

Abstract
Vascular smooth muscle cells exhibit intercellular Ca2+ waves in response to local mechanical or KCl stimulation. Recently, a new type of intercellular Ca2+ wave was observed in vitro in a linear arrangement of smooth muscle cells. The intercellular wave was denominated ultrafast Ca2+ wave and it was suggested to be the result of the interplay between membrane potential and Ca2+ dynamics which depended on influx of extracellular Ca2+, cell membrane depolarization and its intercel- lular propagation. In the present study we measured experimentally the conduction velocity of the membrane depolarization and performed simulations of the ultrafast Ca2+ wave along coupled smooth muscle cells. Numerical results reproduced a wide spectrum of experimental observations, including Ca2+ wave velocity, electrotonic membrane depolarization along the network, effects of inhibitors and independence of the Ca2+ wave speed on the intracellular stores. The numerical data also provided new physiological insights suggesting ranges of crucial model parameters that may be altered experimentally and that could significantly affect wave kinetics allowing the modulation of the wave characteristics experimentally. Numerical and experimental results supported the hypothesis that the propagation of membrane depolarization acts as an intercellular messenger mediating intercellular ultrafast Ca2+ waves in smooth muscle cells.
Communication between vascular smooth muscle cells (SMCs) plays an important role in coordinating vascular function and compromised intercellular signaling may underlie pathological conditions. Continuous electrical and ionic movements take place between coupled cells which affect resting states and enable conduction of signals. Electrical current, inositol 1,4,5-trisphosphate (IP3) and Ca2+ are considered as important mediators of vascular communication. Nevertheless, Ca2+ and IP3 fluxes through gap junctions are small and thus, their passive diffusion should have a limited effect on Ca2+ mobilization at distant sites1. One way of cellular communication is by intercellular Ca2+ waves, the propagation of an increase in intracellular Ca2+ concentration. Such intercellular Ca2+ waves have been induced in vitro by mechanical, electrical or chemical stimuli2,3,4 and classified according to the mechanism involved and the velocity amplitude, denominating the ultrafast Ca2+ wave as an electrically propagated wave5,6. Novel insights have been gained from mathematical models which connect clusters of SMCs7,8,9,10,11. In particular, in ref. 11 the authors confirmed the hypothesis that intercellular Ca2+ waves observed in arterial SMCs12 resulted from electrical coupling. Assuming gap junctional communication by means of electrical coupling, IP3 diffusion, and Ca2+ diffusion these models reproduced experimental observations like asynchronous Ca2+ flashings, recruitment of cells and vasomotion in absence of endothelium13,14,15,16,17. In the present study, we adapted the model presented in ref. 11 to elucidate the mechanisms underlying the ultrafast Ca2+ wave and to investigate the particular conditions for intercellular ultrafast Ca2+ wave to occur as well as the properties of the membrane depolarization. Our study showed the direct interplay between the Ca2+ wave and the spreading of the membrane depolarization. We tested, discussed and demonstrated that an intercellular ultrafast Ca2+ wave is driven by the propagation of cell membrane depolarization and its speed is not dependent on the intracellular Ca2+ stores. Simulations predicted novel results and opened the field for further experimental studies to investigate the effect of electrical coupling and whole-cell conductance on Ca2+ wave velocity and on the propagation speed of membrane depolarization.
Results
Propagation of the induced intercellular ultrafast Ca2+ wave and induced membrane depolarization
For the set of parameters corresponding to the numerical control case (see Methods), the time evolution of the [Ca2+], normalized by the steady state concentration before activation ([Ca2+]0), is depicted in Fig. 1A. Before the stimulation (t < 1 s), all cells were at the same resting state. After the stimulation, we observed a global Ca2+ increase and each cell reached a new steady state with an asymptotic [Ca2+] that decreased exponentially with the distance from the stimulated site. We measured a typical scale of 4,16 cells (R2 = 0,99) (Fig. 1B). Experimentally, we found a similar exponential relationship between the percentage of maximum increase and the distance from the stimulation site with a typical scale of 4,4 cells (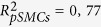 ;
; 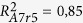 ). A closer look to the short time dynamics just after the stimulation showed a lag in the Ca2+ increase as an indication of an intercellular Ca2+ wave. To estimate the onset times and calculate the wave velocity, we measured for each cell, the time required to reach different thresholds from 1% to 15% of its maximum value of [Ca2+]. For each threshold, we determined the velocity from the difference in the onset times between one particular cell to the following cell and then averaged over all the cells and for all threshold values. We obtained a wave velocity of ~333(±40) cell.s−1.
). A closer look to the short time dynamics just after the stimulation showed a lag in the Ca2+ increase as an indication of an intercellular Ca2+ wave. To estimate the onset times and calculate the wave velocity, we measured for each cell, the time required to reach different thresholds from 1% to 15% of its maximum value of [Ca2+]. For each threshold, we determined the velocity from the difference in the onset times between one particular cell to the following cell and then averaged over all the cells and for all threshold values. We obtained a wave velocity of ~333(±40) cell.s−1.
Figure 1.

(A) Numerical results for the time courses of Ca2+ responses in the simulated control case. Stimulation occurs at 1 s. Stimulated cell (black), 4th cell (red), 8th cell (blue), 12th (green) and 16th cell (pink). [Ca2+]0 is the steady state cytosolic [Ca2+] before stimulation. Inset presents the short time dynamics after stimulation (t0) in lin-log scale. (B) Percentages of maximum F/F0 (npSMCs = 16; nA7r5 = 19) and [Ca2+]/[Ca2+]0 increases, assuming 100% as the maximum level reached by the stimulated cell. Red line fits the exponential decrease with distance from the stimulated site and cell unit is estimated by taking a cell of 150 μm. (npSMCs = 22; nA7r5 = 82).
