

Abstract
Purpose
BCL2 overexpression is a hallmark of chronic lymphocytic leukemia (CLL). The novel BH3 mimetic navitoclax (ABT-263) specifically inhibits BCL2 and related proteins BCL-xl and BCL-w, potently inducing apoptosis of CLL cells in vitro. A phase I trial in patients with CLL was conducted to evaluate the safety, pharmacokinetics, and biologic activity of oral navitoclax.
Patients and Methods
Twenty-nine patients with relapsed or refractory CLL received daily navitoclax for 14 days (10, 110, 200, or 250 mg/d; n = 15) or 21 days (125, 200, 250, or 300 mg/d; n = 14) of each 21-day cycle. Dose escalation decisions were informed by continual reassessment methodology.
Results
Lymphocytosis was reduced by more than 50% in 19 of 21 patients with baseline lymphocytosis. Among 26 patients treated with navitoclax ≥ 110 mg/d, nine (35%) achieved a partial response and seven maintained stable disease for more than 6 months. Median treatment duration was 7 months (range, 1 to ≥ 29 months). Median progression-free survival was 25 months. Activity was observed in patients with fludarabine-refractory disease, bulky adenopathy, and del(17p) CLL. Thrombocytopenia due to BCL-xl inhibition was the major dose-limiting toxicity and was dose-related. Low MCL1 expression and high BIM:MCL1 or BIM:BCL2 ratios in leukemic cells correlated with response. We determined that the navitoclax dose of 250 mg/d in a continuous dosing schedule was optimal for phase II studies.
Conclusion
BCL2 is a valid therapeutic target in CLL, and its inhibition by navitoclax warrants further evaluation as monotherapy and in combination in this disease.
INTRODUCTION
Chronic lymphocytic leukemia (CLL) is characterized by high-level expression of BCL2 in all patients, and the accumulation of mature leukemic lymphocytes is proposed to be a direct consequence of the antiapoptotic effect of BCL2. As such, BCL2 has long been considered a high priority therapeutic target in this disease, but previous attempts to target BCL2 clinically with oblimersen and obataclax have not demonstrated major antileukemic activity.1,2
The closely related compounds navitoclax and ABT-737 are BH3 mimetics, a new class of anticancer therapeutics that depend on inhibition of BCL2 and related antiapoptotic intracellular proteins for their anticancer activity.3–5 Such targeted compounds were developed because of the critical role BCL2 is recognized to play in tumorogenesis6 and chemotherapy resistance7–10 when highly expressed. Both bind BCL2, BCL-xl, and BCL-w, relieving repression of BAX and BAK, which then oligomerize on the mitochondrial outer membrane and trigger apoptosis.3–5 Although much of the early preclinical data were generated by studying ABT-737, an analog with superior oral bioavailability was required for clinical application so navitoclax was developed. These drugs3–5,11 display absolute dependence on the mitochondrial apoptotic pathway for cell killing, show significant single-agent activity against cell lines expressing high levels of BCL2 or BCL-xl,3,12,13 and synergize with chemotherapeutic drugs in preclinical in vivo models of lymphoproliferative diseases.5,14,15
In vitro, primary CLL cells show marked sensitivity to ABT-7373,16,17 and navitoclax,18 including highly chemoresistant CLL cells and cells from patients with del(17p) and del(11q) CLL.19 The modality of death is apoptosis, and this is observed within 4 hours of exposure.17 Given this compelling biologic rationale, evaluation of navitoclax in patients with relapsed or refractory CLL was planned as one of three initial concurrent phase I studies.
Preclinical toxicology studies indicated that the anticipated human toxicities for navitoclax reflected the known essential nonredundant functions of the target proteins as defined by deficiency of BCL-xl,20 BCL2,21 and BCL-w22 in mice (thrombocytopenia, lymphopenia, and impaired spermatogenesis, respectively). Pharmacologic inhibition of BCL-xl in mice20 or dogs23 causes an acute, dose-proportional reduction in platelet count within 1 to 2 days via induction of apoptosis24 and subsequent clearance of platelets from the circulation.20 Megakaryocytes and reticulated platelets are less susceptible, and thus compensatory increased megakaryopoiesis and partial correction of the thrombocytopenia are observed in preclinical models during ongoing dosing.23
The objectives of this study were to define the safety profile, dose-limiting toxicity (DLT), maximum-tolerated dose (MTD), pharmacokinetics, and preliminary efficacy of navitoclax and to determine a recommended dose and schedule for phase II assessment in patients with relapsed or refractory CLL.
PATIENTS AND METHODS
Study Design
Study M06-873 (ClinicalTrials.gov Identifier: NCT00481091) was designed as an open-label, multicenter, dose-escalation phase I/IIA study. Phase I enrolled patients between July 2007 and June 2009 with data cutoff of July 16, 2010, and is the subject of this report.
The study was conducted according to the Declaration of Helsinki and relevant International Conference on Harmonization Good Clinical Practice guidelines and with approval from the local institutional review board, independent ethics committee, or research ethics board of all participating study sites. All participants provided written informed consent before participating in this study.
Patient Eligibility
Patients with relapsed or refractory CLL requiring treatment were considered eligible if they met all of the inclusion criteria and none of the exclusion criteria (Table 1). Patients could not have received any anti-CLL therapy within 14 days (30 days for monoclonal antibody therapy) before the first dose of navitoclax.
Table 1.
Entry Criteria
| Inclusion |
| Informed consent |
| Age > 18 years |
| ECOG performance status of 0 or 1 |
| Adequate hematology |
| Neutrophil count ≥ 1.0 × 109/L |
| Platelet count ≥ 75 × 109/L |
| Hemoglobin concentration ≥ 90 g/L |
| Serum creatinine ≤ 2.0 mg/dL or calculated creatinine clearance ≥ 50 mL/min |
| Adequate liver function |
| AST, ALT ≤ 3 × ULN |
| Bilirubin ≤ 1.5 × ULN |
| Normal coagulation profile |
| Exclusion |
| History of immune thrombocytopenia or refractoriness to platelet transfusion in the last 12 months |
| Ongoing use of an antiplatelet agent or anticoagulant therapy |
| Current bleeding or history of active peptic ulcer disease or erosive gastritis/esophagitis |
| History of significant cardiovascular disease (eg, myocardial infarction, thrombotic event, or thromboembolic event in the last 6 months) |
| Active infection |
| Current pregnancy or breastfeeding |
| Previous stem-cell transplantation |
NOTE. All patients had an original diagnosis of chronic lymphocytic leukemia, but lymphocytosis was not required at study entry.
