

Abstract
Background
Bacillus cereus group isolates that produce diarrheal or emetic toxins are frequently isolated from raw milk and, in spore form, can survive pasteurization. Several species within the B. cereus group are closely related and cannot be reliably differentiated by established taxonomical criteria. While B. cereus is traditionally recognized as the principal causative agent of foodborne disease in this group, there is a need to better understand the distribution and expression of different toxin and virulence genes among B. cereus group food isolates to facilitate reliable characterization that allows for assessment of the likelihood of a given isolate to cause a foodborne disease.
Results
We performed whole genome sequencing of 22 B. cereus group dairy isolates, which represented considerable genetic diversity not covered by other isolates characterized to date. Maximum likelihood analysis of these genomes along with 47 reference genomes representing eight validly published species revealed nine phylogenetic clades. Three of these clades were represented by a single species (B. toyonensis –clade V, B. weihenstephanensis – clade VI, B. cytotoxicus - VII), one by two dairy-associated isolates (clade II; representing a putative new species), one by two species (B. mycoides, B. pseudomycoides – clade I) and four by three species (B. cereus, B. thuringiensis, B. anthracis – clades III-a, b, c and IV). Homologues of genes encoding a principal diarrheal enterotoxin (hemolysin BL) were distributed across all, except the B. cytotoxicus clade. Using a lateral flow immunoassay, hemolysin BL was detected in 13 out of 18 isolates that carried hblACD genes. Isolates from clade III-c (which included B. cereus and B. thuringiensis) consistently did not carry hblACD and did not produce hemolysin BL. Isolates from clade IV (B. cereus, B. thuringiensis) consistently carried hblACD and produced hemolysin BL. Compared to others, clade IV was significantly (p = 0.0001) more likely to produce this toxin. Isolates from clade VI (B. weihenstephanensis) carried hblACD homologues, but did not produce hemolysin BL, possibly due to amino acid substitutions in different toxin-encoding genes.
Conclusions
Our results demonstrate that production of diarrheal enterotoxin hemolysin BL is neither inclusive nor exclusive to B. cereus sensu stricto, and that phylogenetic classification of isolates may be better than taxonomic identification for assessment of B. cereus group isolates risk for causing a diarrheal foodborne disease.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-016-2883-z) contains supplementary material, which is available to authorized users.
Keywords: Bacillus cereus group, WGS, Virulence genes, Dairy, Toxin production, Hemolysin BL
Background
Bacillus cereus was identified as a causative agent in 19 % of foodborne outbreaks that were reported in the United States from 1998 to 2008 [1]. Although the majority of these outbreaks were traced back to rice (50 %) and meat (24 %) [1], B. cereus group isolates have also been frequently isolated from milk e.g., [2, 3]. B. cereus spores can survive high temperature exposure, therefore insufficient cooling or storage of food at temperatures below 60 °C can support their growth in food following thermal treatment. Depending on the strain, growth to high levels may result in food spoilage and present a risk for foodborne illness after ingestion [4, 5].
The B. cereus group consists of eight pathogenic and non-pathogenic bacterial species – B. cereus, B. anthracis, B. thuringiensis, B. cytotoxicus, B. weihenstephanensis, B. mycoides, B. pseudomycoides, and B. toyonensis [6, 7]. This group of bacterial species is also referred to as B. cereus sensu lato with the specific species B. cereus being referred to as B. cereus sensu stricto. Members of the B. cereus group are closely related and cannot always be differentiated based on phenotypic and biochemical characteristics as detailed in the Bergey’s Manual of Determinative Bacteriology [8]. Nevertheless, some specific phenotypic characteristics have traditionally been used to differentiate key species within the B. cereus group. For example, a combination of capsule production, non-motility and inability to cause hemolysis is specific for B. anthracis, the production of crystal proteins is specific for B. thuringiensis, and rhizoid colony morphology is specific for B. mycoides and B. pseudomycoides [8]. In addition, the ability to grow at 7 °C but not at 43 °C is typically considered to be specific for B. weihenstephanensis [8]. Even with molecular methods, species classification of B. cereus group isolates can be challenging. For example, the human pathogens B. cereus and B. anthracis, and the insect pathogen B. thuringiensis cannot be reliably differentiated with most molecular typing methods, including DNA sequence analysis of 16S rDNA, rpoB and MLST loci [3, 9, 10]. Consistent with these findings, studies employing whole genome sequencing have confirmed high genomic similarity of B. cereus, B. thuringiensis and B. anthracis, which explains the difficulties in speciation of B. cereus group isolates [11–14].
Due to the highly similar genomic backbone of B. cereus group isolates, different studies have explored whether characterization of virulence gene presence/absence patterns may provide a better predictor of a strain’s ability to cause anthrax or gastrointestinal disease, compared to traditional phenotypic or molecular taxonomic classification. These methods typically determine the presence/absence of B. cereus group virulence genes encoded on plasmids or within the chromosome, which were previously linked to virulence in humans and/or animals. Among plasmid-encoded virulence genes are those encoding the anthrax toxin (cya, lef and pag) and the poly-γ-D-glutamate capsule biosynthesis genes (capABCDE) [12, 15]. These virulence determinants are encoded on pXO1, pXO2 or similar plasmids, which have been identified among B. anthracis isolates, as well as B. cereus isolates able to cause anthrax or anthrax-like disease in humans and animals [15–20]. The cesABCD operon represents another set of virulence genes encoded on plasmids (e.g., pCERE01, pBCE4810). These genes encode cereulide synthetase necessary for non-ribosomal biosynthesis of the emetic toxin cereulide, which causes a food-borne intoxication in humans [21, 22]. Lastly, strains carrying plasmid-encoded crystal protein genes (cry) are pathogenic to insects, but crystal proteins do not affect humans [23, 24].
In contrast, genes encoding the virulence determinants implicated in the diarrheal type of the foodborne disease are typically chromosomally encoded. Examples include hemolysin BL (encoded by hblABCD), nonhemolytic enterotoxin (nheABC), cytotoxin K (cytK) and cereolysin (cerAB) [24, 25]. Genes encoding diarrheal toxins are distributed across different B. cereus group species, including B. cereus, B. thuringiensis, B. weihenstephanensis, B. mycoides and B. pseudomycoides [26–29]. Expression of these toxins is governed by complex regulation pathways, which are not well understood [29–32]. In addition to these principal virulence genes, multiple other genes exist, which may indirectly contribute to virulence, such as hlyII, entFM and entABC, which encode hemolysin II, enterotoxin FM, and a putative enterotoxin, respectively [25].
Overall, classification into currently defined species within the B. cereus group does not provide for reliable identification of isolates that are or are not likely to cause human foodborne disease. For example, production of diarrheal toxins in the B. cereus group is not species - specific. While some attempts have been made to use single locus (panC) sequence data to develop alternative classification approaches that allow for more accurate assessment of pathogenicity and virulence of B. cereus group isolates [33, 34], these approaches still require significant refinement and further data. In particular, the accuracy of classification into seven phylogenetic groups, as determined based on panC sequences needs to be confirmed using whole genome sequences. The diversity of B. cereus group isolates evaluated to-date by WGS has included only a limited number of food isolates and, to our knowledge, few genome sequences for dairy isolates are publicly available. However, sporeforming pathogens and spoilage organisms are becoming increasingly important in dairy products as reduced post-processing contamination with rapidly growing Gram-negative bacteria provide an opportunity for growth of B. cereus group isolates that would not compete well in the presence of many Gram-negative bacteria introduced by post-processing contamination. Hence, we have carried out a comparative genomic analysis of 22 dairy-associated B. cereus group isolates and 47 B. cereus group isolates with publicly available genome sequences to investigate their (i) phylogenetic clustering based on core genome single nucleotide polymorphisms (SNPs), (ii) clustering based on the virulence gene presence/absence, and (iii) distribution of hemolysin BL and nonhemolytic toxin production among isolates in different phylogenetic clades.
Results and discussion
Identification of multiple novel MLST allelic and sequence types reflects the diversity of dairy-associated B. cereus group isolates
Whole genome sequencing was performed for 22 diverse dairy-associated isolates (Table 1) that had been identified as members of B. cereus group based on rpoB sequence data. The median whole genome sequence (WGS) coverage for the 22 isolates was 57-fold (ranging from 33 to 78-fold), the median number of contigs >1 kb was 128 (ranging from 63 to 846; only one sequence, FSL W8-0767 was on the higher end), and the median draft genome size was 5.75 Mbp.
Table 1.