Similarly as the in vitro experiments reported in ref. 18, numerical results showed that membrane potential increased after stimulation. Maximum of the depolarization was greater for cells close to the stimulated one (Fig. 2A). We calculated the percentage of membrane depolarization using the maximum depolarization value of each cell with respect to the steady state membrane potential before the stimulation. Figure 2B shows that the percentage of membrane depolarization followed an electrotonic behaviour with exponential decrease. We obtained a characteristic lenght scale of 4,03 cells (R2 = 0,98) in good agreement with previously reported experimental data (assuming a cell length of 100–150 μm) for vascular SMCs 4,46–6,13 cells18.
Figure 2.

(A) Numerical results for the time courses of the membrane potential in the simulated control case. Stimulation occurs at t = 1 s. Stimulated cell (black), 4th cell (red), 8th cell (blue), 12th (green) and 16th cell (pink). Inset presents the short time dynamics after stimulation (t0) in lin-log scale. (B) Percentages of membrane depolarization (npSMCs = 16; nA7r5 = 19). The percentages of membrane depolarization were calculated using the maximum depolarization value of each cell and comparing it with the steady state membrane potential before stimulation. Red line fits the exponential decrease with distance from the stimulated site and cell unit is estimated by taking a cell of 150 μm. (npSMCs = 22; nA7r5 = 82).
Role of gap junctions channels and VOCCs in the simulated array of SMCs
Recent studies showed that incubation of the SMCs in vitro3,18 or arterial strips12 with gap junction uncoupler palmitoleic acid (PA) or with VOCCs inhibitor nifedipine totally abolished the Ca2+ waves. In relation to these experimental results, we simulated similar conditions by modification of the parameters governing the dynamics of these channels (g and GCa, see Methods). We first simulated a total inhibition of the gap junctions by setting the electrical coupling coefficient g to 0.
As in in vitro conditions3,18, we observed a total suppression of the Ca2+ and the membrane potential signals under gap junctions inactivation. Only the stimulated cell showed a Ca2+ increase and a membrane depolarization; responses of the other cells of the network were insignificant (dashed line in Fig. 3A,B). We extended the analysis for a wide range of electrical coupling constants (Fig. 3E) and observed that both the speed of the Ca2+ wave and the propagation speed of membrane depolarization increased like the square root of the coupling, in a similar way as in a system of coupled oscillators. Within this range of electrical coupling, we obtained Ca2+ wave speeds from 46 cell.s−1 to 866 cell.s−1 and membrane depolarization conduction speeds from 60 cell.s−1 to 833 cell.s−1.
Figure 3. Role of gap junctions and VOCCs on the Ca2+ dynamics and membrane potential.

(A) Time evolution of the Ca2+ responses and (B) membrane potential variations with inactivated gap junctions. The black dashed line is the response of the other cells but the stimulated one. Gap junctions inhibition was simulated by setting the electrical coupling coefficient (g) to 0 s−1. (C) Time evolution of the Ca2+ response and (D) membrane potential variations with inactivated VOCCs. VOCCs inhibition was simulated by setting the whole-cell conductance to 0 mV−1.s−1. (E) Theoretical Ca2+ wave velocity (red) and propagation speed of membrane depolarization (black) as a function of g. The dashed line represents the scaling g1/2. (F) Ca2+ wave velocity (red) and propagation speed of membrane depolarization (black) as a function of GCa. Inset in (C) is the time evolution of the Ca2+ response and the inset in (D) is membrane potential variations with an impose Ca2+ transient in the first cell.
We simulated VOCCs blockage by imposing a null whole-cell conductance GCa = 0. This condition inhibited any Ca2+ entry through VOCCs anywhere along the SMCs network, including the stimulated cell, resulting in the abolition of the Ca2+ wave (Fig. 3C). Importantly, there was still a membrane depolarization spreading along the network even in zero VOCCs conductance conditions (Fig. 3D). In the simulations, none of the cells had a [Ca2+] response for GCa = 0, whereas in the experiments with nifedipine the stimulated cell had (in response to the activation of other channels such as stretch activated channels). To ensure that in the simulations the abolition of the ultrafast [Ca2+] wave was not a trivial consequence of the absence of [Ca2+] increase of the stimulated cell, we designed a numerical experiment in which we imposed a [Ca2+] profile to the first cell (insets in Fig. 3C,D). In this situation, the other cells did not respond to the imposed [Ca2+] increase whereas a spreading of membrane depolarization still occurred in the system (inset in Fig. 3D). These numerical results were an important milestone of the current study and were crucial to confirm the hypothesis that [Ca2+] increase over the network of SMCs was fundamentally due to the Ca2+ entry through the opening of VOCCs in response to a propagation of membrane depolarization along the SMCs network. Beside the condition of null whole-cell conductance, we also investigated the effect of GCa on the speeds of the [Ca2+] wave and membrane depolarization propagation and observed a non trivial increase with GCa.
Role of the intracellular Ca2+ stores (IP3 and Ryanodine receptors) in the simulated array of SMCs
In previous experimental studies3,18, results obtained using ryanodine showed that the magnitude of the Ca2+ wave was unaffected in linear arrangements of cultured vascular SMCs (see Supplementary Fig. S1E). This suggested that the Ca2+ induced Ca2+ release mechanism (CICR), through the RyRs, did not substantially contribute to the mechanism of ultrafast Ca2+ wave propagation, nor to the rate and amplitude of the [Ca2+] increase. Nevertheless, in the case of a minimal CICR, this event would be subsequent to the onset of the [Ca2+] increase and then would not affect drastically the ultrafast Ca2+ wave velocity. We simulated experiments inhibiting each of the two intracellular Ca2+ sources by modifying F for the Ca2+ release from IP3 sensitive stores and C for the CICR mechanism (Equations 9 and 12). Comparison between simulations under inactive Ryrs (Fig. 4A,B) and numerical control case (Figs 1A and 2B) did not show any significant changes in the Ca2+ and membrane depolarization responses.