Abbreviation: ECOG, Eastern Cooperative Oncology Group; ULN, upper limit of normal.
Treatment
Patients were assigned sequentially to dose-escalation cohorts of at least three. Initially, navitoclax was administered orally, once daily, for 14 days followed by 7 days off drug in each 21-day cycle. Subsequently, patients received a continuous, once daily, dosing schedule after a 7-day lead-in course of navitoclax 100 mg daily. Patients continued treatment in 21-day cycles as long as there was clinical benefit in the absence of significant toxicity. Dose reductions were allowed for toxicity. Patients received prophylaxis against tumor lysis syndrome and also received supportive care per local practice.
Dose Escalation
A continual reassessment method (CRM) was used to efficiently obtain an estimate of the MTD (defined as the dose at which 30% of patients experience a DLT) and to determine the recommended phase II dose.25,26 Once three patients in a dose cohort completed cycle 1, all available data were entered into a responder model of dose and probability of DLT generated by using preclinical and accumulating clinical data. The navitoclax dose for the next cohort was adjusted to the CRM-estimated MTD, provided that dose escalation did not exceed either an increase of more than 100 mg or a 40% increase from the just completed dose level. Dose escalation ceased when the model-estimated MTD was within 10% of the just-completed dose cohort. At this point, the model was considered to have converged and the trial-determined MTD was declared.
Study Assessments
Safety assessments included history, physical examination, vital signs, ECG, 2D echocardiogram, blood chemistry, hematology, and urinalysis. Safety assessments were performed on screening, day 1 of lead-in, day 1 of cycle 1, weekly through cycle 2, day 1 of each subsequent cycle, and at the end of treatment. Platelet counts were intensively monitored at multiple time points throughout the study.
Adverse events (AEs) were graded according to the National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events (CTCAE), Version 3.0.27 AEs that were judged possibly or probably related to navitoclax were considered to be DLTs if they satisfied any of the following criteria: grade 4 thrombocytopenia, grade ≥ 2 bleeding associated with thrombocytopenia, or all other grade ≥ 3 AEs with certain protocol-defined exceptions (grade 3 laboratory tumor lysis syndrome without clinical manifestations; grade 3 or 4 leukopenia, lymphopenia, or afebrile neutropenia; alopecia; and grade 3 nausea, vomiting, and/or diarrhea unless unresponsive to treatment). Each DLT required interruption and possible discontinuation of navitoclax therapy; navitoclax could be reintroduced at a reduced dose providing toxicity resolved to grade ≤ 1 or baseline if grade 2 toxicity was present at study entry. Following a DLT of thrombocytopenia, navitoclax could be reintroduced at a reduced dose if the platelet count returned to ≥ 50 × 109/L (grade ≤ 2 toxicity).
Efficacy
Exploratory efficacy end points included progression-free survival (PFS), overall response rate (ORR), time to disease progression, overall survival, and duration of overall response. Objective response was evaluated by using the 1996 NCI Working Group response criteria for CLL28 at the end of cycles 2 and 4, the end of every third cycle thereafter, and at the final visit. In addition, changes in lymph nodes and spleen were evaluated by serial computed tomography scanning,29 and individual measurements were recorded. Information on pharmacokinetics and biologic studies is included in the Data Supplement.
Statistical Analysis
Pharmacokinetic parameters were determined by using a noncompartmental approach. The method of Kaplan and Meier was used for time to event analyses. Comparisons and correlations between groups were performed by using GraphPad Prism (GraphPad Software, La Jolla, CA). The specific tests used are stated in the relevant figure legends.
Fig 1.

Patient status on study by dose and response. For the intermittent schedule cohort (A) and the continuous schedule cohort (B), the upper horizontal bar graphs indicate the time on study for each patient and the lower tables present a summary of dose-limiting toxicities (DLTs) and dose modifications. For the time on study graph, patients are grouped according to their observed best response, and the color of the bar reflects the navitoclax dose cohort they entered. For patients achieving a partial response (PR), the time on study to reach PR is represented by the lighter blue segment of the bar. When a patient discontinued the study, the reason is indicated on the right end of the colored bar, and for patients still receiving navitoclax, their duration on study in months is similarly indicated. AE, adverse event; De, death; ID, investigator discretion; PD, progressive disease; WC, withdrew consent.
RESULTS
Patient Characteristics
Twenty-nine patients with a median age of 67 years (range, 50 to 79 years) were enrolled. Their key clinical characteristics are summarized in Table 2. Twenty patients had at least one of the following adverse prognostic features: bulky lymphadenopathy, fludarabine resistance, or deletion of 17p13.2 or 11q22.3 by fluorescent in situ hybridization.
Table 2.
Patient Characteristics
| Characteristic | No./Total | % |
|---|---|---|
| Age, years | ||
| Median | 67 | |
| Range | 50-79 | |
| Sex | ||
| Male | 19/29 | 66 |
| Female | 10/29 | 34 |
| Lymphocyte count × 109/L | ||
| Median | 15.5 | |
| Range | 0.8-284.3 | |
| Splenomegaly | 13/29 | 45 |
| Bulky adenopathy | 12/29 | 41 |
| No. prior therapies | ||
| Median | 4.5 | |
| Range | 1-11 | |
| Fludarabine refractory | 9/29 | 31 |
| Unfavorable FISH* | ||
| 17p13.2 del | 11/25 | 44 |
| 11q22.3 del | 7/25 | 28 |
Abbreviation: FISH, fluorescent in situ hybridization.
Reported per Döhner et al30; not all patients were evaluated.