Characteristics and sequence accession numbers for dairy-associated B. cereus group isolates sequenced in this study
| Isolate | Source | BioSample ID | SRA Run | GenBank accession number | PubMLST ID | rpoB allelic type (AT) | MLST STa | Novel MLST AT or STb |
|---|---|---|---|---|---|---|---|---|
| FSL H7-0926 | Pasteurized 2 % milk | SAMN03800014 | SRR2541537 | LOBD00000000 | 1774 | 90 | 667 | |
| FSL H8-0482 | Soil from areas adjacent to barns | SAMN03800015 | SRR2541601 | LOAZ00000000 | 1764 | 129 | 223 | |
| FSL H8-0488 | Water from hose in milking area | SAMN03800016 | SRR2541602 | LOBA00000000 | 1765 | 129 | 111 | |
| FSL K6-0040 | Raw milk | SAMN03800017 | SRR2541603 | LOBE00000000 | 1775 | 362 | 1101 | ilv 245, ST-1101 |
| FSL K6-0043 | Raw milk | SAMN03800018 | SRR2541604 | LOMW00000000 | 1763 | 363 | 1087 | pta 235, ST-1087 |
| FSL K6-0067 | Raw milk | SAMN03800019 | SRR2541605 | LOMN00000000 | 1762 | 365 | 1086 | gmk 138, pur 218, ST-1086 |
| FSL K6-0069 | Raw milk | SAMN03800020 | SRR2541606 | LOBB00000000 | 1756 | 194 | 1080 | glp 224, gmk 135, ilv 237, pta 233, ST-1080 |
| FSL K6-0073 | Raw milk | SAMN03800021 | SRR2541607 | LOMO00000000 | 1773 | 366 | 33 | |
| FSL K6-0267 | Raw milk | SAMN03800022 | SRR2541613 | LOMP00000000 | 1755 | 90 | 1090 | ST-1090 |
| FSL M8-0117 | Raw milk | SAMN03800023 | SRR2541639 | LONG00000000 | 1761 | 308 | 1085 | gmk 137, pyc 186, ST-1085 |
| FSL W8-0003 | Ricotta/mozzarella whey | SAMN03800024 | SRR2541640 | LOMQ00000000 | 1760 | 125 | 1084 | ilv 239, ST-1084 |
| FSL W8-0050 | Condensed product from evaporator | SAMN03800025 | SRR2541641 | LOMR00000000 | 1772 | 380 | 32 | |
| FSL W8-0169 | Raw milk | SAMN03800026 | SRR2541651 | LOBC00000000 | 1757 | 61 | 1081 | gmk 136, pta 234, ST-1081 |
| FSL W8-0268 | Evaporator - liquid | SAMN03800027 | SRR2541662 | LOMS00000000 | 1759 | 92 | 1083 | tpi 185, ST-1083 |
| FSL W8-0275 | Liquid milk whey permeate | SAMN03800028 | SRR2541668 | LOMT00000000 | 1771 | 463 | 1050 | |
| FSL W8-0483 | Liquid raw milk from silo | SAMN03800029 | SRR2541674 | LOMU00000000 | 1758 | 120 | 1082 | ilv 238, ST-1082 |
| FSL W8-0520 | Processed liquid milk | SAMN03800030 | SRR2541680 | LONH00000000 | 1770 | 481 | 1032 | |
| FSL W8-0523 | Liquid from blended whey silo | SAMN03800031 | SRR2541686 | LOMV00000000 | 1769 | 481 | 1032 | |
| FSL W8-0640 | Processed skim milk | SAMN03800032 | SRR2541688 | LOQA00000000 | 1754 | 154 | 1089 | ST-1089 |
| FSL W8-0824 | Finished dairy product | SAMN03800033 | SRR2541693 | LOQB00000000 | 1767 | 92 | 24 | |
| FSL W8-0932 | Processed skim milk | SAMN03800034 | SRR2541715 | LOQC00000000 | 1766 | 120 | 365 | |
| FSL W8-0767 | Milk powder | SAMN03800035 | SRR2541718 | LOQD00000000 | 1768 | 154 | 787 |
WGS data were initially used to extract sequence data for seven genes that are used in the B. cereus PubMLST typing scheme [35]. MLST data differentiated the 22 isolates into 21 different MLST sequence types (STs), providing increased discriminatory power over rpoB sequence typing that identified 16 different rpoB allelic types (ATs). Eleven of these MLST types represented novel STs in the PubMLST database (Table 1) [35]. Two of them were a novel combination of existing ATs and nine of them carried one to four novel allelic types, which were deposited in the PubMLST database. These data show that the isolates selected represent considerable B. cereus group strain diversity that has not been observed among isolates characterized by MLST or WGS to date. These data also demonstrate the importance of investigating populations from diverse sources when characterizing the genomic diversity of different bacterial groups. These initial analyses further justify our investigation of the genomic diversity among dairy-associated B. cereus group isolates, particularly since previous studies focused on clinical sources and food sources most commonly linked to human disease outbreaks (e.g., rice, vegetables) (e.g., [36, 37]).
WGS-based phylogeny reveals clades consistent with rpoB and MLST clustering of isolates
Genome sequence data for the 22 dairy-associated isolates and 47 reference genomes (obtained from NCBI, RefSeq; see Additional file 1: Table S1) of B. cereus group isolates were used to build a core genome kSNP tree (Fig. 1), which was based on 2362 core genome SNPs. The resulting phylogeny was compared to clustering of the 22 dairy-associated isolates that was observed in separate phylogenies constructed with either rpoB or the 7 MLST genes sequence data. WGS revealed nine B. cereus group clades. These clades were named according to previously proposed phylogenetic groups based on panC sequences [33]. Where panC phylogenetic groups could be separated into different WGS clades this was clarified by use of alphabetical subdesignation (e.g., panC group III was resolved into WGS clades III-a, III-b, and III-c). Seven of WGS clades (i.e., clades II to VI) included between two and seven dairy-associated isolates sequenced here. While the 22 dairy-associated isolates also clustered in seven clades in the MLST tree, these isolates represented only 6 phylogenetic clades in the rpoB tree. Specifically, one of the clades in the rpoB tree was resolved into two clades in MLST tree, including one clade with isolates FSL M8-0117 and FSL K6-0067, and one with FSL K6-0069 and FSL W8-0169 (Fig. 2). Clustering of dairy-associated isolates in the rpoB and WGS trees was very similar, but not absolutely congruent, as the rpoB tree did not resolve clades II and III-b (Fig. 2). The WGS phylogeny was congruent with MLST phylogeny, and with the seven phylogenetic groups determined based on panC sequence similarity (see Fig. 1), which were proposed by Guinebretière et al. [33, 34]. Overall, these data suggest that both rpoB and panC single-locus-based characterization allows for classification that is expected to be largely congruent with WGS clades.
Fig. 1.
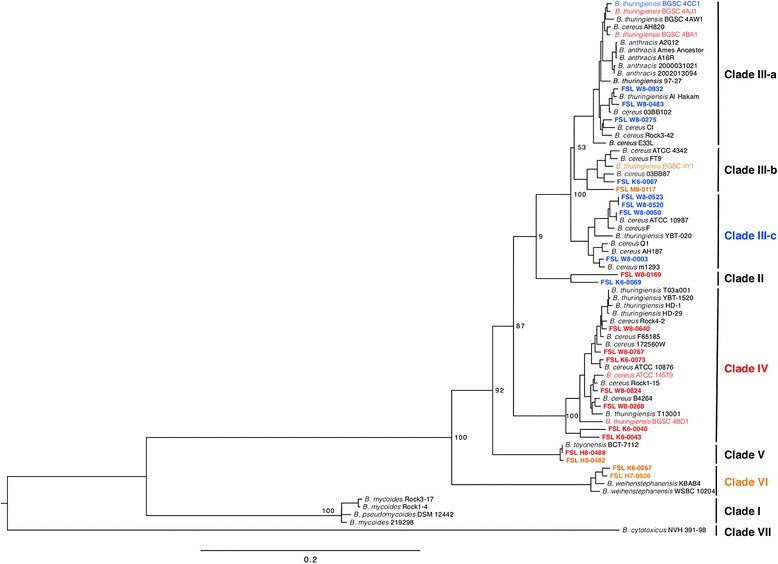
Core genome phylogeny of 69 B. cereus group isolates. Maximum likelihood tree was constructed using core genome SNPs identified with kSNP for 22 dairy-associated isolates and 47 reference isolates. Phylogeny was inferred using RaxML under general time-reversible model with gamma distributed substitution sites, and 1000 bootstrap repetitions. Bar represents 0.2 substitutions per site. WGS revealed nine B. cereus group phylogenetic clades. These clades were named according to previously proposed phylogenetic groups based on panC sequence types [33]. Where panC phylogenetic groups could be separated into different WGS clades this was clarified by the use of alphabetical subdesignation (e.g., panC group III was resolved into WGS clades III-a, III-b, and III-c). Isolates in red carried hblACD genes and produced hemolysin BL, isolates in orange carried hblACD genes, but did not produce hemolysin BL, and isolates in blue did not carry hblACD genes nor did they produce hemolysin BL. Dairy-associated isolates are in bold
Fig. 2.
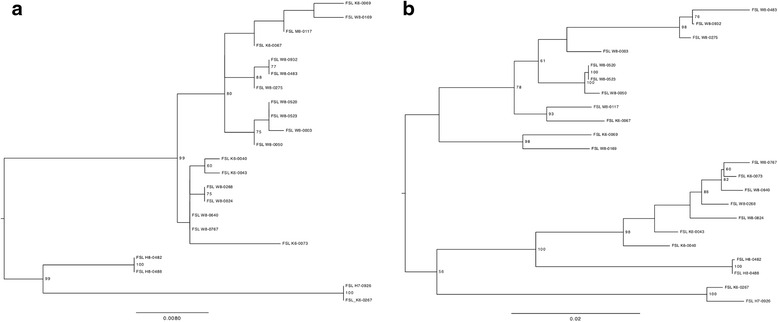
Maximum likelihood phylogenetic tree constructed based on (a) rpoB and (b) 7 MLST loci. Maximum likelihood tree constructed in RaxML under general time-reversible model with gamma distributed and invariant substitution sites, and 1000 bootstrap repetitions, based on (a) rpoB and (b) 7 MLST loci sequences for 22 dairy-associated isolates. Bar represents (a) 0.02 and (b) 0.0080 substitutions per site. Only bootstrap values ≥ 60 are shown on the trees
Genomic sequence data confirm that the B. cereus group includes both monophyletic species, as well as currently designated species that are not consistent with phylogeny
The WGS-based phylogeny constructed here includes whole genome sequence (WGS) data for 47 reference genomes of B. cereus group isolates that had been previously sequenced and identified at the species level by others (i.e., B. cereus, B. anthracis, B. cytotoxicus, B. thuringiensis, B. weihenstephanensis, B. mycoides, B. pseudomycoides, B. toyonensis). Metadata for these reference isolates are available in Additional file 1: Table S1. Among nine clades defined here, all but clade II (representing a putative new species) included at least one reference strain. Three of the WGS clades (V, VI and VII) included reference strains of only a single species, including B. toyonensis (clade V), B. weihenstephanensis (clade VI) and B. cytotoxicus (clade VII). While only one reference strain genome was available for each B. toyonensis and B. cytotoxicus, both B. weihenstephanensis reference genomes analyzed clustered into clade VI. Clades V and VI each, also included dairy-associated isolates. All three of these clades are supported by 100 % bootstrap values. These data suggest that B. toyonensis, B. weihenstephanensis and B. cytotoxicus represent monophyletic species. Consistently with our findings, B. cytotoxicus has been previously classified in a separate phylogenetic group [33].