Figure 4. Effects of RyRs or IP3Rs inhibition in the Ca2+ dynamics and membrane potential in a simulated array of SMCs.

(A) Time evolution of the Ca2+ response and (B) membrane potential variations along the network SMCs with inactive RyRs. RyRs inhibition was simulated by diminishing the CICR rate constant (C) to 0 μm.s−1. (C) Time evolution of the Ca2+ response and (D) membrane potential variations with inactive IP3Rs. IP3Rs inhibition was simulated by diminishing the maximal rate of activation-dependent Ca2+ influx (F) to 0 μm.s−1. Stimulated cell (black), 4th cell (red), 8th cell (blue), 12th cell (green) and 16th cell (pink). (E) Percentages of maximum F/F0 and [Ca2+]/[Ca2+]0 increases with inactive RyRs (npSMCs = 4, nA7r5 = 4) and (F) with inactive IP3Rs (npSMCs = 5, nA7r5 = 5). Red line fits the exponential decrease with distance from the stimulated site.
The other source of Ca2+ release from intracellular Ca2+ stores that could be involved was the IP3Rs path. We investigated whether IP3Rs did play a role in the generation of the ultrafast Ca2+ wave by incubating the two types of vascular SMCs with 30 μM 2-APB for 10 minutes. The inhibition of IP3 receptors with 2-APB caused a significant decrease in the average fluorescence amplitude in the stimulated cell (43,4 ± 9.2%, npSMC = 5; 50 ± 12.5%, nA7r5 = 5), but did not suppress the propagation (see Supplementary Fig. S1F) nor the wave magnitude of the ultrafast Ca2+ wave (91,3 ± 22cell.s−1, npSMCs = 5; 94 ± 22cell.s−1, nA7r5 = 5). This new experimental result supported the postulated hypothesis that the Ca2+ release through IP3Rs was not involved in the ultrafast Ca2+ wave mechanism. Altogether the experimental and theoretical results obtained by blocking the intracellular stores, demonstrated the hypothesis that the CICR did not contribute significantly to the mechanism of the ultrafast Ca2+ wave propagation nor did it to the fast Ca2+ increase process along the SMCs network.
Conduction velocity of the mechanically-induced membrane depolarization
For the first time, we estimated the spreading velocity of the membrane depolarization for two types of vascular SMCs using intracellular membrane potential recording with the microelectrodes technique combined with precise patterning technique. Experimentally, two identical mechanical stimulations in the same cell and two consecutive successful impalements in two different cells in the same network allowed to calculate the conduction velocity of the membrane depolarization along the network (Fig. 5A–D). The experimental conduction velocity obtained in linear networks of cultured vascular SMCs was 98 ± 27,3 cell.s−1 (n = 3) for pSMCs and 94,1 ± 24 cell.s−1 (n = 4) for A7r5 and were not statistically different (P > 0,05). Numerically, we also measured the conduction velocity by extracting the onset times at which the depolarization reached 50% of its maximal level. For the values of the coupling constant g and whole-cell conductance GCa under study, we confirmed the linear relationship between the speed of the ultrafast Ca2+ wave and the conduction speed of the membrane depolarization (Fig. 5E,F). This suggests that in principle under different physiological conditions, it is possible to infer one velocity from the knowledge of the other.
Figure 5. Representative conduction velocity approach in a linear arrangement of SMCs.

Phase contrast images showing the spatial arrangement of the microelectrode (left) and the micropipette (right) in the (A) first and in the (B) second mechanical stimulation attempt. Scale bar: 50 μm. Typical membrane potential recordings in a cell during mechanical stimulation in another cell for the (C) first and for the (D) second attempt. The conduction velocity was calculated by dividing the separation distance between the two impaled cells and the time delayed between the two membrane potential registrations (t1–t2). The arrows in the (C,D) panels indicate the moment when the mechanical stimulation was launched. Theoretical relation between the Ca2+ wave velocity and the propagation speed of membrane depolarization varying g (E) or GCa (F).
Discussion
This study focused on the mechanism of propagation of intercellular ultrafast Ca2+ waves in vascular smooth muscle cells resulting a from single cell membrane depolarization.
Our findings indicated that the propagation of membrane depolarization mediated by the electrical coupling between cells was at the origin of the observed ultrafast Ca2+ waves whereas intracellular and intercellular diffusion mechanisms did not play a significant role. In the numerical control case, the value of the velocity was found within the correct order of magnitude but slightly overestimated compared to experimental results. Different factors may explain the difference between the numerical and experimental wave velocities, like model parameters (that correspond to in vivo conditions whereas experiments were conducted in vitro) or experimental conditions (the temperature might affect biological rate processes up to two times for a change of 10 °C19). Nevertheless, the exact experimental values of the Ca2+ and depolarization velocities were found for smaller electrical coupling (or whole cell conductance) in a region of the parameter space that may correspond to a situation closer to in vitro conditions.
Membrane depolarization resulted from outward chloride currents through Ca2+ -activated chloride channels (Equation 14) and propagated along the SMCs network in an electrotonic manner with an exponential decay of 4 cells ranging within the experimental values for the two types of SMCs18. Most experimental evidences suggested that conducted responses depended primarily on passive electrotonic spread1 and this feature was also found to be essential in intact artery11. The flux of Ca2+ through VOCCs depended on the voltage and decayed exponentially along the network. Consequently, a similar decay for the maximal Ca2+ increase was expected. Numerically and experimentally we found that the space constants for the percentage of depolarization and the level of maximum Ca2+ increase were similar, confirming these two processes to be linked. This behaviour was observed after chemical stimulation in endothelial cells20, local mechanical stimulation in epithelial21 and in primary SMCs3.
