sequence. To conclude, under this new mapping scheme, the tRF CTGTCACGCGGGAGACCGGGGTT will be encoded by the license plate ‘tRF-23-NMEH623K0E.’ The scripts needed to generate a tRF-license plate for a tRF and to decode a tRF-license plate are available at http://cm.jefferson.edu/MINTcodes/.
2.10 MINTsubmit: submit your entries to MINTbase
We designed MINTbase to be a dynamic repository of information pertaining to tRFs. Considering that the ready availability of deep sequencing data will make it simpler for increasing numbers of groups to generate tRF information, MINTbase also provides a conduit for the automated deposition of new tRF expression records. A user interaction with MINTsubmit begins at https://cm.jefferson.edu/MINTsubmit/. The system will subsequently send a verification code to the user-provided email address. The user will then proceed by entering the verification code together with other relevant information that pertains to the sample. In order to ensure fairness of access to the service, users will only be able to submit datasets that are already deposited in a public repository such as NIH’s Gene Expression Omnibus (GEO) and have been described in an article that is already listed on PubMed (and, thus, has a PubMed identifier). As soon as the provided information has been verified, the user’s submitted dataset will continue its progress through the automated pipeline that will incorporate it into MINTbase and make it publicly available.
2.11 Various
MINTbase’s knowledge repository is organized as a MySQL database. Users access the database via a web application, which is written in Java. Every time the search form is submitted or the tables are sorted, a new request is submitted to the application. Each request is handled separately and the results are kept around for 2 h to help accelerate subsequent downloading and page-viewing requests.
The secondary structures for the nuclearly-encoded tRNAs that are highlighted in the ‘tRNA alignment’ vista were obtained from http://gtrnadb2009.ucsc.edu/Hsapi19/Hsapi19-structs.html (Chan and Lowe, 2009). The secondary structures for the mitochondrially-encoded tRNAs were obtained from mitotRNAdb (Juhling et al., 2009). The secondary structure of the eight exact tRNA-lookalikes (Telonis et al., 2014) was borrowed from and mirrors that of their mitochondrial tRNA counterparts.
3 Discussion
We presented MINTbase, a novel framework for studying fragments that arise from mitochondrial and nuclear mature tRNAs. MINTbase is an easy-to-use resource for accessing information on tRFs and interactively studying them from diverse vantage points using one or more of the five available vistas. Each vista provides distinct functionalities and its output is interconnected to some of the other four vistas, which in turn enables exploration of the active results from multiple angles at any time. The output pages provide filtering and sorting capabilities thereby permitting an interactive study and further filtering of the active results by the user. For example, the user can easily group the results to investigate possible connections to a type of tRF, a specific source tRNA, number of instances in tRNA space, etc. Additionally, several output pages also include links to external databases (e.g. the UCSC Genome Browser, PubMed, etc.) aimed at providing the user with other easily accessible information.
MINTbase offers several advantages. First, it addresses the need for a database that includes the most recent discoveries in the field of tRFs and tRNA biology. The discoveries of the new structural type of i-tRFs, of tRFs produced from mitochondrial tRNAs and of SHOT-RNAs, represent recent advances that are already part of MINTbase’s knowledge repository.
Second, three of the five vistas that MINTbase makes available are meant to capture and highlight the inherent complexity of the space of mature tRNAs while illustrating at the same time the ambiguity among parental tRNA genes: the ‘Genomic Loci’ vista provides a detailed perspective on the possible genomic origins of a tRF whereas the ‘RNA Molecule’ and ‘tRF Summary’ vistas summarize the distribution of each tRF molecule across the various anticodons/isodecoders from which it could arise. Thereby, MINTbase allows the user to retrieve information in multiple ways while also keeping at the forefront the complexity of the tRNA space throughout the user’s interactive session.
Third, MINTbase has been built to incorporate the spectrum of all possible tRFs (currently up to 50 nt in length—longer tRFs can be accommodated trivially) that can ever arise from a mature tRNA, whether it is mitochondrially or nuclearly encoded. Our intent was to design a tool that can further promote research activities in the field of tRFs. Thus, in addition to listing molecules with known expression, MINTbase can optionally provide detailed information on fragments that could potentially derive from mature tRNAs and for which no information exists yet in the public domain. Such candidate tRFs can serve as the subject of focused experimental studies by MINTbase’s users.
Lastly, MINTbase permits users to add their own datasets to the repository. This is done easily through the use of a submission conduit that we also provide. Save a mandatory manual verification of the provided information the process is entirely automated.
In summary, we hope MINTbase will prove a useful framework for researchers who study tRNAs and tRFs by providing quick access to the available knowledge, by juxtaposing relevant information in order to enable comparative analyses, by adopting and using both genome-centric and sequence-centric labeling schemes and by enabling web-based exploratory studies through its user-friendly, tRNA-centric interface.
