

Abstract
The cascade of inflammatory pathogenetic mechanisms in multiple sclerosis (MS) has no specific conventional MRI correlates. Clinicians therefore stipulate improved imaging specificity to define the pathological substrates of MS in vivo including mapping of intracellular sodium accumulation. Based upon preclinical findings and results of previous sodium MRI studies in MS patients we hypothesized that the fluid-attenuated sodium signal differs between acute and chronic lesions. We acquired brain sodium and proton MRI data of N = 29 MS patients; lesion type was defined by the presence or absence of contrast enhancement. N = 302 MS brain lesions were detected, and generalized linear mixed models were applied to predict lesion type based on sodium signals; thereby controlling for varying numbers of lesions among patients and confounding variables such as age and medication. Hierarchical model comparisons revealed that both sodium signals average tissue (χ2(1) = 27.89, p < 0.001) and fluid-attenuated (χ2(1) = 5.76, p = 0.016) improved lesion type classification. Sodium MRI signals were significantly elevated in acute compared to chronic lesions compatible with intracellular sodium accumulation in acute MS lesions. If confirmed in further studies, sodium MRI could serve as biomarker for diagnostic assessment of MS, and as readout parameter in clinical trials promoting attenuation of chronic inflammation.
Introduction
Multiple sclerosis (MS) is an inflammation-mediated disease of the CNS1. The genesis of MS is characterized by a cascade of pathologic events, including focal lymphocytic infiltration, microglia activation, demyelination, and axonal degeneration2. A potentially important inflammatory mechanism leading to axonal degeneration is the production of reactive-oxygen species and nitric oxide from activated microglia and infiltrated macrophages3,4. Reactive-oxygen species and nitric oxide promote mitochondrial injury5,6. Impaired neuronal mitochondrial function, in turn, induces additional oxidative stress by increased production of reactive-oxygen species7, and results in reduced ATP production8,9. Concurrently, there is an increased energy demand due to the re-organization of sodium channels, which occurs in response to axonal demyelination10,11. The neuronal energy deficit leads to an intracellular accumulation of sodium ions12, and thereby promotes the sodium-calcium exchanger to operate in reverse13. The resulting intracellular culmination of calcium can contribute to further mitochondrial damage and activation of nitric oxide synthase, proteases as well as lipases3 – a cascade eventually resulting in neuronal cell death9.
The cascade of pathogenetic mechanisms in MS has no specific conventional MRI correlates. Imaging specificity needs to be improved to define the pathological substrates of MS in vivo14. Based on the reported effects of MS on sodium channels, sodium MRI seems promising for this endeavour: Inglese and colleagues15 and Paling and colleagues16 showed that the average tissue sodium signal is elevated in MS lesions compared to normal-appearing white matter, and increased in normal-appearing white matter of MS patients compared to healthy controls. Also, the two groups revealed a correlation between sodium signal strength and disability status. However, both intra- and extracellular sodium ions contribute to this overall signal and therefore changes cannot be attributed to a certain microstructural compartment. Consequently, one long-term goal in understanding the mechanisms of MS is to distinguish intracellular from extracellular sodium ions3. In this study, we propose to include an additional, so-called fluid-attenuated sodium signal (also known as intracellular-weighted or relaxation-weighted sodium signal), which allows further information on the origin of signal alterations. As this technique emphasizes sodium ions with short relaxation times a weighting towards the intracellular sodium compartment is achieved17,18,19. Similar to our approach, Madelin and colleagues recently applied a sodium MR sequence to estimate the intracellular sodium concentration of healthy brain tissue20; sequence details are discussed elsewhere21. Further, it has been shown in brain tumours, that the fluid-attenuated sodium signal adds significant information to the average tissue sodium signal based on a strong correlation between fluid-attenuated sodium signal and the proliferation rate of tumour cells21,22.
On the basis of preclinical work1,4,5,8,9,12,13 and sodium MRI findings in MS15,16,23,24, we postulated that the fluid-attenuated sodium signal should differ between acute and chronic lesions, and, thus, provide additional information on lesion type in MS. Therefore, we measured both average tissue and fluid-attenuated sodium signals of brain tissue lesions in MS, and compared these measures between acute and chronic lesions as defined by the actual reference standard, which is the presence or absence of contrast-enhancement in canonical T1-weighted images. Additionally, we were able to record signals from three patients with acute lesions before and after drug based treatment.
Methods
Ethics statement
The study was approved by the local Medical Ethics Committee (Faculty of Clinical Medicine, University of Heidelberg). All participants provided written informed consent prior to enrolment. The procedures that follow were in accordance with the declaration of Helsinki.
Participants and study design
In this study, we enrolled patients with the diagnosis of MS, and acute and/or chronic brain lesions. Eligible patients had no history of head trauma, no vascular pathologies or medical conditions possibly leading to brain lesion formation other than MS, and no contraindications to ultra-high field MRI. Diagnosis of MS was made according to the 2005 McDonald criteria25. The course of disease was defined according to Lublin and Reingold26: A secondary-progressive MS was diagnosed if a deterioration of clinical disability independent of relapses exists for at least six months following an initial relapsing-remitting MS course. Detailed epidemiologic patient data are shown in Table 1.