Our numerical model allowed to explore the behavior of ultrafast Ca2+ waves beyond standard experimental conditions. Setting the electrical coupling coefficient to zero completely inhibited the wave as in experiments with a gap junction inhibitor3,18. This abolition has also been shown in simulated regenerative Ca2+ waves11 and experimentally in arterial strips12. We predicted the Ca2+ wave speed to increase as the square root of the electrical coupling parameter. This result could be further tested experimentally by increasing the electrical coupling with drugs used for acute opening of gap junctions22 or by imposing growth arrested conditions in cell preparations (that had been reported to enhance gap junctions permeability23). Therefore, we provided potential insights for further research in the field of gap junction proteomics, suggesting a possible modulation of Ca2+ wave velocity probably due to alterations of the connexins expression in the gap junctions. Setting VOCCs conductance to zero completely inhibited the Ca2+ wave; only the electrotonic propagation of membrane potential to neighboring cells was preserved. This confirmed that VOCCs were the main source of Ca2+ influx and the principal actor for the [Ca2+] increase during mechanical or KCl stimulation. Simulations predicted an increase of the velocity of the ultrafast Ca2+ in response to an increase of the VOCCs conductance. Our result was qualitatively in agreement with simulated regeneative Ca2+ waves, where a linear increase of the wave velocity had been observed for moderate values of GCa11. Interestingly, we found a nonlinear increase for higher values of VOCCs conductance. This numerical result was important because it is extremely challenging to modify experimentally the whole-cell conductance of VOCCs. Assuming that the effect of a VOCCs activator (i.e. Bay K8644) would be an increase in whole-cell conductance of VOCCs, we could correlate experiments in the presence of this drug with simulations at an elevated VOCCs conductance. Experimentally, it has been found that the Ca2+ wave was enhanced during Bay K8644 treatment in arterial strips12. In cultured rat mesenteric SMCs the incubation with Bay K8644 resulted in a longer [Ca2+] increase but not in a higher Ca2+ wave velocity3. It may be not suitable experimentally to obtain higher VOCCs conductance values only by the addition of elevated concentrations of Bay K8644. This result could be due to the fact that Bay K8644 may not increase drastically the VOCCs whole-cell conductance and highlighted the difficulty to predict experimentally the consequences of a gradual change of the VOCCs conductance. Therefore, this new numerical result opens the possibility to search and study new mechanisms in which one could not only enhance the Ca2+ wave, but also manipulate and tune the velocity of the ultrafast wave experimentally.
We provided further experimental evidences and confirmed numerically that the ultrafast Ca2+ wave was unaffected, at least in its onset, by the inhibition of each of the two main sources of intracellular Ca2+18. Simulations showed that setting the CICR rate constant to zero did not have a major effect in propagation of the Ca2+ wave. In this way, the in vitro system differed from the ex vivo case, where the CICR was essential for Ca2+ flashes and zero CICR rate constant abolishes the Ca2+ wave11. The other physiologically possible intracellular Ca2+ source is the IP3Rs. In agreement with experiments, the blockade of the IP3 mediated Ca2+ release did not affect the Ca2+ nor the membrane potential responses. In our systemm the stimulation was not based on background agonist. Consequently the IP3 levels remained constant and then no Ca2+ release occurred through this path, indicating that intracellular Ca2+ stores were not involved in the mechanism of the ultrafast Ca2+ wave. Furthermore, according to previous Ca2+ waves description and classification, most of the Ca2+ waves with a speed >10 mm.s−1 were not governed by intracellular Ca2+ processes5,6.
In a tissue made of electrically unexcitable cells, a change in the membrane potential could electrotonically propagate intercellularly with an exponential decrease in amplitude over distance. This coupling can only extend a few millimeters but can be very rapid (a few milliseconds)24. We estimated a conduction velocity that was in the same order of magnitude than previous values obtained for different experimental models such as guinea pig ureter (153 cell.s−1)25 and small intestine (513–586 cell.s−1)26. As suggested, the propagation of the membrane depolarization can be sufficient to coordinate smooth muscle cell relaxation within and among branches of vascular resistance networks27. Although the electrical signal that propagated in the linear arrangements of SMCs is not an action potential, the membrane potential depolarization must pass through the same gap junctions as for action potentials like for spontaneously electrical waves (known as slow waves) in the Gastric Antrum28,29,30,31 and in airway SMCs32. This indicated that the speed of propagation of these two phenomena should be directly correlated and the ultrafast Ca2+ wave was most probably a consequence of the membrane depolarization spreading. Nonetheless, if a delay between the spreading of the depolarization and the initial entry of the Ca2+ through VOCCs was expected, it would be conserved for the whole network without affecting the velocities. Comparison between the velocities of the ultrafast Ca2+ wave and the membrane depolarization propagation revealed no statistical difference for both types of vascular SMCs. Altogether these results suggest that it is likely that the velocity of the ultrafast Ca2+ wave was determined by the propagation speed of the membrane depolarization followed by Ca2+ entry through VOCCs. Similarly to gastrointestinal electrical slow waves propagation31, the wave range of the ultrafast Ca2+ wave in cultured vascular SMCs was not limited by the diffusion of Ca2+ in relation to conduction by CICR. On the contrary, it was mediated by current spread that had more spatial influence compared to the diffusion of second messengers of Ca2+ or IP3. Moreover, the model predicted that the direct relation between those two phenomena held for a wide range of electrical coupling coefficient and whole cell conductance for VOCCs. This fact, may endorse new experimental approaches to explore the physiological consequences in the vasculature.