Patient Flow
The first 15 patients were enrolled in the intermittent 14-/21-day dosing schedule (Fig 1A). Two patients remained on study and 13 discontinued because of disease progression (n = 5), AEs (n = 5), withdrawal of consent (n = 2), or death (n = 1). Five patients required dose reductions, predominantly because of severe thrombocytopenia in cycles 1 or 2. DLTs were observed in three patients in cycle 1: grade 3 tumor lysis syndrome (110 mg) and grade 4 thrombocytopenia (110 mg, 250 mg). All resolved with dose interruption (Data Supplement). By using the CRM methodology, the MTD was estimated to be 200 mg/d with intermittent dosing.
With the aim of avoiding dose-limiting thrombocytopenia, the protocol was amended to evaluate escalating doses on a continuous schedule following a 7-day 100 mg/d lead-in phase. Commencing at 125 mg/d (a dose level approximating the overall exposure of the 200 mg/d for the 14-/21-day regimen), 14 patients were enrolled in the 21-/21-day schedule (Fig 1B), and five remain on study. Reasons for study discontinuation included disease progression (n = 4), AEs (n = 3), withdrawal of consent (n = 1), and investigator discretion (n = 1). Nine patients required dose reductions. Three patients experienced DLTs in cycle 1: grade 4 thrombocytopenia (200 mg, 300 mg) and grade 2 nausea (250 mg). The MTD in the 21-/21-day schedule was estimated to be 250 mg/d, with thrombocytopenia being the DLT.
Navitoclax-Induced Thrombocytopenia
On the 14-/21-day schedule, acute reductions in platelet counts were observed in all patients receiving navitoclax at more than 10 mg/d. Platelet nadirs were transient, typically occurring on days 2 to 5, followed by partial recovery during continued dosing and full recovery during the 7 days off drug. Re-exposure resulted in repetition of this cycle (Fig 2A). In cycle 1, the nadirs (mean ± standard deviation) were related to dose: 10 mg, 111 ± 12 × 109/L; 110 mg, 67 ± 51 ×109/L; 200 mg, 46 ± 9 ×109/L; 250 mg, 26 ± 5 ×109/L. Lead-in dosing of 100 mg/d for 7 days followed by dose escalation reduced the depth of the nadir and therefore the occurrence of grade 4 thrombocytopenia during the first cycle (Fig 2A), allowing escalation to higher doses than achievable on the 14-/21-day schedule. The mean respective nadirs for 125 mg, 200 mg, 250 mg, and 300 mg doses were 79 ± 24, 79 ± 65, 58 ± 5, and 40 ± 13 ×109/L, respectively. Continuous dosing also minimized cyclic variability observed with intermittent dosing. Despite these adjustments, severe thrombocytopenia requiring dose reduction was also observed during later cycles in four of seven patients in the 300 mg and 250 mg continuous dosing cohorts. Grade 4 thrombocytopenia resolved to more than 50 ×109/L within 1 to 2 weeks of dose interruption, thus allowing resumption of navitoclax at a lower dose. Thrombocytopenia was not associated with any clinically significant bleeding during this study.
Fig 2.

Navitoclax reduces platelet counts and chronic lymphocytic leukemia (CLL) burden in peripheral blood (PB) and lymph nodes. (A) Navitoclax scheduling modulates the severity of thrombocytopenia. The platelet counts of patients receiving 250 mg navitoclax daily in either the 14-days-on/7-days-off intermittent dosing cohort or the continuous dosing cohort incorporating a 7-day 100-mg lead-in phase are plotted for the first 50 days. The dotted line represents a platelet count of 25 × 109/L, denoting the grade 4 toxicity level. One patient in the continuous cohort interrupted dosing prematurely for nonthrombocytopenic toxicity. (B) Navitoclax pharmacokinetics: The mean ± standard deviation plasma concentrations of navitoclax are plotted for the hours following initial dosing on day 1 and on day 14 for each dose cohort on the intermittent dosing schedule. (*) On day 14, n = 3 for 110 mg, n = 4 for 200 mg, and n = 2 for 250 mg. (C) Waterfall plot of most favorable changes in PB lymphocytosis. Only patients with lymphocytosis (> 5 × 109/L) at study entry are included. (D) Rapid reduction in PB lymphocytosis. The PB lymphocyte counts (× 109/L) during cycles 1 to 3 for individual patients enrolled in the 250-mg dose cohorts on either the intermittent or continuous schedules are presented. (E) Navitoclax induces apoptosis of CLL cells in vivo. Representative immunoblots of lysates of PB CLL cells from a patient from each of the 10-mg, 200-mg (intermittent), and 300-mg (continuous) dosing cohorts. For each patient, lysates from screening (Scr; pre-exposure), day 14 of cycle 1 (C1), and day 1 of cycle 3 (C3) were electrophoresed, before membranes were probed for caspase 3, cleaved caspase 3, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH). In the 200-mg and 300-mg examples, but not the 10-mg example, the presence of cleaved caspase 3 in the cycle 1 and cycle 3 lanes reflects activation of apoptosis during exposure to navitoclax. GAPDH is reduced in these particular samples because it is known to be degraded during apoptosis. Cleaved caspase 3 was detected in six of nine patients analyzed after receiving ≥ 110 mg/d of navitoclax. (F) Waterfall plot of most favorable change in lymphadenopathy. Asterisks identify patients with bulky lymphadenopathy (> 5 cm diameter masses) at study entry. (G) Representative images from abdominal computed tomography scans of a patient with fludarabine-refractory del(17p) CLL at study entry (left) and after seven cycles of navitoclax 200 mg/d (right). Bulky mesenteric adenopathy has almost completely resolved. A benign left renal cyst has not altered over time. This response has been maintained for more than 2 years.
Safety
Table 3 lists all AEs observed in 10% or more of the patients. The most common emergent AEs were diarrhea, nausea, vomiting, fatigue, thrombocytopenia, and neutropenia. The GI events were mild and were considered likely to be related to navitoclax. Serious AEs other than grade 4 thrombocytopenia considered potentially related to navitoclax included tumor lysis syndrome (n = 1; related), neutropenia (n = 8; related or probably related), polymerase chain reaction–confirmed progressive multifocal leukoencephalopathy (n = 1 heavily pretreated patient; possibly related), and myocardial infarction (n = 1; possibly related). Grade 4 neutropenia was observed both early (cycles 1 to 5) and late (cycles 7 to 14) and was reversible with either dose reduction or administration of granulocyte colony-stimulating factor. Three of four patients in the 300-mg cohort experienced grade 4 neutropenia within the first four cycles.