Table 1. Patient characteristics.
| Patient ID | Age (years) | Gender | Stage | EDSS | MS lesion type | Methyl-prednisolone | Medication other than methyl- prednisolone | Disease duration (months) | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Acute | Chronic | Prior to MRI | At the time of MRI | At the time of MRI | ||||||
| 1 | 27 | Female | RRMS | 2 | X | Interferon beta | 15.05 | |||
| 2 | 42 | Male | RRMS | 2 | X | X | None | 0.30 | ||
| 3 | 20 | Male | RRMS | 0 | X* | Fingolimod | 10.02 | |||
| 4 | 18 | Male | RRMS | 2 | X | Interferon beta | 9.63 | |||
| 5 | 58 | Male | RRMS | 3 | X | Glatiramer ac. | 412.55 | |||
| 6 | 33 | Male | RRMS | 1.5 | X | X | Glatiramer ac. | 44.05 | ||
| 7 | 25 | Male | RRMS | 0 | X* | X | None | 5.55 | ||
| 8 | 50 | Male | RRMS | 0 | X | Natalizumab | 96.62 | |||
| 9 | 50 | Female | RRMS | 4 | X* | X | X | None | 66.06 | |
| 10 | 49 | Male | RRMS | 1.5 | X | Dimethyl fum. | 231.67 | |||
| 11 | 47 | Female | RRMS | 4 | X | Interferon beta | 212.32 | |||
| 12 | 39 | Female | RRMS | 2 | X | X | None | 0.03 | ||
| 13 | 34 | Female | RRMS | NI | X | X | X | NI | NI | |
| 14 | 52 | Female | RRMS | 3 | X | X | Glatiramer ac. | 101.08 | ||
| 15 | 48 | Female | RRMS | 2 | X | X | X | None | 0.03 | |
| 16 | 50 | Female | RRMS | 5 | X | X | None | 0.53 | ||
| 17 | 50 | Female | SPMS | 5.5 | X | Glatiramer ac. | 103.84 | |||
| 18 | 25 | Female | RRMS | 1 | X | Fingolimod | 102.83 | |||
| 19 | 23 | Female | RRMS | 0 | X | Fingolimod | 30.09 | |||
| 20 | 40 | Male | RRMS | 3 | X | Interferon beta | 28.91 | |||
| 21 | 25 | Female | RRMS | 2 | X | X | X | Interferon beta | 34.76 | |
| 22 | 43 | Male | RRMS | 2 | X | X | Interferon beta | 24.80 | ||
| 23 | 27 | Female | RRMS | 2 | X* | X | X | None | 4.07 | |
| 24 | 45 | Female | RRMS | 4 | X | X | Fingolimod | 210.91 | ||
| 25 | 34 | Female | RRMS | 1 | X | X | Interferon beta | 43.36 | ||
| 26 | 37 | Male | RRMS | 2.5 | X | Interferon beta | 73.23 | |||
| 27 | 36 | Male | RRMS | 1 | X | X | None | 0.99 | ||
| 28 | 60 | Male | RRMS | 1.5 | X | None | 49.87 | |||
| 29 | 34 | Male | RRMS | 2 | X | X | X | None | 110.32 | |
RRMS = relapsing-remitting multiple sclerosis; SPMS = secondary progressive multiple sclerosis; *acute lesion of the spinal cord, no brain lesion; ac. = acetate; fum. = fumarate; NI = no information available.
Procedures
MR imaging
Cross-sectional experiment
For lesion detection, clinical routine proton MRI data were acquired using a 3 Tesla whole-body system (Tim Trio 3T, Siemens Healthcare, Erlangen, Germany) with T2-FLAIR, T2-TSE, native and contrast-enhanced T1-3D ultrafast gradient sequences. Sodium MRI was performed using a 7 Tesla whole-body MR system (Magnetom 7T, Siemens Healthcare, Erlangen, Germany) and a double-resonant (1H/23Na) quadrature birdcage coil with an inner coil diameter of 26 cm (Rapid Biomed GmbH, Rimpar, Germany). All sodium MR sequences were based on a 3D density-adapted projection reconstruction technique27 providing an average tissue sodium signal, and a fluid-attenuated sodium signal. Weighting in this context means that extracellular sodium may contribute to the fluid-attenuated signal. There is no quantitative information of how strong the weighting is. Sequence details are shown in Table 2.
Table 2. Sodium MRI sequence details.
| Sodium MR Sequence | ||
|---|---|---|
| Average tissue sodium MRI signal* | Fluid-attenuated sodium MRI signal** | |
| Echo Time (TE) | 0.35 ms | 0.75 ms |
| Repetition Time (TR) | 160 ms | 185 ms |
| Readout Duration (TRO) | 10 ms | 16.7 ms |
| Inversion Time (TI) | — | 41 ms |
| Nominal Spatial Resolution | 3.0 × 3.0 × 3.0 mm3 | 4.4 × 4.4 × 4.4 mm3 |
| Acquisition Time | 10 min 4 s | 9 min 52 s |
*We minimized relaxation weighting by a short TE and a long TR; **We measured the fluid-attenuated sodium signal using an inversion recovery sequence which suppresses signal of sodium ions with long relaxation times as, for example, in cerebro-spinal fluid.
Longitudinal experiment
Three patients (PIDs no. 15, 21 and 29; cf. Table 1) of the cross-sectional experiment were additionally measured after administration of high-dose methylprednisolone using the identical MR imaging protocol. Thus, in these patients acute MS lesions could be longitudinally monitored before and after steroid therapy.