Conclusion
In this work we provided an accurate framework to investigate the behavior of ultrafast Ca2+ wave under various physiological conditions, including extreme physiological conditions where wave properties could only be extrapolated from experiments. Conceptually, the present approach represented a significant advance by combining new theoretical and new experimental data for both Ca2+ and membrane potential dynamics. Numerically, we found that the ultrafast Ca2+ was not regenerated, it had a velocity and a spatial range in agreement with experiments. We validated the electrotonic behavior of the membrane depolarization spread and measured both in experiments and simulations the propagation speed. The model served also as a support for the experimental findings in which the intracellular Ca2+ sources were not involved in the generation of ultrafast Ca2+ wave. In our model, VOCCs were necessary for Ca2+ entry after depolarization. This Ca2+ influx event progressively propagated along the SMCs network with an intensity depending on the amplitude of the membrane depolarization. Importantly, the model predicted that a membrane depolarization was still present even when the VOCCs were inactive. It also allowed us to estimate valid ranges of critical parameters that are challenging to measure experimentally, such as electrical coupling coefficient and whole-cell conductance for VOCCs. Finally, the model reproduced, validated and allowed a better understanding of recently reported experimental data. In the context of the vascular system, analysis and modeling of SMCs mechanisms should ultimately provide new insights into the dynamic coordination of vasculature and thereby contribute to the regulation of tissue perfusion.
Methods
Experimental methods
The cell culture of primary SMCs (pSMCs) and rat aortic SM cell line (A7r5), pattern microfabrication, mechanical stimulation protocols (Fig. 6), Ca2+ fluoroscence and membrane depolarization measurements with microelectrode technics (Fig. 7), were previously detailed in ref. 18.
Figure 6. Local mechanical stimulus was performed with a micropipette (1 μm tip diameter, Eppendorf Femtotips I).
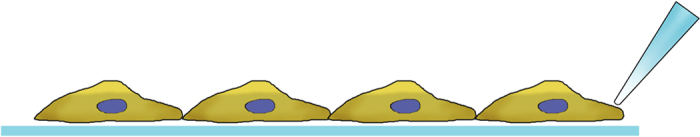
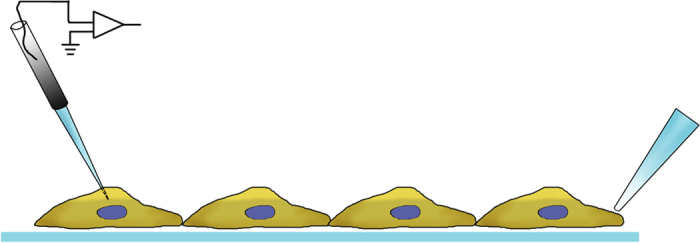
Figure 7. Intracellular recordings of vascular SMCs membrane potential were performed with microelectrodes made from borosilicate glass tubing containing an inner glass filament (World Precision Instruments, Sarasota, FL, USA) and pulled on a horizontal puller P-2000 (Sutter Instruments, Novato, CA, USA).
Chemical and drugs
We used the drug 2-Aminoethoxy-diphenylborate (2-APB) (EMD Millipore, Billerica, MA, USA) that blocks IP3 receptors. 2-APB was first dissolved at 100 mM in DMSO, and then diluted to 30 μM in buffer solution. Cells were allowed to preincubate in 2-APB for 10 minutes.
Ryanodine (20 μM), PA (50 μM), Nifedipine (10 μM).
Velocity measurements of membrane depolarization spreading
One of the important features in the use of the microelectrodes technique in combination with a precise patterned growth of cells and a reproducible way of mechanical stimulation is the possibility to measure the conduction velocity of the membrane depolarization. The local mechanical stimulation was performed using a high speed programmable micromanipulator for cell injection equipped with a micropipette. The electrical recordings were collected through a microelectrode; the conditioned signal was recorded with an analog-to-digital acquisition card (NI-USB 6009, National Instruments Corporation). A custom Matlab program (The MathWorks, Inc.) controlled simultaneously the micromanipulator via the serial interface and the acquisition card, handling the synchronization of the mechanical stimulation and the recording of the running depolarization. Thus, performing twice the mechanical stimulation in the same cell with the same conditions and registering the changes in the membrane potential for two different cells within the cell network, it was possible to estimate the spreading velocity of the membrane depolarization. The spreading velocity is calculated by dividing the difference in the onset times of membrane depolarization for the two different membrane potential registrations (two different cells in the same network) by the separation distance between them. The crucial part that allowed the conduction velocity calculation was the development of a program that allowed us to have a recording system that controls both the moment of stimulation and the registration of electrical events.
Analysis of experimental data
The experimental data presented here are expressed as means ± SEM (n). Statistical significance was tested using Student’s t-test on paired data; P < 0.05 was considered significant; n represents the number of experiments.
Numerical methods
The equations describing the cytosol [Ca2+] and the membrane potential dynamics in SMCs were taken from11. This model takes into account the most relevant cellular mechanisms involved during the generation of Ca2+ increases33. The model equations were solved using a fourth-order Runge-Kutta method and integrated on a one-dimensional line of SMCs comprising 100 cells. Within each SMC, the Ca2+ and the membrane potential dynamics are described by Eqs 1, 2, 3, 4, 5 and each cell was connected with its nearest left and right neighbors via electrical coupling (neglecting intracellular propagation of membrane potential). Local mechanical34 or KCl stimulation4 induced membrane depolarization. We mimicked the single cell stimulation process in the model by changing the reversal potential of K+ and Cl− (vK and vCl) in Eqs 14 and 15 of the first cell. Extracellular KCl changed the Nernst potentials for K+ and Cl−, depolarizing the cell membrane of the stimulated SMC35. We translated the unit mm.s−1 to cells.s−1 assuming a typical length for a single SMC of 100–150 μm, estimated from the cells spatial configuration used in ref. 18. In the simulations, ~16 cells correspond approximatively to the number of cells in the experiments.