Table 3.
All Adverse Events Observed in ≥ 10% of Patients
| Adverse Event | No. | % | Grade |
|||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | |||
| GI | ||||||
| Diarrhea | 22 | 76 | 15 | 7 | 0 | 0 |
| Nausea | 17 | 59 | 11 | 6 | 0 | 0 |
| Vomiting | 9 | 31 | 8 | 0 | 1 | 0 |
| Decreased appetite | 6 | 21 | 5 | 1 | 0 | 0 |
| Dyspepsia | 4 | 14 | 3 | 1 | 0 | 0 |
| Abdominal pain | 4 | 14 | 4 | 0 | 0 | 0 |
| General | ||||||
| Fatigue | 10 | 35 | 4 | 5 | 0 | 1 |
| Peripheral edema | 6 | 21 | 4 | 2 | 0 | 0 |
| Insomnia | 5 | 17 | 2 | 3 | 0 | 0 |
| Pyrexia | 4 | 14 | 2 | 2 | 0 | 0 |
| Dizziness | 4 | 14 | 2 | 2 | 0 | 0 |
| Headache | 4 | 14 | 4 | 0 | 0 | 0 |
| Hypertension | 3 | 10 | 1 | 2 | 0 | 0 |
| Blood | ||||||
| Thrombocytopenia | 8 | 28 | 0 | 0 | 3 | 5 |
| Neutropenia | 8 | 28 | 0 | 0 | 1 | 7 |
| Epistaxis | 5 | 17 | 4 | 1 | 0 | 0 |
| Contusion | 4 | 14 | 3 | 1 | 0 | 0 |
| Respiratory | ||||||
| Cough | 9 | 31 | 6 | 3 | 0 | 0 |
| Upper RTI | 7 | 24 | 2 | 5 | 0 | 0 |
| Sinusitis | 6 | 21 | 2 | 4 | 0 | 0 |
| Lower RTI | 4 | 14 | 0 | 1 | 3 | 0 |
| Pneumonia | 4 | 14 | 0 | 1 | 2 | 1 |
| Productive cough | 4 | 14 | 4 | 0 | 0 | 0 |
| Nasopharyngitis | 4 | 14 | 4 | 0 | 0 | 0 |
| Dyspnea | 3 | 13 | 2 | 1 | 0 | 0 |
| Pain | ||||||
| Arthralgia | 4 | 14 | 2 | 2 | 0 | 0 |
| Oropharyngeal | 3 | 10 | 2 | 1 | 0 | 0 |
| Extremity | 3 | 10 | 0 | 3 | 0 | 0 |
| Skin | ||||||
| Basal cell carcinoma | 4 | 14 | 0 | 3 | 1 | 0 |
| Skin lesion | 4 | 14 | 1 | 2 | 1 | 0 |
| Dry skin | 3 | 10 | 3 | 0 | 0 | 0 |
| Rash | 3 | 10 | 1 | 2 | 0 | 0 |
| Squamous cell carcinoma | 3 | 10 | 0 | 1 | 2 | 0 |
| Infections and other | ||||||
| Herpes simplex | 4 | 14 | 2 | 2 | 0 | 0 |
| Urinary tract | 4 | 14 | 0 | 4 | 0 | 0 |
| Viral | 4 | 14 | 4 | 0 | 0 | 0 |
| Influenza-like illness | 3 | 10 | 2 | 1 | 0 | 0 |
| Stomatitis | 3 | 10 | 1 | 2 | 0 | 0 |
Abbreviation: RTI, respiratory tract infection.
Pharmacokinetics
Twenty-eight patients were included in the pharmacokinetic analysis (Data Supplement). Peak concentrations were observed 6 to 8 hours postdose in both the 14-/21-day (Fig 2B) and 21-/21-day schedules. The interpatient variability in area under the curve was 46%. Navitoclax exposure was dose-proportional between 10 mg and 250 mg in the 14-/21-day cohort. Pharmacokinetic parameters were stable over multiple cycles. No consistent trend was observed in dose-normalized steady-state trough concentrations over time. Navitoclax exposure did not demonstrate any apparent correlation with age, body weight, body surface area, renal function, or total bilirubin levels (data not shown).
Preliminary Efficacy
Peripheral blood lymphocytosis.
Nineteen (90%) of 21 patients with peripheral blood lymphocytosis at study entry achieved at least a 50% reduction from baseline at maximum response (Fig 2C). No apparent relationship was observed between increasing doses of navitoclax over 110 mg/d and reduction in circulating lymphocytes (Data Supplement). Significant reductions in lymphocytosis occurred within days of commencing navitoclax (Fig 2D). These were associated with morphologic (Data Supplement) and biochemical changes of apoptosis in circulating CLL cells (Fig 2E), confirming the mechanism of action of navitoclax. The median number of cycles to maximum reduction was three (range, one to 24).
Splenomegaly and nodal disease.
In five of 13 patients with palpable splenomegaly at study entry, the splenomegaly resolved, and an additional five patients achieved partial reductions in splenic size; improvements were evident within three cycles in nine of these 10 responding patients. Nodal disease was reduced in 21 of 29 patients, with 11 achieving ≥ 50% reduction from baseline in tumor volume as assessed by computed tomography criteria29; 10 of these 11 patients had received ≥ 200 mg/d navitoclax (Fig 2F). The median time to nodal response was 50 days (range, 43 to 548 days).
Overall response.
Nine patients achieved a partial response (PR). Bone marrow biopsies were performed when the patient was considered to have a potential complete response, and residual diffuse CLL involvement was observed in all patients so investigated. The ORR was 31% (nine of 29 patients). Among the 26 patients who received doses of navitoclax sufficient to achieve sustained exposure of biologically active concentrations (≥ 110 mg/d; Fig 2B and Data Supplement), the ORR was 35%. Responses appeared durable, since seven patients had stable disease (SD) features for more than 12 months from commencement of therapy (Fig 1). SD was the best objective response in 18 patients and was sustained for at least 6 months in eight patients (44%). The median PFS and the median time to disease progression were both 25 months (Fig 3A; data not shown).