Image processing
Sodium image reconstruction was performed offline by a custom-written MATLAB script (The Mathworks Inc, Natick, MA, USA). To reduce Gibbs ringing artifacts, a Hamming filter was applied. Contrast-enhanced T1-weighted images (T1 CE) were skull-stripped by the Brain Extraction Tool (BET, part of FMRIB’s Software Library FSL)28 and served as individual reference images. T2-TSE, T2-FLAIR, and sodium images were co-registered to this reference image using an affine registration with 12 degrees of freedom as implemented in FMRIB’s Linear Image Registration Tool (FLIRT, part of FSL)29. Figure 1 shows an example of co-registered sodium and proton MR images. Sodium data were normalized to healthy parenchyma, i.e., to brain tissue without T2 signal alterations and without pathological contrast enhancement in the T1 signal: For every individual and each MR sequence, we performed a voxelwise division of signal values from identified lesions by the respective mean signal of healthy tissue. Normalizing the data aimed at the reduction of inter-individual variance in the sodium MR signal, and at the comparability of imaging data among subjects. All values are in arbitrary units.
Figure 1. Exemplary co-registered sodium and proton images of a patient with acute MS lesions (PID no. 15).
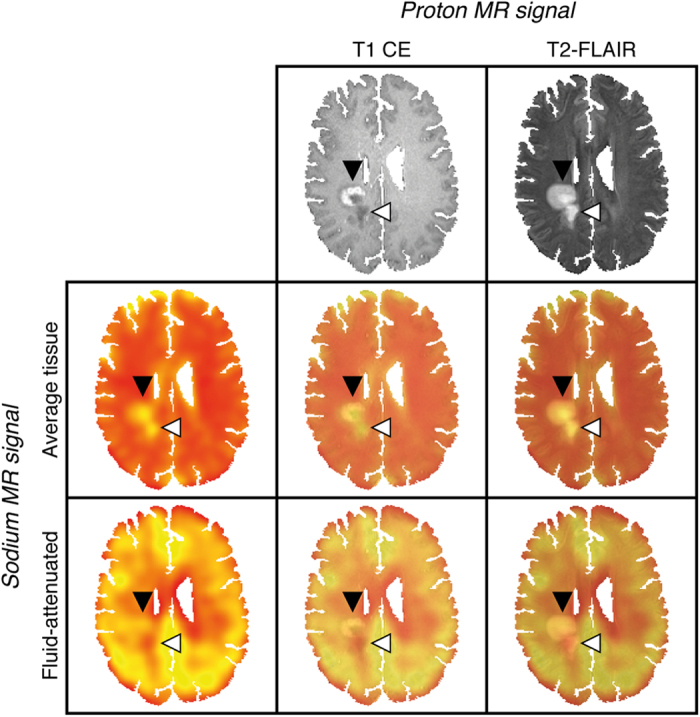
Proton MRI demonstrates two (confluent) right-central white matter lesions. The anterior lesion exhibits contrast-enhancement and corresponding elevated average tissue and fluid-attenuated sodium signals compatible with acute inflammation (black arrowhead). The posterior lesion shows no signs of blood-brain barrier disruption, an increased average tissue sodium signal and a reduced fluid-attenuated sodium signal – a combination consistent with the residuals of brain tissue inflammation (white arrowhead). This representative example demonstrates high intermodal registration accuracy of the applied affine image transformation method (FLIRT); T1 CE, T2-FLAIR and sodium images as well as the corresponding overlays are shown.
Multiple sclerosis lesions
MS lesion volumes of interest were defined by T2-FLAIR signal alterations and, where applicable, by the T1 signal of contrast-enhancing portions. Thereby, the presence (acute lesion) or absence of contrast enhancement (chronic lesion) determined the type of lesion. Lesions less than 30 mm2 in volume, and less than 5 mm in transversal diameter were discarded from further analysis to reduce partial volume effects. All included lesions were located within white matter. Outlier corrections removed data diverging by 2.5 standard deviations from the intra-individual mean value of all lesions. Plausibility checks controlled for T1 CE signals to be higher in acute compared to chronic lesions; lesions not fulfilling these criteria were removed from further analysis.
Statistical analysis
Prediction of lesion type
For each lesion its type (acute vs. chronic) was predicted using generalized linear mixed models with random intercepts, which allow controlling for varying numbers of lesions and lesion types between participants. The predictive value of average tissue sodium signal and fluid-attenuated sodium signal was explored by adding these predictors successively to a base model and evaluating whether these led to an improvement of the model which outweighed the additional number of free parameters using likelihood ratio tests. Several other factors possibly associated with lesion type were controlled for by adding them as predictors to all models. In detail, age, gender, methylprednisolone application, medication other than methylprednisolone, expanded disability status scale (EDSS) score, and disease duration were included in all models. Model parameters were estimated using Laplace approximation as implemented in the lme4 package30 in R. To ease convergence of the algorithm all continuous predictors were scaled, i.e., z-transformed prior to parameter estimation.
Correlation analyses
As for the prediction of lesion type a hierarchical design was used to test for the relationship between sodium and proton MRI signals, thereby, controlling for differences in the number of lesion (types) across participants. To ensure continuity with the previous analysis we calculated linear mixed models, predicting separately native T1, T1 CE, T2-TSE, and T2-FLAIR from average tissue and fluid-attenuated sodium signals. All signals were standardized beforehand, thus, the estimated regression parameters correspond to Pearson’s correlation coefficient. Note that due to the hierarchical design fixed effects parameters are free of inter-subject variability in the strength of the signal, thus, being higher than suggested from a scatterplot across all data points. Therefore, scatterplots are not shown here. Statistical significance of the estimated parameter values was tested using type III Wald chi-square tests.