Model of coupled Smooth Muscle Cells
The Ca2+ dynamics in each SMC (i), is described by five variables: the Ca2+ concentration in the cytosol (ci); the Ca2+ concentration in the sarcoplasmic reticulum (si); the cell membrane potential (vi); the open-state probability of Ca2+ activated potassium channels (wi) and the IP3 concentration (Ii). Cells are connected to their nearest neighbors via gap-junctional electrical coupling.
The mathematical approach for modeling the Ca2+ dynamics and electrophysiology in a SMC is given by the following equations:
 |
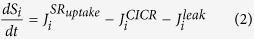 |
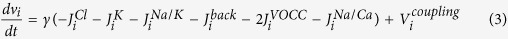 |
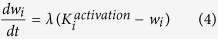 |
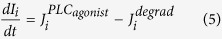 |
The equations (1, 2, 3, 4, 5) are detailed in refs 11 and 33.
JVOCC models the Ca2+ influx through VOCCs:
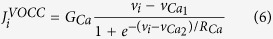 |
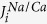 is the Na+/Ca2+ exchange:
is the Na+/Ca2+ exchange:
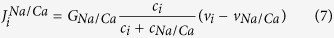 |
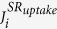 represents the SR uptake:
represents the SR uptake:
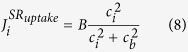 |
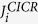 corresponds the Ca2+ induced Ca2+ release (CICR):
corresponds the Ca2+ induced Ca2+ release (CICR):
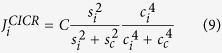 |
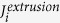 models the Ca2+ extrusion from the SMCs by the Ca2+ -ATPase pumps:
models the Ca2+ extrusion from the SMCs by the Ca2+ -ATPase pumps:
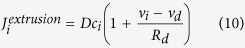 |
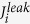 is the leak from SR:
is the leak from SR:
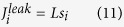 |
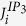 represents the Ca2+ release from IP3-sensitive stores:
represents the Ca2+ release from IP3-sensitive stores:
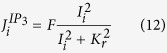 |
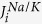 models the Na+-K+-ATPase:
models the Na+-K+-ATPase:
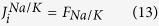 |
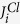 corresponds to the chloride channels and it takes into account that they are Ca2+ -activated;
corresponds to the chloride channels and it takes into account that they are Ca2+ -activated;
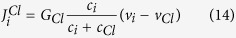 |
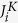 is the K+ efflux:
is the K+ efflux:
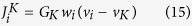 |
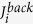 represents the background currents:
represents the background currents:
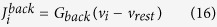 |
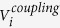 models the electrical coupling:
models the electrical coupling:
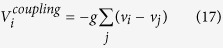 |
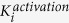 represents the Ca2+ and the voltage activation of the K+ channels:
represents the Ca2+ and the voltage activation of the K+ channels:
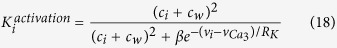 |
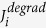 models the IP3 degradation:
models the IP3 degradation:
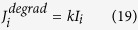 |
Model parameters
Model parameters were taken from11 except the whole-cell conductance for background current Gback and SR uptake constant B values to avoid spontaneous oscillations and ensure the stability of the system during the stimulation.
For the control case we fixed the whole-cell conductance for VOCCs GCa = 0,036195 μ.M.mV−1.s−1 and the value of the electrical coupling coefficient g = 1000 s−1. The latter corresponds to the gap-junctional conductance measured experimentally36,37,38,39. The gap junctional electrical coupling coefficient g is related to the gap junctional conductance G by g = G/Cm, where Cm is the cell membrane capacitance.
To further investigate the effect these two crucial parameters on the wave, we used g from 10 to 5000 s−1 and GCa from 0,026 to 0,0525 μM.mV−1.s−1.
Table 1. List of parameters.
| Parameter | Description | Value |
|---|---|---|
| F | Maximal rate of activation-dependent Ca2+ influx11 | 3,45 μM.s−1 |
| Kr | Half-saturation constant for agonist-dependent Ca2+ entry11 | 1,0 μM |
| GCa | Whole-cell conductance for VOCCs11 | 0,036195 μM.mV−1.s−1 |
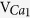 |
Reversal potential for VOCCs | 100,0 mV |
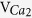 |
Half-point of the VOCC activation sigmoidal11 | −24,0 mV |
| RCa | Maximum slope of the VOCC activation sigmoidal11 | 8,5 mV |
| GNa/Ca | Whole-cell conductance for the Na+/Ca2+ exchanger11 | 0,006 μM.mV−1.s−1 |
| cNa/Ca | Half-point for activation of Na+/Ca2+ exchanger by Ca2+ 11 | 0,5 μM |
| vNa/Ca | Reversal potential for the Na+/Ca2+ exchanger11 | −30,0 mV |
| B | SR uptake rate constant | 2,025 μM.s−1 |
| cb | Half-point of the SR ATPase activation sigmoidal11 | 1,0 μM |
| C | CICR rate constant11 | 1545,0 μM.s−1 |
| sc | Half-point of the CICR Ca2+ efflux sigmoidal11 | 2,0 μM |
| cc | Half-point of the CICR activation sigmoidal11 | 0,9 μM |
| D | Rate constant for Ca2+ extrusion by the ATPase pump11 | 3,6 s−1 |
| vd | Intercept of voltage dependence of extrusion ATPase | −100,0 mV |
| Rd | Slope of voltage dependence of extrusion ATPase11 | 250,0 mV |
| L | Leak from SR rate constant11 | 0,375 s−1 |
| γ | Scaling factor relating net movement of ion fluxes11 | 492,5 mV.μM−1 |
| membrane potential (inversely related to cell capacitance) | ||
| FNa/K | Net whole-cell flux via the Na+-K+-ATPase11 | 0,03 μM.s−1 |
| GCl | Whole-cell conductance for Cl− current11 | 0,6 μM.mV−1.s−1 |
| cCl | Ca2+ sensitivity for Cl− channels11 | 0,7 μM |
| vCl | Reversal potential for Cl− channels11 | −25,0 mV |
| GK | Whole-cell conductance for K+ efflux11 | 0,045 μM.mV−1.s−1 |
| vK | Reversal potential for K+ 11 | −94 mV |
| λ | Rate constant for net KCa channel opening11 | 675 |
| cw | Translation factor for Ca2+ dependence of KCa11 | 0,0 μM |
| channel activation sigmoidal11 | ||
| β | Translation factor for the membrane potential dependence11 | 0,001 μM2 |
| of KCa channel activation sigmoidal11 | ||
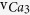 |
Half-point for the KCa channel activation sigmoidal11 | −27,0 mV |
| RK | Maximum slope of the KCa activation sigmoidal11 | 12,0 mV |
| Gback | Whole-cell conductance for background currents40,41 | 0,0045 μM.mV−1.s−1 |
| vrest | Equilibrium potential11 | −55,0 mV |
| k | Rate constant of IP3 degradation11 | 0,1 s−1 |
| g | Electrical coupling coefficient11 | 1000 s−1 |
Additional Information
How to cite this article: Quijano, J. C. et al. Intercellular ultrafast Ca2+ wave in vascular smooth muscle cells: numerical and experimental study. Sci. Rep. 6, 31271; doi: 10.1038/srep31271 (2016).