Fig 3.
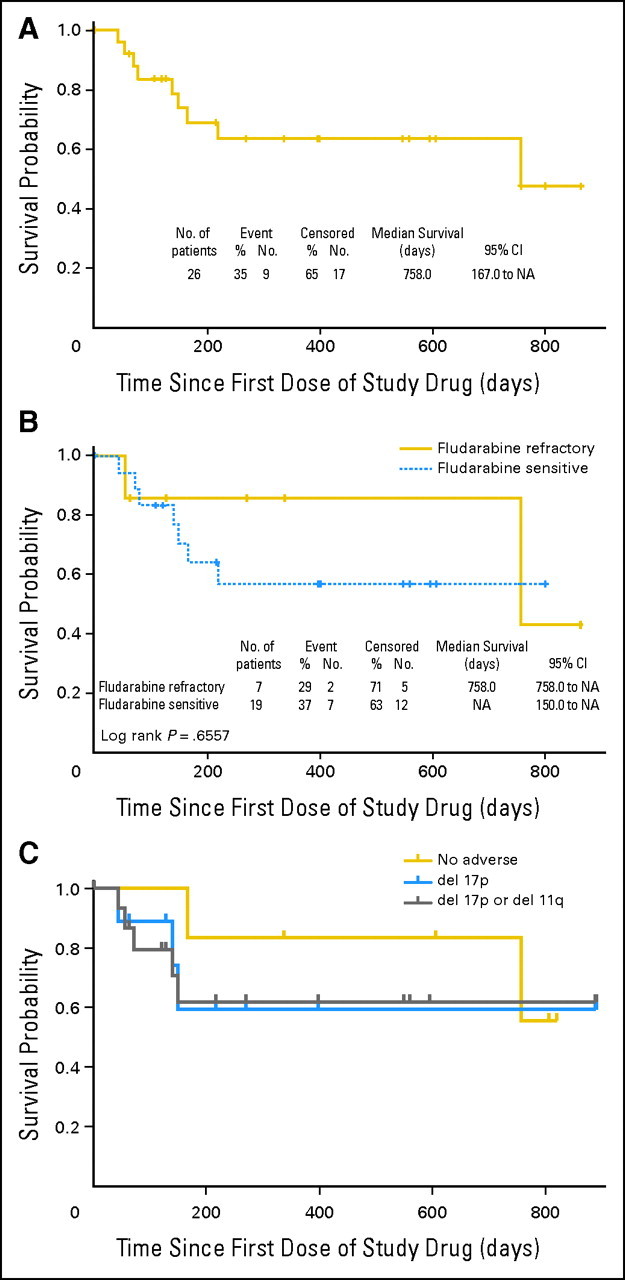
Durability of antileukemic activity of navitoclax. (A) The progression-free survival (PFS) for all patients receiving ≥ 110 mg/d navitoclax from study entry (n = 26) is displayed in a Kaplan-Meier plot. (B) PFS for a subset of these patients with fludarabine-refractory disease. Fluorescent in situ hybridization data were available for 22 of 26 patients receiving ≥ 110 mg/d navitoclax. (C) PFS for patients with del17p13.2 chronic lymphocytic leukemia (n = 9), either del17p13.2 or del11p22.3 (n = 16), or chronic lymphocytic leukemia with neither of these abnormalities (n = 6). NA, not applicable.
Among seven patients with fludarabine-refractory disease receiving ≥ 110 mg/d, one achieved a PR and five had overall SD while demonstrating some antitumor efficacy with either a more than 50% reduction in peripheral blood lymphocytosis and/or a substantial reduction in lymphadenopathy. The median PFS of fludarabine-refractory patients was 25 months (Fig 3B). Among nine patients with bulky lymphadenopathy receiving navitoclax at doses ≥ 110 mg/d, three achieved a PR and six had SD while demonstrating some antitumor efficacy. Similarly, three of nine patients with del(17p) CLL treated with navitoclax at ≥ 110 mg/d achieved a PR, and their median PFS has not been reached (Figs 2G and 3C).
BCL2 Family Proteins as Potential Biomarkers
The levels of expression at baseline of the target proteins, as well as the related prosurvival protein MCL1 that is not inhibited by navitoclax and which can mediate navitoclax resistance in vitro,4,5 and the BH3-only protein BIM, which antagonizes all BCL2-related prosurvival proteins,31 were analyzed in a subset of patients treated at two centers to explore their potential as biomarkers. Neither BCL-w nor BCL-xl were detectable in high concentrations (Fig 4A; data not shown). BCL2 was highly but variably expressed, and MCL1 was expressed in intermediate levels in some samples (Fig 4). No correlation was observed between the level of BCL2 and in vivo response. MCL1 expression was inversely correlated with maximum reduction in lymphocytosis (Fig 4D). Navitoclax is thought to kill MCL1-expressing cells by displacing BIM from a stable complex with BCL2, thereby freeing BIM to antagonize MCL1.16 Consistent with this, high BIM:MCL1 ratios and possibly high BIM:BCL2 ratios were associated with achieving PR (Fig 4).
Fig 4.

High-level BCL2 expression, but not BCL-w or BCL-xl, in peripheral blood chronic lymphocytic leukemia cells and inverse correlation of MCL1 expression with response. The concentrations of BCL2, BCL-w, BCL-xl, BIM, and MCL1 were measured as described in Patients and Methods by using immunoblots. BCL-xl was typically not detected (< 6 fmol/106 cells). (A) Concentrations of BCL2 and BCL-w for individual patients treated at two centers. (B and C) BCL2 concentrations and BIM:BCL2 ratios determined for 10 patients treated with ≥ 110 mg/d navitoclax v best observed clinical response (P = .3 and P = .06, respectively; unpaired two-tailed t test). (D) Shows significant inverse relationship between MCL1 expression in peripheral blood lymphocytes and the maximal reduction in peripheral blood lymphocytosis in the nine patients in whom it could be measured (Spearman coefficient r = −0.63; one-tailed P = .04), as hypothesized. (E and F) MCL1 concentrations and BIM:MCL1 ratios determined for the same patients v best observed clinical response (P = .1 and P =.03, respectively; unpaired two-tailed t test).