Longitudinal data
This analysis aimed at revealing changes in sodium MRI signals after steroid therapy. To this aim, average tissue and fluid-attenuated sodium signals were separately predicted by the timepoint of signal measurement. This analysis compares signal values between pre- and post-medication states while accounting for differences in lesion number and average signal strength in single patients. A statistical model based on data from three patients cannot be representative with regard to the whole patient group. To derive a measure of confidence for parameter estimates from this model we performed 100,000 parametric bootstraps of each model.
Results
Cross-sectional experiment
Lesion segmentation
N = 330 supratentorial parenchymal lesions were detected and thereby classified as acute (n = 82) or chronic (n = 248) depending on the presence or absence of pathologic contrast enhancement. Outlier corrections and plausibility checks (cf. Methods) reduced the number of lesions leaving n = 302 lesions including n = 70 acute and n = 232 chronic lesions for further analysis.
Prediction of lesion type
MS lesions were analysed by generalized linear mixed models, which revealed that both, average tissue and fluid-attenuated sodium signals significantly improved lesion type classification (average tissue sodium signal: χ2(1) = 27.89, p < 0.001; fluid-attenuated sodium signal: χ2(1) = 5.76, p = 0.016). The average, as well as the fluid-attenuated sodium signal of acute MS lesions were thus significantly elevated compared to chronic MS lesions. In Fig. 2, these results are exemplarily visualized for intra- and inter-individual MS lesions.
Figure 2. Exemplary intra- and inter-individual sodium MRI data of chronic and acute MS lesions.

Intra-individual (A; PID no. 29) proton MRI shows a T2-hyperintense chronic lesion of the right-frontal parenchyma (A), left panel; lesion no. 2) without contrast-enhancement (CE) and an acute T2-hyperintense, right-temporal lesion with associated CE (A, right panel; lesion no. 2). Similar, inter-individual (B; PIDs no. 13 and no. 18) proton MRI shows a T2-hyperintense chronic lesion of the left-central parenchyma (B), left panel; lesion no. 2) without contrast-enhancement (CE) and an acute T2-hyperintense, right fronto-mesial lesion with associated CE (B, right panel; lesion no. 5). In acute lesions the (intra- and inter-individual) average tissue and the fluid-attenuated sodium signals are increased compared to chronic lesions. Scatter plots and histograms of both sodium signals visualize intra- and inter-individual examples with ideal separation of acute and chronic lesions by average tissue and fluid-attenuated sodium signals; this is not true for all lesions, that is, scatter plots of MS lesions can be larger, and may be somewhat overlapping between lesion types. This may be, at least in part, owed to the fact that both, T2 signal and contrast enhancement are primarily unspecific14,43. Nevertheless, generalized linear mixed models revealed a significant improvement of lesion type classification by the average as well as the fluid-attenuated sodium signal. Both signals were significantly increased in acute lesions. (All values are in arbitrary units; dashed lines projected onto the zoomed sodium images indicate the morphological outlines as defined by proton MRI; please note however, that these outlines are not necessarily identical to sodium MRI signal alterations of these lesions; thick gray grid lines represent the average tissue (y-axis) and fluid-attenuated sodium signal values (x-axis) of healthy parenchyma)
Correlation analyses of sodium and proton MR signals
Hierarchical analyses of sodium and proton MRI signals in MS lesion volumes of interest revealed a weak correlation of the average tissue sodium signal with the T2-FLAIR signal (β = 0.38 (0.09), χ2 = 18.86, p < 0.001), but indicated strong correlations of the average tissue sodium signal with T2-TSE (β = 0.90 (0.07), χ2 = 147.04, p < 0.001) and native T1 signals (β = 0.92 (0.10), χ2 = 86.75, p < 0.001). There was no correlation between average tissue sodium signal and T1 CE signal.
In addition, statistical analyses detected a correlation of fluid-attenuated sodium signal and T1 CE signal (β = 0.34 (0.08), χ2 = 17.94, p < 0.001). There were no associations of the fluid-attenuated sodium signal with T2-FLAIR, T2-TSE, and native T1 signal. Of note, there was no correlation between average tissue and fluid-attenuated sodium signals.
Longitudinal experiment
Hierarchical comparisons of average tissue and fluid-attenuated sodium signals across pre- and post-medication measurements revealed a decrease of both signals after the application of high-dose methylprednisolone. Average tissue sodium signals decreased by estimated 0.07 a.u. ([−0.18, 0.03]; 95% confidence interval based on parametric bootstrapping). Fluid-attenuated sodium signals even decreased by 1.45 a.u. [−2.51, −0.38] after medication. Longitudinal sodium MRI data are shown in Figs 3 and 4.
Figure 3. Longitudinal sodium MRI data of an acute lesion (PID no. 15).

After application of high-dose methylprednisolone both the average tissue and fluid-attenuated sodium signal decrease. Based upon the cross sectional findings on sodium signal differences between acute and chronic MS lesions, this signal behavior was proposed before. Moreover, it strongly supports the notion that findings of our study are compatible with intracellular sodium accumulation in acute inflammatory MS lesions. Please note, that due to the pharmacological intervention changes in tissue sodium concentration of healthy parenchyma could not be ruled out. Thus, for the longitudinal observations, normalization referred to transmitter amplitudes instead of the sodium signal of healthy parenchyma.
Figure 4. Longitudinal monitoring of all acute MS lesions (PIDs no. 15, 21 and 29).