Clade I included three genomes previously identified as B. mycoides, as well as the B. pseudomycoides type strain genome. It has previously been shown that B. mycoides and B. pseudomycoides isolates (identified based on rhizoid colony morphology) cluster in two MLST clades – one including the B. pseudomycoides type strain, and the other including B. weihenstephanensis and B. mycoides type strains [38]. Clustering of previously identified (reference) B. mycoides genomes with genome of B. pseudomycoides type strain in our study suggests that these B. mycoides isolates were originally misclassified, and should be re-classified as B. pseudomycoides. Rhizoid colony morphology is typical for both B. mycoides and B. pseudomycoides and is therefore not an appropriate differentiating characteristic. Classification based on panC sequence analysis is more reliable, as B. pseudomycoides cluster in panC group I, and B. mycoides in panC group VI [33].
Each of the remaining four clades (III-a, III-b, III-c and IV) included both B. cereus and B. thuringiensis reference genomes. In addition to B. cereus and B. thuringiensis, clade III-a also included all 5 B. anthracis reference genomes from our isolate set. These results are consistent with MLST studies that have also shown (i) that B. cereus and B. thuringiensis cluster together (e.g., [9]) and (ii) that B. anthracis isolates cluster with B. cereus (e.g., [39]). Overall, our data confirmed the polyphyletic nature of the species B. cereus and B. thuringiensis by demonstrating their clustering in multiple “multi-species” clades, similar to what was previously reported based on the WGS analyses [11–14]. This shows that WGS phylogeny does not support the current taxonomical classification of B. cereus, B. thuringiensis and B. anthracis, and suggests that SNP-based phylogenetic clustering, even at the core genome level, does not allow for identification of these species the way they are defined now. Similar to our findings, previous MLST studies have shown that clustering of B. cereus group isolates does not yield clades that are consistent with species designation [9, 38, 39]. There is thus increasing and consistent evidence that supports the need for a re-alignment of the B. cereus group taxonomy to be consistent with the WGS phylogeny. The data generated here can form the basis for future development of SNP-based assays that will allow for rapid identification of major B. cereus group clades and subtypes.
Insecticidal crystal protein genes were not detected in all isolates previously identified as B. thuringiensis, and were detected in isolates representing other species
In order to further characterize the genomes analyzed here, we determined the presence/absence of 36 virulence genes (Additional file 2: Table S2 and Additional file 3: Table S3) in the whole genome sequences of the 69 B. cereus group genomes using protein BLAST. The presence of 8 of these 36 virulence genes was confirmed by performing PCR on 28 B. cereus group isolates that were physically available to us (Table 2). While standard protein BLAST approach was generally successful in identifying the genomic presence of virulence genes (see below for details), cry, which encodes the insecticidal crystal protein typically linked to B. thuringiensis, showed substantially higher amino acid variability than other virulence-associated genes [40]. As this increases the chances of false negative results using protein BLAST, we initially searched for cry using an Endotoxin_M hidden Markov model (HMM) extracted from Pfam database (PF00555). This HMM search identified cry homologues in just 4 out of 14 B. thuringiensis genomes. To improve the sensitivity of the HMM search, we thus constructed a custom Hidden Markov model based on ten cry variants representing different 1° domains available in the B. thuringiensis nomenclature database [41]. This new model identified cry genes in 13 isolates (Additional file 3: Table S3), including the same 4 isolates that were positive for cry with the Enterotoxin_M HMM search. These 13 isolates represented B. thuringiensis (10/14 reference genomes), B. weihenstephanensis (1 reference genome), and two dairy-associated B. cereus group isolates. All genomes were also annotated with RAST [42], which identified cry genes in four B. thuringiensis isolates, three dairy-associated isolates and a single B. weihenstephanensis isolate. Overall, cry genes were detected in 16 isolates by at least one of the approaches used (Table 3, Additional file 3: Table S3). The challenges with detection of cry genes are consistent with previous reports that these genes are highly diverse (e.g., [23]) and demonstrate a need for further development of improved tools for detection of cry genes based on WGS data. Interestingly, isolates identified as carrying cry genes were distributed across clades III-a (n = 3), III-b (n = 2), III-c (n = 1), IV (n = 8), V (n = 1) and VI (n = 1), indicating that cry homologs are widely distributed across different clades of B. cereus group isolates.
Table 2.
Distribution of key virulence genes among B. cereus group species and WGS clades
| % Isolates that carry virulence genesa | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Species | cry | cya + lef + pag | capA | capB | capC | capD | capE | cesA | cesB | cesC | cesD | cytK | entFM | hblA | hblB | hblC | hblD | nheABC |
| B. anthracis (n = 5) | 60 | 80 | 80 | 80 | 80 | 80 | 100 | 100 | ||||||||||
| B. cereus (n = 20) | 15 | 15 | 5 | 15 | 15 | 10 | 5 | 5 | 5 | 5 | 70 | 95 | 50 | 45 | 45 | 50 | 100 | |
| B. cytotoxicus (n = 1) | 100 | 100 | 100 | |||||||||||||||
| B. mycoides (n = 3) | 33 | 100 | 100 | 67 | 100 | 100 | 100 | |||||||||||
| B. pseudomycoides (n = 1) | 100 | 100 | 100 | 100 | ||||||||||||||
| B. thuringiensis (n = 14) | 71 | 7 | 7 | 7 | 79 | 100 | 86 | 86 | 86 | 86 | 100 | |||||||
| B. toyonensis (n = 1) | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | ||||||||||
| B. weihenstephanensis (n = 2) | 50 | 100 | 100 | 50 | 100 | 100 | 100 | |||||||||||
| Dairy-associated isolates (n = 22) | 9 | 68 | 100 | 59 | 50 | 59 | 59 | 100 | ||||||||||
| WGS clade | ||||||||||||||||||
| Clade I (n = 4) | 100 | 100 | 75 | 75 | 75 | 100 | ||||||||||||
| Clade II (n = 2) | 100 | 100 | 50 | 50 | 50 | 50 | 100 | |||||||||||
| Clade III-a (n = 19) | 16 | 26 | 37 | 26 | 37 | 37 | 32 | 47 | 100 | 26 | 26 | 26 | 26 | 100 | ||||
| Clade III-b (n = 6) | 33 | 17 | 83 | 83 | 67 | 67 | 67 | 67 | 100 | |||||||||
| Clade III-c (n = 10) | 10 | 10 | 10 | 10 | 10 | 50 | 100 | 10 | 10 | 10 | 10 | 100 | ||||||
| Clade IV (n = 20) | 40 | 5 | 5 | 5 | 95 | 100 | 100 | 95 | 95 | 100 | 100 | |||||||
| Clade V (n = 3) | 33 | 33 | 33 | 100 | 100 | 100 | 100 | 100 | 100 | |||||||||
| Clade VI (n = 4) | 25 | 100 | 100 | 25 | 100 | 100 | 100 | |||||||||||
| Clade VII (n = 1) | 100 | 100 | 100 | |||||||||||||||
a Virulence genes were identified in whole genome sequences using BLAST
Table 3.
Toxin gene presence, hemolysin BL (HBL) and nonhemolytic enterotoxin (NHE) production and hemolytic potential
| Isolate | Virulence genes presence [PCR]a | Virulence genes presence [WGS]a, b | Production of toxina, c | Hemolysisd | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| hblA | hlbC | hblD | nheA | nheB | nheC | ctyK | entFM | hblA | hblC | hblD | nheA | nheB | nheC | ctyK | entFM | HBL | NHE | ||
| FSL H7-0926 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 1 | 0 | 1 | B |
| FSL H8-0482 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 1 | 0 | 1 | B |
| FSL H8-0488 | 1 | 1 | 1 | 1 | 0 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 1 | 1 | 1 | B |
| FSL K6-0040 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | B |
| FSL K6-0043 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | B |
| FSL K6-0067 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 0 | 1 | - |
| FSL K6-0069 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 0 | 1 | B |
| FSL K6-0073 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | A |
| FSL K6-0267 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 1 | 0 | 1 | A |
| FSL M8-0117 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 1 | A |
| FSL W8-0003 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 1 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 1 | 0 | 1 | - |
| FSL W8-0050 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 0 | 1 | B |
| FSL W8-0169 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | B |
| FSL W8-0268 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | B |
| FSL W8-0275 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 0 | 1 | B |
| FSL W8-0483 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 1 | 0 | 1 | B |
| FSL W8-0520 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 0 | 1 | B |
| FSL W8-0523 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 0 | 1 | B |
| FSL W8-0640 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | B |
| FSL W8-0824 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | B |
| FSL W8-0932 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 1 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 1 | 0 | 1 | A |
| FSL W8-0767 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | A |
| B. thuringiensis BGSC 4Y1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 1 | B |
| B. thuringiensis BGSC 4CC1 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 0 | 1 | B |
| B. thuringiensis BGSC 4AJ1 | 1 | 1 | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | B |
| B. thuringiensis BGSC 4BD1 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 1 | 1 | 1 | B |
| B. thuringiensis BGSC 4BA1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | B |
| B. cereus ATCC 14579 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | B |
a 0 indicates absence of a gene; 1 indicates presence of a gene
b WGS whole genome sequence
c HBL hemolysin BL, NHE nonhemolytic enterotoxin
d “-“ indicates non-hemolytic; “A” indicates α hemolysis, “B” indicates β hemolysis
Anthrax toxin genes were found in isolates previously identified as B. anthracis, as well as B. cereus
Protein BLAST identified the anthrax toxin genes cya, lef and pagA in (i) 3 of the 5 B. anthracis reference genomes (strains 2000031021 and 2002013094 did not have these genes) and, (ii) 3 out of 20 B. cereus reference genomes (03BB102, CI and 03BB87). Strain CI was previously designated as B. cereus biovar anthracis because of the presence of anthrax virulence genes [15]. The complete capABCDE operon, which encodes the components for poly-γ-D-glutamate capsule biosynthesis, was found in (i) 4 of the 5 B. anthracis reference genomes (strain A16R did not have these genes) and, (ii) in 1 of the 20 B. cereus genomes (B. cereus biovar anthracis strain Cl). Hence, B. cereus strain CI carried a full complement of B. anthracis virulence genes. In addition, an incomplete capABCDE operon, characterized by the absence of capB, was detected in B. cereus strains 03BB102 and F65185, as well as in B. thuringiensis BGSC 4AJ1. B. toyonensis BCT-7112 carried only capE (Additional file 3: Table S3). These findings are consistent with previous reports of B. cereus strains isolated from patients with anthrax-like symptoms [16–18]. Interestingly, two B. cereus strains that carried anthrax genes (isolates 03BB102, CI) were found in clade III-a, which contains all B. anthracis reference genomes, while B. cereus 03BB87 was found in sister clade III-b.