DISCUSSION
Navitoclax demonstrated significant single-agent activity against circulating, nodal, and splenic disease in patients with CLL. Antileukemic activity was evident within days, particularly in circulating lymphocyte counts, with maximum clinical responses typically observed within the first 4 months. Although no CRs were observed, durable PRs were seen in 35% and SD for more than 6 months in 27% of patients receiving continuing treatment with doses ≥ 110 mg/d. The depth of response was not clearly related to dose in these patients. Durable responses were observed in patients with fludarabine-refractory disease, bulky adenopathy, and del(17p) CLL.
This initial clinical experience with navitoclax, confirmed in the parallel phase I study in lymphoma,32 provides the first convincing clinical validation of BCL2 as a useful therapeutic target in CLL. Previous attempts to target BCL2 have not resulted in the substantial antitumor activity seen in this study. Oblimersen, an antisense oligodeoxyribonucleotide designed to reduce BCL2 translation displayed minimal single-agent activity in a phase I/II study, with transient PRs achieved in only two of 26 patients.2 Similarly, a phase I study of obataclax, a small molecule with inhibitory activity against BCL2 and related prosurvival proteins, reported only one PR in 26 patients with CLL.1 Unlike with navitoclax, the mechanism of cell killing by obataclax in vitro is not exclusively via the BCL2 family–regulated mitochondrial apoptosis pathway,33 and its lack of single-agent activity may be a consequence of inadequate inhibition of BCL2 family proteins.
In sharp contrast, prior preclinical evaluation has defined the mechanism of action of navitoclax as being dependent on binding and inhibition of BCL2, BCL-w, or BCL-xl.3,4 In the absence of significant BCL-xl or BCL-w expression in CLL, BCL2 is the key antiapoptotic protein inhibited by navitoclax. The single-agent activity of navitoclax in vivo reflects the previously reported potent in vitro activity of ABT-737 and navitoclax3,16,18,19 in this disease. Consistent with the rapid induction of apoptosis in vitro,17 circulating CLL cells decreased rapidly in patients treated with navitoclax.
However, responses in patients were more heterogeneous than predicted by in vitro data, raising the possibility that insufficient BCL2 inhibition is being achieved in vivo. Peak concentrations of navitoclax observed were between 1 and 10 μmol/L in patients receiving doses ≥ 110 mg. Although these concentrations were within the predicted efficacy range estimated from preclinical animal studies5 and whole blood assays for CLL cytoxicity,18 it remains uncertain if they provide maximal antileukemic activity because the DLT of thrombocytopenia prevented exploration of higher doses. Development of more selective BH3 mimetics that inhibit BCL2 but not BCL-xl may be required to allow the full potential of BCL2 inhibitory therapy to be explored.
No significant correlation between level of expression of BCL2 and response was observed in this study. This suggests that BCL2-independent survival factors are operating, and these could provide an alternative explanation for the persistence of disease in vivo during ongoing therapy. MCL1 is a recognized resistance factor in vitro,4,5 and this study provides the first clinical data supporting its role in determining response of tumor cells to navitoclax. Higher levels of MCL1 before therapy were associated with lesser reductions in lymphocytosis and best clinical response when adjusted for the level of BIM expression. The clinical utility of measuring MCL1 and BIM:MCL1 or BIM:BCL2 ratios as biomarkers requires formal assessment in future clinical trials. Resistance to BCL2 inhibition warrants exploration of combination therapy. In preclinical in vivo models, both ABT-737 and navitoclax display synergy with cytotoxic agents and targeted therapies against other lymphoproliferative diseases5,14 and malignancies.3,34 Along with the promising single-agent activity observed in this study of heavily pretreated patients, such data underpin the rationale for recently commenced clinical trials of combination therapies of navitoclax with current standard-of-care drugs in CLL.
Supplementary Material
Acknowledgment
We thank Saul Rosenberg and Steve Elmore for insightful discussions; Raymond Knight, Melissa Shah, Julianne Dziubinski, Michelle Pedersen, Diane D'Amico, Michael Dawson, and Renee Greco for operational support; Di Li and Joseph Beason for statistical analyses; and Ai Lockard for editorial assistance. All are employees of Abbott Laboratories.
Footnotes
Supported by Abbott Laboratories and Genentech. Additional support for correlative studies was provided by National Health and Medical Research Council of Australia, Victorian Cancer Agency, Leukaemia Foundation Australia, Australian Cancer Research Foundation, and the Leukemia and Lymphoma Society.
Presented in part at the 44th Annual Meeting of the American Society of Clinical Oncology (ASCO), Chicago, IL, May 30-June 3, 2008; the 45th Annual Meeting of ASCO, Orlando, FL, May 29-June 2, 2009; 50th American Society of Hematology (ASH) Annual Meeting and Exposition, San Francisco, CA, December 6-9, 2008; 51st ASH Annual Meeting and Exposition, New Orleans, LA, December 5-8, 2009; and 14th Congress of the European Hematology Association, Berlin, Germany, June 4-7, 2009.
Authors' disclosures of potential conflicts of interest and author contributions are found at the end of this article.
Clinical trial information can be found for the following: NCT00481091.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Although all authors completed the disclosure declaration, the following author(s) indicated a financial or other interest that is relevant to the subject matter under consideration in this article. Certain relationships marked with a “U” are those for which no compensation was received; those relationships marked with a “C” were compensated. For a detailed description of the disclosure categories, or for more information about ASCO's conflict of interest policy, please refer to the Author Disclosure Declaration and the Disclosures of Potential Conflicts of Interest section in Information for Contributors.