Sodium signal changes after administration of high-dose methylprednisolone support findings of the cross-sectional analyses. Hierarchical comparisons of average tissue and fluid-attenuated sodium signals across pre- and post-medication measurements revealed a decrease of both signals after the application of high-dose methylprednisolone (cf. Methods and Results section for details).
Discussion
Our study confirms that both, the average tissue sodium signal and the fluid-attenuated sodium signal significantly differ between acute and chronic MS lesions of human brain parenchyma. Additional longitudinal sodium data from before and after application of high-dose methyprednisolone demonstrate that these differences are MS specific. Therefore, the detected increase in fluid-attenuated sodium signal is compatible with the intracellular sodium accumulation observed in acute MS lesions. This finding is in excellent agreement with previous reports on increased expression of sodium channels, and intracellular sodium accumulation occurring as pathophysiological event in experimental autoimmune encephalomyelitis (EAE) and MS11,12,31. Moreover, consistent with results of our study, Petracca and colleagues32 demonstrated sodium signals at a brain regional level that might reflect neuro-axonal metabolic dysfunction in MS.
In Fig. 5, our results and recent preclinical findings are put into context. There is strong evidence that sodium channels play an important role in immune cell function in EAE and MS11,31. Nav1.6 is the predominant sodium channel expressed in microglia and macrophages33,34. Its expression is up regulated on activation of these cells in EAE and acute MS lesions35,36. In MS lesions, activated microglia and infiltrated macrophages4 produce reactive-oxygen species and nitric oxide3, which impair mitochondrial function in neurons37,38. Thereby, ATP production and energy supply is reduced. Concurrently, there is an increased neuronal energy demand due to the redistribution, as well as the co-localization of Nav1.6 and the sodium-calcium exchanger along demyelinated axons in EAE31 and in acute MS lesions11. This mismatch between energy supply and demand thereby creates a state of virtual hypoxia39, and leads to an accumulation of intracellular sodium ions12 similar to the pathogenic cascade following ischemic stroke40. The ionic balance changes promote the sodium-calcium pump to operate in reverse13, i.e. to import calcium into the cell. This leads to injurious calcium levels, and eventually to the activation of proteases, lipases, and nitric oxide-synthase as well as to mitochondrial damage, and ultimately to neuronal cell death9. The role of sodium channels in immune cell function is further emphasized by the fact that blocking sodium channels with phenytoin reduces inflammatory cell infiltration in MS lesions35. Also, with safinamide and flecainide, two other sodium channel blockers, activation of microglia and macrophages is suppressed and symptoms are ameliorated in EAE36.
Figure 5. Overview of the pathological mechanisms in MS that might contribute to sodium MRI signal elevation in acute lesions.

The pathogenesis of MS involves a cascade of pathological events, including focal lymphocytic infiltration, microglia and macrophage activation, demyelination, and axonal degeneration2. Nav1.6 is the predominant sodium channel expressed in microglia and macrophages, and is up regulated upon activation of these cells in EAE and acute MS lesions35,36. Activated microglia and macrophages produce reactive-oxygen species4 and nitric oxide3 and thus induce mitochondrial dysfunction in neurons37,38 with subsequently reduced energy supply. Concurrently, re-distribution of Nav1.6 along demyelinated axons in EAE31 and acute MS lesions11 increases energy demand. The mismatch between reduced energy supply and elevated demand creates a virtual hypoxia39, which also impairs the sodium-potassium pump9, and leads to an intracellular accumulation of sodium ions12. This ionic imbalance promotes the sodium-calcium exchanger to operate in reverse13. Consecutive calcium import leads to injurious intracellular calcium levels with deleterious sequelae to the neuron9. Mechanisms of MS associated with intracellular sodium accumulation that possibly contribute to elevated average and fluid-attenuated sodium MRI signals are marked in blue. Also, hyper-cellularity itself might cause an increased fluid-attenuated signal. In fact, it may well be that both, intracellular sodium accumulation and hyper-cellularity contribute to the increased sodium signals observed in acute MS lesions as both are part of the same pathophysiologic mechanism.
Theoretically, longitudinal relaxation times of sodium ions within the cell resemble those in hyper-cellular environment. Therefore, increased fluid-attenuated sodium signals of acute MS lesions might as well originate, at least in part, from hyper-cellularity related to inflammation; that is to accumulating immune cells like activated microglia or infiltrated macrophages. In fact, it may well be that both, intracellular sodium accumulation and hyper-cellularity contribute to the increased sodium signals observed in acute MS lesions as both are part of the same pathophysiologic mechanism.
Inglese and colleagues15 and Paling and colleagues16 showed that the average tissue sodium signal is elevated in MS lesions compared to normal-appearing white matter, and increased in normal-appearing white matter of MS patients compared to healthy controls. In addition to the average tissue sodium signal, in the study presented here we acquired a fluid-attenuated sodium signal. Correlation analyses revealed an association between fluid-attenuated sodium and T1 CE signals, but not between average tissue sodium and T1 CE signals; the average tissue sodium signal strongly correlated with the T2-TSE and native T1 signals whereas the fluid-attenuated sodium signal did not. In addition, there was no correlation between average tissue and fluid-attenuated sodium signals – facts that underline the additional information of the fluid-attenuated sodium signal. The fluid-attenuated signal provides new biological information on MS lesions. Thereby, it allows for a more detailed interpretation of the average tissue sodium signal increase previously reported in MS lesions, which might be owed to increased extracellular space by neuroaxonal loss41 or vasogenic oedema42, or to intra-axonal sodium accumulation12.