Diarrheal toxin genes were identified across multiple B. cereus group species
A protein BLAST was used to identify genes previously reported as encoding (i) metabolic pathways for production of cereulide linked to emetic illness and (ii) toxins linked to diarrheal illness. The cereulide synthetase gene cluster (cesABCD) was found in only one isolate, B. cereus AH187 from WGS clade III-c. Genes encoding toxins linked to diarrheal illness were found in a larger number of genomes. These genes included (i) hblABCD operon, (ii) nheABC operon and (iii) cytK.
-
(i)
Genes encoding hemolysin BL (hblABCD)
Two different hbl operons have been identified previously [43]. One operon (hbl-I) consists of two genes encoding hemolysin BL binding component (hblA – 1125 nt, and hblB – 1398 nt), one gene encoding lytic component L2 (hblC), and one gene encoding lytic component L1 (hblD). The second operon (hbl-II) consists of a single gene encoding a binding component (hblA) and the two genes encoding lytic components (hblC and hblD) of hemolysin BL. Both of these operons have been previously reported in B. cereus strain ATCC 10876 [43]. We have identified hblACD genes encoding hemolysin BL in 10/20 B. cereus isolates (including one that did not carry hblC), 12/14 B. thuringiensis isolates, all 3 B. mycoides isolates, both B. weihenstephanensis isolates, the only B. toyonensis isolate and 13 dairy-associated isolates (Table 2). The B. pseudomycoides isolate carried only a hblA. Thirty-seven out of 42 isolates that carried a gene encoding the hemolysin BL binding component hblA, also carried a longer variant of binding component-encoding gene, hblB. Four Hbl-encoding genes were found across all except clade VII (B. cytotoxicus). For the 28 isolates screened by PCR, PCR results were consistent with identification of hblACD genes by BLAST (Table 3). While one study has reported presence of hblACD genes in all 70 tested isolates from ready-to-eat vegetables in South Korea [36], another study has found one or more hbl genes in 23.5 – 70.6 % of B. cereus group isolates from food products in Brazil [44]. The hblA has been detected in all 57 B. cereus isolates from raw vegetable samples collected in Mexico City [45]. While hblA and hblD have been detected in 72 % B. cereus isolates (n = 88) from retail spices in USA, hblC has been detected in 71 % of these isolates [28]. The high prevalence of the genes in the hblACD operon (up to 100 %) has also been reported in rice B. cereus isolates obtained in South Korea [37]. On the other hand, the hblACD genes have been found less frequently (34.5 % of 87) in B. cereus group isolates from fermented Korean soybean products [46]. While at least one hbl gene has been detected in 58.7 % of 63 B. cereus isolates from milk and dairy products in Brazil, all three hbl genes have been detected in 36.5 % of these isolates [47]. The fact that genes encoding all three toxin components are not always detected in a given isolate indicates that these genes either do not always co-exist or that false negative PCR reactions occur, e.g., due to the mismatches in primer annealing regions. Similar to our study, the hblACD genes have been detected in all tested B. thuringiensis isolates from rice in Korea [37]. On the other hand, the hblACD genes have been detected in only 67 % of the B. thuringiensis isolates from retail spices in the USA [28]. These results, however, are not always directly comparable due to the differences in PCR methods used across the studies. Overall, our data support that hblACD genes are found across B. cereus group species and phylogenetic clades.
-
(ii)
Genes encoding nonhemolytic enterotoxin (nheABC)
The nheABC genes encoding the nonhemolytic enterotoxin were identified by BLAST for all isolates (Table 2). The identification of nheABC genes by PCR was in agreement with BLAST results for all but 2 cases (FSL H8-0488, BGSC 4AJ1; Table 3). In these two cases BLAST search identified a significant hit, but nheB PCR produced a negative result. The nheB in these two isolates showed 1 nt mismatch with the nheB forward primer annealing region, however this mismatch was found also in two other isolates with positive nheB PCR reaction, suggesting that the sequence divergence in this region was unlikely to be the cause of false negative PCR results. Previously reported prevalence of nheABC genes among B. cereus group isolates has been generally higher (above 97 %) compared to the prevalence of hblACD genes in most studies, regardless of the strain source [28, 36, 44, 46]. The exception is the Korean study that reported lower prevalence of nheABC genes among B. cereus isolated from white rice (83.8 - 86.5 % of 37) [24].
-
(iii)
Genes encoding cytotoxin K (cytK-1 and cytK-2)
The gene encoding cytotoxin K (cytK) was detected by BLAST in approximately half of the isolates from clades III-a and III-c, and the majority of isolates from clades III-b, II, IV and VII (Table 2). The variant cytK-1, which is specific for B. cytotoxicus, was detected only in the B. cytotoxicus reference genome, while cytK-2 was also detected in other genomes. Results obtained by BLAST were consistent with results obtained by PCR for 26 out of 28 tested isolates. In two cases (FSL H7-0926 and FSL K6-0267) where discrepancies were observed (Table 3), the PCR produced a positive result, while BLAST did not detect any significant hits in WGS (Additional file 3: Table S3). This may be due to the inability to detect the gene located in the region of the genome that these two draft assemblies did not cover. Fewer studies have previously evaluated the presence of cytK; these studies reported cytK in 64.7 % of 17 dairy isolates and 76.8 % of 155 B. cereus food isolates from Brazil [44], 70.4 % of 125 B. cereus isolated from milk and cereal products in Thailand [48] and 57.5 % of 87 B. cereus group isolates from Korean fermented soybean products [46].
Gene encoding putative cell wall peptidase (entFM)
The gene encoding a putative cell wall peptidase (entFM) was identified by BLAST in all isolates except B. cereus FT9. The entFM was also detected in all 28 isolates tested by PCR (Table 3). The product of this gene is a putative virulence factor, reported to be involved in bacterial shape, motility, adhesion to epithelial cells, biofilm formation, and vacuolization of macrophages [49].
Virulence gene profiles among dairy-associated isolates
The 22 dairy-associated isolates did not carry anthrax-associated virulence genes, nor genes encoding emetic toxin biosynthetic pathway. Thirteen out of 22 dairy-associated strains screened by PCR carried all eight tested diarrheal toxin genes as well as entFM (Table 3). The same toxin profile was most commonly identified among B. cereus group strains isolated from pasteurized milk and cereal products in Thailand [48]. Importantly, hblACD, nheABC, cytK and entFM were also found in two dairy-associated isolates, FSL K6-0267 and FSL H7-0926, which clustered in B. weihenstephanensis clade VI. The hblACD, nheABC and entFM genes were present in two reference B. weihenstephanensis isolates as well, initially suggesting that this psychrotolerant species may present not only a food spoilage risk, but that some strains of this species may also pose a public health concern. Subsequent experiments described in detail below, however found that an antibody-based assay did not detect hemolysin BL in these two B. weihenstephanensis isolates, indicating that further characterization of these isolates is needed to fully assess their pathogenicity.
Anthrax toxin genes, diarrheal toxin genes and crystal protein-encoding genes are not exclusively associated with B. anthracis, B. cereus, and B. thuringiensis species, respectively
To test for potential association of different B. cereus group species with certain toxin gene profiles, we analyzed the virulence gene presence/absence data by principal component analysis (PCA). The first 5 and 10 principal components (PCs) cumulatively described 69.9 % and 91.0 % of variance in the data, respectively. Clustering of isolates based on virulence gene presence/absence was visualized by plotting eigenvectors associated with PC 1 (x axis), PC 4 (y axis) and PC 3 (size of the data points) (Fig. 3). PC 2 was omitted, as it specifically characterized a single isolate carrying cereulide synthetase gene cluster (B. cereus AH187).
Fig. 3.
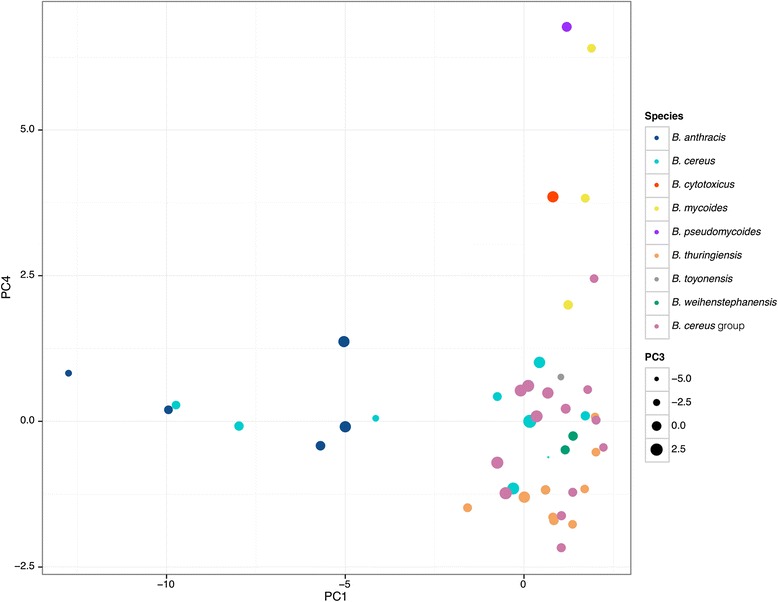
PCA clustering of B. cereus group isolates based on virulence gene presence/absence. PCA analysis was carried out using the data on presence/absence of 30 virulence genes that had variable presence across the analyzed 69 isolates. Reference isolates are color-coded according to the previously identified species, and all 22 dairy-associated isolates are labeled as “B. cereus group”. The figure demonstrates clustering of analyzed isolates based on the PC 1 (x axis), PC 4 (y axis) and PC 3 (dot size). PC 2 was omitted, as it specifically characterized a single isolate carrying genes that encode the cereulide biosynthetic pathway. The B. cereus and B. anthracis and B. cereus isolates carrying anthrax toxin and poly-γ-D-glutamate capsule genes form a clearly separated group on the bottom left side of the figure, while environmental isolates and B. cytotoxicus tend to cluster on the upper right side. Isolates carrying genes associated with diarrheal foodborne disease cluster on the bottom right side
B. cereus group isolates formed three PCA clusters (Fig. 3) that each included isolates of multiple species. The first cluster is comprised of B. anthracis and B. cereus isolates carrying anthrax and/or capsular biosynthesis genes (negative PC1, close to neutral PC 4), the second one is comprised of nonpathogenic B. mycoides and B. pseudomycoides, as well as B. cytotoxicus (positive PC1 and PC4), while the majority of B. cereus, B. thuringiensis and B. weihenstephanensis remained unresolved in the third cluster associated with diarrheal toxin genes (close to neutral PC1 and PC4).