Employment or Leadership Position: Hao Xiong, Abbott Laboratories (C); Yue Cui, Abbott Laboratories (C); Todd A. Busman, Abbott Laboratories (C); Evelyn M. McKeegan, Abbott Laboratories (C); Andrew P. Krivoshik, Astellas Pharma Global Development (C), Abbott Laboratories (C); Sari H. Enschede, Abbott Laboratories (C); Rod Humerickhouse, Abbott Laboratories (C) Consultant or Advisory Role: Jennifer R. Brown, Calistoga Pharmaceuticals (C), Celgene (C), Pharmacyclics (C); William G. Wierda, Genentech (C), Celgene (C), Micromet (C); David C.S. Huang, Abbott Laboratories (U), Genentech (U) Stock Ownership: Hao Xiong, Abbott Laboratories; Yue Cui, Abbott Laboratories; Todd A. Busman, Abbott Laboratories; Evelyn M. McKeegan, Abbott Laboratories; Andrew P. Krivoshik, Abbott Laboratories; Sari H. Enschede, Abbott Laboratories; Rod Humerickhouse, Abbott Laboratories Honoraria: None Research Funding: Andrew W. Roberts, Abbott Laboratories; Jennifer R. Brown, Celgene, Genzyme; William G. Wierda, GlaxoSmithKline, Abbott Laboratories; Thomas J. Kipps, Abbott Laboratories Expert Testimony: None Other Remuneration: Dennis A. Carney, Abbott Laboratories
AUTHOR CONTRIBUTIONS
Conception and design: Andrew W. Roberts, David C.S. Huang, Hao Xiong, Todd A. Busman, Evelyn M. McKeegan, Andrew P. Krivoshik, Sari H. Enschede, Rod Humerickhouse
Financial support: Rod Humerickhouse
Provision of study materials or patients: Andrew W. Roberts, John F. Seymour, Jennifer R. Brown, Thomas J. Kipps, Dennis A. Carney, Simon Z. He, David C.S. Huang
Collection and assembly of data: Andrew W. Roberts, John F. Seymour, Jennifer R. Brown, William G. Wierda, Thomas J. Kipps, Seong Lin Khaw, Dennis A. Carney, Simon Z. He, Todd A. Busman, Evelyn M. McKeegan, Andrew P. Krivoshik
Data analysis and interpretation: Andrew W. Roberts, John F. Seymour, Jennifer R. Brown, William G. Wierda, Seong Lin Khaw, Simon Z. He, David C.S. Huang, Hao Xiong, Yue Cui, Todd A. Busman, Evelyn M. McKeegan, Andrew P. Krivoshik, Sari H. Enschede, Rod Humerickhouse
Manuscript writing: All authors
Final approval of manuscript: All authors
Affiliations
Andrew W. Roberts and Simon Z. He, The Royal Melbourne Hospital; Andrew W. Roberts, Seong Lin Khaw, Simon Z. He, and David C.S. Huang, The Walter and Eliza Hall Institute of Medical Research; Andrew W. Roberts and David C.S. Huang, Australian Cancer Research Foundation Centre for Therapeutic Target Discovery; Andrew W. Roberts, John F. Seymour, Seong Lin Khaw, Dennis A. Carney, Simon Z. He, and David C.S. Huang, University of Melbourne, Parkville; John F. Seymour and Dennis A. Carney, Peter MacCallum Cancer Center, Victoria, Australia; Jennifer R. Brown, Dana-Farber Cancer Institute, Boston, MA; William G. Wierda, MD Anderson Cancer Center, The University of Texas, Houston, TX; Thomas J. Kipps, Moores University of California at San Diego Cancer Center, La Jolla, CA; Hao Xiong, Yue Cui, Todd A. Busman, Evelyn M. McKeegan, Andrew P. Krivoshik, Sari H. Enschede, and Rod Humerickhouse, Abbott Laboratories, Abbott Park, IL.
REFERENCES
- 1.O'Brien SM, Claxton DF, Crump M, et al. Phase I study of obatoclax mesylate (GX15-070), a small molecule pan-Bcl-2 family antagonist, in patients with advanced chronic lymphocytic leukemia. Blood. 2009;113:299–305. doi: 10.1182/blood-2008-02-137943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.O'Brien SM, Cunningham CC, Golenkov AK, et al. Phase I to II multicenter study of oblimersen sodium, a Bcl-2 antisense oligonucleotide, in patients with advanced chronic lymphocytic leukemia. J Clin Oncol. 2005;23:7697–7702. doi: 10.1200/JCO.2005.02.4364. [DOI] [PubMed] [Google Scholar]
- 3.Oltersdorf T, Elmore SW, Shoemaker AR, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 4.van Delft MF, Wei AH, Mason KD, et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell. 2006;10:389–399. doi: 10.1016/j.ccr.2006.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tse C, Shoemaker AR, Adickes J, et al. ABT-263: A potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68:3421–3428. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- 6.Vaux DL, Cory S, Adams JM. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 1988;335:440–442. doi: 10.1038/335440a0. [DOI] [PubMed] [Google Scholar]
- 7.Strasser A, Huang DC, Vaux DL. The role of the bcl-2/ced-9 gene family in cancer and general implications of defects in cell death control for tumourigenesis and resistance to chemotherapy. Biochim Biophys Acta. 1997;1333:F151–F178. doi: 10.1016/s0304-419x(97)00019-x. [DOI] [PubMed] [Google Scholar]
- 8.Schmitt CA, Lowe SW. Bcl-2 mediates chemoresistance in matched pairs of primary E(mu)-myc lymphomas in vivo. Blood Cells Mol Dis. 2001;27:206–216. doi: 10.1006/bcmd.2000.0372. [DOI] [PubMed] [Google Scholar]
- 9.Kitada S, Takayama S, De Riel K, et al. Reversal of chemoresistance of lymphoma cells by antisense-mediated reduction of bcl-2 gene expression. Antisense Res Dev. 1994;4:71–79. doi: 10.1089/ard.1994.4.71. [DOI] [PubMed] [Google Scholar]
- 10.Miyashita T, Reed JC. bcl-2 gene transfer increases relative resistance of S49.1 and WEHI7.2 lymphoid cells to cell death and DNA fragmentation induced by glucocorticoids and multiple chemotherapeutic drugs. Cancer Res. 1992;52:5407–5411. [PubMed] [Google Scholar]