In a longitudinal experiment, we monitored the sodium signals before and after administration of high-dose methylprednisolone and found a significant decrease in average tissue and fluid-attenuated sodium signal of acute lesions following anti-inflammatory pharmacologic intervention (Figs 3 and 4). Thereby, the longitudinal findings support the cross-sectional results of this study. They indicate that sodium signal differences between acute and chronic MS lesions are indeed attributable to inflammatory mechanisms. Moreover, results are in agreement with the cascade of those pathobiological events in MS that are associated with intracellular sodium increase, and eventually lead to lesion formation.
The spatial resolution of sodium MRI is limited, and therefore partial volume effects may have lowered sensitivity in detecting signal changes especially in very small inflammatory lesions. Still, statistical analyses indicate sodium signal changes large enough to predict lesion type. In future, technical advances like iterative image reconstruction techniques and multi-channel array coils will improve the spatial resolution, and, thereby facilitate the analysis of small lesions.
In this study, MS lesions were identified by their T2 signal; they were then defined as acute or chronic depending on the presence and absence of contrast enhancement in T1-weighted images, respectively. Although this approach is gold standard in diagnostic imaging of MS to date it most probably introduces uncertainty into sodium MRI based lesion type prediction because lesion definition relies on T2 signal as well as contrast enhancement, and both are principally unspecific14,43; that is, for example, T2-hyperintense lesions may represent acute inflammation although no contrast enhancement is detectable44. Despite this possible unexplained variance in sodium MRI data, again, significant sodium signal differences between acute and chronic MS lesions could be detected in the study presented here. Invasive studies would be necessary to confirm whether sodium imaging is able to classify lesions not classified by proton MRI.
To our knowledge, this is the first in vivo study that discloses significant sodium signal differences between acute and chronic MS lesions of the brain. The fluid-attenuated sodium signal reveals new biological information on lesion evolution, which is attributable to inflammatory mechanisms and compatible with those pathogenetic events of MS leading to intracellular sodium accumulation, hyper-cellularity and neurodegeneration. If confirmed in other studies, the average tissue and the fluid-attenuated sodium signal could be considered as neuroimaging biomarkers for diagnostic assessment and readout parameter for therapy monitoring, and complement clinical outcome measures. Together with the excellent spatial tissue characterization of proton MRI, the biological tissue information by sodium MRI might promote the endeavors to enhance imaging specificity for defining the pathological substrates of MS in vivo – a long-term goal as suggested by experts in the field of neurology3,14,45. In addition, the combination of proton (lesion identification) and sodium (lesion differentiation) MRI appears to be a potential alternative to contrast media administration in the assessment of MS; avoiding gadolinium-based contrast agents is advocated with respect to unwanted gadolinium accumulation in brain tissue46,47,48,49. This is especially relevant for MS patients as they rely on continuous follow-up MRI including gadolinium administration.
Additional Information
How to cite this article: Biller, A. et al. Sodium MRI in Multiple Sclerosis is Compatible with Intracellular Sodium Accumulation and Inflammation-Induced Hyper-Cellularity of Acute Brain Lesions.. Sci. Rep. 6, 31269; doi: 10.1038/srep31269 (2016).
Acknowledgments
This work was funded in part by the Helmholtz Alliance ICEMED (Imaging and Curing Environmental Metabolic Diseases) (A.M.N.).
Footnotes
Author Contributions A.B. was responsible for concept and design. A.B., I.P., J.J., N.B. and A.M.N. acquired the data. A.B. and S.B. were responsible for data analysis. A.B., R.D., B.W. and J.K. interpreted the data. A.B. wrote the paper. All authors reviewed and approved the paper.
References
- Lassmann H., Bruck W. & Lucchinetti C. F. The immunopathology of multiple sclerosis: an overview. Brain pathology (Zurich, Switzerland) 17, 210–218, doi: 10.1111/j.1750-3639.2007.00064.x (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compston A. & Coles A. Multiple sclerosis. Lancet 372, 1502–1517, doi: 10.1016/s0140-6736(08)61620-7 (2008). [DOI] [PubMed] [Google Scholar]
- Ciccarelli O. et al. Pathogenesis of multiple sclerosis: insights from molecular and metabolic imaging. The Lancet. Neurology 13, 807–822, doi: 10.1016/s1474-4422(14)70101-2 (2014). [DOI] [PubMed] [Google Scholar]