Supplementary Material
Acknowledgments
This work was supported by the Swiss National Science Foundation grant FN 310300-127122. We are grateful to C. Gabella and C. Labouesse for useful discussions.
Footnotes
Author Contributions J.C.Q. conceived and conducted experiments, D.N. and F.R. designed the numerical implementation of the model, F.R. conducted numerical experiments, J.C.Q. and F.R. analyzed the data, J.C.Q. and F.R. wrote the paper. J.J.M. supervised the work. All authors reviewed the manuscript.
References
- Nagaraja S., Kapela A. & Tsoukias N. M. Intercellular communication in the vascular wall: a modeling perspective. Microcirculation (New York, N.Y.: 1994) 19, 391–402 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young S. H., Ennes H. S. & Mayer E. A. Propagation of calcium waves between colonic smooth muscle cells in culture. Cell Calcium 20, 257–271 (1996). [DOI] [PubMed] [Google Scholar]
- Halidi N., Boittin F. X., Bény J. L. & Meister J. J. Propagation of fast and slow intercellular Ca2+ waves in primary cultured arterial smooth muscle cells. Cell Calcium 50, 459–467 (2011). [DOI] [PubMed] [Google Scholar]
- Kalthof B., Bechem M., Pott L. & Schramm N. Monitoring of ca2+-transients in electrically stimulated a7r5 vascular smooth muscle cells fills the experimental gap between kcl- induced depolarization and patch-clamp studies. J. Pharmacol. Toxicol. Methods. 30, 143–147 (1993). [DOI] [PubMed] [Google Scholar]
- Jaffe L. F. Classes and mechanisms of calcium waves. Cell Calcium 14, 736–745 (1993). [DOI] [PubMed] [Google Scholar]
- Jaffe L. F. The propagation speeds of calcium action potentials are remarkably invariant (2003). [DOI] [PubMed]
- Sneyd J., Keizer J. & Sanderson M. J. Mechanisms of calcium oscillations and waves: a quantitative analysis. The FASEB journal: official publication of the Federation of American Societies for Experimental Biology 9, 1463–1472 (1995). [DOI] [PubMed] [Google Scholar]
- Höfer T., Politi A. & Heinrich R. Intercellular Ca2+ Wave Propagation through Gap-Junctional Ca2+ Diffusion: A Theoretical Study. Biophysical Journal 80, 75–87 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höfer T., Venance L. & Giaume C. Control and plasticity of intercellular calcium waves in astrocytes: a modeling approach. J. Neurosci. 22, 4850–4859 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenigsberger M., Sauser R., Lamboley M., Bény J.-L. & Meister J.-J. Ca2+ dynamics in a population of smooth muscle cells: modeling the recruitment and synchronization. Biophysical journal 87, 92–104 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenigsberger M., Seppey D., Bény J. L. & Meister J. J. Mechanisms of propagation of intercellular calcium waves in arterial smooth muscle cells. Biophysical Journal 99, 333–343 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seppey D., Sauser R., Koenigsberger M., Bény J.-L. & Meister J.-J. Intercellular calcium waves are associated with the propagation of vasomotion along arterial strips. American journal of physiology. Heart and circulatory physiology 298, H488–H496 (2010). [DOI] [PubMed] [Google Scholar]
- Hamada H., Damron D. S., Hong S. J., Van Wagoner D. R. & Murray P. A. Phenylephrine-induced Ca$^{2+}$ oscillations in canine pulmonary artery smooth muscle cells. Circulation research 81, 812–823 (1997). [DOI] [PubMed] [Google Scholar]
- Peng H., Matchkov V., Ivarsen A., Aalkjaer C. & Nilsson H. Hypothesis for the initiation of vasomotion. Circulation research 88, 810–815 (2001). [DOI] [PubMed] [Google Scholar]
- Sell M., Boldt W. & Markwardt F. Desynchronising effect of the endothelium on intracellular Ca2+ concentration dynamics in vascular smooth muscle cells of rat mesenteric arteries. Cell Calcium 32, 105–120 (2002). [DOI] [PubMed] [Google Scholar]
- Haddock R. E., Hirst G. D. S. & Hill C. E. Voltage independence of vasomotion in isolated irideal arterioles of the rat. The Journal of Physiology 540, 219–229 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamboley M., Schuster A., Bény J.-L. & Meister J.-J. Recruitment of smooth muscle cells and arterial vasomotion. American journal of physiology. Heart and circulatory physiology 285, H562–H569 (2003). [DOI] [PubMed] [Google Scholar]
- Quijano J. C., Vianay B., Bény J. L. & Meister J. J. Ultrafast Ca2+ wave in cultured vascular smooth muscle cells aligned on a micropatterned surface. Cell Calcium 54, 436–445 (2013). [DOI] [PubMed] [Google Scholar]
- Prosser C. L. Comparative animal physiology (Philadelphia-London: Saunders, 1973), 3 edn. [Google Scholar]