The first plotted principal component (PC 1) was associated with the presence of genes coding for diarrheal toxins hemolysin BL (hblACD), cereolysin A and B (cerAB), cytotoxin (cytK), crystal protein (cry), phosphatidylcholine-specific phospholipase C (plcB) and enterotoxic protein (bceT). Additionally, PC 1 was associated with absence of genes encoding anthrax toxin (cya, lef, pagA), poly-γ-D-glutamate capsule biosynthesis genes (capABCDE), global transcription regulator (atxA), hyaluronic acid synthase (hasA), hemolysin II (hlyII) and hemolysin II regulator (hlyIIR). The second plotted principal component (PC 4) was associated with presence of enterotoxic protein gene (bceT), and absence of genes encoding (clo), cytotoxin K (cytK), hemolysin II (hlyII) and hemolysin II regulator (hlyIIR), enterotoxic protein (bceT), phosphatidylinositol-specific phospholipase C (PI-PLC; plcA) and crystal proteins (cry).
Due to the inability to resolve the B. cereus, B. thuringiensis and B. weihenstephanensis based on the presence/absence of virulence genes, we further investigated the production of hemolysin BL and nonhemolytic enterotoxin and their association with WGS phylogenetic clades and sequence polymorphism on the 28 strains that were physically available for phenotypic tests. Details of this analysis are discussed in the following sections.
Presence of hblACD and nheABC is not sufficient for production of detectable hemolysin BL and nonhemolytic enterotoxin Nhe
To assess the congruence of hblACD and nheABC presence with toxin production, and to identify gene polymorphisms responsible for potential inconsistencies, we examined the 22 dairy-associated B. cereus group isolates sequenced in this study, five B. thuringiensis reference isolates and B. cereus ATCC 14579, for detectable levels of hemolysin BL and nonhemolytic enterotoxin Nhe using Duopath Cereus Enterotoxins kit (Table 3). All tested isolates carried the nheABC genes, and all of them also produced the Nhe toxin at detectable levels (Table 3). In contrast, just 18 out of 28 isolates carried hblACD genes. Hemolysin BL was detected in only 13 of the 18 isolates that had the hblACD genes present, and in none of the isolates that did not have hblACD. Future studies assessing transcription of hblACD and using other phenotypic methods (e.g., tissues culture assays; ELISA assays with other [e.g., polyclonal antibodies]) will be needed to explain why some isolates that carry the hblACD do not express the HBL toxin.
Previous studies have confirmed the presence of nheABC in 83.8-86.5 % B. cereus and B. thuringiensis isolates from Korean white rice samples, but only 64.9 % of them produced NheA [37]. The hblACD genes were present in 83.8-94.6 % of these white rice isolates; yet only 78.4 % of them produced HblC [37]. Reis et al. [47] have reported 23 out of 63 isolated strains carrying all three hbl genes, but only 20 of these producing the hemolysin BL toxin. Data from these studies, as well as the present study suggest that the presence of hblACD genes may not be sufficient for production of detectable hemolysin BL. The hemolysin BL toxin may not be detected because i) the hblC is not transcribed, ii) transcribed mRNA is not translated, iii) protein is not exported, or iv) the protein was not in the configuration detectable by monoclonal antibody from Duopath Cereus Enterotoxins kit. These results demonstrate that presence of hblACD genes alone does not predict the production of hemolysin BL, suggesting that it may not be possible to accurately predict the risk for a B. cereus group isolate to cause a diarrheal disease based on the gene presence alone.
Isolates with a truncated hblA variant or specific amino acid substitutions in hblC or hblD genes do not express detectable hemolysin BL
In order to explain the discrepancies between hblABC gene presence and hemolysin BL production in five isolates (FSL H7-0926, FSL H8-0482, FSL K6-0267, FSL M8-0117 and B. thuringiensis BGSC 4Y1), we have examined the hbl genes and individual gene amino acid sequence variability. The 466 aa long hblB gene (characteristic for hbl-I operon [43]) was not identified in two dairy-associated B. weihenstephanensis isolates (assigned to this species based on WGS phylogeny) that carried hblACD genes, but did not produce detectable hemolysin BL. Absence of hblB and presence of hblACD was also observed in B. weihenstephanensis KBAB4, B. cereus 17256 W and B. mycoides 219298. Another B. weihenstephanensis isolate (WSBC 10204) carried all four hbl genes, including hblA and hblB. The hblB is located after the stem loop structure in hbl operon, which may regulate its co-transcription with the rest of the genes in the hbl operon [43]. The role of hblB is not well understood yet, however, a mutant lacking hbl-I operon has previously been demonstrated to be less cytotoxic compared to a wild type strain carrying both operons [43]. The three B. weihenstephanensis isolates that lacked hblB also had two unique 2-aa insertions (on positions 34-35 and 246-247) and a 2-aa deletion (on position 234-235) in hblA. The fourth B. weihenstephanensis isolate (WSBC 10204), however, did not have these insertions or deletion in hblA. Overall, these observations provide a possible mechanistic explanation (i.e., absence of hblB and/or unique mutations in hblA) for why some B. weihenstephanensis may not show production of hemolysin BL detectable by the Duopath kit. Further studies, including creation of appropriate mutant strains, will be needed to provide more definitive insights.
Specific hblACD mutations resulting in amino acid substitutions were observed in the dairy isolate FSL M8-0117 and in B. thuringiensis BGSC 4Y1, which both carry the hblACD genes, but did not produce detectable hemolysin BL. For isolate FSL M8-0117 the threonine at the 410th position of hblC (lytic component L2) was substituted with alanine. The polar threonine is commonly found in catalytic sites; therefore its substitution with hydrophobic alanine may impact protein functionality [50]. Similarly, a threonine at the 358th position of B. thuringiensis BGSC 4Y1 hblC gene was replaced with an electrically charged lysine. A non-neutral substitution was also identified in hblD (lytic component L1) of B. thuringiensis BGSC 4Y1, where a histidine at the 27th position was replaced with glutamine. The electrically charged histidine does not substitute well with any other amino acid and is often part of binding or active sites [50]. Its replacement with polar glutamine may therefore affect the toxin assembly and activity. Neither of these substitutions was observed in isolates producing detectable hemolysin BL toxin. Single amino acid substitutions have been previously shown to abolish monoclonal antibody reactivity with toxin components in other organisms, such as B. anthracis [51]. Substitutions in specific asparagine and proline residues of anthrax protective antigen have been demonstrated to reduce the cytotoxicity and antibody binding, while specific lysine, leucine and tyrosine residues have affected only the antibody binding [51]. It is not yet understood whether observed mutations abolish fully functioning toxin expression, or just its detection by the monoclonal antibodies used in Duopath kit [52]. Future studies are needed to address these questions in order to allow for sequence-based prediction of the toxicity associated with different hbl gene variants.
WGS clade IV is associated with hemolysin BL toxin production
Isolates that were tested for toxin production were distributed among clades II to VI. All tested isolates from clades III-b, III-c and VI did not produce a detectable hemolysin BL toxin. Clades II, III-a and V all included isolates with and without production of hemolysin BL; 2 of 6, 1 of 2, and 1 of 2 tested isolates, respectively, produced hemolysin BL. All tested isolates from phylogenetic clade IV tested positive for hemolysin BL production. Hemolysin BL production was significantly (p = 0.0001) associated with this clade, which is consistent with a previous report [34].
Conclusions
WGS-based phylogenetic classification of B. cereus group isolates showed better association with presence of toxin genes and toxin production than traditional taxonomic species classification. Clade classification along with the identification of virulence gene presence/absence and allelic variation in hemolysin BL-encoding genes thus appears to provide for better characterization of isolates with regard to their likelihood of causing diarrheal foodborne and other diseases. Overall, our findings support previous studies (e.g. [33, 34]) that have emphasized the need to re-evaluate the taxonomy of the B. cereus group, specifically of the species B. cereus and B. thuringiensis, which both were found in multiple clades. In the meantime, our data support a previous proposal that B. cereus group isolate nomenclature should include species and phylogenetic clade. The clade classification can be achieved by WGS as well as rpoB, panC or MLST sequencing, albeit at a lower level of phylogenetic resolution compared to WGS.
Methods
Bacterial isolates
Twenty-two B. cereus group isolates from dairy-associated sources were selected for WGS, from an existing Cornell Food Safety Laboratory strain collection that includes 3608 Bacillus isolates (as of 28 March 2016) that have been classified, as previously described [53, 54], by sequencing of a 632 nt internal fragment of rpoB, which encodes the β-subunit of RNA polymerase. Detailed data for all isolates are available in the Food Microbe Tracker (FMT) database [54]. Overall, isolates were selected to represent different combinations of rpoB AT, toxin gene profiles, and hemolytic potential (Table 3), based on initial screen of 114 isolates that had been classified as B. cereus group based on rpoB sequence data [55] (Table 3). Two selected isolates (FSL W8-0520 and FSL W8-0523, both rpoB AT481) shared the same profiles. Isolate sources included raw milk, dairy processing facility environments, and other dairy sources (Table 3).