- Frischer J. M. et al. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain: a journal of neurology 132, 1175–1189, doi: 10.1093/brain/awp070 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahad D., Ziabreva I., Lassmann H. & Turnbull D. Mitochondrial defects in acute multiple sclerosis lesions. Brain: a journal of neurology 131, 1722–1735, doi: 10.1093/brain/awn105 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahad D. J. et al. Mitochondrial changes within axons in multiple sclerosis. Brain: a journal of neurology 132, 1161–1174, doi: 10.1093/brain/awp046 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassmann H., van Horssen J. & Mahad D. Progressive multiple sclerosis: pathology and pathogenesis. Nature reviews. Neurology 8, 647–656, doi: 10.1038/nrneurol.2012.168 (2012). [DOI] [PubMed] [Google Scholar]
- Smith K. J. Sodium channels and multiple sclerosis: roles in symptom production, damage and therapy. Brain pathology (Zurich, Switzerland) 17, 230–242, doi: 10.1111/j.1750-3639.2007.00066.x (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman S. G. Mechanisms of disease: sodium channels and neuroprotection in multiple sclerosis-current status. Nature clinical practice. Neurology 4, 159–169, doi: 10.1038/ncpneuro0735 (2008). [DOI] [PubMed] [Google Scholar]
- Black J. A., Newcombe J., Trapp B. D. & Waxman S. G. Sodium channel expression within chronic multiple sclerosis plaques. Journal of neuropathology and experimental neurology 66, 828–837, doi: 10.1097/nen.0b013e3181462841 (2007). [DOI] [PubMed] [Google Scholar]
- Craner M. J. et al. Molecular changes in neurons in multiple sclerosis: Altered axonal expression of Nav1.2 and Nav1.6 sodium channels and Na+/Ca2+ exchanger. Proceedings of the National Academy of Sciences of the United States of America 101, 8168–8173, doi: 10.1073/pnas.0402765101 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp B. D. & Stys P. K. Virtual hypoxia and chronic necrosis of demyelinated axons in multiple sclerosis. The Lancet. Neurology 8, 280–291, doi: 10.1016/s1474-4422(09)70043-2 (2009). [DOI] [PubMed] [Google Scholar]
- Stys P. K., Waxman S. G. & Ransom B. R. Ionic mechanisms of anoxic injury in mammalian CNS white matter: role of Na+ channels and Na(+)-Ca2+ exchanger. The Journal of neuroscience: the official journal of the Society for Neuroscience 12, 430–439 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippi M. et al. Association between pathological and MRI findings in multiple sclerosis. The Lancet Neurology 11, 349–360, doi: http://dx.doi.org/10.1016/S1474-4422 (12)70003-0 (2012). [DOI] [PubMed] [Google Scholar]
- Inglese M. et al. Brain tissue sodium concentration in multiple sclerosis: a sodium imaging study at 3 tesla. Brain: a journal of neurology 133, 847–857, doi: 10.1093/brain/awp334 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paling D. et al. Sodium accumulation is associated with disability and a progressive course in multiple sclerosis. Brain: a journal of neurology 136, 2305–2317, doi: 10.1093/brain/awt149 (2013). [DOI] [PubMed] [Google Scholar]
- Nagel A. M. et al. 3 Tesla sodium inversion recovery magnetic resonance imaging allows for improved visualization of intracellular sodium content changes in muscular channelopathies. Investigative radiology 46, 759–766, doi: 10.1097/RLI.0b013e31822836f6 (2011). [DOI] [PubMed] [Google Scholar]
- Stobbe R. & Beaulieu C. In vivo sodium magnetic resonance imaging of the human brain using soft inversion recovery fluid attenuation. Magn Reson Med 54, 1305–1310, doi: 10.1002/mrm.20696 (2005). [DOI] [PubMed] [Google Scholar]
- Winter P. M. & Bansal N. TmDOTP(5-) as a (23)Na shift reagent for the subcutaneously implanted 9L gliosarcoma in rats. Magn Reson Med 45, 436–442 (2001). [DOI] [PubMed] [Google Scholar]
- Madelin G., Kline R., Walvick R. & Regatte R. R. A method for estimating intracellular sodium concentration and extracellular volume fraction in brain in vivo using sodium magnetic resonance imaging. Sci Rep 4, 4763, doi: 10.1038/srep04763 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel A. M. et al. The potential of relaxation-weighted sodium magnetic resonance imaging as demonstrated on brain tumors. Investigative radiology 46, 539–547, doi: 10.1097/RLI.0b013e31821ae918 (2011). [DOI] [PubMed] [Google Scholar]
- Biller A. et al. Improved Brain Tumor Classification by Sodium MR Imaging: Prediction of IDH Mutation Status and Tumor Progression. AJNR Am J Neuroradiol 37, 66–73, doi: 10.3174/ajnr.A4493 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maarouf A. et al. Topography of brain sodium accumulation in progressive multiple sclerosis. Magma (New York, N.Y.), doi: 10.1007/s10334-013-0396-1 (2013). [DOI] [PubMed] [Google Scholar]
- Zaaraoui W. et al. Distribution of brain sodium accumulation correlates with disability in multiple sclerosis: a cross-sectional 23Na MR imaging study. Radiology 264, 859–867, doi: 10.1148/radiol.12112680 (2012). [DOI] [PubMed] [Google Scholar]
- Polman C. H. et al. Diagnostic criteria for multiple sclerosis: 2005 revisions to the “McDonald Criteria”. Annals of neurology 58, 840–846, doi: 10.1002/ana.20703 (2005). [DOI] [PubMed] [Google Scholar]
- Lublin F. D. & Reingold S. C. Defining the clinical course of multiple sclerosis: results of an international survey. National Multiple Sclerosis Society (USA) Advisory Committee on Clinical Trials of New Agents in Multiple Sclerosis. Neurology 46, 907–911 (1996). [DOI] [PubMed] [Google Scholar]