- Domenighetti A. A., Bény J. L., Chabaud F. & Frieden M. An intercellular regenerative calcium wave in porcine coronary artery endothelial cells in primary culture. The Journal of physiology 513 (Pt 1, 103–116 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchill G. C., Atkinson M. M. & Louis C. F. Mechanical stimulation initiates cell-to-cell calcium signaling in ovine lens epithelial cells. Journal of cell science 109 (Pt 2, 355–65 (1996). [DOI] [PubMed] [Google Scholar]
- Salameh A. & Dhein S. Pharmacology of Gap junctions. New pharmacological targets for treatment of arrhythmia, seizure and cancer?(2005). [DOI] [PubMed]
- Kurjiaka D. T., Olsen M. V. & Burt J. M. Gap junctions (Werner R. Editor. IOS Press, 1998). [Google Scholar]
- Bény J.-L. Information networks in the arterial wall. News Physiol. Sci. 14, 68–73 (1999). [DOI] [PubMed] [Google Scholar]
- Tsuchiya T. & Takei N. Pressure responses and conduction of peristaltic wave in guinea-pig ureter. Jpn. J. Physiol. 40, 139–149 (1990). [DOI] [PubMed] [Google Scholar]
- Stevens R. J., Publicover N. G. & Smith T. K. Propagation and neural regulation of calcium waves in longitudinal and circular muscle layers of guinea pig small intestine. Gastroenterology 118, 892–904 (2000). [DOI] [PubMed] [Google Scholar]
- Emerson G. G., Neild T. O. & Segal S. S. Conduction of hyperpolarization along hamster feed arteries: augmentation by acetylcholine. American journal of physiology. Heart and circulatory physiology 283, H102–9 (2002). [DOI] [PubMed] [Google Scholar]
- Ward S. M., Dixon R. E., de Faoite A. & Sanders K. M. Voltage-dependent calcium entry underlies propagation of slow waves in canine gastric antrum. J Physiol 561, 793–810 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayguinov O., Ward S. M., Kenyon J. L. & Sanders K. M. Voltage-gated Ca2+ currents are necessary for slow-wave propagation in the canine gastric antrum. Am J Physiol Cell Physiol 293, C1645–59 (2007). [DOI] [PubMed] [Google Scholar]
- David Hirst G. S., Pilar Garcia-Londoño A. & Edwards F. R. Propagation of slow waves in the guinea-pig gastric antrum. J Physiol 5711, 165–177 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Helden D. F., Laver D. R., Holdsworth J. & Imtiaz M. S. The generation and propagation of gastric slow waves. Clin. Exp. Pharmacol. Physiol. 37, 516–524 (2009). [DOI] [PubMed] [Google Scholar]
- Janssen L. J., Hague C. & Nana R. Ionic mechanisms underlying electrical slow waves in canine airway smooth muscle. AJP - Lung Cellular and Molecular Physiology 275, L516–L523 (1998). [DOI] [PubMed] [Google Scholar]
- Parthimos D., Edwards D. H. & Griffith T. M. Minimal model of arterial chaos generated by coupled intracellular and membrane Ca2+ oscillators. The American journal of physiology 277, H1119–H1144 (1999). [DOI] [PubMed] [Google Scholar]
- Meininger G. A. & Davis M. J. Cellular mechanisms involved in the vascular myogenic response. Am J Physiol 263, H647–59 (1992). [DOI] [PubMed] [Google Scholar]
- Nelson M. T., Patlak J. B., Worley J. F. & Standen N. B. Calcium channels, potassium channels, and voltage dependence of arterial smooth muscle tone. The American journal of physiology 259, C3–C18 (1990). [DOI] [PubMed] [Google Scholar]
- Moore L. K., Beyer E. C. & Burt J. M. Characterization of gap junction channels in a7r5 vascular smooth muscle cells. Am. J. Physiol. Cell Physiol 260, 975–1981 (1991). [DOI] [PubMed] [Google Scholar]
- Moore L. K. & Burt J. M. Gap junction function in vascular smooth muscle: influence of serotonin. The American journal of physiology 269, H1481–9 (1995). [DOI] [PubMed] [Google Scholar]
- Li X. & Simard J. M. Multiple connexins form gap junction channels in rat basilar artery smooth muscle cells. Circ Res 84, 1277–1284 (1999). [DOI] [PubMed] [Google Scholar]
- Yamamoto Y., Klemm M. F., Edwards F. R. & Suzuki H. Intercellular electrical communication among smooth muscle and endothelial cells in guinea-pig mesenteric arterioles. Journal of Physiology 535, 181–195 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen J. C. B. et al. Activation of a cGMP-sensitive calcium-dependent chloride channel may cause transition from calcium waves to whole cell oscillations in smooth muscle cells. American journal of physiology. Heart and circulatory physiology 293, H215–28 (2007). [DOI] [PubMed] [Google Scholar]
- Jacobsen J. C. B. et al. A model of smooth muscle cell synchronization in the arterial wall. American journal of physiology. Heart and circulatory physiology 293, H229–H237 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.