Supplementary Material
Acknowledgements
The authors thank Eric Londin, Shozo Honda, Keisuke Morichika, Megumi Shigematsu and Yohei Kirino for beta testing and feedback on an early version of the interface.
Funding
This work has been supported by a William M. Keck Foundation grant award (I.R.) and by Institutional Funds.
Conflict of Interest: none declared.
References
- Barciszewska M.Z. et al. (2016) tRNA - the golden standard in molecular biology. Mol. BioSyst. 12, 12–17. [DOI] [PubMed] [Google Scholar]
- Casas E. et al. (2015) Characterization of circulating transfer RNA-derived RNA fragments in cattle. Front. Genet., 6, 271.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan P.P., Lowe T.M. (2009) GtRNAdb: a database of transfer RNA genes detected in genomic sequence. Nucleic Acids Res., 37, D93–D97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole C. et al. (2009) Filtering of deep sequencing data reveals the existence of abundant Dicer-dependent small RNAs derived from tRNAs. RNA, 15, 2147–2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emara M.M. et al. (2010) Angiogenin-induced tRNA-derived stress-induced RNAs promote stress-induced stress granule assembly. J. Biol. Chem., 285, 10959–10968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebetsberger J., Polacek N. (2013) Slicing tRNAs to boost functional ncRNA diversity. RNA Biol., 10, 1798–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodarzi H. et al. (2015) Endogenous tRNA-derived fragments suppress breast cancer progression via YBX1 displacement. Cell, 161, 790–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusfield D. Algorithms on Strings, Trees, and Sequences: Computer Science and Computational Biology. Cambridge, UK: Cambridge University Press, 1997. [Google Scholar]
- Hirose Y. et al. (2015) Precise mapping and dynamics of tRNA-derived fragments (tRFs) in the development of Triops cancriformis (tadpole shrimp). BMC Genet., 16, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda S. et al. (2015) Sex hormone-dependent tRNA halves enhance cell proliferation in breast and prostate cancers. Proc. Natl. Acad. Sci. USA, 112, E3816–E3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov P. et al. (2011) Angiogenin-induced tRNA fragments inhibit translation initiation. Mol. Cell, 43, 613–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juhling F. et al. (2009) tRNAdb 2009: compilation of tRNA sequences and tRNA genes. Nucleic Acids Res., 37, D159–D162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaiskos S. et al. (2015) Age-driven modulation of tRNA-derived fragments in Drosophila and their potential targets. Biol. Direct, 10, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozomara A., Griffiths-Jones S. (2011) miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res., 39, D152–15D157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P. et al. (2014) Meta-analysis of tRNA derived RNA fragments reveals that they are evolutionarily conserved and associate with AGO proteins to recognize specific RNA targets. BMC Biol., 12, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P. et al. (2015) tRFdb: a database for transfer RNA fragments. Nucleic Acids Res., 43, D141–D145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappalainen T. et al. (2013) Transcriptome and genome sequencing uncovers functional variation in humans. Nature, 501, 506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillai M.M. et al. (2014) HITS-CLIP reveals key regulators of nuclear receptor signaling in breast cancer. Breast Cancer Res. Treat., 146, 85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selitsky S.R. et al. (2015) Transcriptomic analysis of chronic Hepatitis B and C and liver cancer reveals MicroRNA-mediated control of cholesterol synthesis programs. mBio, 6, e01500–e01515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigematsu M. et al. (2014) Tranfer RNA as a source of small functional RNA. J. Mol. Biol. Mol. Imag., 1, 8. [PMC free article] [PubMed] [Google Scholar]
- Shigematsu M., Kirino Y. (2015) tRNA-Derived Short Non-coding RNA as Interacting Partners of Argonaute Proteins. Gene Regul. Syst. Biol., 9, 27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobala A., Hutvagner G. (2011) Transfer RNA-derived fragments: origins, processing, and functions. Wiley Interdiscipl. Rev. RNA, 2, 853–862. [DOI] [PubMed] [Google Scholar]
- Telonis A.G. et al. (2014) Nuclear and mitochondrial tRNA-lookalikes in the human genome. Front. Genet., 5, 344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telonis A.G. et al. (2015) Mitochondrial tRNA-lookalikes in nuclear chromosomes: Could they be functional? RNA Biol., 12, 375–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telonis A.G. et al. (2015) Dissecting tRNA-derived fragment complexities using personalized transcriptomes reveals novel fragment classes and unexpected dependencies. Oncotarget, 6, 24797–24822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telonis A.G. et al. (2016) Consequential considerations when mapping tRNA fragments. BMC Bioinformatics, 17, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Network (2012) Comprehensive molecular portraits of human breast tumours. Nature, 490, 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.