- Nagel A. M. et al. Sodium MRI using a density-adapted 3D radial acquisition technique. Magn Reson Med 62, 1565–1573, doi: 10.1002/mrm.22157 (2009). [DOI] [PubMed] [Google Scholar]
- Smith S. M. Fast robust automated brain extraction. Human brain mapping 17, 143–155, doi: 10.1002/hbm.10062 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkinson M. & Smith S. A global optimisation method for robust affine registration of brain images. Med Image Anal 5, 143–156 (2001). [DOI] [PubMed] [Google Scholar]
- Bates D., Maechler M., Bolker B. M. & Walker S. C. Fitting Linear Mixed-Effects Models using lme4. J Stat Softw (2015). [Google Scholar]
- Craner M. J., Hains B. C., Lo A. C., Black J. A. & Waxman S. G. Co-localization of sodium channel Nav1.6 and the sodium-calcium exchanger at sites of axonal injury in the spinal cord in EAE. Brain: a journal of neurology 127, 294–303, doi: 10.1093/brain/awh032 (2004). [DOI] [PubMed] [Google Scholar]
- Petracca M. et al. Brain intra- and extracellular sodium concentration in multiple sclerosis: a 7 T MRI study. Brain: a journal of neurology, doi: 10.1093/brain/awv386 (2016). [DOI] [PubMed] [Google Scholar]
- Black J. A. & Waxman S. G. Sodium channels and microglial function. Experimental neurology 234, 302–315, doi: 10.1016/j.expneurol.2011.09.030 (2012). [DOI] [PubMed] [Google Scholar]
- Ellwardt E. & Zipp F. Molecular mechanisms linking neuroinflammation and neurodegeneration in MS. Experimental neurology 262PA, 8–17, doi: 10.1016/j.expneurol.2014.02.006 (2014). [DOI] [PubMed] [Google Scholar]
- Craner M. J. et al. Sodium channels contribute to microglia/macrophage activation and function in EAE and MS. Glia 49, 220–229, doi: 10.1002/glia.20112 (2005). [DOI] [PubMed] [Google Scholar]
- Morsali D. et al. Safinamide and flecainide protect axons and reduce microglial activation in models of multiple sclerosis. Brain: a journal of neurology 136, 1067–1082, doi: 10.1093/brain/awt041 (2013). [DOI] [PubMed] [Google Scholar]
- Bolanos J. P. et al. Nitric oxide-mediated mitochondrial damage in the brain: mechanisms and implications for neurodegenerative diseases. J Neurochem 68, 2227–2240 (1997). [DOI] [PubMed] [Google Scholar]
- Zielasek J. et al. Inhibition of brain macrophage/microglial respiratory chain enzyme activity in experimental autoimmune encephalomyelitis of the Lewis rat. Neurosci Lett 184, 129–132 (1995). [DOI] [PubMed] [Google Scholar]
- Stys P. K. Axonal degeneration in multiple sclerosis: is it time for neuroprotective strategies? Annals of neurology 55, 601–603, doi: 10.1002/ana.20082 (2004). [DOI] [PubMed] [Google Scholar]
- Wetterling F. et al. Sodium-23 magnetic resonance imaging has potential for improving penumbra detection but not for estimating stroke onset time. J Cereb Blood Flow Metab 35, 103–110, doi: 10.1038/jcbfm.2014.174 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perier O. & Gregoire A. Electron microscopic features of multiple sclerosis lesions. Brain: a journal of neurology 88, 937–952 (1965). [DOI] [PubMed] [Google Scholar]
- Bruck W. et al. Inflammatory central nervous system demyelination: correlation of magnetic resonance imaging findings with lesion pathology. Annals of neurology 42, 783–793, doi: 10.1002/ana.410420515 (1997). [DOI] [PubMed] [Google Scholar]
- Simon J. H. MRI outcomes in the diagnosis and disease course of multiple sclerosis. Handbook of clinical neurology 122, 405–425, doi: 10.1016/b978-0-444-52001-2.00017-0 (2014). [DOI] [PubMed] [Google Scholar]
- Hochmeister S. et al. Dysferlin is a new marker for leaky brain blood vessels in multiple sclerosis. Journal of neuropathology and experimental neurology 65, 855–865, doi: 10.1097/01.jnen.0000235119.52311.16 (2006). [DOI] [PubMed] [Google Scholar]
- Filippi M. et al. Ultra-high-field MR imaging in multiple sclerosis. Journal of neurology, neurosurgery, and psychiatry 85, 60–66, doi: 10.1136/jnnp-2013-305246 (2014). [DOI] [PubMed] [Google Scholar]
- Kanda T. et al. Gadolinium-based Contrast Agent Accumulates in the Brain Even in Subjects without Severe Renal Dysfunction: Evaluation of Autopsy Brain Specimens with Inductively Coupled Plasma Mass Spectroscopy. Radiology 276, 228–232, doi: 10.1148/radiol.2015142690 (2015). [DOI] [PubMed] [Google Scholar]
- Kanda T. et al. High Signal Intensity in Dentate Nucleus on Unenhanced T1-weighted MR Images: Association with Linear versus Macrocyclic Gadolinium Chelate Administration. Radiology 275, 803–809, doi: 10.1148/radiol.14140364 (2015). [DOI] [PubMed] [Google Scholar]
- Kanda T., Ishii K., Kawaguchi H., Kitajima K. & Takenaka D. High Signal Intensity in the Dentate Nucleus and Globus Pallidus on Unenhanced T1-weighted MR Images: Relationship with Increasing Cumulative Dose of a Gadolinium-based Contrast Material. Radiology 270, 834–841, doi: doi: 10.1148/radiol.13131669 (2014). [DOI] [PubMed] [Google Scholar]
- Errante Y. et al. Progressive increase of T1 signal intensity of the dentate nucleus on unenhanced magnetic resonance images is associated with cumulative doses of intravenously administered gadodiamide in patients with normal renal function, suggesting dechelation. Investigative radiology 49, 685–690, doi: 10.1097/rli.0000000000000072 (2014). [DOI] [PubMed] [Google Scholar]