- 11.Deng J, Carlson N, Takeyama K, et al. BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer Cell. 2007;12:171–185. doi: 10.1016/j.ccr.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 12.Tahir SK, Yang X, Anderson MG, et al. Influence of Bcl-2 family members on the cellular response of small-cell lung cancer cell lines to ABT-737. Cancer Res. 2007;67:1176–1183. doi: 10.1158/0008-5472.CAN-06-2203. [DOI] [PubMed] [Google Scholar]
- 13.Tahir SK, Wass J, Joseph MK, et al. Identification of expression signatures predictive of sensitivity to the Bcl-2 family member inhibitor ABT-263 in small cell lung carcinoma and leukemia/lymphoma cell lines. Mol Cancer Ther. 2010;9:545–557. doi: 10.1158/1535-7163.MCT-09-0651. [DOI] [PubMed] [Google Scholar]
- 14.Mason KD, Vandenberg CJ, Scott CL, et al. In vivo efficacy of the Bcl-2 antagonist ABT-737 against aggressive Myc-driven lymphomas. Proc Natl Acad Sci U S A. 2008;105:17961–17966. doi: 10.1073/pnas.0809957105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ackler S, Mitten MJ, Foster K, et al. The Bcl-2 inhibitor ABT-263 enhances the response of multiple chemotherapeutic regimens in hematologic tumors in vivo. Cancer Chemother Pharmacol. 2010;66:869–880. doi: 10.1007/s00280-009-1232-1. [DOI] [PubMed] [Google Scholar]
- 16.Del Gaizo Moore V, Brown JR, Certo M, et al. Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT-737. J Clin Invest. 2007;117:112–121. doi: 10.1172/JCI28281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vogler M, Dinsdale D, Sun XM, et al. A novel paradigm for rapid ABT-737-induced apoptosis involving outer mitochondrial membrane rupture in primary leukemia and lymphoma cells. Cell Death Differ. 2008;15:820–830. doi: 10.1038/cdd.2008.25. [DOI] [PubMed] [Google Scholar]
- 18.Vogler M, Furdas SD, Jung M, et al. Diminished sensitivity of chronic lymphocytic leukemia cells to ABT-737 and ABT-263 due to albumin binding in blood. Clin Cancer Res. 2010;16:4217–4225. doi: 10.1158/1078-0432.CCR-10-0777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mason KD, Khaw SL, Rayeroux KC, et al. The BH3 mimetic compound, ABT-737, synergizes with a range of cytotoxic chemotherapy agents in chronic lymphocytic leukemia. Leukemia. 2009;23:2034–2041. doi: 10.1038/leu.2009.151. [DOI] [PubMed] [Google Scholar]
- 20.Mason KD, Carpinelli MR, Fletcher JI, et al. Programmed anuclear cell death delimits platelet life span. Cell. 2007;128:1173–1186. doi: 10.1016/j.cell.2007.01.037. [DOI] [PubMed] [Google Scholar]
- 21.Nakayama K, Nakayama K, Negishi I, et al. Targeted disruption of Bcl-2 alpha beta in mice: Occurrence of gray hair, polycystic kidney disease, and lymphocytopenia. Proc Natl Acad Sci U S A. 1994;91:3700–3704. doi: 10.1073/pnas.91.9.3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Print CG, Loveland KL, Gibson L, et al. Apoptosis regulator bcl-w is essential for spermatogenesis but appears otherwise redundant. Proc Natl Acad Sci U S A. 1998;95:12424–12431. doi: 10.1073/pnas.95.21.12424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang H, Nimmer PM, Tahir SK, et al. Bcl-2 family proteins are essential for platelet survival. Cell Death Differ. 2007;14:943–951. doi: 10.1038/sj.cdd.4402081. [DOI] [PubMed] [Google Scholar]
- 24.Schoenwaelder SM, Yuan Y, Josefsson EC, et al. Two distinct pathways regulate platelet phosphatidylserine exposure and procoagulant function. Blood. 2009;114:663–666. doi: 10.1182/blood-2009-01-200345. [DOI] [PubMed] [Google Scholar]
- 25.Storer BE. An evaluation of phase I clinical trial designs in the continuous dose-response setting. Stat Med. 2001;20:2399–2408. doi: 10.1002/sim.903. [DOI] [PubMed] [Google Scholar]
- 26.Potter DM. Adaptive dose finding for phase I clinical trials of drugs used for chemotherapy of cancer. Stat Med. 2002;21:1805–1823. doi: 10.1002/sim.1141. [DOI] [PubMed] [Google Scholar]
- 27.Cancer Therapy Evaluation Program. Common Terminology Criteria for Adverse Events (CTCAE) Version 3.0. http://ctep.cancer.gov/protocolDevelopment/electronic_applications/ctc.htm.
- 28.Cheson BD, Bennett JM, Grever M, et al. National Cancer Institute-sponsored Working Group guidelines for chronic lymphocytic leukemia: Revised guidelines for diagnosis and treatment. Blood. 1996;87:4990–4997. [PubMed] [Google Scholar]
- 29.Hallek M, Cheson BD, Catovsky D, et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: A report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood. 2008;111:5446–5456. doi: 10.1182/blood-2007-06-093906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Döhner H, Stilgenbauer S, Benner A, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med. 2000;343:1910–1916. doi: 10.1056/NEJM200012283432602. [DOI] [PubMed] [Google Scholar]
- 31.Chen L, Willis SN, Wei A, et al. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell. 2005;17:393–403. doi: 10.1016/j.molcel.2004.12.030. [DOI] [PubMed] [Google Scholar]
- 32.Wilson WH, O'Connor OA, Czuczman MS, et al. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: A phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol. 2010;11:1149–1159. doi: 10.1016/S1470-2045(10)70261-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vogler M, Weber K, Dinsdale D, et al. Different forms of cell death induced by putative BCL2 inhibitors. Cell Death Differ. 2009;16:1030–1039. doi: 10.1038/cdd.2009.48. [DOI] [PubMed] [Google Scholar]
- 34.Konopleva M, Contractor R, Tsao T, et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell. 2006;10:375–388. doi: 10.1016/j.ccr.2006.10.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.