An additional 47 reference genomes of B. cereus group isolates from human clinical cases, environment, and food or plant origin were extracted from NCBI Genome database and included in the analysis to represent the phylogenetic diversity of B. cereus group species (Additional file 1: Table S1).
rpoB allelic typing, virulence gene PCR screening and hemolysis
The molecular typing of isolates in the FMT database had been performed by PCR amplification and sequencing of a 632 nt long internal fragment of rpoB, which has been used for rpoB allelic (AT) assignment, as described previously [53, 55]. Novel rpoB ATs were assigned to every new variant of the 632 nt rpoB gene fragment that differed from any rpoB sequence in the existing database by at least 1 nt. The 632 nt rpoB sequences of the 22 FSL isolates were used for construction of a maximum likelihood phylogenetic tree in RaxML version 8 using general time-reversible (GTR) model with gamma distributed and invariant sites (GAMMAI), and 1000 bootstrap repetitions [56]. The phylogenetic tree was edited in FigTree v.1.4.2, and deposited on Figshare (https://figshare.com/s/7f5965d498bd33ccaac4).
Isolates were further characterized by PCR screening for the presence of genes encoding hemolysin BL (hblA, hblC, hblD), the nonhemolytic enterotoxin (nheA, nheB, nheC), and cytotoxin K (cytK) and FM toxin (entFM) [57, 58].
The ability of isolates to induce hemolysis was evaluated by plating overnight cultures onto Trypticase soy agar (TSA) supplemented with 5 % sheep blood (BBL, New Jersey, USA), followed by incubation at 35 °C for 24 h according to the FDA’s Bacteriological Analytical Manual. Alpha hemolysis was determined based on the dark green agar discoloration around and under the colonies, and beta hemolysis based on the transparent zones around and under the colonies (Table 3).
Detection of hemolysin BL and nonhemolytic toxin production
Hemolysin BL and nonhemolytic toxin (Nhe) production was detected using Duopath Cereus Enterotoxins immunological lateral flow assay (Merck) for all 22 FSL B. cereus group isolates, B. cereus ATCC 14579, and five B. thuringiensis isolates (BGSC 4Y1, BGSC 4CC1, BGSC 4BD1, BGSC 4BA1). Duopath test kits were used as specified by manufacturer’s instructions, to detect toxin production in bacterial cultures grown in Brain-heart infusion (BHI) broth at 37 °C for 20-24 h. The Duopath Cereus Enterotoxins kit is able to detect the NheB component of the Nhe enterotoxin at concentration of ≥6 ng/ml and Hbl-L(2) component of hemolysin BL at concentrations ≥20 ng/ml [52]. Results reported represent two biological replicates. Associations of hemolysin BL production with WGS phylogenetic clades were tested using Fisher’s exact test in R version 3.2.2 [59].
DNA extraction, library preparation and Illumina sequencing
Frozen cultures (-80 °C) stored in 15 % v/v glycerol-BHI media were streaked on BHI agar and incubated for 24 h at 32 °C. The DNA of isolates was extracted using QIAamp DNA MiniKit (Qiagen, Valencia, CA) with a 30 min Gram positive pre-treatment in 80 μl of 20 mg/ml lysozyme at 56 °C, and longer centrifugation times at lower centrifugal force as previously reported by our group [60]. DNA was eluted in 50 μl Tris-HCl (pH 8.0), and purity was assessed spectrophotometrically using a Nanodrop. Double-stranded DNA (dsDNA) was quantified with Picogreen (Invitrogen, Palsley, UK), normalized to a concentration of 0.2 ng/μl, and used to prepare a sequencing library with Nextera XT DNA Sample Preparation kit and Nextera XT Index Kit with 96 indices (Illumina, Inc. San Diego, CA). Subsequently, samples were pooled and sequenced in 2 lanes of a HiSeq 2500 rapid run with 2 x 100 bp paired-end sequencing with estimated 67x coverage (Genomics Facility of the Cornell University Institute of Biotechnology).
Genome assembly
Trimmomatic v0.32 [61] was used to trim Illumina adapters, as well as low quality bases and reads, according to the program’s quick start parameters for paired-end reads. Short read quality was evaluated using FastQC v0.11.2 [62]. Processed sequence reads were assembled with SPAdes v3.0.0 with the suggested k-mer set for bacterial genome assembly and the best assembly was used in the analysis [63]. Quality of assemblies was verified by QUAST [64] and the average genome coverage was determined using SAMtools [65].
Multilocus sequence typing (MLST)
MLST sequence types were determined for 22 FLS isolates by running SRST2 v0.1.6 [66] on short read fastq.gz files, using the B. cereus allele and sequence type definition database obtained from the PubMLST database [35] using a getmlst.py script. Unidentified allelic types and sequence types were extracted from the whole genome sequences using BLAST and submitted to the PubMLST database [35]. The concatenated sequences of 7 MLST loci for the 22 FSL isolates were used for construction of a maximum likelihood phylogenetic tree in RaxML version 8 using general time-reversible (GTR) model with gamma distributed and invariant sites (GAMMAI), and 1000 bootstrap repetitions [56]. The phylogenetic tree was edited in FigTree v.1.4.2, and deposited on Figshare (https://figshare.com/s/7f5965d498bd33ccaac4).
Phylogenetic classification based on panC sequence
The panC gene encoding pantoate-beta-alanine ligase was extracted from analyzed set of WGS by BLAST using B. cereus ATCC 14579 pantoate-beta-alanine ligase gene (NCBI Gene ID: 1203890) as a reference sequence. Extracted panC sequences were used for classification of isolates into seven phylogenetic groups proposed by Guinebretière et al. [33, 34] using their online classification tool [67]. The identified phylogenetic groups, I to VII, were associated with WGS clades identified in our study (Fig. 1).
Single nucleotide polymorphism (SNP) calling and phylogenetic analysis
The 22 draft genomes sequenced in this study and 47 closed genomes extracted from NCBI Genome were used to build a SNP-based phylogeny with kSNP v2 using kmer size 21, as determined by Kchooser [68]. The core SNPs were aligned in MEGA v6.06 [69] with MUSCLE algorithm and used to build the maximum likelihood phylogeny in RaxML under general time-reversible model with gamma distributed sites (GTRGAMMA) and 1000 bootstrap repetitions. The phylogenetic tree was edited in FigTree v.1.4.2, and deposited on Figshare (https://figshare.com/s/7f5965d498bd33ccaac4). The absolute core SNP distance matrix was calculated using PAUP 4.0a147 (Additional file 4: Table S4).
Virulence gene presence/absence and PCA analysis
Thirty-five reference virulence protein sequences (Additional file 2: Table S2) were queried against the whole genome sequence database (n = 69) assembled here, using tblastn function of standalone BLAST to investigate the presence/absence of virulence gene homologues. The R script was used to filter BLAST hits with >70 % alignment length, >50 % amino acid sequence identity and <10-5 e-value. The 50 % amino acid identity cut-off was determined based on the congruence of the BLAST search with results of the PCR screening (Fig. 2). Due to the high degree of evolutionary divergence of crystal protein (cry) genes of B. thuringiensis, these were identified using an Endotoxin_M hidden Markov model (HMM) extracted from Pfam database (PF00555), a custom-built hidden Markov model (HMM) using 10 cry variants representing the different 1° domains available in the B. thuringiensis nomenclature database [41], and RAST [42]. To build a custom HMM, amino acid sequences were aligned in MEGA v6.06 using MUSCLE algorithm, and were used to run hmmbuild in HMMER v3.1b2 [70]. The assembled chromosomal and plasmid sequences were joined as pseudochromosomes using a shell script, and translated in all 6 reading frames by running transeq in EMBOSS v6.5.7 [71]. Translated genomes were used as a subject database in hmmsearch (HMMER v3.1b2). To ensure the specificity, the conservative threshold e-value and HMM score were determined based on the output of the HMM search against the aligned cry variants that were used for building the model (Additional file 5: HMM_cry_log.sh).
The binary matrix capturing the presence/absence of 36 virulence genes was reduced to 30 genes with variable presence/absence data, and this reduced matrix was used in PCA analysis (R package “stats”; routine “prcomp”). The principal components were plotted using R package “ggplot2”.
NCBI accession numbers
Trimmed WGS reads and assembled genomes were submitted to the SRA and GenBank, under the BioProject ID PRJNA288461. Accession numbers are listed in Table 1.
Acknowledgements
The authors thank to Steven Warchocki for genomic DNA preparation.
Funding
Funding for this project was provided by the New York State Dairy Promotion Advisory Board through the New York State Department of Agriculture and Markets.
Availability of data and materials
Whole genome sequence data are available on NCBI under BioProject ID PRJNA288461 (SRR runs and GenBank accession numbers are listed in Table 1). The rpoB sequence data are available in Food Microbe Tracker database (http://www.foodmicrobetracker.com). The MLST data are available on PubMLST (PubMLST IDs listed in Table 1). Maximum likelihood tree files are available on Figshare (https://figshare.com/s/7f5965d498bd33ccaac4).
Authors’ contributions
JK, LMC and DJK performed computational and statistical data analyses; RAM, SMB and JJ performed experimental analyses. JK and MW co-wrote the manuscript and conceived the study. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Additional files
List of publicly available closed genomes included in this study. (XLSX 43 kb)
Virulence-associated genes included in the comparative genomic analysis of Bacillus cereus group isolates. (XLSX 33 kb)
Presence/absence of 36 virulence genes in whole genome sequences of 69 B. cereus group isolates. (XLSX 19 kb)
Absolute core genome SNP distance for 69 B. cereus group isolates. (XLSX 61 kb)
HMM_cry_log.sh. (SH 932 bytes)
Contributor Information
Jasna Kovac, Email: jk2739@cornell.edu.
Rachel A. Miller, Email: ram524@cornell.edu
Laura M. Carroll, Email: lmc297@cornell.edu
David J. Kent, Email: dk657@cornell.edu
Jiahui Jian, Email: jj528@cornell.edu.
Sarah M. Beno, Email: smb489@cornell.edu
Martin Wiedmann, Email: martin.wiedmann@cornell.edu.
References
- 1.Bennet SD, Walsh KA, Gould LH. Foodborne disease outbreaks caused by Bacillus cereus, Clostridium perfringens, and Staphylococcus aureus—United States, 1998–2008. Clin Infect Dis. 2013;57(3):425–433. doi: 10.1093/cid/cit244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stenfors Arnesen LP, Fagerlund A, Granum PE. From soil to gut: Bacillus cereus and its food poisoning toxins. FEMS Microbiol Rev. 2008;32:579–606. doi: 10.1111/j.1574-6976.2008.00112.x. [DOI] [PubMed] [Google Scholar]
- 3.Miller RA, Kent DJ, Watterson MJ, Boor KJ, Martin NH, Wiedmann M. Spore populations among bulk tank raw milk and dairy powders are significantly different. J Dairy Sci. 2015;98:8492–8504. doi: 10.3168/jds.2015-9943. [DOI] [PubMed] [Google Scholar]
- 4.Schmidt VS, Kaufmann V, Kulozik U, Scherer S, Wenning M. Microbial biodiversity, quality and shelf life of microfiltered and pasteurized extended shelf life (ESL) milk from Germany, Austria and Switzerland. Int J Food Microbiol. 2012;154:1–9. doi: 10.1016/j.ijfoodmicro.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 5.Lücking G, Stoeckel M, Atamer Z, Hinrichs J, Ehling-Schulz M. Characterization of aerobic spore-forming bacteria associated with industrial dairy processing environments and product spoilage. Int J Food Microbiol. 2013;166:270–279. doi: 10.1016/j.ijfoodmicro.2013.07.004. [DOI] [PubMed] [Google Scholar]
- 6.Guinebretière MH, Auger S, Galleron N, Contzen M, De Sarrau B, De Buyser ML, et al. Bacillus cytotoxicus sp. nov. is a novel thermotolerant species of the Bacillus cereus group occasionally associated with food poisoning. Int J Syst Evol Microbiol. 2013;63:31–40. doi: 10.1099/ijs.0.030627-0. [DOI] [PubMed] [Google Scholar]
- 7.Jiménez G, Urdiain M, Cifuentes A, López-López A, Blanch AR, Tamames J, et al. Description of Bacillus toyonensis sp. nov., a novel species of the Bacillus cereus group, and pairwise genome comparisons of the species of the group by means of ANI calculations. Syst Appl Microbiol. 2013;36:383–391. doi: 10.1016/j.syapm.2013.04.008. [DOI] [PubMed] [Google Scholar]
- 8.Schleifer K, Whitman WB. Bergey’s manual of systematic bacteriology: The Firmicutes. 2. New York: Springer; 2009. [Google Scholar]
- 9.Cardazzo B, Negrisolo E, Carraro L, Alberghini L, Patarnello T, Giaccone V. Multiple-locus sequence typing and analysis of toxin genes in Bacillus cereus food-borne isolates. Appl Environ Microbiol. 2008;74:850–860. doi: 10.1128/AEM.01495-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Caamaño-Antelo S, Fernández-No IC, Böhme K, Ezzat-Alnakip M, Quintela-Baluja M, et al. Genetic discrimination of foodborne pathogenic and spoilage Bacillus spp. based on three housekeeping genes. Food Microbiol. 2015;46:288–298. doi: 10.1016/j.fm.2014.08.013. [DOI] [PubMed] [Google Scholar]
- 11.Liu Y, Lai Q, Göker M, Meier-Kolthoff JP, Wang M, Sun Y, et al. Genomic insights into the taxonomic status of the Bacillus cereus group. Sci Rep. 2015;16:14082. doi: 10.1038/srep14082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zwick ME, Joseph SJ, Didelot X, Chen PE, Bishop-Lilly KA, Stewart AC, et al. Genomic characterization of the Bacillus cereus sensu lato species: backdrop to the evolution of Bacillus anthracis. Genome Res. 2012;22:1512–1524. doi: 10.1101/gr.134437.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Varghese NJ, Mukherjee S, Ivanova N, Konstantinidis KT, Mavrommatis K, et al. Microbial species delineation using whole genome sequences. Nucleic Acids Res. 2015;43:6761–6771. doi: 10.1093/nar/gkv657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang A, Ash GJ. Whole genome phylogeny of Bacillus by Feature Frequency Profiles (FFP) Sci Rep. 2015;5:13644. doi: 10.1038/srep13644. [DOI] [Google Scholar]
- 15.Klee SR, Brzuszkiewicz EB, Nattermann H, Brüggemann H, Dupke S, Wollherr A, et al. The genome of a Bacillus isolate causing anthrax in chimpanzees combines chromosomal properties of B. cereus with B. anthracis virulence plasmids. PLoS One. 2010;5:e10986. doi: 10.1371/journal.pone.0010986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hoffmaster AR, Ravel J, Rasko DA, Chapman GD, Chute MD, Marston CK, et al. Identification of anthrax toxin genes in a Bacillus cereus associated with an illness resembling inhalation anthrax. Proc Natl Acad Sci U S A. 2004;101:8449–8454. doi: 10.1073/pnas.0402414101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoffmaster AR, Hill KK, Gee JE, Marston CK, De BK, Popovic T, et al. Characterization of Bacillus cereus isolates associated with fatal pneumonias: strains are closely related to Bacillus anthracis and harbor B. anthracis virulence genes. J Clin Microbiol. 2006;44:3352–3360. doi: 10.1128/JCM.00561-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oh SY, Budzik JM, Garufi G, Schneewind O. Two capsular polysaccharides enable Bacillus cereus G9241 to cause anthrax-like disease. Mol Microbiol. 2011;80:455–470. doi: 10.1111/j.1365-2958.2011.07582.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilson MK, Vergis JM, Alem F, Palmer JR, Keane-Myers AM, Brahmbhatt TN, et al. Bacillus cereus G9241 makes anthrax toxin and capsule like highly virulent B. anthracis Ames but behaves like attenuated toxigenic nonencapsulated B. anthracis Sterne in rabbits and mice. Infect Immun. 2011;79:3012–3019. doi: 10.1128/IAI.00205-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnson SL, Minogue TD, Teshima H, Davenport KW, Shea AA, Miner HL, et al. Finished genome sequence of Bacillus cereus strain 03BB87, a clinical isolate with B. anthracis virulence genes. Genome Announc. 2015;3:e01445–14. doi: 10.1128/genomeA.01446-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoton FM, Andrup L, Swiecicka I, Mahillon J. The cereulide genetic determinants of emetic Bacillus cereus are plasmid-borne. Microbiology. 2005;151:2121–2124. doi: 10.1099/mic.0.28069-0. [DOI] [PubMed] [Google Scholar]
- 22.Ehling-Schulz M, Fricker M, Grallert H, Rieck P, Wagner M, Scherer S. Cereulide synthetase gene cluster from emetic Bacillus cereus: structure and location on a mega virulence plasmid related to Bacillus anthracis toxin plasmid pXO1. BMC Microbiol. 2006;6:20. doi: 10.1186/1471-2180-6-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schnepf E, Crickmore N, Van Rie J, Lereclus D, Baum J, Feitelson J, et al. Bacillus thuringiensis and its pesticidal crystal proteins. Microbiol Mol Biol Rev. 1998;62:775–806. doi: 10.1128/mmbr.62.3.775-806.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim MJ, Han JK, Park JS, Lee JS, Lee SH, et al. Various enterotoxin and other virulence factor genes widespread among Bacillus cereus and Bacillus thuringiensis strains. J Microbiol Biotechnol. 2015;25:872–879. doi: 10.4014/jmb.1502.02003. [DOI] [PubMed] [Google Scholar]
- 25.Clair G, Roussi S, Armengaud J, Duport C. Expanding the known repertoire of virulence factors produced by Bacillus cereus through early secretome profiling in three redox conditions. Mol Cell Proteomics. 2010;9:1486–1498. doi: 10.1074/mcp.M000027-MCP201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Swiecicka I, Van der Auwera GA, Mahillon J. Hemolytic and nonhemolytic enterotoxin genes are broadly distributed among Bacillus thuringiensis isolated from wild mammals. Microb Ecol. 2006;52:544–551. doi: 10.1007/s00248-006-9122-0. [DOI] [PubMed] [Google Scholar]
- 27.Réjasse A, Gilois N, Barbosa I, Huillet E, Bevilacqua C, Tran S, et al. Temperature-dependent production of various PlcR-controlled virulence factors in Bacillus weihenstephanensis strain KBAB4. Appl Environ Microbiol. 2012;78:2553–2561. doi: 10.1128/AEM.07446-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hariram U, Labbé R. Spore prevalence and toxigenicity of Bacillus cereus and Bacillus thuringiensis isolates from U.S. retail spices. J Food Prot. 2015;78:590–596. doi: 10.4315/0362-028X.JFP-14-380. [DOI] [PubMed] [Google Scholar]
- 29.Prüss BM, Dietrich R, Nibler B, Märtlbauer E, Scherer S. The hemolytic enterotoxin HBL is broadly distributed among species of the Bacillus cereus group. Appl Environ Microbiol. 1999;65:5436–5442. doi: 10.1128/aem.65.12.5436-5442.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moravek M, Dietrich R, Buerk C, Broussolle V, Guinebretière MH, Granum PE, et al. Determination of the toxic potential of Bacillus cereus isolates by quantitative enterotoxin analyses. FEMS Microbiol Lett. 2006;257:293–298. doi: 10.1111/j.1574-6968.2006.00185.x. [DOI] [PubMed] [Google Scholar]
- 31.Wang J, Ding T, Oh DH. Effect of temperatures on the growth, toxin production, and heat resistance of Bacillus cereus in cooked rice. Foodborne Pathog Dis. 2014;11(2):133–137. doi: 10.1089/fpd.2013.1609. [DOI] [PubMed] [Google Scholar]
- 32.Jeßberger N, Krey VM, Rademacher C, Böhm ME, Mohr AK, Ehling-Schulz M, et al. From genome to toxicity: a combinatory approach highlights the complexity of enterotoxin production in Bacillus cereus. Front Microbiol. 2015;6:560. doi: 10.3389/fmicb.2015.00560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guinebretière MH, Thompson FL, Sorokin A, Normand P, Dawyndt P, Ehling-Schulz M, et al. Ecological diversification in the Bacillus cereus Group. Environ Microbiol. 2008;10:851–865. doi: 10.1111/j.1462-2920.2007.01495.x. [DOI] [PubMed] [Google Scholar]
- 34.Guinebretière MH, Velge P, Couvert O, Carlin F, Debuyser ML, Nguyen-The C. Ability of Bacillus cereus group strains to cause food poisoning varies according to phylogenetic affiliation (groups I to VII) rather than species affiliation. J Clin Microbiol. 2010;48:3388–3391. doi: 10.1128/JCM.00921-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.PubMLST: Bacillus cereus database. University of Oxford, UK. http://www.pubmlst.org/bcereus. Accessed 4 April 2016.
- 36.Chon JW, Yim JH, Kim HS, Kim DH, Kim H, et al. Quantitative prevalence and toxin gene profile of Bacillus cereus from ready-to-eat vegetables in South Korea. Foodborne Pathog Dis. 2015;12:795–799. doi: 10.1089/fpd.2015.1977. [DOI] [PubMed] [Google Scholar]
- 37.Kim B, Bang J, Kim H, Kim Y, Kim BS, et al. Bacillus cereus and Bacillus thuringiensis spores in Korean rice: prevalence and toxin production as affected by production area and degree of milling. Food Microbiol. 2014;42:89–94. doi: 10.1016/j.fm.2014.02.021. [DOI] [PubMed] [Google Scholar]
- 38.Drewnowska JM, Swiecicka I. Eco-Genetic Structure of Bacillus cereus sensu lato populations from different environments in Northeastern Poland. PLoS One. 2013;8:e80175. doi: 10.1371/journal.pone.0080175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hoffmaster AR, Novak RT, Marston CK, Gee JE, Helsel L, et al. Genetic diversity of clinical isolates of Bacillus cereus using multilocus sequence typing. BMC Microbiol. 2008;8:191. doi: 10.1186/1471-2180-8-191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Crickmore N, Zeigler DR, Feitelson J, Schnepf E, Van Rie J, Lereclus D, et al. Revision of the nomenclature for the Bacillus thuringiensis pesticidal crystal proteins. Microbiol Mol Biol Rev. 1998;62:807–813. doi: 10.1128/mmbr.62.3.807-813.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bt toxin nomenclature. www.btnomenclature.info Accessed 4 Apr 2016.
- 42.Brettin T, Davis JJ, Disz T, Edwards RA, Gerdes S, Olsen GJ, et al. RASTtk: a modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci Rep. 2015;5:8365. doi: 10.1038/srep08365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sastalla I, Fattah R, Coppage N, Nandy P, Crown D, et al. The Bacillus cereus Hbl and Nhe tripartite enterotoxin components assemble sequentially on the surface of target cells and are not interchangeable.”. PLoS One. 2013;8:e76955. doi: 10.1371/journal.pone.0076955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aragon-Alegro LC, Palcich G, Lopes GV, Ribeiro VB, Landgraf M, Destro MT. Enterotoxigenic and genetic profiles of Bacillus cereus strains of food origin in Brazil. J Food Prot. 2008;71:2115–2118. doi: 10.4315/0362-028x-71.10.2115. [DOI] [PubMed] [Google Scholar]
- 45.Flores-Urbán KA, Natividad-Bonifacio I, Vázquez-Quiñones CR, Vázquez-Salinas C, Quiñones-Ramírez EI. Detection of toxigenic Bacillus cereus strains isolated from vegetables in Mexico City. J Food Prot. 2014;77:2144–2147. doi: 10.4315/0362-028X.JFP-13-479. [DOI] [PubMed] [Google Scholar]
- 46.Yim JH, Kim KY, Chon JW, Kim DH, Kim HS, Choi DS, et al. Incidence, antibiotic susceptibility, and toxin profiles of Bacillus cereus sensu lato isolated from Korean fermented soybean products. J Food Sci. 2015;80:M1266–M1270. doi: 10.1111/1750-3841.12872. [DOI] [PubMed] [Google Scholar]
- 47.Reis AL, Montanhini MT, Bittencourt JV, Destro MT, Bersot LS. Gene detection and toxin production evaluation of hemolysin BL of Bacillus cereus isolated from milk and dairy products marketed in Brazil. Braz J Microbiol. 2013;44:1195–1198. doi: 10.1590/S1517-83822013000400024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chitov T, Dispan R, Kasinrerk W. Incidence and diarrhegenic potential of Bacillus cereus in pasteurized milk and cereal products in Thailand. J Food Saf. 2008;28:467–481. doi: 10.1111/j.1745-4565.2008.00125.x. [DOI] [Google Scholar]
- 49.Tran SL, Guillemet E, Gohar M, Lereclus D, Ramarao N. CwpFM (EntFM) is a Bacillus cereus potential cell wall peptidase implicated in adhesion, biofilm formation, and virulence. J Bacteriol. 2010;192:2638–2642. doi: 10.1128/JB.01315-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Betts MJ, Russell, RB. Amino Acid Properties and Consequences of Substitutions. In Barnes MR, Gray IC editors. Bioinformatics for Geneticists. Chichester, UK: John Wiley & Sons, Ltd; 2003. doi: 10.1002/0470867302.ch14.
- 51.Rosovitz MJ, Schuck P, Varughese M, Chopra AP, Mehra V, Singh Y, et al. Alanine-scanning mutations in domain 4 of anthrax toxin protective antigen reveal residues important for binding to the cellular receptor and to a neutralizing monoclonal antibody. J Biol Chem. 2003;278:30936–30944. doi: 10.1074/jbc.M301154200. [DOI] [PubMed] [Google Scholar]
- 52.Krause N, Moravek M, Dietrich R, Wehrle E, Slaghuis J, Märtlbauer E. Performance characteristics of the Duopath® cereus enterotoxins assay for rapid detection of enterotoxinogenic Bacillus cereus strains. Int J Food Microbiol. 2010;144:322–326. doi: 10.1016/j.ijfoodmicro.2010.10.008. [DOI] [PubMed] [Google Scholar]
- 53.Ivy RA, Ranieri ML, Martin NH, den Bakker HC, Xavier BM, Wiedmann M, Boor KJ. Identification and characterization of psychrotolerant sporeformers associated with fluid milk production and processing. Appl Environ Microbiol. 2012;78:1853–1864. doi: 10.1128/AEM.06536-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Food Microbe Tracker. Cornell University, Ithaca. http://www.foodmicrobetracker.com. Accessed 3 Feb 2014.
- 55.Hervert CJ, Miller RA, den Bakker HC, Martin MH, Boor KJ, Wiedmann M. Characterization of toxin genes among Bacillus cereus group isolates. New Orleans: Poster session presented at: The Institute of Food Technologists (IFT) 2014 Annual Meeting & Food Expo; 2014. [Google Scholar]
- 56.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ngamwongsatit P, Buasri W, Pianariyanon P, Pulsrikarn C, Ohba M, et al. Broad distribution of enterotoxin genes (hblCDA, nheABC, cytK, and entFM) among Bacillus thuringiensis and Bacillus cereus as shown by novelprimers. Int J Food Microbiol. 2008;121:352–356. doi: 10.1016/j.ijfoodmicro.2007.11.013. [DOI] [PubMed] [Google Scholar]
- 58.Kim J, Forghani F, Kim J, Park Y, Park M, Wang J, et al. Improved multiplex PCR assay for simultaneous detection of Bacillus cereus emetic and enterotoxic strains”. Food Sci Biotech. 2012;21:1439–1444. doi: 10.1007/s10068-012-0189-8. [DOI] [Google Scholar]
- 59.R Core Team . R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing; 2016. [Google Scholar]
- 60.Stasiewicz MJ, Oliver HF, Wiedmann M, den Bakker HC. Whole-Genome Sequencing allows for improved identification of persistent Listeria monocytogenes in food-associated environments. Appl Environ Microbiol. 2015;81:6024–6037. doi: 10.1128/AEM.01049-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Babraham Bioinformatics. FastQC: A quality tool for high throughput sequence data. http://bioinformatics.babraham.ac.uk/projects/fastqc. Accessed 4 Apr 2016.
- 63.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013;29:1072–1075. doi: 10.1093/bioinformatics/btt086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Inouye M, Dashnow H, Raven LA, Schultz MB, Pope BJ, Tomita T, et al. SRST2: Rapid genomic surveillance for public health and hospital microbiology labs. Genome Med. 2014;6:90. doi: 10.1186/s13073-014-0090-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Guinebretière MH. Affiliation to phylogenetic groups (I to VII) into the Bacillus cereus group. https://www.tools.symprevius.org/bcereus/english.php. Accessed 4 Apr 2016.
- 68.Gardner SN, Hall BG. When whole-genome alignments just won’t work: kSNP v2 software for alignment-free SNP discovery and phylogenetics of hundreds of microbial genomes. PLoS One. 2013;8:e81760. doi: 10.1371/journal.pone.0081760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Finn RD, Clements J, Arndt W, Miller BL, Wheeler TJ, Schreiber F, et al. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2015: doi: 10.1093/nar/gkv397. [DOI] [PMC free article] [PubMed]
- 71.Rice P, Longden I, Bleasby A. EMBOSS: the European molecular biology open software suite. Trends Genet. 2000;16:276–277. doi: 10.1016/S0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Whole genome sequence data are available on NCBI under BioProject ID PRJNA288461 (SRR runs and GenBank accession numbers are listed in Table 1). The rpoB sequence data are available in Food Microbe Tracker database (http://www.foodmicrobetracker.com). The MLST data are available on PubMLST (PubMLST IDs listed in Table 1). Maximum likelihood tree files are available on Figshare (https://figshare.com/s/7f5965d498bd33ccaac4).